

Abstract
We have previously reported that the α1 subunit of sodium potassium adenosine triphosphatase (Na/K-ATPase), acts as a receptor and an amplifier for reactive oxygen species, in addition to its distinct pumping function. On this background, we speculated that blockade of Na/K-ATPase-induced ROS amplification with a specific peptide, pNaKtide, might attenuate the development of steatohepatitis. To test this hypothesis, pNaKtide was administered to a murine model of NASH: the C57Bl6 mouse fed a “western” diet containing high amounts of fat and fructose. The administration of pNaKtide reduced obesity as well as hepatic steatosis, inflammation and fibrosis. Of interest, we also noted marked improvement in mitochondrial fatty acid oxidation, insulin sensitivity, dyslipidemia and aortic streaking in this mouse model. To further elucidate the effects of pNaKtide on atherosclerosis, similar studies were performed in ApoE knockout mice also exposed to the western diet. In these mice, pNaKtide not only improved steatohepatitis, dyslipidemia, and insulin sensitivity, but also ameliorated significant aortic atherosclerosis. Collectively, this study demonstrates that the Na/K-ATPase/ROS amplification loop contributes significantly to the development and progression of steatohepatitis and atherosclerosis. And furthermore, this study presents a potential treatment, the pNaKtide, for the metabolic syndrome phenotype.
Subject terms: Metabolic syndrome, Obesity, Experimental models of disease, Translational research
Introduction
Metabolic syndrome represents a cluster of metabolic abnormalities including obesity, hypertension, dyslipidemia, and insulin resistance with obesity and insulin resistance recognized as the causative factors1–3. Together, these derangements present a significant risk for nonalcoholic steatohepatitis, atherosclerosis, and diabetes, all of which lead to cardiovascular complications. Cardiovascular disease is still the leading cause of mortality in Western countries, with approximately 30% of deaths in the United States attributed to it refs 2 and 4.
Recent studies have demonstrated that oxidative stress and inflammation underlie the pathogenesis of metabolic syndrome and, by extension, NASH and atherosclerosis5–7. Redox imbalance, accompanied by insulin resistance, dyslipidemia and increased circulating FFA levels, are features that usually accompany clinical NASH6. Additionally, deficient hepatic mitochondrial fatty acid oxidation contributes to triglyceride accumulation and steatosis8. It is believed that NASH predisposes to atherosclerosis through the systemic release of inflammatory and oxidative-stress mediators9. Atherosclerosis, itself, is caused by oxidative stress, as oxidized lipids appear to play a pathogenic role10, 11.
We have previously noted that Na/K-ATPase signaling can serve as a feed-forward amplifier for oxidant signaling12–15. Our group has developed a cell permeant NaKtide (pNaKtide) from the N domain of the α1 subunit of Na/K-ATPase. This peptide inhibits Na/K-ATPase-ROS amplification and blocks downstream Src activation16–19. We have recently shown that pNaKtide, by restoring redox balance in adipocytes, attenuates obesity in mice fed a high fat diet20. Given the aforementioned importance of oxidative stress in the pathophysiology of NASH, we chose to examine whether pNaKtide might be effective in ameliorating NASH in C57Bl6 mice fed a western diet. As the effects of pNaKtide detailed in this report extended to the vascular system, additional studies were performed in the ApoE knockout mouse.
Results
Effect of pNaKtide on body weight, visceral and subcutaneous fat content and liver weight in mice fed a western diet
C57Bl6 mice were fed a western diet for 4 weeks and then administered pNaKtide at a dose of 25 mg/kg intraperitoneal (I.P.) every week while the western diet was continued for an additional 8 weeks. The administration of pNaKtide reduced body weight, visceral fat, and subcutaneous content in these mice significantly (Table 1) as we have previously reported20. Further, our results demonstrated that liver weight, which is indicative of fatty liver, was also significantly reduced by pNaKtide administration (Table 1). In parallel experiments, rhodamine-labeled peptide accumulated in hepatic tissue (Supplementary Fig. 1A). Previous studies documented that ApoE−/− mice are relatively resistant to weight gain until fed a western diet for approximately 6 months21. In line with these reports, our results showed that body weight, visceral fat, subcutaneous fat, and liver weight were not significantly increased in ApoE−/− mice fed a western diet as compared to mice on a standard chow diet (Table 1).
Table 1.
Effect of pNaKtide on body weight; visceral fat, subcutaneous fat and liver weight; energy expenditure; locomotor activity and oxygen consumption in C57Bl/6 and ApoE−/− mice fed a western diet.
| C57Bl6 | ApoE−/− | |||||
|---|---|---|---|---|---|---|
| CTR | WD | WD + pNaKtide | CTR | WD | WD + pNaKtide | |
| BW (g, Baseline) | 22.5 ± 0.7 | 21.4 ± 0.4 | 20.1 ± 0.7 | 20.6 ± 0.4 | 20.7 ± 0.6 | 21.2 ± 0.6 |
| BW (g, 4 week) | 24.7 ± 1.08 | 25.6 ± 0.7 | 24.9 ± 0.9 | 24.4 ± 0.6 | 25.8 ± 0.7 | 26.9 ± 1.04 |
| BW (g, 12 week) | 28.2 ± 1.1 | 40.7 ± 0.8** | 32.9 ± 0.9**,## | 28.5 ± 1.04 | 29.9 ± 1.06 | 30.4 ± 1.5 |
| Visceral fat (g) | 0.5 ± 0.05 | 3.1 ± 0.1** | 1.9 ± 0.1**,## | 0.5 ± 0.02 | 0.5 ± 0.06 | 0.5 ± 0.02 |
| SC Fat (g) | 0.4 ± 0.03 | 1.9 ± 0.1** | 0.9 ± 0.06**,## | 0.3 ± 0.02 | 0.3 ± 0.02 | 0.3 ± 0.02 |
| Liver weight (g) | 1.8 ± 0.04 | 2.7 ± 0.2** | 1.8 ± 0.08## | 1.7 ± 0.07 | 1.7 ± 0.04 | 1.7 ± 0.03 |
| Energy expenditure (kcal/kg/hr) | 15.2 ± 0.3 | 12.6 ± 0.6* | 15.1 ± 0.6# | 18.8 ± 0.4 | 16.1 ± 0.3** | 18.06 ± 0.4# |
| Locomotor Activity (Ambulatory count/24 hours) | 19565.2 ± 2188.2 | 11702 ± 660.05** | 24438.1 ± 2678.4# | 23670.5 ± 1500.6 | 18757 ± 483.4* | 26301.8 ± 1603.3# |
| Oxygen consumption (ml/kg/hr) | 3083.01 ± 72.1 | 2622.05 ± 138.5* | 3155 ± 129.06# | 3799.7 ± 75.3 | 3342.2 ± 93.9* | 3704.5 ± 82.5# |
Results are means ± SEM, n = 6/group. *p < 0.05, **p < 0.01 vs. control; #<0.05, ##p < 0.01 vs WD.
Effect of pNaKtide on energy expenditure and locomotor activity in mice fed a western diet
Energy expenditure, determined as heat production (kcal/kg/hour), was significantly decreased in both strains of mice fed a western diet compared to mice fed a normal chow diet (Table 1). The administration of pNaKtide significantly increased energy expenditure suggesting that these groups utilized more calories compared to mice fed a western diet (Table 1). Also oxygen consumption was increased in both C57Bl6 and ApoE−/− mice fed a western diet and treated with pNaKtide (Table 1). Similarly, treatment with pNaKtide significantly increased locomotor activity, as determined by the ambulatory count over a 24-hour period (Table 1).
Effect of pNaKtide on fatty acid oxidation genes in adipose tissue of mice fed a western diet
Since adipose mitochondria participate in energy expenditure through fatty acid oxidation, quantitative PCR was done to measure fatty acid oxidation genes in adipose tissue. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is a major regulator of mitochondrial oxidative metabolism22. Our results showed that PGC-1α mRNA expression was significantly decreased in adipose tissue of C57Bl6 or ApoE−/− mice fed a western diet compared to the control group (Table 2). Administration of pNaKtide significantly increased PGC-1α mRNA expression in these mice. Carnitinepalmitoyltransferase-1 (CPT-1) is a rate-limiting enzyme of mitochondrial fatty acid β-oxidation23. CPT-1 mRNA expression was decreased in both C57Bl6 and ApoE−/− mice fed a western diet compared to control mice and this decrease was reversed by treatment with pNaKtide (Table 2). The UCP 1 and 2 are markers of energy metabolism and its uncoupled energy is used for heat production in fat tissue24. Our results demonstrated that both strains of mice fed a western diet exhibited decreased expression of UCP 1 and 2 in adipose tissue in comparison to mice fed a normal diet. Our results showed that UCP1 and 2 mRNA expressions were significantly increased by pNaKtide treatment in adipose tissue compared to mice fed a western diet in both strains of mice (Table 2).
Table 2.
Effect of pNaKtide on adipose tissue fat oxidation genes in C57Bl6 and ApoE−/− mice fed a western diet.
| C57Bl6 | ApoE−/− | |||||
|---|---|---|---|---|---|---|
| CTR | WD | WD + pNaKtide | CTR | WD | WD + pNaKtide | |
| PGC1α mRNA | 0.9 ± 0.003 | 0.5 ± 0.02* | 1.9 ± 0.03*,## | 0.9 ± 0.003 | 0.4 ± 0.06** | 1.4 ± 0.1*,## |
| CPT1 mRNA | 0.9 ± 0.003 | 0.3 ± 0.03* | 1.3 ± 0.2## | 0.9 ± 0.003 | 0.4 ± 0.06* | 1.9 ± 0.3*,## |
| UCP1 mRNA | 0.9 ± 0.03 | 0.1 ± 0.01* | 1.3 ± 0.1## | 0.9 ± 0.003 | 0.3 ± 0.04* | 2.2 ± 0.6# |
| UCP2 mRNA | 0.9 ± 0.003 | 0.2 ± 0.05* | 2.1 ± 0.3**,## | 0.9 ± 0.003 | 0.2 ± 0.04* | 1.3 ± 0.3## |
Relative mRNA expression of adipose tissue genes. Results are means ± SEM, n = 8/group. *p < 0.05, **p < 0.01 vs. control; #<0.05, ##p < 0.01 vs WD.
Effect of pNaKtide on hepatic lipogenesis and inflammation in mice fed a western diet
Histological sections of liver tissue from C57Bl6 mice fed a western diet showed significant inflammation and a large number of vacuoles in the liver. C57Bl6 mice treated with pNaKtide exhibited decreased lipid and inflammatory cell infiltration as seen by H&E staining (Fig. 1A).
Figure 1.
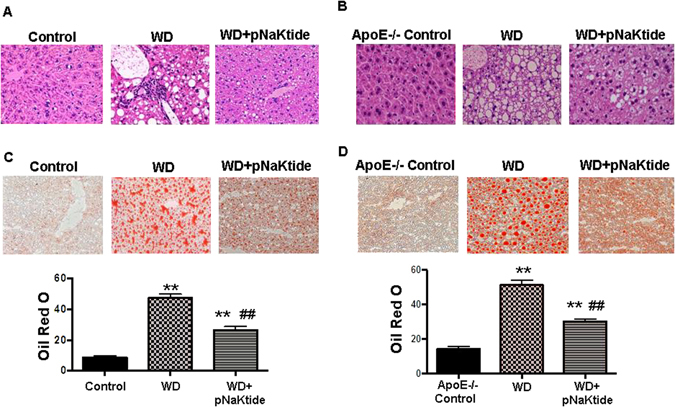
Effect of pNaKtide on liver histology and hepatic lipid accumulation and steatohepatitis in C57Bl6 (Panel A) and APOE−/− (Panel B) fed a western diet (defined in methods). Representative H&E staining in liver in C57Bl6 (A) and ApoE−/− (B) mice, representative Oil Red O slides in C57Bl6 (C) and ApoE−/− (D) mice and quantification of Oil Red O stain shown in bottom. Quantification displayed as mean ± SEM, n = 8–10/group, *p < 0.05 vs control, #p < 0.05 vs WD, **p < 0.01 vs control, ##p < 0.01 vs WD.
H&E staining of liver tissues from ApoE−/− mice fed a western diet also had a significant increase in lipid and inflammatory cell infiltration compared to control ApoE−/− mice. pNaKtide decreased lipid and inflammatory cell infiltration compared to mice fed a western diet (Fig. 1B). Both C57Bl6 and ApoE−/− mice fed a western diet had significantly increased lipid accumulation in the liver compared to mice fed a normal chow diet (Fig. 1C,D) as demonstrated by Oil red O staining and administration of pNaKtide decreased this. Increased fat accumulation in hepatocytes is mostly attributed to enhanced uptake and synthesis of fatty acids and triglycerides25. Our results showed that hepatic triglycerides and FFA levels were significantly increased in C57Bl6 and ApoE−/− mice fed a western diet compared to control mice (Table 3). As expected, pNaKtide treatment significantly decreased hepatic triglycerides and FFA levels compared to mice fed a western diet.
Table 3.
Effect of pNaKtide on hepatic lipid accumulation markers, inflammatory, fibrotic markers, and mitochondrial fatty acid oxidation genes in C57Bl/6 and ApoE−/− mice fed a western diet.
| C57Bl6 | ApoE−/− | |||||
|---|---|---|---|---|---|---|
| CTR | WD | WD + pNaKtide | CTR | WD | WD + pNaKtide | |
| Lipid Accumulation Markers | ||||||
| Liver triglycerides (mg TG/mg protein) | 0.1 ± 0.01 | 0.4 ± 0.04** | 0.2 ± 0.01## | 0.8 ± 0.03 | 2.05 ± 0.1** | 0.9 ± 0.08## |
| Liver FFA (nmol/100 mg) | 18.3 ± 1.2 | 30.4 ± 2.5** | 17.3 ± 0.8*,## | 7.5 ± 1.06 | 27.4 ± 4.7** | 11.09 ± 1.8# |
| FAS mRNA | 0.9 ± 0.003 | 3.2 ± 0.1** | 1.4 ± 0.3## | 0.9 ± 0.003 | 2.8 ± 0.5** | 1.3 ± 0.1## |
| CD36 mRNA | 0.9 ± 0.003 | 4.1 ± 0.3** | 1.9 ± 0.5## | 0.9 ± 0.003 | 2.6 ± 0.1** | 1.1 ± 0.1## |
| Fatty Acid Oxidation Markers | ||||||
| PGC1α mRNA | 1.01 ± 0.02 | 0.5 ± 0.09* | 1.2 ± 0.01## | 1.01 ± 0.02 | 0.6 ± 0.03** | 1.1 ± 0.02## |
| CPT1 mRNA | 1.01 ± 0.02 | 0.6 ± 0.03* | 1.4 ± 0.1## | 1.01 ± 0.02 | 0.4 ± 0.1* | 1.3 ± 0.08## |
| LCAD mRNA | 1.006 ± 0.02 | 0.4 ± 0.03** | 1.7 ± 0.2*,## | 1.006 ± 0.02 | 0.4 ± 0.07* | 1.2 ± 0.07## |
| Oxidative Stress Markers | ||||||
| TBARS (umol/mg protein) | 19.4 ± 1.04 | 31.2 ± 1.3** | 18.1 ± 2.8## | 21.6 ± 3.5 | 48.5 ± 7.1** | 28.1 ± 1.4# |
| Inflammatory Markers | ||||||
| F4/80 mRNA | 0.9 ± 0.003 | 3.1 ± 0.3** | 1.3 ± 0.1## | 0.9 ± 0.003 | 2.1 ± 0.2** | 1.3 ± 0.1## |
| MCP-1 mRNA | 0.9 ± 0.003 | 2.4 ± 0.2** | 1.3 ± 0.1## | 0.9 ± 0.003 | 3.9 ± 0.2** | 2.2 ± 0.5## |
| IL-6 mRNA | 0.9 ± 0.003 | 3.5 ± 0.8** | 1.2 ± 0.1# | 0.9 ± 0.003 | 3.5 ± 0.8* | 1.2 ± 0.1# |
| Fibrosis Markers | ||||||
| Fibronectin mRNA | 0.9 ± 0.003 | 2.4 ± 0.2** | 0.5 ± 0.04## | 0.9 ± 0.003 | 2.5 ± 0.3** | 0.9 ± 0.06## |
| MMP-13 mRNA | 0.9 ± 0.003 | 2.8 ± 0.1** | 1.08 ± 0.1## | 0.9 ± 0.003 | 3.3 ± 0.7* | 2.3 ± 0.9 |
| MMP-9 mRNA | 0.9 ± 0.004 | 3.5 ± 0.1** | 1.02 ± 0.1## | 0.9 ± 0.004 | 2.08 ± 0.3** | 1.2 ± 0.2## |
Results are means ± SEM, n = 6–8/group. *p < 0.05, **p < 0.01 vs. control; #<0.05, ##p < 0.01 vs WD.
Fatty acid synthase (FAS) expression, an important marker of lipogenesis, was increased in both strains of mice fed a western diet and this elevated level was decreased to control levels with pNaKtide treatment (Table 3). CD36, a fatty acid transport protein, contributes to the progression of inflammation in NASH5, 26. Our results showed that CD36 mRNA expression was significantly increased in both strains of mice fed a western diet compared to control mice, and this increase was negated by treatment with pNaKtide (Table 3). Similarly, F4/80, MCP-1 and IL-6 mRNA expression, markers of macrophage/kupffer cell infiltration and inflammation27, were also increased in both strains of mice fed a western diet compared to control mice (Table 3). These changes were significantly attenuated with pNaKtide treatment.
Effect of pNaKtide on hepatic fibrosis in mice fed a western diet
Masson’s trichrome staining of liver sections from both strains of mice fed a western diet demonstrated focal portal fibrosis (blue staining) compared to control mice and this increase was negated by treatment with pNaKtide (Fig. S3A,B). Excess collagen synthesis in the liver is a sensitive indicator of NASH and fibrosis28. Type 1-collagen levels were significantly increased in both strains of mice fed a western diet, and this was significantly decreased to control levels with pNaKtide treatment (Fig. S3C,D). Similarly, mRNA expression of hepatic fibronectin, MMP9 and MMP13, genes related to fibrogenesis29, 30, were also elevated in both strains of mice fed a western diet, and these increases were also attenuated with pNaKtide treatment (Table 3).
Effect of pNaKtide on hepatic p-Src, ERK, and α1 carbonylation in mice fed a western diet
Protein carbonyl groups are widely used markers for oxidative stress15. We have shown previously that reactive oxygen species (ROS) initiates carbonylation of the α1 Na/K-ATPase subunit which in turn activates the Src signaling cascade with downstream modulation of extracellular signal-regulated kinase 1/2 (ERK1/2)20. Our results showed that carbonylation of the α1 Na/K-ATPase subunit was increased in C57Bl6 and ApoE−/− mice fed a western diet, and this increase was negated by pNaKtide treatment (Fig. 2A and B). Treatment with pNaKtide also blocked Na/K-ATPase-regulated Src and ERK 1/2 activation in both strains of mice fed a western diet (Fig. 2C–F). Further our results showed that TBARS, another marker of oxidative stress, was decreased by pNaKtide treatment compared to mice fed a western diet (Table 3), demonstrating pNaKtide-mediated abrogation of NASH and oxidative stress.
Figure 2.
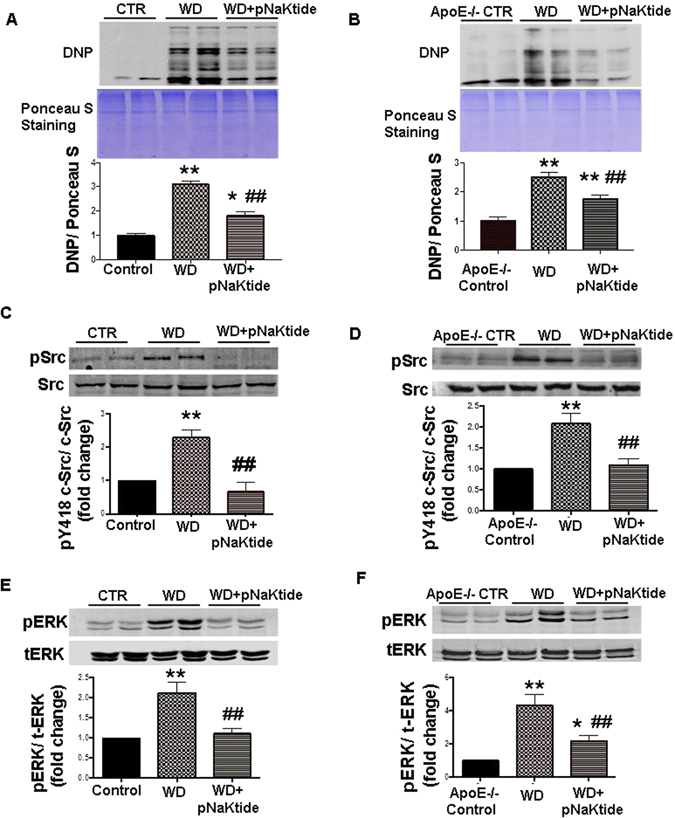
Effect of pNaKtide on α1 carbonylation and Na/K-ATPase/Src/ROS amplification signaling pathway in livers of C57Bl6 and ApoE−/− mice. Whole cell lysates were prepared with Nonidet P-40 buffer and Western blot analysis was performed to determine protein carbonylation with Ponceau S staining as a loading control in C57Bl6 (A) and ApoE−/− (B) mice, activation of c-Src in C57Bl6 (C) and ApoE−/− (D) and activation of ERK 1/2 in C57Bl658 and ApoE−/− (F) with mean band density normalized to total Src and total ERK respectively. Results are expressed as means ± SE, n = 6–8/group, **p < 0.01 vs control, ##p < 0.01 vs WD.
Effect of pNaKtide on hepatic mitochondrial fatty acid oxidation in mice fed a western diet
Mitochondria play an important role in energy metabolism, being the primary site for fatty acid oxidation in the liver31–33. Our results showed that hepatic PGC1α levels were decreased in both strains of mice fed a western diet compared to the control groups, and pNaKtide treatment significantly increased hepatic PCG1α expression (Fig. 3A and B). In the mitochondrial extract of both mice strains fed a western diet, the hepatic expression of CPT-1 was less compared to the control group (Fig. 3C and D). Administration of pNaKtide increased CPT-1 levels significantly compared to mice fed a western diet. Long-chain acyl-CoA dehydrogenase (LCAD), is involved in the first reaction of mitochondrial fatty acid oxidation and is important for energy homeostasis23. Similar to previous studies34, 35, our results demonstrated that LCAD levels were significantly increased by pNaKtide treatment in liver mitochondrial extracts compared to mice fed a western diet (Fig. 3E and F). Quantitative PCR was also done for the above genes and our results followed the same trend as the Western blot analysis. As expected, PCG1α, CPT-1 and LCAD mRNA expression were significantly increased by pNaKtide treatment in both C57Bl6 and ApoE−/− mice fed a western diet (Table 3).
Figure 3.
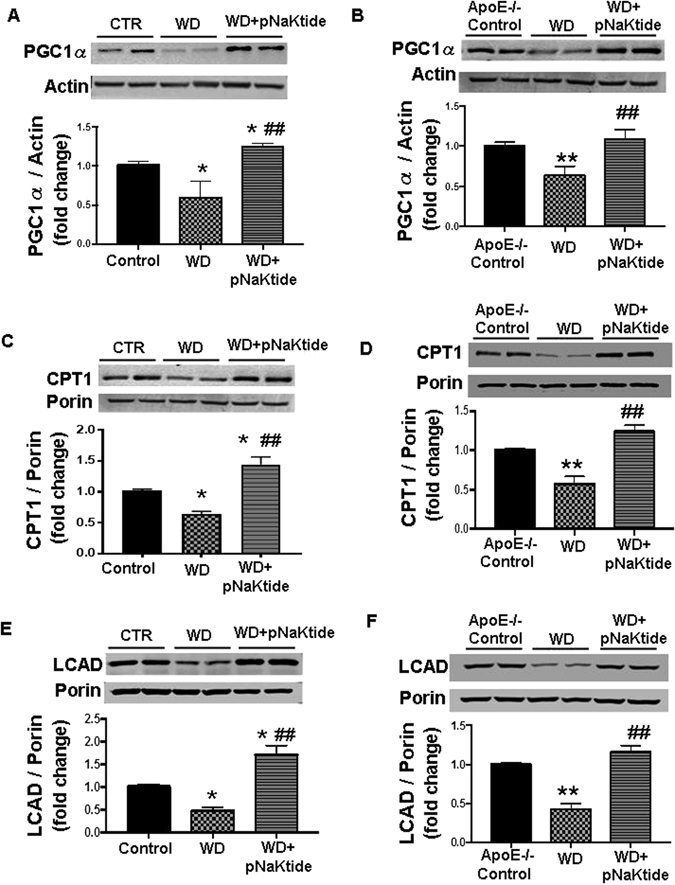
Effect of pNaKtide on hepatic mitochondrial fatty acid oxidation in C57Bl6 and ApoE−/− mice fed a western diet. PGC1α Western blot analysis of liver homogenates, with data shown as mean band density normalized to actin in C57Bl6 (A) and ApoE−/− (B) mice, CPT-1 Western blot analysis of mitochondrial isolates from liver homogenates, with data shown as mean band density normalized to porin in C57Bl6 (C) and ApoE−/− (D) mice, and LCAD Western blot analysis of mitochondrial isolates from liver homogenates, with data shown as mean band density normalized to porin in C57Bl6 (E) and ApoE−/− (F) mice. Results are means ± SE, n = 6/group, *p < 0.05 vs control, #p < 0.05 vs. WD, **p < 0.01 vs control, ##p < 0.01 vs WD.
Effect of pNaKtide on metabolic profile in mice fed a western diet
The insulin resistance phenotype in both strains of mice fed a western diet was reversed by the administration of pNaKtide. This was characterized by improved glucose tolerance (Fig. 4) and a significant improvement in the homeostasis model assessment of insulin resistance (HOMA-IR) score (Table 4). Increased plasma ALT, FFA and triglycerides levels are strongly associated with insulin resistance and NASH. The administration of pNaKtide significantly attenuated the diet induced increase in plasma ALT, triglyceride, and FFA levels in C57Bl6 and ApoE−/− mice fed a western diet (Table 4). Treatment of pNaKtide also significantly decreased plasma LDL levels in C57Bl6 and ApoE−/− mice fed a western diet (Table 4). Further, our results showed that both strains of mice fed a western diet had decreased plasma HDL levels (Table 4) and this decrease was reversed by pNaKtide. Generation of ROS has been implicated in the pathogenesis of NASH and atherosclerosis. Our results showed that administration of pNaKtide decreased plasma ROS levels in both strains of mice fed a western diet (Table 4).
Figure 4.
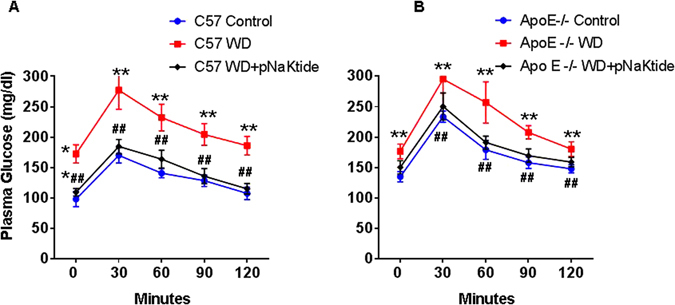
Effect of pNaKtide on glucose tolerance test (GTT) in C47Bl6 (A) and ApoE−/− (B) mice fed a western diet. Data displayed as mean ± SEM, n = 8–10/group, *p < 0.05 vs control, #p < 0.05 vs WD, **p < 0.01 vs control, ##p < 0.01 vs WD.
Table 4.
Effect of pNaKtide on metabolic profile in plasma of C57Bl6 and ApoE−/− mice fed a western diet.
| C57Bl6 | ApoE−/− | |||||
|---|---|---|---|---|---|---|
| CTR | WD | WD + pNaKtide | CTR | WD | WD + pNaKtide | |
| HOMA-IR | 0.8 ± 0.08 | 2.5 ± 0.3** | 1.09 ± 0.03## | 1.3 ± 0.06 | 5.6 ± 0.4** | 2.1 ± 0.1## |
| ALT (U/L) | 3.5 ± 0.9 | 7.9 ± 0.6** | 4.9 ± 0.4# | 3.3 ± 0.6 | 6.9 ± 0.8** | 4.2 ± 0.5# |
| Triglycerides (mg/dl) | 24.3 ± 1.7 | 39.8 ± 4.8** | 26.8 ± 1.6# | 94.02 ± 4.2 | 147.6 ± 3.1** | 116 ± 9.05## |
| FFA (mmol/l) | 0.2 ± 0.02 | 0.9 ± 0.07** | 0.6 ± 0.06*,# | 0.4 ± 0.04 | 1.03 ± 0.08** | 0.6 ± 0.08# |
| LDL (mg/dl) | 10.2 ± 0.7 | 42.6 ± 3.1** | 23.5 ± 3.1**,## | 101.4 ± 4.9 | 239.5 ± 16.2** | 158.3 ± 7.3**,## |
| HDL (mg/dl) | 48.4 ± 0.7 | 14.5 ± 1.03** | 45.7 ± 2.9## | 32.9 ± 2.01 | 8.1 ± 2.6** | 28.3 ± 3.2## |
| ROS (mmol) | 1.01 ± 0.1 | 4.6 ± 0.7** | 1.5 ± 0.3## | 1.4 ± 0.4 | 6.6 ± 0.06** | 3.3 ± 0.6## |
Results are means ± SEM, n = 6/group. *p < 0.05, **p < 0.01 vs. control; #<0.05, ##p < 0.01 vs WD.
Effect of pNaKtide on aorta architecture and plaques in mice fed a western diet
Bright field images of whole and open aortas were taken and Sudan IV staining of open aortas was performed in C57Bl6 and as expected, there was no gross evidence of atherosclerotic plaques (data not shown). Cross-sections of the C57Bl6 aortas were stained with Oil Red O to estimate lipid deposition. Lipid accumulation in the aorta was increased in mice fed a western diet compared to the control group (Fig. S4). Treatment with pNaKtide significantly decreased lipid staining compared to mice fed a western diet.
The purpose of using the ApoE−/− mouse was to examine the atherosclerosis process. Bright field images of dissected aortas were taken to visualize and quantify lesion size in ApoE−/− mice. In both the whole closed aorta and open aorta images (Fig. 5A and B respectively), ApoE−/− mice fed a western diet had increased plaque size. This was significantly decreased by pNaKtide treatment. These data were confirmed with Sudan IV staining for lipid deposition (Fig. 6A). The Sudan IV staining was significantly decreased in pNaKtide treated ApoE−/− mice, which demonstrated decreased lipid deposition. Experiments with rhodamine-labeled pNaKtide did not demonstrate pNaKtide localization to aortic plaques (Supplementary Fig. 1B). Oil Red O staining of the aorta demonstrated increased lipid accumulation in ApoE−/− mice fed a western diet compared to the control group, and treatment with pNaKtide significantly decreased this lipid deposition (Fig. 6B). Furthermore, H&E staining of the aorta revealed a significant increase in cross-sectional lesion area with intimal thickening, acellular matrix of lipid pool, mixed inflammatory cell infiltrate, and necrotic area in ApoE−/− mice fed a western diet (Fig. 6C). All of these outcomes- lesion size, necrotic core area, inflammatory cell infiltration, and intimal thickness improved with pNaKtide treatment (Fig. 6C).
Figure 5.
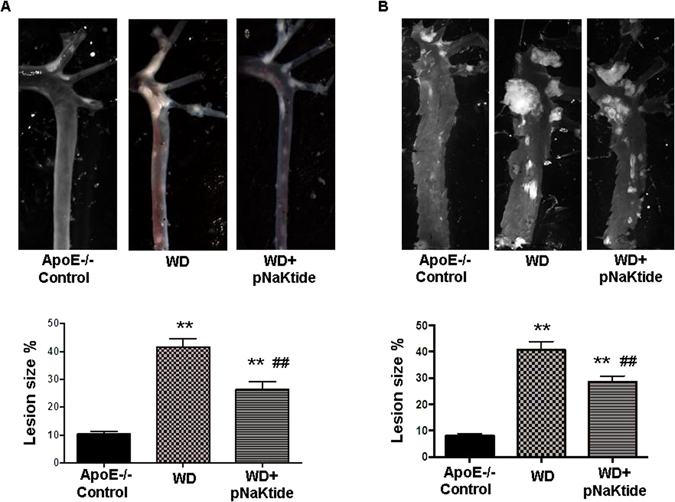
Effect of pNaKtide on atherosclerotic plaque size in ApoE−/− mice aortas fed a western diet. Bright field images of whole closed aortas (A) and open aortas (B) in ApoE−/− mice fed a western diet. The amount of lesion formation in each animal was expressed as a ratio of plaque integral density to total surface area of the aorta. Results are shown as means ± SEM, n = 8/group, **p < 0.01 vs control, ##p < 0.01 vs. WD.
Figure 6.
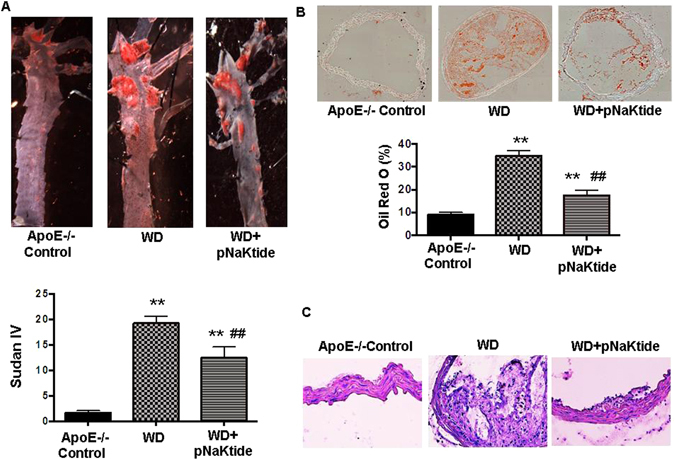
Effect of pNaKtide on atherosclerotic plaque size in ApoE−/− mice aortas fed a western diet. Sudan IV staining for lipid deposition in open aortas (A), oil red O staining for lipid accumulation in aorta cross-sectional images (B), and aorta H&E staining (C). Quantified results shown as mean ± SEM, n = 8/group, *p < 0.05 vs control, **p < 0.01 vs control, ##p < 0.01 vs. WD.
Discussion
This study demonstrates that Na/K-ATPase amplification of oxidant signaling is important in the pathophysiology of NASH and atherosclerosis in the murine models studied. We report that the metabolic syndrome phenotype, present in both C57Bl6 and ApoE−/− mice fed a western diet, improved with pNaKtide treatment which attenuated Na/K-ATPase induced oxidant amplification20. Obesity, as measured by body weight and visceral fat weight, was significantly reduced in C57Bl6 mice but not in ApoE−/− mice treated with the pNaKtide. We suggest that this is a consequence of the ApoE−/− mouse being relatively resistant to weight gain with the western diet21. In order to determine the mechanism by which pNaKtide affected adiposity, we utilized the CLAMS system to measure metabolic rate. We observed that both energy expenditure and activity level were significantly increased in pNaKtide treatment groups compared to animals fed a western diet, suggesting that increased energy expenditure is at least partly responsible for reduced adiposity in mice fed a western diet. Adipose mitochondria regulate whole-body energy metabolism via dissipating energy and generating heat in adipose tissue8. PGC1α and CPT-1 are essential for adipose tissue fatty acid oxidation8, 22, 36. UCP-1 and 2 play important roles in the control of energy expenditure by uncoupling respiration from ATP synthesis, thereby dissipating energy as heat and affecting energy metabolism efficiency37, 38. In our study, we have shown that C57BL6 and ApoE−/− mice fed a WD have decreased CPT-1, UCP-1, UCP-2, and PGC1α mRNA expression levels in adipose tissue. This was significantly improved with pNaKtide treatment. Increased CPT-1 and PGC1α expression with pNaKtide may be related to an increase in fatty acid oxidation8. Given the established relationship between UCP-1 and 2 and thermogenesis, pNaKtide may increase metabolic rate and energy metabolism in adipose tissue20. Overall, we suggest that pNaKtide increases fat utilization by increasing mitochondrial fatty acid oxidation in adipose tissue, oxygen consumption, and metabolic rate leading to an overall decrease in both body weight and visceral adiposity.
Since obesity is closely associated with NASH, the effect of pNaKtide on steatohepatitis was evaluated. According to the two-hit hypothesis, hepatic steatosis appears to be a prerequisite for subsequent events that lead to NASH39. Obesity and insulin resistance result in increased release of FFAs from adipocytes and increased hydrolysis of TGs, which contribute to elevated plasma levels of FFA40. Furthermore, increased FFAs induce oxidative stress via ROS generation25, which may initiate the Na/K-ATPase/ROS amplification pathway to propagate steatosis and NASH. Treatment with pNaKtide showed significant attenuation of hepatic steatosis as evident by decreased lipid staining, FAS expression, and TG and FFA concentrations in the livers of C57Bl6 and ApoE−/− mice fed a western diet. Also, pNaKtide attenuated CD36 expression, which we have previously linked to Na/K-ATPase signaling41. In addition, ALT, a well-established marker of liver injury, was also decreased by pNaKtide treatment42.
Oxidative stress and inflammation are known to underlie the etiopathogenesis of NASH43, 44 and can be categorized as the second hit with regard to the two hit hypothesis. pNaKtide decreased hepatic α1 carbonylation, p-Src, and downstream p-ERK expression to attenuate Na/K-ATPase-induced ROS amplification. Mitochondria play a central role in ATP production and energy expenditure through fatty acid oxidation31–33. Our results showed that pNaKtide treatment increased hepatic expression of genes involved in fatty acid oxidation including CPT-1, PGC1α and LCAD in the setting of caloric excess, to ameliorate the NASH phenotype. Collectively, it appears that pNaKtide, through its mitigation of ROS, has a favorable effect on mitochondrial biogenesis and fatty acid oxidation in the setting of a WD. It promotes increased substrate utilization, decreased lipid storage, and most importantly minimizes the impact ROS has on these processes, therefore limiting the impact of a second hit.Consequences of oxidative stress (ROS) include lipid peroxidation, which can cause direct damage to hepatocytes and induce an inflammatory response with upregulation of inflammatory cytokines such as MCP-1 and IL-645. We have shown TBARS (a marker of oxidative stress), MCP-1 and IL-6 are all decreased with pNaKtide. Further, our results show that administration of pNaKtide significantly attenuated WD-induced inflammatory cell infiltration in hepatic tissue. This close interaction of ROS and inflammatory cytokines may be how pNaKtide (via the Na/K-ATPase signaling cascade) is able to mitigate the inflammatory component of the second hit,necessary for the development of NASH. With regard to fibrosis, prior studies have shown that the Na/K-ATPase signal cascade, once initiated, activates PKCδ through ROS amplification. PKCδ then phosphorylates Fli-1, which in turn, increases collagen expression16–19. We have similarly shown an increase in hepatic collagen expression in WD fed mice, and pNaKtide significantly decreased collagen deposition. Overall, there was significant attenuation of NASH in C57Bl6 and ApoE−/− mice fed a WD by pNaKtide, as evidenced by decreased lipid accumulation, inflammatory cell infiltration, and trichrome staining for fibrosis. This is supported by findings of decreased markers of lipogenesis (FAS, liver TG/FFAs, CD36), inflammation (IL-6, MCP-1, F4/80), and fibrosis (fibronectin, MMP9, MMP13, collagen-1). We propose that pNaKtide treatment, by decreasing ROS through the Na/K-ATPase pathway, was sufficient to suppress the oxidative stress and inflammation necessary to attenuate steatohepatitis.
NASH is associated with insulin resistance (IR), which results in hypertriglyceridemia and hyperglycemia, which in turn, increases plasma FFAs in the circulation. Increased levels of FFAs in plasma induce oxidative stress via ROS generation46. Dyslipidemia is frequently observed in patients with NASH, and treatment of dyslipidemia plays a critical role in the overall management of these patients. pNaKtide improved insulin sensitivity and dyslipidemia, which prompted an examination of how pNaKtide affected vascular disease. Increased levels of circulating FFAs and oxidative stress lead to increased lipid deposition within the vasculature, atherosclerosis, and ultimately cardiovascular complications46. Elevated LDL levels have been associated with an increase in the amount of cholesterol entering the plaque47, 48. More importantly, an increase in serum HDL may contribute to active LDL removal from the vessel wall and from macrophage or foam cells49. In the C57Bl6 mouse fed a western diet, fatty streaking was markedly reduced by pNaKtide. We further saw attenuation of aortic atherosclerosis in the ApoE−/− mouse fed a western diet. We suggest that pNaKtide’s beneficial effect on atherosclerosis was secondary to a favorable systemic lipid profile. The pNaKtide significantly reduced plasma LDL and TG levels and increased HDL50. Whether the plaque decrease observed in this study will translate into significant reductions in clinical events is yet to be determined, however modifying the levels of harmful and protective lipids should still be an important clinical objective. Taken together, this study demonstrates that pNaKtide improves the metabolic syndrome phenotype, specifically obesity, NASH, insulin resistance, dyslipidemia, and atherosclerosis, effects that are mediated by blockage of the Na/K-ATPase/ROS amplification loop. Unfortunately, we were only able to study male animals in the current study. Certainly, the effects of gender and female sex hormones on the development of oxidant stress and obesity may be extremely important.
At this juncture, we would offer a disclaimer that there are a number of interventions that have been shown to be effective in improving the perturbations associated with obesity in these animal models, and some of these have even been found to be effective in clinical practice. However, we would argue that our approach focusing on Na/K-ATPase induced oxidant amplification is novel and potentially presents a new and effective target for therapeutic intervention in the treatment of obesity and its related manifestation dyslipidemia, NASH and atherosclerosis. It should not be forgotten that obesity, metabolic syndrome, diabetes and cardiovascular disease present an increasing burden on healthcare systems and cost in the world today. The approach outlined in this report offers a portal to decreasing the increasing burden that obesity places on society.
In conclusion, we noted that oxidant stress characterized the metabolic syndrome, NASH and vascular abnormalities induced by a western diet in both C57Bl6 and ApoE−/− mice. We further noted that addressing Na/K-ATPase signaling associated oxidant amplification with pNaKtide ameliorated these abnormalities. Should these data be confirmed in humans, strategies like pNaKtide, which focus on this oxidant amplification loop, may potentially augment our clinical approach to these problems.
Material and Methods
Experimental Design
All animal studies were approved by the Marshall University Animal Care and Use Committee in accordance with the National Institutes of Health Guidelines for Care and Use of Laboratory Animals. C57Bl6 mice and ApoE knockout mice (6–8 weeks old, male) were purchased from Jackson Laboratory. Upon arrival to the Byrd Biotechnology Center, ARF, Animal Research Facility, mice were placed in cages and were fed normal chow diet and had access to water ad libitum or were fed Western Diet (WD) and had ad libitum access to high fructose solution. Western diet containing fructose is a well-known model of diet induced metabolic syndrome, and it contributes to chronic metabolic imbalance and to the development of NASH and atherosclerosis51–53. WD was purchased commercially from Envigo (Indianapolis, IN). WD contained 42% fat, 42.7% carbohydrate, and 15.2% protein yielding 4.5 KJ/g. Fructose was purchased commercially from Alfa Aesar (Ward Hill, MA). Fructose was made at a concentration of 42 g/L, yielding 0.168 KJ/mL. The animals were randomly divided into 4 groups (8–10 mice per group) as follows: (1) Control, (2) Control + pNaKtide, (3) WD, and (4) WD + pNaKtide. The number of animals in each group was determined by power analysis following assumptions derived from our previous study with mice54, using a variance of 0.2 within the groups, power of 0.80, and alpha error of 0.05. After 4 weeks of control or WD diet respectively, group 2 and group 4 were injected with pNaKtide for 8 weeks (dissolved in saline and injected I.P. at a dose of 25-mg/kg-body weight every 7 days. The body weight was measured every week. At the end of the 12-week period, mice were placed on an 8-hour fast, anesthetized with sodium pentobarbital (65 mg/kg, I.P.) and blood was obtained from the tail vein for measurement of glucose using a glucometer and measurement of insulin using ELISA assay kit (Abcam, Cambridge, MA). At the time of sacrifice, body weight, visceral fat content, and liver weight of all mice were measured. Liver tissue and aortas were flash frozen in liquid nitrogen and maintained at −80 °C until assayed. Blood samples were collected for lipid profile measurement. Our results showed that there was no significant difference between the Control and Control + pNaKtide C57Bl6 group with regard to body weight (Supplementary Fig. S2A), glucose tolerance (Supplementary Fig. S2B), triglycerides (Supplementary Fig. S2C), HDL (Supplementary Fig. S2D), and LDL (Supplementary Fig. S2E). Therefore we proceeded without the Control + pNaKtide group in subsequent C57BL6 and ApoE knockout mouse studies.
Indirect calorimetry and locomotor activity
At the end of the 12-week experimental period, energy expenditure and locomotor activity were measured using an eight-chamber CLAM (Columbus Instruments, Columbus, OH, USA). In this system, total oxygen consumption (VO2) and carbon dioxide production (VCO2) were measured, and VO2 was converted to individual heat production (kcal/hour) by Columbus software. This software calculates the heat production by multiplying the calorific value CV = 3.815 + (1.232 × RER) by the observed VO2 (Heat = CV × VO2). The energy expenditure is then calculated as a ratio of heat produced divided by body mass. A system of infrared beams detects movement of animals in CLAMS, and locomotor activity was determined as ambulatory count, the number of times different beams were broken in either the x- or y-axes during an interval. All mice were acclimatized to monitoring cages for 24 hours prior to an additional 48 hours of recordings under the regular 12-hour light–dark cycle.
Blood measurement of LDL, HDL, Triglycerides and Free Fatty Acids (FFA)
Plasma LDL and HDL levels were determined using an ELISA Assay according to manufacturer protocols (Abcam, Cambridge, MA). Triglycerides and FFA levels were quantified using a Quantification Assay Kit according to manufacturer protocols (Abcam, Cambridge, MA).
Blood ROS and hepatic TBARS measurement
Plasma ROS levels were determined using the OxiSelectTMIn Vitro ROS Assay Kit according to manufacturer’s protocols (Cell Biolabs, Inc, San Diego, CA). Hepatic oxidative injury was measured as TBARS using an assay kit according to manufacturer’s protocol (Cayman Chemical, Ann Arbor, MI). Liver samples were homogenized in a buffer solution containing 50 mM tris-HCL (pH 7.4) and 1.15% KCL and then centrifuged. The supernatant was used for the assay. Data were normalized to total protein and presented as micromoles per milligram of protein.
Glucose tolerance test
Glucose clearance was determined using an intraperitoneal glucose tolerance test before termination of the experiment. Mice were fasted for 8 h, after which a glucose solution (2 g/kg body weight, injected as a 10% solution) was injected into the peritoneal cavity. Samples were taken from the tail vein at 0, 30, 60, 90, and 120 min after glucose injection. Blood glucose was measured using the Accutrend Sensor glucometer.
Determination of homeostasis model assessment of insulin resistance
The HOMA-IR was calculated from mouse blood using glucose and insulin concentrations obtained after 8 hours of food withdrawal, using the following formula:
Distribution of rhodamine B labeled pNaKtide
Mice were injected (by i.p.) without (as control) or with rhodamine B labeled pNaKtide (25 mg/Kg body weight). Three hours after injection, the mice were sacrificed and the hepatic tissues were imaged and analyzed. The images were taken at laser power 5% (emission readings for rhodamine B are 580–650 nm) with a Leica SP5 TCS II equipped with coherent chameleon multiphoton vision II (IR) laser and analyzed by Leica LAS/AF software.
Determination of Triglyceride and FFA levels in hepatic tissue
For triglyceride measurement, liver samples were homogenized in ice-cold phosphate-buffered saline (PBS). Tissue lipids were extracted with methanol/chloroform (1:2), dried, and resuspended in 5% fat-free bovine serum albumin. Triglyceride levels were determined using a commercially available kit according to the manufacture’s protocol (Abcam, Cambridge, MA). For FFA measurement, liver tissue was homogenized in 1% (w/v) Triton X-100 in chloroform solution. After centrifugation of samples, the lower organic phase was collected and dried to remove chloroform. The dried lipids were dissolved in fatty acid assay buffer and FFA levels were determined using a commercially available kit according to the manufacturer’s protocol (Sigma-Aldrich, St. Louis, MO).
Histopathological examination in hepatic tissue
Liver samples from each group were fixed in 4% paraformaldehyde, dehydrated, embedded in paraffin, and sectioned. Formalin-fixed, paraffin-embedded sections were cut (6 μm thick) and mounted on glass slides. The sections were deparaffinized in xylene and hematoxylin and eosin and Masson’s trichrome staining were performed for histological analysis.
Oil Red-O staining
For Oil Red O staining, liver and aorta tissues, which were frozen in OCT compounds, were cut into 6 μm sections, mounted on slides and allowed to dry for 1–2 hours. The sections were fixed with 4% paraformaldehyde (PFA) for 1 hour and then the slides were rinsed with PBS (pH 7.4). After air-drying, the slides were stained in 0.3% Oil Red O solution for 3 minutes. The slides were rinsed with distilled water 2 times and mounted with 87% glycerol. The microphotographs were taken on a Nikon Eclipse 80i microscope equipped with a Nikon camera head DS-Fi1 (Nikon, Japan). For quantitative analysis, the total area of red pixels on the Oil-Red-O stained tissue section was measured by using the Image J software provided by NIH. The data were expressed as the mean ± SEM of percentage of the Oil-Red-O stained areas with respect to total area.
Aorta Dissection and Sudan IV staining
Aorta Harvesting
The aorta was dissected and harvested under a stereomicroscope from the iliac bifurcation to the aortic root, including the brachiocephalic trunk, left common carotid artery, and left subclavian artery. The adventitial fat was removed. The aortic trunk was washed with cold PBS and pinned onto a black wax surface.
Whole Aorta Imaging
Bright field images of the dissected aorta were taken with a Leica stereomicroscope (Leica MZ10F; Leica Microsystems, Buffalo Grove, IL) attached to a QImaging Camera (Q26644; Surrey, Canada) at a total magnification of 8x (objective lens was 0.8x and eye piece lens was 10x).
Open Aorta Imaging
Whole aortas were opened longitudinally from the aortic root to the iliac bifurcation and pinned to a black wax surface. Bright field mages were acquired using a Leica stereomicroscope attached to a QImaging Camera at a total magnification of 8x.
Sudan IV Staining
To identify lipid rich intraluminal lesions, the aortas were stained with commercially available Sudan IV (Sigma, St Louis, MO). A 0.5% Sudan IV solution was made using acetone and ethanol at an equal ratio. Tissues were treated with 70% ethanol for 5 minutes followed by Sudan IV for 15 minutes. They were then washed with 80% ethanol for 3 minutes and rinsed with PBS for 3 minutes. Finally tissues were treated with 0.4% PFA for storage and imaging.
Haematoxylin and Eosin Staining
The aorta, stored in OCT, was cut into 6 μm sections and stained with haematoxylin and eosin for histological analysis.
Quantification of Aortic Lesions
Quantification of atherosclerotic lesions was done using Image J 1.49 V (NIH). The amount of lesion formation in each animal was expressed as a ratio of plaque integral density to total surface area of the aorta.
Measurement of c-Src and ERK1/2 phosphorylation
Whole cell lysates from hepatic tissue were prepared with NP-40 buffer and activation of c-Src and ERK 1/2 was determined as previously described by Yan et al.15, 55. After immunoblotting for phospho-c-Src and phospho- ERK1/2, the same membrane was stripped and immunoblotted for total c-Src and total ERK 1/2. Activation of c-Src and ERK1/2 was expressed as the ratios of phospho-c-Src/total Src and phospho-ERK1/2/total ERK 1/2, respectively with both measurements normalized to 1.0 for the control samples.
Assessment of protein carbonylation
Whole-cell lysates from hepatic tissues were prepared with NP-40 buffer and western blotting for protein carbonylation assay was done55. The signal density values of control samples were normalized to 1.0 with Ponceau S staining as a loading control.
Western blot analysis
Liver was pulverized with liquid nitrogen and placed in a homogenization buffer. Homogenates were centrifuged, the supernatant was isolated, and immunoblotting was performed. The supernatant was used for the determination of Collagen-1 and PGC1α as previously reported36, 56. Loading conditions were controlled for using tubulin (collagen1) and actin (PGC1α). Mitochondrial extraction was performed on hepatic tissue by using a mitochondrial isolation kit (Abcam, Cambridge, MA). In brief, liver tissues were minced, and placed in pre-chilled dounce homogenizer. The homogenate was centrifuged at 1000 g for 10 mins at 4 degrees and the supernatant was saved. The supernatant was spun at 12000 g for 15 mins at 4 degrees and the pellet was saved. The pellet was washed and resuspended in isolation buffer and protease inhibitor, centrifuged at 12000 g for 15 mins. The pellet was again collected and the process was repeated. The pellet was resuspended in isolation buffer and protease inhibitor and used immediately. This mitochondrial homogenate was used for the determination of CPT-1 and LCAD and Porin was used as a loading control.
RNA extraction and real-time PCR
Total RNA was extracted from liver and adipose tissue using RNeasy Protect Mini kit (QIAGEN, Maryland, USA) according to manufacturer’s instructions. Total RNA (1 μg) was transcribed into cDNA using GeneAmp kit (Applied Biosystems, Branchburg, NJ, USA) reverse transcription reagents. Total RNA was analyzed by a quantitative real time polymerase chain reaction57. Real-time PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) on a 7500 HT Fast Real-Time PCR System (Applied Biosystems). Specific primers used were FAS, CD36, IL-6, F4/80, MCP-1, MMP9, MMP13, fibronectin, UCP-1, UCP-2, CPT-1, LCAD, PGC1α and GADPH. Each reaction was performed in triplicate. The comparative threshold cycle (Ct) method was used to calculate the fold amplification as specified by the manufacturer. All experimental samples were normalized using actin as an internal control and normalization was performed in separate reactions.
Statistical analyses
Data were tested for normality and then subjected to parametric analysis. When more than two groups were compared, one-way ANOVA was performed prior to comparison of individual groups, and the post-hoc t-tests were adjusted for multiple comparisons using the Tukey-Kramer correction. Data are presented as mean ± SEM.
Electronic supplementary material
Acknowledgements
This work was supported by National Institutes of Health Grants HL109015 (to J.I.S. and Z.X.), HL071556 and HL105649 (to J.I.S.), and HL55601 and HL34300 (to N.G.A.), by the Brickstreet Foundation (to J.I.S. and N.G.A.) and by the Huntington Foundation, Inc.
Author Contributions
Komal Sodhi: Designed and performed the experiments and wrote the manuscript. Krithika Srikanthan: Performed the experiments and wrote the manuscript. Perrine Goguet-Rubio: Performed the experiments. Alexandra Nichols: Performed the experiments. Amrita Mallick: Performed the experiments. Athar Nawab: Performed the experiments. Rebecca Martin: Performed the experiments. Preeya T. Shah: Performed the experiments. Muhammad Chaudhry: Performed the experiments. Saroj Sigdel: Edited the manuscript. Mehiar El-Hamdani: Edited the manuscript. Jiang Liu: Performed the experiments. Zijian Xie: Designed the experiments and edited the manuscript. Nader G. Abraham: Designed the experiments and edited the manuscript. Joseph I. Shapiro: Conceived and designed the experiments and wrote the manuscript.
Competing Interests
Several of the authors (Komal Sodhi, Jiang Liu, Nader Abraham and Joseph I. Shapiro) have patents awarded and/or patent applications, which may ultimately constitute a competing financial interest.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1038/s41598-022-13458-w"
Change history
5/30/2022
This article has been retracted. Please see the Retraction Notice for more detail: 10.1038/s41598-022-13458-w
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-00306-5
References
- 1.Ritchie SA, Connell JM. The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutrition, metabolism, and cardiovascular diseases: NMCD. 2007;17:319–326. doi: 10.1016/j.numecd.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Srikanthan K, Feyh A, Visweshwar H, Shapiro JI, Sodhi K. Systematic Review of Metabolic Syndrome Biomarkers: A Panel for Early Detection, Management, and Risk Stratification in the West Virginian Population. International journal of medical sciences. 2016;13:25–38. doi: 10.7150/ijms.13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanwar P, Kowdley KV. The Metabolic Syndrome and Its Influence on Nonalcoholic Steatohepatitis. Clinics in liver disease. 2016;20:225–243. doi: 10.1016/j.cld.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Mozaffarian D, et al. Executive Summary: Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:447–454. doi: 10.1161/CIR.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 5.Polimeni L, et al. Oxidative stress: New insights on the association of non-alcoholic fatty liver disease and atherosclerosis. World journal of hepatology. 2015;7:1325–1336. doi: 10.4254/wjh.v7.i10.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haas JT, Francque S, Staels B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annual review of physiology. 2016;78:181–205. doi: 10.1146/annurev-physiol-021115-105331. [DOI] [PubMed] [Google Scholar]
- 7.Tarantino G, Finelli C. Pathogenesis of hepatic steatosis: the link between hypercortisolism and non-alcoholic fatty liver disease. World journal of gastroenterology: WJG. 2013;19:6735–6743. doi: 10.3748/wjg.v19.i40.6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogge MM. The role of impaired mitochondrial lipid oxidation in obesity. Biol Res Nurs. 2009;10:356–373. doi: 10.1177/1099800408329408. [DOI] [PubMed] [Google Scholar]
- 9.Sumida Y, Niki E, Naito Y, Yoshikawa T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free radical research. 2013;47:869–880. doi: 10.3109/10715762.2013.837577. [DOI] [PubMed] [Google Scholar]
- 10.Vogiatzi G, Tousoulis D, Stefanadis C. The role of oxidative stress in atherosclerosis. Hellenic journal of cardiology: HJC=Hellenike kardiologike epitheorese. 2009;50:402–409. [PubMed] [Google Scholar]
- 11.Li H, Horke S, Forstermann U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis. 2014;237:208–219. doi: 10.1016/j.atherosclerosis.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Liu J, et al. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. The Journal of biological chemistry. 2000;275:27838–27844. doi: 10.1074/jbc.M002950200. [DOI] [PubMed] [Google Scholar]
- 13.Xie Z, et al. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. The Journal of biological chemistry. 1999;274:19323–19328. doi: 10.1074/jbc.274.27.19323. [DOI] [PubMed] [Google Scholar]
- 14.Liang M, et al. Identification of a pool of non-pumping Na/K-ATPase. The Journal of biological chemistry. 2007;282:10585–10593. doi: 10.1074/jbc.M609181200. [DOI] [PubMed] [Google Scholar]
- 15.Yan Y, et al. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. The Journal of biological chemistry. 2013;288:34249–34258. doi: 10.1074/jbc.M113.461020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, et al. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. The Journal of biological chemistry. 2009;284:21066–21076. doi: 10.1074/jbc.M109.013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J, Kennedy DJ, Yan Y, Shapiro JI. Reactive Oxygen Species Modulation of Na/K-ATPase Regulates Fibrosis and Renal Proximal Tubular Sodium Handling. International journal of nephrology. 2012;2012:381320. doi: 10.1155/2012/381320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai F, et al. Identification of a mutant alpha1 Na/K-ATPase that pumps but is defective in signal transduction. The Journal of biological chemistry. 2013;288:13295–13304. doi: 10.1074/jbc.M113.467381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Z, et al. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. The Journal of biological chemistry. 2011;286:32394–32403. doi: 10.1074/jbc.M110.207597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sodhi K, et al. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Science advances. 2015;1:e1500781. doi: 10.1126/sciadv.1500781. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Kennedy AJ, Ellacott KL, King VL, Hasty AH. Mouse models of the metabolic syndrome. Disease models & mechanisms. 2010;3:156–166. doi: 10.1242/dmm.003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes. 2005;54:1392–1399. doi: 10.2337/diabetes.54.5.1392. [DOI] [PubMed] [Google Scholar]
- 23.Nassir F, Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease. International journal of molecular sciences. 2014;15:8713–8742. doi: 10.3390/ijms15058713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abraham NG, et al. CYP2J2 targeting to endothelial cells attenuates adiposity and vascular dysfunction in mice fed a high-fat diet by reprogramming adipocyte phenotype. Hypertension. 2014;64:1352–1361. doi: 10.1161/HYPERTENSIONAHA.114.03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes. 1997;46:3–10. doi: 10.2337/diab.46.1.3. [DOI] [PubMed] [Google Scholar]
- 26.Collot-Teixeira S, Martin J, McDermott-Roe C, Poston R, McGregor JL. CD36 and macrophages in atherosclerosis. Cardiovascular research. 2007;75:468–477. doi: 10.1016/j.cardiores.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 27.Zhu ML, Shi JP. [Roles of kupffer cells activation in pathogenesis and progression of non-alcoholic fatty liver disease] Zhonghua gan zang bing za zhi=Zhonghua ganzangbing zazhi=Chinese journal of hepatology. 2012;20:718–720. [PubMed] [Google Scholar]
- 28.Schierwagen R, et al. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Scientific reports. 2015;5:12931. doi: 10.1038/srep12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu T, Wang X, Karsdal MA, Leeming DJ, Genovese F. Molecular serum markers of liver fibrosis. Biomarker insights. 2012;7:105–117. doi: 10.4137/BMI.S10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okazaki I, et al. Fibrogenesis and Carcinogenesis in Nonalcoholic Steatohepatitis (NASH): Involvement of Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinase (TIMPs) Cancers (Basel) 2014;6:1220–1255. doi: 10.3390/cancers6031220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamuta M, et al. Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. International journal of molecular medicine. 2005;16:631–635. [PubMed] [Google Scholar]
- 32.Kim E, Choi Y, Jang J, Park T. Carvacrol Protects against Hepatic Steatosis in Mice Fed a High-Fat Diet by Enhancing SIRT1-AMPK Signaling. Evidence-based complementary and alternative medicine: eCAM. 2013;2013:290104. doi: 10.1155/2013/290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohjima M, et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. International journal of molecular medicine. 2007;20:351–358. [PubMed] [Google Scholar]
- 34.Zou X, et al. Mitochondrial dysfunction in obesity-associated nonalcoholic fatty liver disease: the protective effects of pomegranate with its active component punicalagin. Antioxidants & redox signaling. 2014;21:1557–1570. doi: 10.1089/ars.2013.5538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahn J, Lee H, Jung CH, Ha T. Lycopene inhibits hepatic steatosis via microRNA-21-induced downregulation of fatty acid-binding protein 7 in mice fed a high-fat diet. Mol Nutr Food Res. 2012;56:1665–1674. doi: 10.1002/mnfr.201200182. [DOI] [PubMed] [Google Scholar]
- 36.Singh SP, et al. PGC-1 alpha regulates HO-1 expression, mitochondrial dynamics and biogenesis: Role of epoxyeicosatrienoic acid. Prostaglandins & other lipid mediators. 2016;125:8–18. doi: 10.1016/j.prostaglandins.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahadik SR, Lele RD, Saranath D, Seth A, Parikh V. Uncoupling protein-2 (UCP2) gene expression in subcutaneous and omental adipose tissue of Asian Indians: Relationship to adiponectin and parameters of metabolic syndrome. Adipocyte. 2012;1:101–107. doi: 10.4161/adip.19671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chevillotte E, Giralt M, Miroux B, Ricquier D, Villarroya F. Uncoupling protein-2 controls adiponectin gene expression in adipose tissue through the modulation of reactive oxygen species production. Diabetes. 2007;56:1042–1050. doi: 10.2337/db06-1300. [DOI] [PubMed] [Google Scholar]
- 39.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. The Journal of clinical investigation. 2004;114:147–152. doi: 10.1172/JCI200422422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wahren J, Sato Y, Ostman J, Hagenfeldt L, Felig P. Turnover and splanchnic metabolism of free fatty acids and ketones in insulin-dependent diabetics at rest and in response to exercise. The Journal of clinical investigation. 1984;73:1367–1376. doi: 10.1172/JCI111340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kennedy DJ, et al. CD36 and Na/K-ATPase-alpha1 form a proinflammatory signaling loop in kidney. Hypertension. 2013;61:216–224. doi: 10.1161/HYPERTENSIONAHA.112.198770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verma S, Jensen D, Hart J, Mohanty SR. Predictive value of ALT levels for non-alcoholic steatohepatitis (NASH) and advanced fibrosis in non-alcoholic fatty liver disease (NAFLD) Liver international: official journal of the International Association for the Study of the Liver. 2013;33:1398–1405. doi: 10.1111/liv.12226. [DOI] [PubMed] [Google Scholar]
- 43.Fon Tacer K, Rozman D. Nonalcoholic Fatty liver disease: focus on lipoprotein and lipid deregulation. Journal of lipids. 2011;2011:783976. doi: 10.1155/2011/783976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lanthier N. Targeting Kupffer cells in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: Why and how? World journal of hepatology. 2015;7:2184–2188. doi: 10.4254/wjh.v7.i19.2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitsuyoshi H, Itoh Y, Okanoue T. [Role of oxidative stress in non-alcoholic steatohepatitis] Nihon Rinsho. 2006;64:1077–1082. [PubMed] [Google Scholar]
- 46.Dowman JK, Tomlinson JW, Newsome PN. Pathogenesis of non-alcoholic fatty liver disease. QJM: monthly journal of the Association of Physicians. 2010;103:71–83. doi: 10.1093/qjmed/hcp158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rabbani R, Topol EJ. Strategies to achieve coronary arterial plaque stabilization. Cardiovascular research. 1999;41:402–417. doi: 10.1016/S0008-6363(98)00279-X. [DOI] [PubMed] [Google Scholar]
- 48.Armstrong ML, Megan MB. Lipid depletion in atheromatous coronary arteries in rhesus monkeys after regression diets. Circulation research. 1972;30:675–680. doi: 10.1161/01.RES.30.6.675. [DOI] [PubMed] [Google Scholar]
- 49.Fuster V. Elucidation of the role of plaque instability and rupture in acute coronary events. The American journal of cardiology. 1995;76:24C–33C. doi: 10.1016/S0002-9149(99)80467-6. [DOI] [PubMed] [Google Scholar]
- 50.Inaba T, et al. Cholesterol reduction and atherosclerosis inhibition by bezafibrate in low-density lipoprotein receptor knockout mice. Hypertens Res. 2008;31:999–1005. doi: 10.1291/hypres.31.999. [DOI] [PubMed] [Google Scholar]
- 51.Lalloyer F, et al. Peroxisome proliferator-activated receptor-alpha gene level differently affects lipid metabolism and inflammation in apolipoprotein E2 knock-in mice. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1573–1579. doi: 10.1161/ATVBAHA.110.220525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nature reviews. Gastroenterology & hepatology. 2010;7:251–264. doi: 10.1038/nrgastro.2010.41. [DOI] [PubMed] [Google Scholar]
- 53.Machado MV, et al. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One. 2015;10:e0127991. doi: 10.1371/journal.pone.0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kennedy DJ, et al. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am J Physiol Renal Physiol. 2008;294:F450–454. doi: 10.1152/ajprenal.00472.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan Y, et al. Ouabain-stimulated trafficking regulation of the Na/K-ATPase and NHE3 in renal proximal tubule cells. Molecular and cellular biochemistry. 2012;367:175–183. doi: 10.1007/s11010-012-1331-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haller ST, et al. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am J Hypertens. 2012;25:690–696. doi: 10.1038/ajh.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barron N, et al. Engineering CHO cell growth and recombinant protein productivity by overexpression of miR-7. Journal of biotechnology. 2011;151:204–211. doi: 10.1016/j.jbiotec.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 58.A DM, A H, A D, F S, K S. Effective Factors on Patients’ Satisfaction with Emergency Care Services using Factor Analysis: A Cross Sectional Study. Journal of clinical and diagnostic research: JCDR. 2014;8:XC01–XC04. doi: 10.7860/JCDR/2014/8236.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.