

A potentially universal method for the de novo solution of the crystal structures of membrane proteins is described.
Keywords: Membrane protein, x-ray crystallography, experimental phasing, serial crystallography, SAD, iodide
Abstract
We describe a fast, easy, and potentially universal method for the de novo solution of the crystal structures of membrane proteins via iodide–single-wavelength anomalous diffraction (I-SAD). The potential universality of the method is based on a common feature of membrane proteins—the availability at the hydrophobic-hydrophilic interface of positively charged amino acid residues with which iodide strongly interacts. We demonstrate the solution using I-SAD of four crystal structures representing different classes of membrane proteins, including a human G protein–coupled receptor (GPCR), and we show that I-SAD can be applied using data collection strategies based on either standard or serial x-ray crystallography techniques.
INTRODUCTION
Membrane proteins (MPs) are the key functional components of cell membranes. They carry out the main functions of cells including ion and solute transport and energy and signal transduction. MPs represent roughly one-third of the proteins encoded in any genome, including the human one (1). However, although they are also extremely important drug targets [up to 60% of all existing drug targets are MPs (2)], they still remain poorly structurally characterized compared to soluble proteins, mainly because their production and crystallization are two major bottlenecks of structural biology. Structural investigations by x-ray crystallography thus remain a significant challenge. This challenge becomes even more demanding if the crystal structure cannot be solved by molecular replacement (MR) methods. In these cases, the most popular approaches are the use of heavy atom derivatization (3), the substitution of methionine with selenomethionine (4), and native sulfur single-wavelength anomalous diffraction (SAD) phasing (5, 6). However, these techniques can be expensive and hazardous and are often not very efficient (6–8). A reliable, fast, and easy method of preparing these derivatives would thus be a huge step forward in the production of MP crystal structures.
Several reports have shown halide-SAD to be a promising method for phasing the crystal structures of water-soluble proteins (9, 10), and more recently, iodide-SAD (I-SAD) has been used to solve the structure of an MP (7). The distribution of positively charged amino acids at, or close to, the membrane hydrophobic-hydrophilic interface is considered to be a major determinant of the transmembrane topology of integral MPs (11) and is one of their most universal features. In this context, the “positive-inside” rule suggests that regions of polytopic (multispanning) MPs facing the cytoplasm are enriched with arginine, lysine, and, to a lesser extent, histidine amino acid residues (11–13). In addition, there are also observations of an enrichment of tryptophan and tyrosine residues at the membrane surface interfaces (14). From an electrostatic point of view, the existence of uncompensated charge in close proximity to a medium with lower dielectric constant (for example, the membrane hydrophobic-hydrophilic interface) is unfavorable, and to compensate for this, the positively charged residues placed at the interface are likely to interact particularly strongly with bulk halide ions. Thus, we hypothesized that halide-SAD might be a universal phasing method in x-ray MP crystallography.
To test this hypothesis and to develop protocols for halide-SAD phasing, we selected four target representatives for different large classes of MPs varying in the sizes of their membrane and soluble components: the recently discovered and characterized light-driven sodium pump from the marine bacterium Krokinobacter eikastus (KR2) (15), the light-driven proton pump from marine actinobacterial clade rhodopsins (MACRs) (16), a fragment of histidine protein kinase NarQ from Escherichia coli (17), and a human adenosine A2A G protein–coupled receptor (GPCR) (18). We attempted to solve their crystal structures by preparing iodide or bromide derivatives via the cryo-soaking of native crystals in solutions of either NaI (I-SAD) or NaBr (Br-SAD). The results we report here suggest that I-SAD is an efficient, fast, nontoxic, and potentially universal technique for the de novo (that is, not MR-based) solution of MP crystal structures that will help to improve throughput in MP structure determination.
RESULTS
Iodide–single-wavelength anomalous diffraction
Diffraction data sets were collected from several different crystals of each of the target proteins after first soaking them in precipitant solution supplemented with 0.5 M NaI (Table 1). In all cases, diffraction data were collected using x-rays of λ = 1.85 Å, where f″ for iodine/iodide = 9.2 e− (6.7 keV; all values of f″ quoted are taken from http://skuld.bmsc.washington.edu/scatter/). For each target, several individual data sets were merged to obtain a single high-redundancy data set for use in structure solution (Table 1). In all four cases, I-SAD phasing of the crystal structure was straightforward (Fig. 1 and Table 2), providing experimental phases that allowed the automated building of initial models, which could be further refined. By including native data sets, we could also test the efficiency of I-SIRAS (single isomorphous replacement with anomalous scattering) phasing of all four crystal structures. This approach (Fig. 2) provided even higher contrast solutions than I-SAD protocols, suggesting that introduction of iodide ions into the crystals had not resulted in significant non-isomorphism. In addition to the above methods, in experiments based on the serial x-ray crystallography MeshAndCollect pipeline (19), we also collected a succession of partial data sets from a series of iodide-soaked crystals of KR2, each with a maximum dimension of ~10 to 15 μm (Table 3 and Fig. 3). Here, structure solution was also successful using either I-SAD or I-SIRAS (Fig. 3), suggesting that large crystals are not necessary for the collection of high-quality I-SIRAS/I-SAD diffraction data for the solution of MP crystal structures.
Table 1. I-SAD/I-SIRAS data collection statistics.
Entries in parentheses represent values for the highest-resolution bin. All statistics and data quality indicators are calculated, treating Friedel’s pairs as separate reflections. Linear merging R value ; redundancy-independent merging R value ; CC1/2—correlation coefficient between intensities from random half–data sets; .
|
Protein/ space group |
Crystal/ data set |
Unit cell dimensions (Å, o) |
Resolution range (Å) |
Multiplicity |
Completeness (%) |
Rmerge (%) |
Rmeas (%) |
<I/σ(I)> |
CC1/2 (%) |
SigAno |
CCanom (%) |
SigAnoinner |
CCanom_inner (%) |
|||||
| a | b | c | α | β | γ | |||||||||||||
| NarQ F222 |
Native | 57.58 | 73.87 | 235.56 | 90 | 90 | 90 | 60–2.0 (2.1–2.0) |
3.61 (3.50) | 99.8 (99.7) | 8.8 (79.7) |
10.4 (94.2) |
10.58 (1.58) |
99.9 (62.3) |
0.76 (0.73) |
−3 (−4) | 0.68 | −4 |
| 1 | 57.27 | 73.64 | 236.15 | 90 | 90 | 90 | 60−2.9 (3.1−2.9) |
6.69 (5.99) | 99.7 (99.1) | 15.8 (77.1) |
17.2 (84.6) |
10.23 (2.45) |
99.8 (87.5) |
1.24 (0.88) |
47 (26) | 4.12 | 92 | |
| 2 | 57.40 | 73.79 | 237.02 | 90 | 90 | 90 | 60−3.3 (3.5−3.3) |
6.73 (6.78) | 99.7 (99.8) | 20.8 (75.9) |
22.6 (82.2) |
7.33 (2.36) |
99.6 (86.1) |
1.15 (0.76) |
41 (8) | 3.17 | 90 | |
| 3 | 57.22 | 73.59 | 236.42 | 90 | 90 | 90 | 60−3.1 (3.3−3.1) |
6.74 (6.53) | 99.7 (99.7) | 14.2 (62.0) |
15.4 (67.5) |
9.87 (3.20) |
99.8 (88.8) |
1.43 (1.08) |
57 (32) | 4.03 | 93 | |
| 4 | 56.92 | 73.20 | 236.25 | 90 | 90 | 90 | 60−2.9 (3.1−2.9) |
6.69 (6.69) | 99.7 (99.8) | 11.3 (50.3) |
12.3 (54.6) |
12.37 (3.30) |
99.8 (93.3) |
1.57 (0.78) |
64 (5) | 5.82 | 96 | |
| 5 | 57.21 | 73.52 | 236.54 | 90 | 90 | 90 | 60−2.7 (2.9−2.7) |
6.69 (6.59) | 99.8 (99.8) | 11.4 (73.3) |
12.4 (79.6) |
11.93 (2.47) |
99.9 (85.3) |
1.51 (0.82) |
61 (16) | 6.03 | 97 | |
| 6 | 57.12 | 73.34 | 236.74 | 90 | 90 | 90 | 60−2.9 (3.1−2.9) |
6.72 (6.52) | 99.4 (99.5) | 11.3 (67.4) |
12.3 (73.2) |
12.20 (2.67) |
99.9 (87.1) |
1.53 (0.80) |
60 (1) | 5.85 | 96 | |
| 7 | 56.99 | 73.12 | 236.07 | 90 | 90 | 90 | 60−2.9 (3.1−2.9) |
6.73 (6.68) | 98.9 (98.4) | 11.1 (71.4) |
12.0 (77.4) |
12.56 (2.51) |
99.7 (82.5) |
1.57 (0.80) |
62 (8) | 6.00 | 97 | |
| 8 | 57.24 | 73.66 | 236.43 | 90 | 90 | 90 | 60−2.7 (2.9−2.7) |
6.73 (6.68) | 99.8 (99.8) | 12.6 (71.2) |
13.6 (77.4) |
11.03 (2.67) |
99.9 (86.8) |
1.41 (0.94) |
54 (16) | 5.15 | 96 | |
| Merged | 57.17 | 73.48 | 236.45 | 90 | 90 | 90 | 60−2.7 (2.9−2.7) |
42.95 (13.26) | 99.9 (99.8) | 17.5 (76.0) |
17.7 (79.1) |
20.68 (3.12) |
99.8 (91.7) |
2.26 (0.85) |
78 (17) | 10.14 | 98 | |
| KR2 I222 |
Native | 40.62 | 83.66 | 234.46 | 90 | 90 | 90 | 60−2.5 (2.65−2.5) |
6.58 (6.45) | 99.7 (99.6) | 9.6 (85.9) |
10.5 (93.7) |
13.73 (2.10) |
99.8 (81.2) |
0.84 (0.81) |
15 (19) | 1.42 | 44 |
| 1 | 40.50 | 83.66 | 234.13 | 90 | 90 | 90 | 60−2.8 (3.0−2.8) |
6.85 (6.72) | 99.7 (99.8) | 13.5 (83.0) |
14.6 (89.9) |
11.87 (2.44) |
99.8 (80.7) |
1.22 (0.80) |
41 (10) | 6.76 | 97 | |
| 2 | 40.52 | 83.63 | 233.80 | 90 | 90 | 90 | 60−2.8 (3.0−2.8) |
6.82 (6.71) | 99.8 (99.7) | 13.7 (77.0) |
14.8 (83.5) |
11.92 (2.60) |
99.6 (87.2) |
1.24 (0.79) |
44 (7) | 7.26 | 98 | |
| 3 | 40.51 | 83.75 | 234.10 | 90 | 90 | 90 | 60−2.5 (2.65−2.5) |
6.73 (6.60) | 99.9 (100) | 11.3 (75.4) |
12.3 (82.0) |
11.95 (2.35) |
99.9 (79.7) |
1.22 (0.76) |
42 (0) | 7.64 | 98 | |
| 4 | 40.72 | 83.99 | 234.21 | 90 | 90 | 90 | 60−2.65 (2.8−2.65) |
6.71 (6.40) | 99.8 (99.7) | 10.6 (64.0) |
11.5 (69.7) |
12.70 (2.66) |
99.6 (89.0) |
1.25 (0.77) |
46 (5) | 6.96 | 98 | |
| Merged | 40.56 | 83.76 | 234.06 | 90 | 90 | 90 | 60−2.5 (2.65−2.5) |
22.06 (6.60) | 100 (100) | 13.7 (75.1) |
14.0 (81.6) |
19.43 (2.30) |
99.9 (80.0) |
1.64 (0.75) |
60 (1) | 13.23 | 99 | |
| MACR P1 |
Native | 40.76 | 56.85 | 57.65 | 63.6 | 79.0 | 80.1 | 60−2.0 (2.2−2.0) |
1.05 (1.07) | 76.2 (73.1) | 7.3 (26.7) |
10.3 (37.9) |
6.43 (1.81) |
99.5 (86.2) |
n/a | n/a | n/a | n/a |
| 1 | 40.77 | 56.89 | 57.28 | 63.8 | 78.6 | 80.2 | 60−2.2 (2.4−2.2) |
1.86 (1.81) | 70.1 (23.0) | 5.1 (27.2) |
7.2 (38.5) |
10.40 (2.61) |
99.5 (87.0) |
1.16 (0.88) |
43 (8) | 3.61 | 91 | |
| 2 | 40.75 | 56.87 | 57.32 | 63.6 | 78.4 | 80.2 | 60−2.4 (2.6−2.4) |
1.85 (1.79) | 72.5 (20.0) | 6.4 (44.9) |
9.0 (63.4) |
7.86 (1.55) |
99.5 (72.4) |
1.07 (0.87) |
36 (22) | 3.14 | 89 | |
| 3 | 40.79 | 56.77 | 57.29 | 63.6 | 78.5 | 80.2 | 60−2.0 (2.2−2.0) |
1.88 (1.84) | 81.9 (72.3) | 5.1 (38.3) |
7.2 (54.2) |
8.64 (1.86) |
99.7 (70.4) |
1.07 (0.76) |
35 (−5) | 3.49 | 90 | |
| Merged | 40.77 | 56.84 | 57.30 | 63.7 | 78.5 | 80.2 | 60−2.0 (2.2−2.0) |
3.68 (1.84) | 89.4 (71.8) | 7.9 (38.7) |
9.0 (54.8) |
11.03 (1.81) |
99.7 (69.5) |
1.22 (0.75) |
46 (−5) | 4.85 | 95 | |
| A2AAR-BRIL-ΔC C2221 |
Native | 39.62 | 179.54 | 139.71 | 90 | 90 | 90 | 60−2.4 (2.6−2.4) |
2.94 (2.98) | 99.6 (99.8) | 9.4 (41.1) |
11.5 (50.2) |
9.60 (2.74) |
99.5 (80.4) |
0.77 (0.77) |
−4 (−1) | 0.74 | −14 |
| 1 | 39.63 | 179.66 | 139.57 | 90 | 90 | 90 | 60−3.4 (3.6−3.4) |
6.93 (6.96) | 100 (100) | 24.8 (73.6) |
26.8 (79.5) |
7.33 (2.56) |
99.1 (81.2) |
0.96 (0.79) |
17 (0) | 1.94 | 71 | |
| 2 | 39.65 | 179.79 | 139.81 | 90 | 90 | 90 | 60−2.8 (3.0−2.8) |
6.87 (6.87) | 99.7 (99.0) | 18.5 (78.1) |
20.0 (84.5) |
8.83 (2.29) |
99.4 (76.0) |
0.98 (0.76) |
18 (−5) | 2.57 | 82 | |
| 3 | 39.40 | 179.87 | 139.52 | 90 | 90 | 90 | 60−3.6 (3.8−3.6) |
6.67 (6.54) | 99.9 (99.8) | 23.5 (72.1) |
25.5 (78.5) |
8.45 (3.21) |
99.2 (82.8) |
1.13 (1.08) |
46 (41) | 1.96 | 72 | |
| 4 | 39.57 | 179.76 | 139.99 | 90 | 90 | 90 | 60−3.8 (4.0−3.8) |
6.83 (6.79) | 99.9 (99.9) | 30.7 (84.7) |
33.3 (91.9) |
6.67 (2.97) |
98.6 (72.0) |
0.96 (0.91) |
19 (8) | 1.59 | 63 | |
| 5 | 39.60 | 179.10 | 139.42 | 90 | 90 | 90 | 60−3.0 (3.2−3.0) |
3.02 (2.98) | 98.9 (99.0) | 14.5 (65.7) |
17.6 (80.1) |
7.06 (1.67) |
98.7 (56.6) |
0.93 (0.75) |
16 (−3) | 2.06 | 74 | |
| 6 | 39.50 | 179.46 | 139.40 | 90 | 90 | 90 | 60−3.4 (3.6−3.4) |
6.85 (6.52) | 99.9 (100) | 19.9 (69.1) |
21.5 (75.2) |
9.61 (3.20) |
99.4 (80.6) |
1.07 (0.89) |
27 (5) | 2.46 | 81 | |
| 7 | 39.63 | 179.65 | 139.96 | 90 | 90 | 90 | 60−3.2 (3.4−3.2) |
6.27 (5.83) | 99.9 (99.7) | 21.1 (68.3) |
23.1 (75.1) |
8.86 (2.61) |
98.9 (84.4) |
1.02 (0.92) |
24 (12) | 2.06 | 76 | |
| Merged | 39.57 | 179.61 | 139.67 | 90 | 90 | 90 | 60−2.8 (3.0−2.8) |
27.10 (6.88) | 99.8 (99.1) | 24.5 (77.6) |
24.9 (83.9) |
14.55 (2.25) |
99.8 (76.1) |
1.24 (0.73) |
35 (−3) | 4.83 | 94 | |
Fig. 1. I-SAD phasing of MP crystal structures.
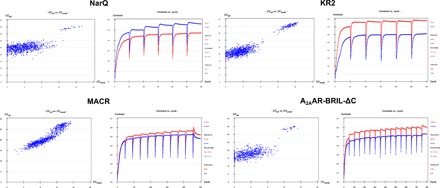
Heavy atom substructure determination (SHELXD, left) and phase calculation and extension (SHELXE, right) for each of the four targets investigated. In all four cases, plots of CCall versus CCweak (1000 trials) from SHELXD show a bimodal distribution indicative of a correct iodide ion substructure solution, whereas the difference in contrast (SHELXE) between the two possible hands (red, original; blue, inverted) indicates successful structure solution.
Table 2. I-SAD structure solution and refinement statistics.
| Protein | ||||||||
| NarQ | KR2 | MACR | A2AAR-BRIL-ΔC | |||||
| Structure solution and initial model building | ||||||||
| Data | Eight anomalous + native |
Data set #8 + native |
Four anomalous + native |
Three anomalous + native |
Seven anomalous + native |
|||
| Method | I-SAD | I-SIRAS | I-SAD | I-SIRAS | I-SAD | I-SIRAS | I-SAD | I-SIRAS |
| Initial resolution range (Å) | 60−2.7 | 60−2.7 | 60−2.5 | 60−2.5 | 60−2.0 | 60−2.0 | 60−2.8 | 60−2.8 |
| Number of residues in the protein |
237 | 288 | 220 | 447 | ||||
| Substructure | 14·I | 10·I | 16·I | 17·I | 26·I† 14·I |
16·I | 8·I | 10·I |
| Resolution cutoff for substructure search (Å) |
3.0 | 2.8 | 2.8 | 2.8 | 2.5 | 2.9 | 3.7 | 3.9 |
| Resolution extension by native data (Å) |
1.95 | 1.95 | 2.5 | 2.5 | 2.0 | 2.0 | 2.4 | 2.4 |
| CCall/CCweak | 40/15.4 | 31.8/18.1 | 42.6/20.7 | 37.7/25.1 | 28.8/15.3 | 29.8/19.6 | 35.0/14.8 | 36.7/21.4 |
| Solvent content used in SHELXE (%) |
47 | 47 | 58 | 59 | 50 | 50 | 49 | 49 |
| CCmap, highest-resolution shell (%) | 74 | 77 | 67 | 84 | 87 | 81 | 73 | 74 |
| Number of Ala residues traced by SHELXE |
178 | 187 | 212 | 208 | 190 | 272 | 155 | 250 |
| Rwork/Rfree after initial model building (%) |
23.0/28.5* | 24.8/32.9* | 22.4/30.1* | 22.6/29.3* | 24.2/29.2†‡ 29.4/32.7‡ | 29.7/46.2* 29.9/32.7‡ | 30.7/32.9‡ | 29.5/45.8* |
| Number of residues built in initial model building |
215* | 216* | 235* | 265* | 343†‡ 300‡ | 69* 319‡ | 277‡ | 168* |
| Structure refinement | ||||||||
| Data set (see Table 1) | 8 | 3 | 3 | 2 | ||||
| Rwork/Rfree final (%) | 19.0/26.2 | 19.0/22.6 | 17.7/21.9 | 23.4/29.4 | ||||
| Number in the ASU (asymmetric unit) | ||||||||
| Protein residues | 223 | 268 | 431 | 388 | ||||
| Water molecules | 60 | 49 | 78 | 64 | ||||
| Iodide ions | 12 | 20 | 22 | 6 | ||||
| Averaged B-factors (Å2) | ||||||||
| Protein residues | 43 | 40 | 38 | 38 | ||||
| Water molecules | 34 | 44 | 47 | 27 | ||||
| Iodide ions | 70 | 66 | 71 | 50 | ||||
| Ramachandran plot | ||||||||
| Preferred | 216 (99.1%) | 259 (97.4%) | 419 (98.4%) | 373 (97.9%) | ||||
| Allowed | 2 (0.9%) | 6 (2.2%) | 7 (1.6%) | 7 (1.8%) | ||||
| Outliers | 0 | 1 (0.4%) | 0 | 1 (0.3%) | ||||
*Initial model built by ARP-wARP.
†Phasing carried out using phenix.autosol (all others used SHELXC/D/E).
‡Initial model built by phenix.autobuild.
Fig. 2. I-SIRAS phasing of MP crystal structures.
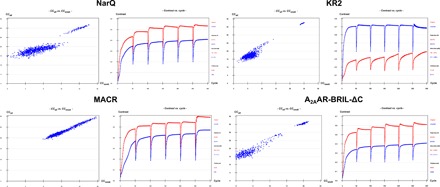
Heavy atom substructure determination (SHELXD, left) and phase calculation and extension (SHELXE, right) for I-SIRAS phasing for each of the four targets investigated. As might be expected, I-SIRAS produces both clearer substructure solutions and better contrast in electron density maps phased using the two possible hands (see Fig. 1 for a comparison).
Table 3. Data collection, structure solution, and initial model building statistics resulting from I-SAD/I-SIRAS phasing of the crystal structure of KR2 using diffraction data collected using serial methods.
| I-SAD | I-SIRAS | I-SAD | I-SIRAS | |||||
| Data collection |
No. of crystals | 136 | 39 | Structure solution and Refinement |
Initial resolution range (Å) |
60–2.8 | 60–2.9 | |
| Method | SAD | SIRAS | Number of residues in the protein |
288 | 288 | |||
| Space group | I222 | I222 | Substructure | 16·I | 12·I | |||
| Unit cell | a | 40.6 | 41.0 | Resolution cutoff for substructure search (Å) |
3.9 | 3.4 | ||
| b | 83.6 | 84.0 | Resolution extension by native data (Å) |
2.5 | 2.5 | |||
| c | 233.5 | 234.3 | CCall/CCweak | 38.7/11.0 | 26.7/9.6 | |||
| No. of frames | 13,600 | 3900 | Solvent content (%) |
63 | 60 | |||
| Oscillation range (°) |
0.1 | 0.1 | CCmap, highest-resolution shell (%) |
47 | 67 | |||
| Resolution range (Å) |
60–2.8 (3.0–2.8) | 60–2.9 (3.1–2.9) | Number of Ala residues traced by SHELXE |
122 | 165 | |||
| Measured reflections |
469,213 (85,461) | 124,613 (22,610) |
Rwork/Rfree after initial model building (%) |
29.6/35.7† | 24.2/31.6* 29.9/35.4† |
|||
| Multiplicity | 24.79 (24.13) | 7.19 (7.13) | Number of residues built in initial model building |
207† | 248* 196† |
|||
| Completeness (%) | 100 (100) | 99.8 (100) | ||||||
| Rmerge (%) | 61.7 (220.7) | 23.8 (154.6) | ||||||
| Rmeas (%) | 63.0 (225.4) | 25.6 (166.6) | ||||||
| <I/σ(I)> | 10.64 (2.27) | 7.71 (1.69) | ||||||
| CC1/2 (%) | 99.8 (70.1) | 99.3 (56.2) | ||||||
| SigAno | 1.21 (0.78) | 1.03 (0.73) | ||||||
| CCanom (%) | 39 (0) | 29 (−1) |
*Model built by ARP-wARP.
†Model built by phenix.autobuild.
Fig. 3. I-SAD/I-SIRAS phasing of the crystal structure of KR2 using diffraction data collected using serial methods.
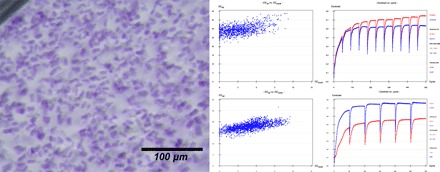
Left: Photograph of crystals, 10 to 15 μm in maximum dimensions, of KR2 in its monomeric blue form. Top right: Heavy atom substructure determination (SHELXD, left) and phase calculation and extension (SHELXE, right) for I-SAD phasing using data collected via serial methods. Bottom right: Heavy atom substructure determination (SHELXD, left) and phase calculation and extension (SHELXE, right) for I-SIRAS phasing using data collected via serial methods.
The positions of the iodide binding sites for all four target proteins are shown in Fig. 4. Iodide binding sites (12, 20, 22, and 6), reproduced in most of the individual data sets for each target (fig. S1), are seen for NarQ, KR2, MACR, and A2AAR-BRIL-ΔC, respectively. As can be seen, the highest-occupancy (as estimated from anomalous difference Fourier maps) sites are clustered close to the predicted membrane interfaces. In the same way as was previously observed in the I-SAD crystal structure solution of soluble proteins (9, 10), the iodide ions interact with a wide variety of positively charged amino acid residues, ordered water molecules, and hydrophobic side chains (Fig. 4 and fig. S3).
Fig. 4. Cartoon representations of the crystal structures of the four classes of MP solved using the I-SAD/I-SIRAS technique.
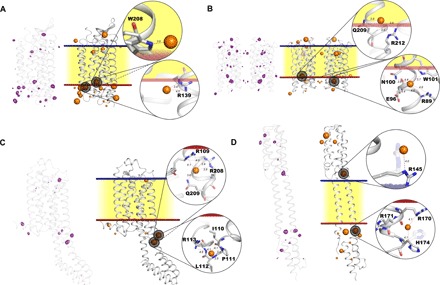
(A) KR2. (B) MACR (noncrystallographic dimer). (C) A2AAR-BRIL-ΔC. (D) NarQ. On the left of each representation, peaks in I-SAD anomalous difference Fourier maps [purple chicken wire, highest-resolution I-SAD data sets for each target (Table 1), contoured at the 3.5 × r.m.s. (root mean square) level] are shown superposed on the Cα backbone of the protein. On the right of each panel, the blue and red lines represent outer and inner lipidic membrane surfaces, respectively, with the hydrophobic region of the lipidic membrane represented in yellow. Iodide ions are shown as orange spheres, divided into three sizes based on the height of anomalous difference map peaks. Two iodide binding sites per crystal structure are highlighted to illustrate their environment. In (D), it is clear that protein residues may change their side-chain conformation upon binding of iodide (conformation in native structures shown as shadows).
To examine the potential universality of the I-SAD method in the de novo solution of MP crystal structures, we examined the availability at the membrane interface of five amino acid residues, namely, arginine, lysine, histidine, tryptophan, and tyrosine in 445 unique transmembrane protein structures deposited in the Protein Data Bank [PDB; www.rcsb.org (20); the database of unique MP structures: http://blanco.biomol.uci.edu/mpstruc/]. According to our analysis (Table 4), 96% of the structures contain aromatic amino acids close to the membrane interface, and 100% of the structures contain positively charged amino acids in this region. This, coupled with the recent report of the crystal structure of the outer membrane β-barrel assembly machinery complex, solved by I-SAD phasing (7), suggests that I-SAD could be a universal technique for the solution of MP crystal structures.
Table 4. The analysis of presence at the membrane hydrophobic-hydrophilic interface of arginine (R), lysine (K), histidine (H), tryptophan (W), and tyrosine (Y).
As outlined in the main text, 445 unique transmembrane protein crystal structures deposited in the PDB were analyzed with, in each case, the membrane surfaces represented by two parallel planes calculated by Lomize et al. (42). Amino acid residues were considered to be present at the membrane interfaces if its Cα atom was not more than 7 Å from the membrane surfaces defined above.
| R/K/H | R/K | W/Y | R | K | H | W | Y | |
| Number of entries where the amino acid residues are present at the membrane interface |
445 (100%) |
442 (99.3%) |
428 (96.2%) |
429 (96.4%) |
414 (93.0%) |
353 (79.3%) |
393 (88.3%) |
422 (94.8%) |
Bromide–single-wavelength anomalous diffraction
To test whether derivatization with bromide might be an alternative method to the I-SAD/I-SIRAS approach described above, we soaked crystals of NarQ and KR2 in precipitant solutions supplemented with 0.5 M NaBr and collected diffraction data sets as presented in Table 5. For both proteins, despite constructing merged data sets with multiplicities greater than 20, all attempts at Br-SAD/Br-SIRAS structure solution were unsuccessful regardless of whether diffraction data were collected far from (NarQ, λ = 0.873 Å; E = 14.2 keV; f″ = 3.4 e−) or close to the peak of the Br K-absorption edge (KR2, λ = 0.920 Å; E = 13.476 keV; f″ = 3.8 e−).
Table 5. Br-SAD data collection statistics.
Entries in parentheses represent values for the highest-resolution bin. All statistics and data quality indicators are calculated treating Friedel’s pairs as separate reflections. Linear merging R value ; redundancy-independent merging R value ; CC1/2—correlation coefficient between intensities from random half–data sets; .
|
Protein/ space group |
Crystal/ data set |
λ (Å) | Unit cell dimensions (Å, o) | ||||||||||||||||
| a | b | c | α | β | γ |
Resolution (Å) |
Multiplicity |
Completeness (%) |
Rmerge (%) |
Rmeas (%) |
<I/σ(I)> |
CC1/2 (%) |
SigAno |
CCanom (%) |
SigAnoinner |
CCanom_inner (%) |
|||
| NarQ F222 |
1 | 0.8729 | 57.87 | 74.79 | 237.85 | 90 | 90 | 90 | 60–2.8 (3.0–2.8) |
2.09 (2.11) | 83.2 (45.2) | 14.5 (105.4) |
18.2 (132.8) |
5.53 (1.21) |
99.4 (47.7) |
0.82 (0.71) |
6 (10) | 1.12 | 37 |
| 2 | 0.8729 | 57.70 | 74.34 | 237.70 | 90 | 90 | 90 | 60–2.8 (3.0–2.8) |
4.03 (4.12) | 98.2 (92.6) | 18.1 (106.9) |
20.8 (122.7) |
6.99 (1.36) |
99.5 (58.3) |
0.83 (0.74) |
8 (3) | 1.56 | 55 | |
| 3 | 0.8729 | 58.21 | 73.57 | 237.42 | 90 | 90 | 90 | 60–2.8 (3.0–2.8) |
4.81 (4.87) | 99.2 (99.2) | 21.3 (105.1) |
24.0 (117.9) |
5.85 (1.43) |
99.5 (65.2) |
0.82 (0.67) |
13 (1) | 1.27 | 45 | |
| 14 | 0.8729 | 57.53 | 73.91 | 237.23 | 90 | 90 | 90 | 60–2.6 (2.8–2.6) |
4.31 (4.06) | 93.2 (68.4) | 15.7 (101.1) |
17.9 (116.5) |
7.38 (1.26) |
99.5 (60.1) |
0.86 (0.71) |
11 (3) | 1.50 | 61 | |
| 15 | 0.8729 | 57.52 | 74.44 | 237.76 | 90 | 90 | 90 | 60–2.8 (3.0–2.8) |
2.58 (2.40) | 93.5 (87.6) | 14.0 (59.6) |
17.5 (74.6) |
5.83 (1.47) |
98.9 (66.1) |
0.85 (0.74) |
18 (−2) | 1.14 | 56 | |
| 16 | 0.8729 | 57.76 | 74.22 | 238.68 | 90 | 90 | 90 | 60–2.6 (2.8–2.6) |
2.89 (2.98) | 97.4 (98.4) | 10.8 (73.5) |
13.0 (88.8) |
8.13 (1.55) |
99.6 (59.7) |
0.87 (0.75) |
9 (2) | 1.80 | 74 | |
| 24 | 0.8729 | 57.51 | 74.20 | 237.26 | 90 | 90 | 90 | 60–2.2 (2.4–2.2) |
2.31 (2.22) | 91.6 (76.8) | 11.4 (125.6) |
14.5 (160.1) |
5.85 (0.62) |
99.6 (69.2) |
0.83 (0.65) |
12 (4) | 1.69 | 79 | |
| Merged | 57.73 | 74.21 | 237.70 | 90 | 90 | 90 | 60–2.8 (3.0–2.8) |
22.3 (21.3) | 94.2 (100) | 19.7 (85.9) |
20.1 (88.0) |
17.3 (5.0) |
99.9 (96.6) |
0.98 (0.75) |
20 (3) | 2.77 | 90 | ||
| KR2 I222 |
32 | 0.91968 | 40.67 | 83.73 | 234.46 | 90 | 90 | 90 | 60–2.8 (3.0–2.8) |
6.68 (6.45) | 89.4 (44.7) | 24.0 (176.7) |
26.1 (191.6) |
7.44 (1.11) |
99.6 (49.4) |
0.97 (0.65) |
43 (3) | 1.75 | 67 |
| 33 | 0.91968 | 40.78 | 83.62 | 234.38 | 90 | 90 | 90 | 60–3.0 (3.2–3.0) |
6.82 (7.05) | 100 (100) | 26.3 (159.7) |
28.6 (172.5) |
7.43 (1.33) |
99.6 (47.5) |
0.92 (0.70) |
21 (−8) | 1.87 | 71 | |
| 37 | 0.91968 | 40.67 | 83.32 | 234.10 | 90 | 90 | 90 | 60–2.8 (3.0–2.8) |
6.77 (6.64) | 99.5 (99.2) | 16.1 (118.5) |
17.4 (128.7) |
9.85 (1.61) |
99.8 (78.1) |
0.88 (0.72) |
17 (7) | 1.69 | 67 | |
| 38 | 0.91968 | 40.58 | 83.31 | 234.04 | 90 | 90 | 90 | 60–2.8 (3.0–2.8) |
6.85 (7.05) | 99.6 (99.4) | 14.4 (103.8) |
15.6 (112.3) |
10.06 (1.73) |
99.8 (84.9) |
0.90 (0.76) |
19 (7) | 1.75 | 69 | |
| 39 | 0.91968 | 40.56 | 83.46 | 233.45 | 90 | 90 | 90 | 60–3.0 (3.2–3.0) |
6.74 (6.54) | 96.1 (79.3) | 17.0 (121.3) |
18.4 (131.7) |
9.52 (1.35) |
99.9 (74.9) |
0.90 (0.73) |
21 (14) | 1.65 | 61 | |
| Merged | 40.65 | 83.49 | 234.09 | 90 | 90 | 90 | 60–2.8 (3.0–2.8) |
31.56 (23.48) | 100 (100) | 23.9 (166.0) |
24.3 (169.7) |
16.72 (2.49) |
99.9 (85.7) |
1.03 (0.74) |
23 (0) | 2.63 | 80 | ||
DISCUSSION
The results outlined above suggest that I-SAD is a potentially universal method for the de novo solution of MP crystal structures. Further advantages of I-SAD are speed, nontoxicity, and simplicity because soaking can be easily done at the crystal-harvesting stage and does not require any additional equipment or safety precautions. In addition, iodide soaking does not, at least in the experiments described here, appreciably disturb crystal quality. Another—not insignificant—advantage lies in the anomalous scattering properties of iodide itself. Although anomalous signals in I-SAD experiments would be optimized by targeting the peak of the L1 absorption edge of iodine (f″ ~ 13.4 e−; λ = 2.4 Å; E = 5.19 keV), routinely accessing the photon energy required is not always possible even on tuneable synchrotron macromolecular crystallography beamlines. However, at λ = 1.85 Å, a wavelength usually accessible on these beamlines, the anomalous scattering properties of iodide are still significant (f″ = 9.2 e−), and in many cases, particularly because our studies suggest the presence of several iodide binding sites in each of the different classes of MPs (Fig. 4), experiments at, or close to, this wavelength will be more than sufficient for structure solution. Therefore, we carried out our experiments at λ = 1.85 Å, using a multicrystal approach to produce the highly redundant data (Table 1), which is often required for SAD structure solution (21). Our experiments also show that I-SAD phasing of MP crystal structures using data collected in a serial fashion from many microcrystals mounted on a so-called fixed target is also possible. Considering that often it is hard to grow MP crystals of sufficient size for standard data collection, this latter conclusion is very important, suggesting that serial I-SAD data collection methods might also be successful for MP crystal structure solution using diffraction data collected at x-ray free-electron lasers (22) or at next-generation diffraction-limited storage ring synchrotron sources.
A notable result of the investigations described here is that Br-SAD does not appear to be a routinely viable method for the de novo solution of the crystal structures of MPs. This is intriguing, particularly because Br-SAD is a relatively well-used technique for the solution of the crystal structures of soluble proteins (9). Subsequent analysis of NarQ and KR2 Br-SAD anomalous difference Fourier maps (Fig. 5) revealed peaks indicating both that Br− binds to these proteins and, moreover, that many of the Br− binding sites observed coincide with the sites observed for binding of I− in the corresponding I-SAD experiments. Thus, Br-SAD/Br-SIRAS phasing of MP crystals structures should be possible. However, a major disadvantage of Br-SAD experiments is that the maximum f″ that can be induced around the Br K-absorption edge is ~4 e−. This compares to f″ ~9.2 e− for I-SAD experiments carried out at λ = 1.85 Å. Anomalous signals in Br-SAD experiments will thus be significantly lower than for long-wavelength I-SAD (see Fig. 6 for an illustration of this). Successful MP Br-SAD experiments will thus demand higher-accuracy data than I-SAD. This will generally require higher multiplicity measurements and/or the merging of more individual data sets, and Br-SAD will thus be less conducive to high-throughput MP structure determination.
Fig. 5. Comparison of peaks in anomalous difference Fourier maps in I-SAD and Br-SAD experiments.
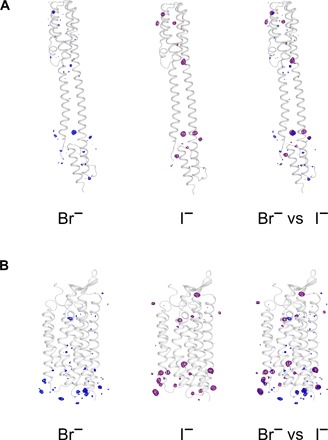
(A) Peaks in anomalous difference Fourier maps in Br-SAD (left) and I-SAD (middle) experiments for NarQ, with a superposition of the two maps shown on the right. (B) Peaks in anomalous difference Fourier maps in Br-SAD (left) and I-SAD (middle) experiments for KR2, with a superposition of the two maps shown on the right. Only the regions close to the proteins are shown, and the maps are contoured at the 3.5 × r.m.s. level. For both NarQ and KR2, the anomalous difference Fourier maps shown are those calculated using anomalous differences in the individual I-SAD data sets of the highest resolution, as detailed in Table 1, and in the merged Br-SAD data sets, as detailed in Table 5.
Fig. 6. Comparison of anomalous signal in I-SAD and Br-SAD diffraction data sets for NarQ (A) and KR2 (B).
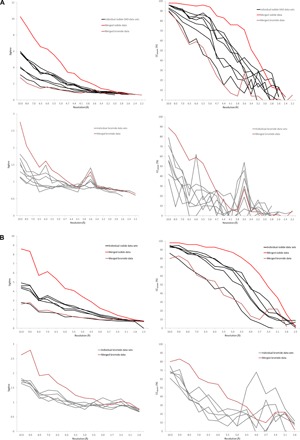
(A) Comparisons of SigAno (left) and CCanom (right) in I-SAD (top panel) and Br-SAD (bottom) panel data sets measured from derivatized NarQ crystals. (B) Comparisons of SigAno (left) and CCanom (right) in I-SAD (top panel) and Br-SAD (bottom) panel data sets measured from KR2 crystals. Individual and merged data sets for I-SAD experiments are as detailed in Table 1, whereas those for Br-SAD are as detailed in Table 5.
MATERIALS AND METHODS
Protein expression and purification
Expression and purification of the sodium-pumping rhodopsin KR2 (UniProt ID N0DKS8) and the chimeric protein A2AAR-BRIL-ΔC of human A2A adenosine receptor (A2AAR) with a thermostabilized apocytochrome b562RIL (UniProt IDs P0ABE7 and P29274) in complex with ZM241385 were performed as described by Gushchin et al. (23) and Liu et al. (24), respectively.
Expression and purification of the nitrate/nitrite sensor kinase NarQ
The nucleotide sequence encoding residues 1 to 230 of the nitrate/nitrite sensor kinase NarQ (17) (UniProt ID P27896) was cloned from E. coli strain BL21 (DE3) and introduced into the pSCodon1.2 expression vector (StabyCodon T7, Eurogentec) via Nde I and Xho I restriction sites. Consequently, the construct harbored a C-terminal His6 tag. NarQ was then expressed in E. coli strain SE1 (StabyCodon T7, Eurogentec). Cells were cultured in shaking baffled flasks in ZYP-5052 auto-inducing medium (25) containing ampicillin (100 mg/liter). After incubating for 2 hours at 37°C, the temperature was decreased to 30°C, and incubation continued overnight. Harvested cells were disrupted in M-110P Lab Homogenizer (Microfluidics) at 25,000 psi in a phosphate-buffered saline buffer with addition of deoxyribonuclease (DNase) I (50 mg/liter; Sigma-Aldrich) and EDTA-free protease inhibitor cocktail (cOmplete, Roche). The membrane fraction of cell lysate was isolated by ultracentrifugation at 90,000g for 1 hour at 4°C. The pellet was resuspended in a buffer containing 50 mM NaH2PO4/Na2HPO4 (pH 8.0), 0.3 M NaCl, and 2% n-dodecyl-β-d-maltoside (DDM) (Anatrace, Affymetrix) and stirred overnight for solubilization. The insoluble fraction was then removed by ultracentrifugation at 90,000g for 1 hour at 4°C. The supernatant was loaded on a gravity flow column containing Ni-NTA resin (Qiagen), and the protein was eluted in a buffer containing 20 mM tris-HCl (pH 8.0), 0.3 M NaCl, 0.2 M imidazole, and 0.1% DDM. Imidazole was then removed by dialysis against 20 mM tris-HCl (pH 8.0), 0.3 M NaCl, and 0.1% DDM for 3 hours. The eluate was subjected to size-exclusion chromatography on a 125-ml Superdex 200 PG column (GE Healthcare Life Sciences) in a buffer containing 20 mM tris-HCl (pH 8.0), 0.3 M NaCl, and 0.1% DDM. Protein-containing fractions were pooled and concentrated to 30 mg/ml for crystallization.
Expression and purification of proton-pumping rhodopsin MACR
The nucleotide sequence encoding Candidatus Actinomarina minuta opsin gene (MACR, UniProt ID S5DM51) (16) was cloned from metagenomic fosmid MedDCM-OCT-S44-C50 without any optimization and was introduced into the pSCodon1.2 vector via Xba I and Bam HI restriction sites. Consequently, the expressed construct harbored an additional C-terminal tag with a sequence PGGGSHHHHHH. E. coli strain SE1 cells were transformed with the pSC-MACR-His6 plasmid. The cells were grown at 37°C in shaking baffled flasks in ZYP-5052 auto-inducing medium containing ampicillin (100 mg/liter). After the glucose level in the growing bacterial culture dropped below 10 mg/liter, 10 μM all-trans-retinal (Sigma-Aldrich) was added, the temperature was reduced to 20°C, and incubation continued overnight. Collected cells were disrupted using the M-110P Lab Homogenizer at 25,000 psi in a buffer containing 20 mM tris-HCl (pH 8.0), 5% glycerol, 0.5% Triton X-100 (Sigma-Aldrich), and DNase I (50 mg/liter). The membrane fraction of cell lysate was obtained by ultracentrifugation at 90,000g for 1 hour at 4°C. The pellets were resuspended in a buffer containing 50 mM NaH2PO4/Na2HPO4 (pH 8.0), 0.1 M NaCl, and 1% DDM. The mixture was left overnight for solubilization. The insoluble fraction was removed by ultracentrifugation at 90,000g for 1 hour at 4°C. The supernatant was loaded on a Ni-NTA column, and the His-tagged protein was eluted in a buffer containing 50 mM NaH2PO4/Na2HPO4 (pH 7.5), 0.1 M NaCl, 0.5 M imidazole, and 0.2% DDM. The eluate was subjected to size-exclusion chromatography on a 125-ml Superdex 200 PG column in a buffer containing 50 mM NaH2PO4/Na2HPO4 (pH 7.5), 0.1 M NaCl, and 0.01% DDM. Protein-containing colored fractions were collected and concentrated to 40 mg/ml for crystallization.
Protein crystallization and iodide soaking
All crystals were grown using the in meso approach used in some of our previous work (23, 24, 26, 27). NarQ, MACR, and KR2 in their respective crystallization buffers (see above) were added to the monoolein-formed lipidic phase (Nu-Chek Prep), whereas the solubilized A2AAR-BRIL-ΔC/ZM241385 complex was mixed with monoolein containing 10% (w/w) cholesterol. The protein-LCP (lipid cubic phase) mixtures (40- to 50-nl aliquots in the case of A2AAR-BRIL-ΔC and 100-nl aliquots in all other cases) were spotted on 96-well LCP glass sandwich plates (Marienfeld) and covered with 800 nl of precipitant solution (see below) using the NT8-LCP crystallization robot (Formulatrix). Crystals were grown at 20°C and reached their final sizes within 2 to 8 weeks.
The best crystals of NarQ were obtained using 0.6 M KH2PO4/Na2HPO4 (pH 4.6) (Qiagen) and 5 mM NaNO3 as the precipitant solution and grew to 50 to 100 μm in size. For KR2, the best crystals were obtained using 2.0 M sodium malonate (pH 4.3) (Hampton Research) as a precipitant solution and grew to 70 to 100 μm in size. The best crystals of MACR were obtained using 2.6 M (NH4)2SO4 and 0.1 M sodium acetate pH 5.2 (Qiagen) as the precipitant and were 100 to 150 μm in size. The best crystals of the A2AAR-BRIL-ΔC/ZM241385 complex were obtained using a precipitant solution comprising 32% polyethylene glycol 400, 25 mM NaSCN, and 0.1 M sodium acetate (pH 5.0) and grew to 40 to 60 μm in size. For serial crystallography SAD experiments, we obtained crystals of KR2 (10 to 15 μm in size) (Fig. 3) using 2.5 M sodium malonate (pH 4.3) (Hampton Research) as the precipitant.
Once crystals reached their final size, crystallization wells were opened as described elsewhere (28), and drops containing the protein-mesophase mixture were covered with 50 μl of the respective precipitant solution. For native data collection (Table 1), harvested crystals were incubated for ~5 min in the respective precipitant solutions. In the case of NarQ, this was supplemented with 20% glycerol as a cryoprotectant. For I-SAD experiments, the incubation solution was supplemented with 0.5 M NaI. For Br-SAD experiments, the incubation solution was supplemented with 0.5 M NaBr. After incubation, crystals were loaded onto MicroMounts and MicroMeshes (MiTeGen), flash-cooled, and stored in liquid nitrogen.
X-ray crystallography
X-ray diffraction data were collected on the European Synchrotron Radiation Facility (ESRF) beamlines ID23-1 (29) or ID23-2 (30) equipped with Dectris pixel array detectors. For I-SAD experiments, diffraction data from crystals soaked in NaI were collected at λ = 1.85 Å. The x-ray beam size at the sample position was varied from 10 to 50 μm in maximum dimension depending on the crystal size. Experimental parameters for optimal data collection were designed using the program BEST (31). Diffraction images were processed with XDS (32) and XSCALE (32), which was also used to merge and scale different data sets (see Table 1 for details). Structure factor amplitudes, anomalous differences (DANO), and FreeR labels were then generated using the CCP4 programs POINTLESS and AIMLESS (33).
The KR2 I-SAD serial data collection was carried out as described by Zander et al. (19) using x-rays of λ = 1.85 Å. Here, 171 partial data sets were successfully processed, and hierarchical cluster analysis (34) was then used to select 136 of these for merging/scaling to produce the final data set. Structure solution and refinement were then carried out as described below for data sets obtained in a “standard” fashion. For the serial crystallography I-SIRAS experiments, the native data set was collected in a standard way, and only 39 of the I-SAD partial data sets were merged to provide the derivative data set.
For Br-SAD experiments, diffraction data from crystals of NarQ soaked in 0.5 M NaBr were collected at λ = 0.872 Å, whereas for KR2 crystals soaked in 0.5 M NaBr, diffraction data were collected at λ = 0.9198 Å. Data processing, merging, and reduction were then carried out as described above for I-SAD experiments (see Table 5 for details). The native data sets were required for both I-SIRAS and Br-SIRAS structure solution protocols and were recorded at either λ = 1.85 Å (KR2 only) or λ = 0.972 Å, with data processing and reduction carried out as described above for I-SAD experiments (see Table 1 for details).
Structure solution and refinement
I-SAD and I-SIRAS phasing protocols were carried out using the SHELXC/D/E pipeline as implemented in HKL2MAP (1000 trials for substructure determination) (35, 36). ARP-wARP (37) or phenix.autobuild (38) was then used for automated model building. Final structural models (Table 2) were obtained by alternating cycles of manual building in Coot (39) with refinement in REFMAC5 (40). Figures illustrating the final models obtained were prepared using PyMOL (41). Anomalous difference Fourier maps were calculated with fast Fourier transform (FFT) (CCP4) using DANO and αcalc + 90o as coefficients (αcalc = calculated phases from the final refined models) and were then used to verify the number, position, and occupancy of iodide ions in the individual and the merged I-SAD data sets (Tables 1 and 2 and fig. S1), with, for each target, only those common to at least half of the data sets included in the final models.
Br-SAD and Br-SIRAS de novo phasing protocols for NarQ and KR2 were also carried out using the SHELXC/D/E pipeline as implemented in HKL2MAP (10,000 trials for substructure determination). However, none of our attempts resulted in successful structure solution. This being the case, structure refinement based on the model obtained from native crystals was carried out using REFMAC5 and Coot. Anomalous difference Fourier maps were then calculated with FFT (CCP4) using DANO and αcalc + 90o as coefficients (αcalc = calculated phases from the final refined models) and were used to verify the number, position, and occupancy of bromide ions in each of the individual and the merged Br-SAD data sets.
Supplementary Material
Acknowledgments
Funding: The work on protein expression, purification and crystallization was supported by the program “Chaires d’excellence” edition 2008 of ANR France, CEA(IBS) – HGF(FZJ) STC 5.1 specific agreement, and used the platforms of the Grenoble Instruct Centre (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). The work on photoactive proteins was supported by Ministry of Education and Science of the Russian Federation (RFMEFI58716X0026). The work on GPCR expression, purification, and crystallization was supported by the Russian Science Foundation (project no. 16-14-10273). I.M. is the recipient of an ESRF PhD Studentship. Author contributions: A.P. and V.G. conceived the research. I.M., V.G., and A.P. prepared the manuscript with the contribution of V.P., G.A.L., and all other authors. K.K., I.G., A.A., and M.S. expressed and purified the proteins. V.S., A.M., and V.C. oversaw the expression and purification. V.P., I.M., and M.S. crystallized the proteins. V.C. supervised GPCR crystallization and data analysis. V.B. and A.M. oversaw the crystallization. I.M., V.P., V.B., M.S., and A.P. collected and analyzed the diffraction data. I.M. and A.P. solved the structures. I.G., A.A., K.K., M.S., and V.C. helped with structure analysis. F.R.-V. contributed to the work on MACR protein production. A.P., G.A.L., and V.G. oversaw the research. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. All diffraction data and refined models have been deposited in the PDB: 5JGP (NarQ), 5JRF (KR2), 5JSI (MACR), and 5JTB (A2AAR-BRIL-ΔC).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602952/DC1
fig. S1. Comparison of peaks in anomalous difference Fourier maps calculated from individual data sets in I-SAD experiment.
fig. S2. The distribution of positively charged and aromatic residues in the crystal structures obtained in I-SAD experiment.
fig. S3. The bound iodide ions and their environment.
REFERENCES AND NOTES
- 1.Wallin E., Von Heijne G., Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 7, 1029–1038 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Overington J. P., Al-Lazikani B., Hopkins A. L., How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Morth J. P., Sørensen T. L., Nissen P., Membrane’s eleven: Heavy-atom derivatives of membrane-protein crystals. Acta Crystallogr. D Biol. Crystallogr. 62, 877–882 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Doublié S., Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 276, 523–530 (1997). [PubMed] [Google Scholar]
- 5.Weinert T., Olieric V., Waltersperger S., Panepucci E., Chen L., Zhang H., Zhou D., Rose J., Ebihara A., Kuramitsu S., Li D., Howe N., Schnapp G., Pautsch A., Bargsten K., Prota A. E., Surana P., Kottur J., Nair D. T., Basilico F., Cecatiello V., Pasqualato S., Boland A., Weichenrieder O., Wang B.-C., Steinmetz M. O., Caffrey M., Wang M., Fast native-SAD phasing for routine macromolecular structure determination. Nat. Methods 12, 131–133 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Li D., Pye V. E., Caffrey M., Experimental phasing for structure determination using membrane-protein crystals grown by the lipid cubic phase method. Acta Crystallogr. D Biol. Crystallogr. 71, 104–122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu Y., Li H., Dong H., Zeng Y., Zhang Z., Paterson N. G., Stansfeld P. J., Wang Z., Zhang Y., Wang W., Dong C., Structural basis of outer membrane protein insertion by the BAM complex. Nature 531, 64–69 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Guo Y., Kalathur R. C., Liu Q., Kloss B., Bruni R., Ginter C., Kloppmann E., Rost B., Hendrickson W. A., Structure and activity of tryptophan-rich TSPO proteins. Science 347, 551–555 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dauter Z., Dauter M., Rajashankar K. R., Novel approach to phasing proteins: Derivatization by short cryo-soaking with halides. Acta Crystallogr. D Biol. Crystallogr. 56, 232–237 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Abendroth J., Gardberg A. S., Robinson J. I., Christensen J. S., Staker B. L., Myler P. J., Stewart L. J., Edwards T. E., SAD phasing using iodide ions in a high-throughput structural genomics environment. J. Struct. Funct. Genomics 12, 83–95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Heijne G., Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature 341, 456–458 (1989). [DOI] [PubMed] [Google Scholar]
- 12.Heijne G. V., Membrane proteins: From sequence to structure. Annu. Rev. Biophys. Biomol. Struct. 23, 167–192 (1994). [DOI] [PubMed] [Google Scholar]
- 13.von Heijne G., Membrane protein structure prediction: Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225, 487–494 (1992). [DOI] [PubMed] [Google Scholar]
- 14.Yau W.-M., Wimley W. C., Gawrisch K., White S. H., The preference of tryptophan for membrane interfaces. Biochemistry 37, 14713–14718 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Inoue K., Ono H., Abe-Yoshizumi R., Yoshizawa S., Ito H., Kogure K., Kandori H., A light-driven sodium ion pump in marine bacteria. Nat. Commun. 4, 1678 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Ghai R., Mizuno C. M., Picazo A., Camacho A., Rodriguez-Valera F., Metagenomics uncovers a new group of low GC and ultra-small marine Actinobacteria. Sci. Rep. 3, 2471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stewart V., Nitrate- and nitrite-responsive sensors NarX and NarQ of proteobacteria. Biochem. Soc. Trans. 31, 1–10 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Jaakola V.-P., Griffith M. T., Hanson M. A., Cherezov V., Chien E. Y. T., Lane J. R., IJzerman A. P., Stevens R. C., The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zander U., Bourenkov G., Popov A. N., de Sanctis D., Svensson O., McCarthy A. A., Round E., Gordeliy V., Mueller-Dieckmann C., Leonard G. A., MeshAndCollect: An automated multi-crystal data-collection workflow for synchrotron macromolecular crystallography beamlines. Acta Crystallogr. D Biol. Crystallogr. 71, 2328–2343 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E., The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dauter Z., Adamiak D. A., Anomalous signal of phosphorus used for phasing DNA oligomer: Importance of data redundancy. Acta Crystallogr. D Biol. Crystallogr. 57, 990–995 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Batyuk A., Galli L., Ishchenko A., Han G. W., Gati C., Popov P. A., Lee M.-Y., Stauch B., White T. A., Barty A., Aquila A., Hunter M. S., Liang M., Boutet S., Pu M., Liu Z.-j., Nelson G., James D., Li C., Zhao Y., Spence J. C. H., Liu W., Fromme P., Katritch V., Weierstall U., Stevens R. C., Cherezov V., Native phasing of x-ray free-electron laser data for a G protein-coupled receptor. Sci. Adv. 2, e1600292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gushchin I., Shevchenko V., Polovinkin V., Kovalev K., Alekseev A., Round E., Borshchevskiy V., Balandin T., Popov A., Gensch T., Fahlke C., Bamann C., Willbold D., Büldt G., Bamberg E., Gordeliy V., Crystal structure of a light-driven sodium pump. Nat. Struct. Mol. Biol. 22, 390–395 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Liu W., Chun E., Thompson A. A., Chubukov P., Xu F., Katritch V., Han G. W., Roth C. B., Heitman L. H., IJzerman A. P., Cherezov V., Stevens R. C., Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Studier F. W., Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Gordeliy V. I., Labahn J., Moukhametzianov R., Efremov R., Granzin J., Schlesinger R., Büldt G., Savopol T., Scheidig A. J., Klare J. P., Engelhard M., Molecular basis of transmembrane signalling by sensory rhodopsin II–transducer complex. Nature 419, 484–487 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Gordeliy V. I., Schlesinger R., Efremov R., Büldt G., Heberle J., Crystallization in lipidic cubic phases: A case study with bacteriorhodopsin. Methods Mol. Biol. 228, 305–316 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Li D., Boland C., Aragao D., Walsh K., Caffrey M., Harvesting and cryo-cooling crystals of membrane proteins grown in lipidic mesophases for structure determination by macromolecular crystallography. J. Vis. Exp. 2012, e4001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nurizzo D., Mairs T., Guijarro M., Rey V., Meyer J., Fajardo P., Chavanne J., Biasci J.-C., McSweeney S., Mitchell E., The ID23-1 structural biology beamline at the ESRF. J. Synchrotron Radiat. 13, 227–238 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Flot D., Mairs T., Giraud T., Guijarro M., Lesourd M., Rey V., van Brussel D., Morawe C., Borel C., Hignette O., Chavanne J., Nurizzo D., McSweeney S. and Mitchell E., The ID23-2 structural biology microfocus beamline at the ESRF. J. Synchrotron Radiat. 17, 107–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bourenkov G. P., Popov A. N., Optimization of data collection taking radiation damage into account. Acta Crystallogr. D Biol. Crystallogr. 66, 409–419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans P., Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Giordano R., Leal R. M. F., Bourenkov G. P., McSweeney S., Popov A. N., The application of hierarchical cluster analysis to the selection of isomorphous crystals. Acta Crystallogr. D Biol. Crystallogr. 68, 649–658 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Pape T., Schneider T. R., HKL2MAP: A graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Cryst. 37, 843–844 (2004). [Google Scholar]
- 36.Sheldrick G. M., Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langer G., Cohen S. X., Lamzin V. S., Perrakis A., Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L.-W., Read R. J., Adams P. D., Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC.
- 42.Lomize M. A., Pogozheva I. D., Joo H., Mosberg H. I., Lomize A. L., OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1602952/DC1
fig. S1. Comparison of peaks in anomalous difference Fourier maps calculated from individual data sets in I-SAD experiment.
fig. S2. The distribution of positively charged and aromatic residues in the crystal structures obtained in I-SAD experiment.
fig. S3. The bound iodide ions and their environment.