

Abstract
Palmitoylethanolamide (PEA) has emerged as a potential nutraceutical, because this compound is naturally produced in many plant and animal food sources, as well as in cells and tissues of mammals, and endowed with important neuroprotective, anti‐inflammatory and analgesic actions. Several efforts have been made to identify the molecular mechanism of action of PEA and explain its multiple effects both in the central and the peripheral nervous system. Here, we provide an overview of the pharmacology, efficacy and safety of PEA in neurodegenerative disorders, pain perception and inflammatory diseases. The current knowledge of new formulations of PEA with smaller particle size (i.e. micronized and ultra‐micronized) when given alone or in combination with antioxidant flavonoids (i.e. luteolin) and stilbenes (i.e. polydatin) is also reviewed.
Linked Articles
This article is part of a themed section on Principles of Pharmacological Research of Nutraceuticals. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.11/issuetoc
Abbreviations
- 2‐AG
2‐arachidonoyl‐glycerol
- AD
Alzheimer's disease
- AEA
anandamide (N‐arachidonoyl‐ethanolamine)
- ALIA
Autacoid Local Inflammation Antagonism
- ALS
amyotrophic lateral sclerosis
- CAD
contact allergic dermatitis
- CCI
chronic constriction injury
- FAAH
fatty acid amide hydrolase
- GPR55
orphan GPCR 55
- Lut
luteolin
- m‐PEA
micronized PEA
- MS
multiple sclerosis
- NAAA
N‐acylethanolamine acid amidase
- NAPE‐PLD
N‐acyl‐phosphatidylethanolamine‐specific phospholipase D
- PD
Parkinson's disease
- PEA
palmitoylethanolamide
- SCI
spinal cord injury
- TBI
traumatic brain injury
- TRPV1
transient receptor potential vanilloid type‐1 channel
- um‐PEA
ultra‐micronized PEA
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes d |
| CB1 receptor | DAGL, diacylglycerol lipase |
| CB2 receptor | FAAH, fatty acid amide hydrolase |
| GPR55 | NAAA, N‐acylethanolamine acid amidase |
| Voltage‐gated ion channels b | NAPE‐PLD, N‐acyl‐phosphatidylethanolamine‐specific phospholipase D |
| TRPV1 channel | |
| Nuclear hormone receptors c | |
| PPAR‐α |
| LIGANDS |
|---|
| 2‐AG, 2‐arachidonoyl‐glycerol |
| AEA, anandamide |
| OEA, oleoylethanolamide |
| PEA, palmitoylethanolamide |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,dAlexander et al., 2015a,b,c,d).
Introduction
Palmitoylethanolamide (PEA) is a lipid mediator used in the clinic for its neuroprotective, anti‐neuroinflammatory and analgesic properties (Re et al., 2007; Esposito and Cuzzocrea, 2013; Mattace Raso et al., 2014; Skaper et al., 2015; Iannotti et al., 2016). It was isolated for the first time from purified lipid fractions of soybeans, egg yolk and peanut meal (Coburn et al., 1954; Ganley et al., 1958) and was then found in a wide variety of food sources (Schuel et al., 2002; Venables et al., 2005; Kilaru et al., 2007; Gouveia‐Figueira and Nording, 2014) (Table 1), cells (Berdyschev et al., 2000; Stella and Piomelli, 2001; Walter et al., 2002; Muccioli and Stella, 2008; De Filippis et al., 2010; Petrosino et al., 2010a), tissues (Epps et al., 1979; Baker et al., 2001; Capasso et al., 2001; Chen et al., 2005; Murillo‐Rodriguez et al., 2006; Petrosino et al., 2007; Abramo et al., 2014) and body fluids (Giuffrida and Piomelli, 1998; Schuel et al., 2002; Schreiber et al., 2007; Richardson et al., 2008; Lam et al., 2010) of several animal species and human subjects. In animals, the biosynthesis of PEA occurs through the hydrolysis of its direct phospholipid precursor, N‐palmitoyl‐phosphatidyl‐ethanolamine, by the action of N‐acyl‐phosphatidyl‐ethanolamine‐selective phospholipase D (NAPE‐PLD) (Okamoto et al., 2004) (Figure 1A). The degradation of PEA to palmitic acid and ethanolamine occurs by the action of two different hydrolytic enzymes, that is, fatty acid amide hydrolase (FAAH) (Cravatt et al., 1996) and, more specifically, N‐acylethanolamine‐hydrolyzing acid amidase (NAAA) (Ueda et al., 2001) (Figure 1A). Interestingly, the biosynthesis and degradation of PEA, as well as other N‐acylethanolamines, in plants, where these compounds exert quite different physiological functions, seem to occur via identical routes and often similar enzymes (Blancaflor et al., 2014).
Table 1.
Food sources that contain PEA
| Food source | Concentration of PEA (ng·g−1 fresh weight) | Reference |
|---|---|---|
| Bovine milk | 0.25 | Gouveia‐Figueira and Nording, 2014 |
| Elk milk | 1.81 | Gouveia‐Figueira and Nording, 2014 |
| Human breast milk | 8.98 ± 3.35 nmol·L−1 | Lam et al., 2010 |
| Human breast milk (110 ± 32.3 lactation days) | 23.4 ± 7.2 nmol·L−1 | Schuel et al., 2002 |
| Common bean (Phaseoulus vulgaris) | 53.5 | Venables et al., 2005 |
| Garden pea (Pisum sativum) | 100 | Venables et al., 2005; Kilaru et al., 2007 |
| Southern or blackeyed peas (Vigna unguiculata) | 138 | Venables et al., 2005 |
| Tomato | 100 | Kilaru et al., 2007 |
| Medicago sativa | 1150 | Venables et al., 2005 |
| Corn | 200 | Kilaru et al., 2007 |
| Soybean (Glycine max) | 6700 | Venables et al., 2005; Kilaru et al., 2007 |
| Soy lecithin | 950 000 | Kilaru et al., 2007 |
| Peanut (Arachis hypogaea) | 3730 | Venables et al., 2005; Kilaru et al., 2007 |
Figure 1.
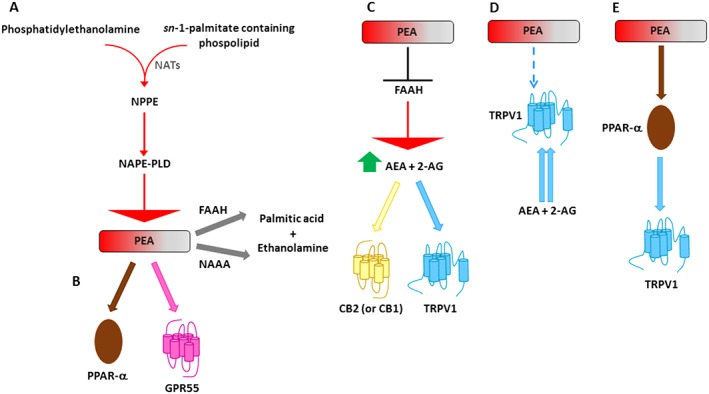
Metabolic pathways and molecular targets of PEA. (A) PEA is biosynthesized from a membrane phospholipid, N‐palmitoylphosphatidylethanolamine (NPPE), via several routes, the most investigated of which is through the direct hydrolysis by NAPE‐PLD. PEA can be then degraded to palmitic acid and ethanolamine by either FAAH or NAAA (Iannotti et al., 2016). (B) PEA can directly activate PPAR‐α (Lo Verme et al., 2005b) or, more controversially, GPR55 (Ryberg et al., 2007). (C) PEA, for example through the inhibition of the expression of FAAH, may increase the endogenous levels of AEA and 2‐AG, which directly activate CB2 (or CB1) receptors and TRPV1 channels (entourage effect) (Di Marzo et al., 2001; Petrosino et al., 2016a). (D) PEA, possibly through an allosteric modulation of TRPV1 channels, potentiates the activation and desensitization by AEA and 2‐AG of TRPV1 channels (entourage effect) (De Petrocellis et al., 2001; Di Marzo et al., 2001; Ho et al., 2008; Petrosino et al., 2016a). (E) PEA may also activate TRPV1 channels via PPAR‐α (Ambrosino et al., 2013, 2014). NAT, N‐acyl‐transferase.
Following the discovery of PEA as a naturally occurring anti‐allergic and anti‐inflammatory compound (Kuehl et al., 1957) and of endogenous metabolic pathways for PEA (Di Marzo et al., 1996; Bisogno et al., 1997; Tsuboi et al., 2005), investigations have been carried out to identify the molecular mechanism of action through which PEA exerts its pharmacological effects. This research has revealed that PEA can act via multiple mechanisms (Iannotti et al., 2016) (Figure 1B–E). The first mechanism of action for PEA was proposed by Rita Levi‐Montalcini's research group, who suggested that PEA acts via ‘Autacoid Local Injury Antagonism (ALIA)’ to down‐regulate mast cell activation (Aloe et al., 1993; Levi‐Montalcini et al., 1996). Later, the existence of a ‘direct receptor‐mediated mechanism’ was proposed, and several studies demonstrated that PEA can act via direct activation of at least two different receptors: the PPAR‐α (Lo Verme et al., 2005a) and the orphan GPCR 55 (GPR55) (Ryberg et al., 2007). It was originally thought that PEA could also be a CB2 receptor agonist (Facci et al., 1995), but subsequent studies revealed that PEA has only very weak affinity for this receptor (Sugiura et al., 2000), explaining why some of its anti‐inflammatory effects are not blocked by CB2 receptor antagonists (Costa et al., 2002). As a result, the theory of the ‘entourage’ effect was put forward to raise the possibility that PEA could produce indirect receptor‐mediated effects (De Petrocellis et al., 2001; Di Marzo et al., 2001; Ho et al., 2008). For example, PEA, through the inhibition of the expression of FAAH, the enzyme responsible for the degradation of the endogenous cannabinoid receptor ligand (or endocannabinoid), anandamide (AEA) (Di Marzo et al., 2001), may indirectly activate CB2 and CB1 receptors (Di Marzo et al., 2001; Petrosino et al., 2016a). Likewise, PEA can indirectly activate the transient receptor potential vanilloid receptor type 1 (TRPV1) channels, which are also targets for the endocannabinoids (Zygmunt et al., 1999, 2013). In addition, PEA is also able to increase AEA‐ or 2‐AG‐induced TRPV1 activation and desensitization (De Petrocellis et al., 2001; Di Marzo et al., 2001; Ho et al., 2008; Petrosino et al., 2016a). More recently, it has also been demonstrated that PEA can activate TRPV1 channels or increase the expression of CB2 receptors via PPAR‐α receptors (Ambrosino et al., 2013; Ambrosino et al., 2014; Guida F, Maione S and Di Marzo V, personal communication). In summary, these results suggest that PEA does not operate through just one main mechanism of action. Instead, synergistic interactions among several mechanisms often seem necessary so that PEA can produce its important therapeutic effects, both in the central and the peripheral nervous system.
In this review, we discuss the molecular targets of PEA, its pharmacological activity in neurological disorders, pain perception and inflammatory diseases, as well as the current evidence on the effectiveness of micronized and ultra‐micronized formulations of PEA when given alone or in combination with the antioxidant flavonoids, such as luteolin, or stilbenes, such as polydatin.
Direct molecular targets of PEA
PPAR‐α
PPAR‐α is a nuclear receptor protein that belongs to the family of PPARs and acts as transcription factor regulating gene expression (Issemann and Green, 1990). PPAR‐α is expressed in many organs and tissues, such as the intestine, heart, liver, kidney, muscle and adipose tissue, and also in several cells of the immune system (Braissant et al., 1996; Daynes and Jones, 2002). Its presence in the latter cells allows it to be implicated in the control of inflammatory processes (Daynes and Jones, 2002; Sheu et al., 2002). The first evidence of PEA as a potential agonist of PPAR‐α was reported by Lo Verme et al. (2005a), who demonstrated that the lipid directly activates the nuclear receptor with an EC50 of 3 μM (Lo Verme et al., 2005a), a potency comparable to that of the synthetic PPAR‐α agonist Wy‐14 643, which produced strong anti‐inflammatory actions (Sheu et al., 2002). Accordingly, it was hypothesized that, like all PPAR agonists, the binding of PEA to PPAR‐α also induces a heterodimerization event with the retinoic acid receptor (RXR), thereby forming the activated receptor complex, which translocates to the nucleus to bind to a peroxisome proliferator response element and reduce the transcription of pro‐inflammatory genes (Lo Verme et al., 2005b).
GPR55
The orphan GPR55 receptor belongs to the large family of GPCRs and, although showing a low homology with CB1 and CB2 receptors, has been suggested to be activated by the main psychoactive constituent of Cannabis sativa, Δ9‐tetrahydrocannabinol, and by the endocannabinoids AEA and 2‐AG (Pertwee, 2007; Sharir et al., 2012). Low concentrations of PEA also seem to directly activate GPR55 with an EC50 of 4 nM (Ryberg et al., 2007), although this issue is still controversial. Accordingly, GPR55 has recently emerged as a putative target for the treatment of inflammation (Yang et al., 2016). It is expressed in almost all brain areas, including the cortex, cerebellum, forebrain, hippocampus and striatum, whereas at the peripheral level, it is amply expressed in the gastrointestinal tract (Ryberg et al., 2007). Although its pharmacology is not completely clear yet, it has been reported that GPR55 utilizes a variety of downstream signalling events including the increase in intracellular calcium via Gq, G12, RhoA, actin, PLC and IP(3)R‐gated stores (Lauckner et al., 2008). ERK1/2, MAPK and the induction of transcriptional regulators such as nuclear factor of activated T‐cells (NFAT), NF‐κB and cAMP response element binding protein have also been shown to be coupled to GPR55 activation (Henstridge et al., 2010).
Indirect molecular targets of PEA
CB1 and CB2 receptors
The CB1 and CB2 receptors, similar to the orphan GPR55 receptor, also belong to the large family of GPCRs (Matsuda et al., 1990; Munro et al., 1993). In the brain, the CB1 receptor is often expressed in presynaptic terminals, and thanks to this localization, its activation usually inhibits neurotransmitter release (Katona et al., 1999). The CB1 receptor is also found in the peripheral nervous system and in almost all mammalian tissue and organs such as adipose tissue, skeletal muscle, bone, skin, heart, liver, gastrointestinal tract, lungs and male and female reproductive systems (Pertwee, 1997). It is usually coupled to Gi/o proteins, whereby its activation inhibits adenylate cyclase activity with the subsequent reduction of intracellular levels of cAMP, or stimulates MAPK activity (Turu and Hunyady, 2010). However, the CB1 receptor can be also be coupled to Gs or Gq proteins (Turu and Hunyady, 2010), as well as to other types of intracellular signals, including the PKB (Akt), phosphoinositide 3‐kinase and PLC/inositol 1,4,5‐trisphosphate/PKC (PLCβ/IP3/PKC) pathways (Gómez del Pulgar et al., 2000; Sanchez et al., 2003).
The CB2 receptor is largely expressed in cells (such as monocytes, macrophages, B‐ and T‐cells, mast cells and keratinocytes) and peripheral organs (such as the spleen, tonsils, thymus gland, gastrointestinal tract and skin) that play a role in the immune response (Izzo, 2004; Pertwee, 2007; Campora et al., 2012; Iannotti et al., 2016). Instead, the expression of CB2 receptors in the brain is very low and is observed particularly in activated astrocytes and microglia (Stella, 2010). Accordingly, the main function of the CB2 receptor seems to be the control of inflammatory and nociceptive responses (Whiteside et al., 2007; Basu and Dittel, 2011). Similar to the CB1 receptor, the CB2 receptor is also coupled to Gi/Go proteins, and as a result, its activation inhibits adenylate cyclase activity and promotes MAPK activity (Demuth and Molleman, 2006).
CB1 and CB2 receptors are not direct targets of PEA, but they can be indirectly activated by PEA through the aforementioned mechanisms of the entourage effect (Sugiura et al., 2000; Di Marzo et al., 2001; Petrosino et al., 2010b, 2016a).
TRPV1
The TRPV1 channel, also known as the capsaicin receptor, belongs to a subfamily of TRP channels, that is, the TRPV channels, with six transmembrane domains and an intramembrane loop linking the fifth and sixth transmembrane domain and forming the pore channel region (Caterina et al., 1997). TRPV1 is a non‐selective ion channel, permeable to mono‐ and divalent cations (i.e. Mg2 +, Ca2 +, Na+), and activated by both physical and mechanical stimuli (i.e. high temperatures, low pH, osmotic changes) as well as by exogenous or endogenous chemical compounds (i.e. capsaicin, AEA, cannabinoids) (Caterina et al., 1997; Di Marzo and De Petrocellis, 2010; Iannotti et al., 2014). TRPV1 is mainly found in dorsal root ganglia and sensory nerve fibres of the Aδ and C‐type. However, it is also expressed in brain neurons, keratinocytes and other cell types (Cristino et al., 2006; Starowicz et al., 2008; Petrosino et al., 2010a; Julius, 2013; Edwards, 2014). The function of TRPV1 is dependent on changes in its phosphorylation state induced by regulatory proteins, including ATP, PKA, PKC, phosphoinositide‐binding protein (PIRT) and phosphatidylinositol 4,5‐bisphosphate (PIP2) (Cortright and Szallasi, 2004; Iannotti et al., 2016). Phosphorylation seems to be a requisite for the activation/sensitization of TRPV1, contributing to pain transmission, inflammation and neurotoxicity (Julius, 2013; Edwards, 2014; Nagy et al., 2014). Conversely, the increase in intracellular Ca2 + following the stimulation of TRPV1 channels activates: (a) proteins, such as calmodulin, that render the channel stable in a locked conformational state; or (b) Ca2 +‐dependent phosphatases, such as calcineurin, which dephosphorylate the TRPV1 channel and again inactivate it (Cortright and Szallasi, 2004; Iannotti et al., 2016). This process of TRPV1 inactivation, also known as ‘desensitization’, contributes to the analgesic and anti‐inflammatory actions of TRPV1 agonists (Nagy et al., 2014; Iannotti et al., 2016).
Two different mechanisms have been suggested for the action of PEA at TRPV1 channels. The first mechanism proposes that PEA can indirectly activate TRPV1 through the so‐called entourage effect. In particular, PEA, possibly through allosteric effects, is able to increase AEA‐ or 2‐AG‐induced activation and desensitization at TRPV1 channels (De Petrocellis et al., 2001; Ho et al., 2008; Petrosino et al., 2016a). The second mechanism proposes that PEA can indirectly activate TRPV1 channels via PPAR‐α. In particular, because the TRPV1 and PPAR‐α antagonists inhibited PEA‐induced intracellular Ca2 + increase (Ambrosino et al., 2013), a direct biochemical interaction was hypothesized to occur between TRPV1 channels and PPAR‐α. In fact, in cells co‐transfected with TRPV1 and PPAR‐α, the latter receptors were detected in TRPV1‐immunoprecipitated fractions, and PPAR‐α agonists activated and desensitized closely‐associated TRPV1 channels (Ambrosino et al., 2014).
Pharmacokinetics of PEA
There are currently only few data available in the literature on the pharmacokinetics and bioavailability of PEA. The first study was published by Zhukov (1999), who investigated the distribution of N‐[1‐14C]‐PEA in rat tissues after i.p. administration. The results indicated the following rank order of radioactivity: adrenal >> diaphragm > spleen > kidney > testis > lung > liver > heart > brain > plasma > erythrocytes (Zhukov, 1999). The regional distribution in the rat brain of orally administered PEA (~100 mg·kg−1) has been investigated by the use of N‐[9,10‐3H]‐PEA by Artamonov et al. (2005) and recently revised by Gabrielsson et al. (2016). The authors found that N‐[9,10‐3H]‐PEA mainly accumulated in the hypothalamus, pituitary and adrenal glands, 20 min after oral administration (Artamonov et al., 2005). The presence of the labelled PEA in the brain (~98 ng·mg−1 of brain tissue) demonstrated the ability of the compound to penetrate, although in small amounts, through the blood–brain barrier (Artamonov et al., 2005). These results were not surprising if we consider that PEA is a poorly water‐soluble substance, which can limit its oral absorption and bioavailability, but they may also suggest a short‐lived action of PEA, in agreement with the fact that this compound is degraded by two different hydrolases, that is, NAAA and FAAH. Later, Grillo et al. (2013) investigated the tissue distribution of PEA formulated as an emulsion in corn oil and administered s.c. to young DBA/2 mice (10 mg·kg−1) (Grillo et al., 2013; Gabrielsson et al., 2016). The authors found that PEA could be successfully emulsified into an oil depot injection through which it effectively reaches tissues such as the retina, heart, brain and blood, both 24 and 48 h after administration (Grillo et al., 2013). More recently, it has been found that after oral administration of PEA (in a corn oil suspension administered to rats by gastric gavage, at dose of 100 mg·kg−1), the highest plasma concentration was achieved after 15 min corresponding to a 20‐fold increase in its basal values. PEA plasma levels dropped 2 h after administration to concentrations very close to the basal ones (Vacondio et al., 2015). Finally, we recently published preliminary data on the bioavailability of two new formulations of PEA, that is, micronized and ultra‐micronized (m‐PEA and um‐PEA), in human volunteers and beagle dogs, respectively (Petrosino et al., 2016a). Our results showed that, in beagle dogs, plasma PEA levels were increased up to sixfold 1 and 2 h after the oral administration of um‐PEA (30 mg·kg−1), and at the same time points, the plasma 2‐AG levels were also increased by up to ∼20‐fold (Petrosino et al., 2016a). In human volunteers, following a ∼twofold peak in plasma PEA levels at 2 h, the plasma 2‐AG levels were increased by up to ~twofold 4 and 6 h after the oral administration of m‐PEA (300 mg) (Petrosino et al., 2016a). This smaller increase was proportionate to the smaller peak of PEA levels detected in human volunteers compared with beagle dogs. This difference, in turn, can be caused by the lower total amount of PEA acutely administered to human volunteers (about ∼5 vs. 30 mg·kg−1 in dogs) and to the fact that this was a micronized formulation as opposed to the ultra‐micronized formulation administered to dogs. Furthermore, the human volunteers engaged for this study were healthy, whereas the dogs were allergic subjects, and the bioavailability of PEA might change during pathological conditions. In summary, these results suggest that, although their bioavailability has not, so far, been compared in the same study with that of ‘normal’ PEA, formulations of this compound with smaller particle size might be a useful alternative to overcome its solubility problems encountered particularly following oral administration. It is well known, in fact, that the solubility of a drug is intrinsically related to the particle size. Particle size reduction by various means, such as jet mill, leads to an increase in the specific surface area with enhanced solubility and potentially higher bioavailability (Rasenack and Müller, 2004). While m‐PEA and um‐PEA seem to have a reasonably good oral bioavailability, complete pharmacokinetics studies are necessary to assess the exact tissue exposure and site of metabolism of PEA when administered through these formulations.
PEA and neurological disorders
Neurodegenerative diseases
Neurodegenerative diseases such as Alzheimer's disease (ad), Parkinson's disease (PD), multiple sclerosis (MS) and amyotrophic lateral sclerosis (ALS) are characterized by gradual and selective neuronal cell death that causes the slow and progressive loss of one or more functions of the nervous system. Depending on the type of disease, neuronal damage can lead to cognitive deficits, dementia, behavioural disorders, motor abnormalities or paralysis.
The potential neuroprotective effects of PEA have been demonstrated in several experimental models of AD. In a mouse model, the s.c. administration of the compound reduced the behavioural impairments, lipid peroxidation, inducible NO synthase (iNOS) induction and caspase‐3 activation induced by i.c.v. injection of amyloid‐β 25‐35 (Aβ25‐35) peptide (D'Agostino et al., 2012). In addition, GW7647, an agonist of PPAR‐α, produced similar effects to PEA, whereas PEA did not protect against Aβ25‐35‐induced memory deficits in PPAR‐αKO mice (D'Agostino et al., 2012). In a different in vivo model, performed in adult male rats and consisting of the intrahippocampal injection of amyloid‐β 1‐42 (Aβ1‐42) peptide, the systemic administration of PEA counteracted the increased transcription and expression of proteins typical of activated astrocytes (GFAP and S100β), as well as the increased expression of amyloidogenic (BACE1 and APP) and phosphorylated τ proteins (Scuderi et al., 2014). PEA also restored the altered expression of microtubule‐associated protein (MAP‐2) and cognitive functions induced by Aβ1‐42 peptide (Scuderi et al., 2014). Also in this case, the involvement of PPAR‐α was confirmed by co‐administration of PEA with GW6471, a selective antagonist of PPAR‐α, which completely abolished PEA‐induced effects (Scuderi et al., 2014). These data are in agreement with previous in vitro studies on rat mixed neuroglial cultures and organotypic hippocampal slices, challenged with Aβ1‐42 peptide, where PEA treatment blunted Aβ1‐42‐induced astrocyte activation and improved neuronal survival, and with the effects of PEA being reversed by another PPAR‐α antagonist, MK886 (Scuderi et al., 2011, 2012). The neuroprotective benefits of the new composite co‐ultraPEALut, consisting of a mixture of PEA and the antioxidant flavonoid, luteolin, in a mass ratio of 10:1, subjected to ultra‐micronization, were first shown both in in vitro and ex vivo organotypic models of ad by using differentiated SH‐SY5Y neuroblastoma cells and hippocampal slice cultures, respectively, stimulated with Aβ1‐42 peptide (Paterniti et al., 2014). Pretreatment with co‐ultraPEALut significantly reduced iNOS, glial fibrillary acidic protein expression and apoptosis, and restored neuronal NO synthase as well as brain‐derived neurotrophic factor (BDNF; Paterniti et al., 2014).
Neuroprotective actions of PEA were also demonstrated in an animal PD model that consists of injecting i.p. the neurotoxin 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) (Esposito et al., 2012). In particular, chronic treatment with PEA (i.p.) counteracted the loss of nigrostriatal neurons, altered expression of microtubule‐associated proteins (MAP‐2 and S100β), activation of astrocytes and expression of iNOS protein induced by MPTP in mice (Esposito et al., 2012). In addition, PEA reduced MPTP‐associated behavioural impairments and motor dysfunctions (Esposito et al., 2012). These effects were dependent on the activation of PPAR‐α because genetic ablation of this receptor exacerbated MPTP‐induced neurotoxicity (Esposito et al., 2012). The same beneficial effects were recently observed in this PD model after the administration of the aforementioned composite co‐ultraPEALut (Siracusa et al., 2015a). Treatment with co‐ultraPEALut was able both to reduce the neuroinflammatory response and to increase the autophagy process caused by MPTP intoxication in mice (Siracusa et al., 2015a).
These findings, taken together, help identify the molecular mechanism through which PEA is able to modulate the altered expression of proteins associated with ad or PD and to down‐regulate activation of pro‐apoptotic markers and pro‐inflammatory factors, which lead to the loss of neurons in the cerebral cortex and hippocampus for ad or in the substantia nigra for PD. Thus, PPAR‐α emerged as the primary target for the neuroprotective effects of PEA in the central nervous system, and this evidence is supported by the use of PPAR‐α agonists and antagonists, which mimic or block PEA effects, respectively.
Therapeutic effects of PEA have also been reported in several chronic models of MS, such as chronic relapsing experimental autoimmune encephalomyelitis (CREAE), Theiler's murine encephalomyelitis virus‐induced demyelinating disease (TMEV‐IDD) and myelin oligodendrocyte glycoprotein‐induced experimental autoimmune encephalomyelitis (MOG‐EAE). Initially, it was observed that the endogenous levels of PEA increased in the spinal cord of mice with CREAE or TMEV‐IDD (Baker et al., 2001; Loria et al., 2008) and also in the plasma of relapsing–remitting and secondary‐progressive MS patients (Jean‐Gilles et al., 2009) (Table 2). In addition, it was demonstrated that the exogenous administration of PEA (i.p.) ameliorated spasticity or motor deficits in mice with CREAE or TMEV‐IDD, respectively, (Baker et al., 2001; Loria et al., 2008) and reduced the severity of neurobehavioral scores in mice with MOG‐induced EAE (Rahimi et al., 2015). Furthermore, the expression of inflammatory cytokines was greatly reduced by PEA both in TMEV‐IDD and MOG‐induced EAE mice (Loria et al., 2008; Rahimi et al., 2015), and these effects were accompanied by decreased demyelination and axonal damage in the latter model (Rahimi et al., 2015). These data suggest that exogenous PEA might be helpful to compensate or amplify the endogenous defence mechanism deployed by the cells or tissues to counteract neurodegenerative and neuro‐inflammatory processes. However, the molecular target(s) through which PEA exerts these effects has(have) not been investigated yet.
Table 2.
Some examples of altered endogenous PEA levels during pathological conditions
| Disease | Type of study | Area | PEA levels | Reference |
|---|---|---|---|---|
| Multiple sclerosis | CREAE in mice | Spinal cord | ↑ | Baker et al., 2001 |
| TMEV‐IDD in mice | Spinal cord | ↑ | Loria et al., 2008 | |
| RR‐SP in patients | Plasma | ↑ | Jean‐Gilles et al., 2009 | |
| Ischaemic stroke | HS in a patient | Ischaemic lesion | ↑ | Schabitz et al., 2002 |
| FCI in mice | Cerebral cortex | ↑ | Franklin et al., 2003 | |
| MCAO in rats | Infarcted brain areas | ↑ | Berger et al., 2004 | |
| Pain | CCI in rats | Spinal cord | ↓ | Petrosino et al., 2007 |
| Rostral ventral medulla | ↓ | |||
| Dorsal raphe magnus | ↓ | |||
| Carrageenan‐induced inflammatory pain in rats | Hind paw | ↓ | Jhaveri et al., 2008 | |
| Moderate to‐severe dysmenorrhea and dyspareunia in human patients | Blood | ↑ | Sanchez et al., 2016 | |
| Chronic neck shoulder pain in women | Trapezius muscle (microdialysis) | ↑ | Ghafouri et al., 2011, 2013, 2014 | |
| Inflammatory nature | Croton oil‐induced chronic intestinal inflammation in mice | Small intestine | ↓ | Capasso et al., 2001 |
| Chronic inflammation in mice (implant of sterile polyethylene sponges instilled with carrageenan under the dorsal skin) | Infiltrating leukocytes | ↓ | Solorzano et al., 2009 | |
| Chronic inflammation in rats (implantation of carrageenan‐soaked sponges on the back) | Newly formed granuloma | ↓ | De Filippis et al., 2010 | |
| Ulcerative colitis in human patients | Colon | ↑ | Darmani et al., 2005 | |
| CdCl2‐administered rat testis | Testis | ↑ | Kondo et al., 1998 | |
| DNFB‐induced CAD in mice | Ear | ↑ | Petrosino et al., 2010 | |
| Atopic dermatitis in dogs | Skin | ↑ | Abramo et al., 2014 | |
| Cyclophosphamide‐induced cystitis in female rats | Bladder | ↑ | Pessina et al., 2015 | |
| Acrolein‐induced cystitis in female rats | Bladder | ↑ | Merriam et al., 2011 | |
| Allergy in rats (experimentally induced airway hyper‐responsiveness) | Brainstem (nucleus solitary tract) | ↑ | Spaziano et al., 2015 | |
| Osteoarthritis and rheumatoid arthritis in human patients | Knee synovial fluid | ↓ | Richardson et al., 2008 | |
| Glaucoma in human patients | Ciliary and choroid body | ↓ | Chen et al., 2005 | |
| Cirrhotic human patients | Blood and liver | ↑ | Caraceni et al., 2010 | |
| Cancer | Local and metastatic syngeneic melanoma in mouse | Blood, paw and metastasis sites | ↓ | Sailler et al., 2014 |
RR‐SP, relapsing–remitting and secondary‐progressive; HS, hemispheric stroke; FCI, focal cerebral ischaemia; MCAO, middle cerebral artery occlusion; CdCl2, cadmium chloride; ↑, increase; ↓, decrease.
The first clinical experience on the efficacy of ultra‐micronized PEA (um‐PEA) was obtained in a single patient with ALS, who received 600 mg um‐PEA p.o. twice daily (Clemente, 2012). The study showed that um‐PEA improved respiration, as noticed by the patient, and the clinical condition as measured by electromyography analysis (Clemente, 2012). More recently, a second clinical study was conducted in 64 patients with ALS randomly assigned to one of two groups: 28 patients received 50 mg riluzole (a drug used to treat ALS and delay the onset of ventilator‐dependence or tracheotomy) plus 600 mg um‐PEA (PEA‐treated patients) twice daily, and 36 patients received riluzole only (untreated patients) (Palma et al., 2016). ALS patients treated with um‐PEA showed a slowdown in the worsening of respiratory function, as measured by a lower reduction in their forced vital capacity over time compared with untreated ALS patients (Palma et al., 2016). Moreover, death and tracheotomy occurred more frequently in untreated than in PEA‐treated patients, suggesting that the disease progressed more slowly in subjects receiving um‐PEA (Palma et al., 2016). The authors also demonstrated that um‐PEA reduced the desensitization of acetylcholine‐evoked currents after repetitive neurotransmitter application, in Xenopus oocytes transplanted with muscle membranes from selected ALS patients (Palma et al., 2016). These results, taken together, suggest that PEA might contribute to the conservation of muscle excitability and be beneficial to ALS as an add‐on treatment (Palma et al., 2016).
Ischaemic stroke and traumatic brain injury
PEA also plays a protective role in neurological disorders caused by ischaemic stroke and traumatic brain injury (TBI). Ischaemic stroke is a condition characterized by reduced blood flow in the brain leading to limited oxygen supply and, as a result, to the death of brain tissue. Depending on the region of the brain affected, a stroke can cause paralysis, speech impairment and loss of memory. As previously described in patients with MS, it was observed that PEA levels also increased in tissue surrounding the primary ischaemic lesion, in a patient with hemispheric stroke (Schabitz et al., 2002), as well as in the mouse cerebral cortex after focal cerebral ischaemia (Franklin et al., 2003) (Table 2). Moreover, it was later reported that, in the blood of acute stroke patients, PEA levels significantly correlate with NIH stroke scale scores (Naccarato et al., 2010). In consideration of these findings, the neuroprotective potential of PEA has been investigated in ischaemic stroke experimental models. In particular, it was observed that when PEA was exogenously administered (i.p.) after transient middle cerebral artery occlusion (tMCAO), an animal model of acute stroke, it reduced infarct size in cortical and total infarct areas compared with controls (Schomacher et al., 2008; Ahmad et al., 2012a), blocked infiltration and activation of astrocytes, reduced pro‐inflammatory marker expression and amended neurobehavioral functions as determined by monitoring motor deficits (Ahmad et al., 2012a). More recently, it was reported that the co‐ultraPEALut, at a lower dose compared with PEA alone, was able to produce the same neuroprotective effects after tMCAO (Caltagirone et al., 2016). Importantly, the administration of co‐ultraPEALut to a cohort of 250 stroke patients was able to improve all clinical indices (such as neurological status, the degree of spasticity, cognitive abilities, pain and independence in daily living activities) after 30 days of treatment (Caltagirone et al., 2016).
These studies suggest that the higher levels of PEA often associated with neurological impairments may represent an adaptive protective mechanism and that the exogenous administration of PEA, alone or in combination with luteolin, might provide a therapeutic alternative to counteract such impairments through as yet uninvestigated molecular mechanisms.
TBI is a condition produced by a violent trauma to the head that causes damage to the brain. The effects on an individual can be balance problems, headache, dizziness, behavioural impairments and loss of memory. In a controlled cortical impact, an adult mouse model of TBI, it was shown that PEA treatment (i.p.) reduced oedema and the size of lesion, blocked the infiltration of astrocytes and decreased the expression of chymase, tryptase and iNOS (Ahmad et al., 2012b). PEA also improved neurobehavioral functions as evaluated by behavioural tests (Ahmad et al., 2012b). Lately, it has been observed that co‐ultraPEALut produces similar effects in the TBI model, but at a lower dose compared with PEA alone (Cordaro et al., 2016), confirming, as suggested above, that the new composite might improve their ability to counteract neurodegeneration and neuroinflammation.
The effects of PEA and co‐ultraPEALut have also been evaluated on secondary damage induced by experimental spinal cord injury (SCI) in mice. In particular, in mice with SCI induced by the application of vascular clips to the dura mater via a four‐level T(5)–T(8) laminectomy, repeated PEA administration (i.p.) reduced the degree of spinal cord inflammation and tissue injury, the infiltration of neutrophils, the expression of pro‐inflammatory cytokines and iNOS, as well as the activation of NF‐κB (Genovese et al., 2008). Moreover, PEA significantly ameliorated the recovery of motor limb function (Genovese et al., 2008). All these neuroprotective effects were absent in PPAR‐α KO mice (Paterniti et al., 2013a). The administration of co‐ultraPEALut (i.p.), at a lower dose compared with PEA alone, also reduced the severity of trauma induced by compression, improved locomotor activity (Paterniti et al., 2013b) and reduced the expression of protein promoter of autophagy (Siracusa et al., 2015b). Interestingly, co‐ultraPEALut stimulated the expression of neurotrophic factors such as BDNF, glial cell‐derived neurotrophic factor, nerve growth factor and neurotrophin‐3, suggesting that this composite exerts a prominent effect on the management of survival and differentiation of new neurons and spine maturation (Crupi et al., 2016). Nevertheless, the exact mechanism by which the composite co‐ultraPEALut seems more efficacious than PEA has not yet been investigated. A possible explanation may be that the administration of this composite, which consists of a mixture of an anti‐inflammatory mediator (i.e. PEA) and an antioxidant compound (i.e. luteolin), produces complementary and synergistic effects by acting simultaneously on two phenomena – inflammation and formation of reactive oxygen species – that independently feed neuronal death. Moreover, chemical–physical considerations argue for higher activity of the composite compared with the single physical mixture. Observation by scanning electron microscopy, in fact, has showed an intimate intermixing of the two components of the composite, and data obtained from differential scanning calorimeter and X‐ray diffraction documented the transformation into a new crystalline form different from the original two, possibly representing a ‘higher energy content’ form (Paterniti et al., 2013b). Based on such findings, one could hypothesize that co‐micronization results in decreased particle‐particle agglomeration and electrostatic attraction compared with PEA in its micronized state, in agreement with data obtained following co‐micronization of different compounds (Spence et al., 2005). This would in turn result in enhanced substance solubility and/or dispersion, a crucial factor for the absorption from the gastrointestinal fluids.
PEA and pain perception
The first studies showing the ability of PEA to produce analgesia and anti‐nociceptive effects date back to 1998, when it was demonstrated that the local administration of PEA is able to inhibit nociception evoked in mice by intraplantar injection of formalin, acetic acid, kaolin or magnesium sulfate (Calignano et al., 1998, 2001), as well as hyperalgesia following turpentine‐induced urinary bladder inflammation in the rat (Jaggar et al., 1998). The analgesia produced by PEA was thought to be mediated by peripheral CB2 receptors because it was reversed by administration of the CB2 receptor antagonist, SR144528 (Calignano et al., 1998, 2001; Farquhar‐Smith and Rice, 2001).
Later, PPAR‐α agonists were proposed as a new class of analgesics because GW7647 was found to be efficacious, like PEA, at reducing pain behaviours elicited in mice by intraplantar injection of formalin or magnesium sulfate, as well as hyperalgesic responses in the chronic constriction injury (CCI) model of neuropathic pain or in the complete Freund's adjuvant and carrageenan models of inflammatory pain (Lo Verme et al., 2006; D'Agostino et al., 2009; Di Cesare Mannelli et al., 2013). The role of PPAR‐α in mediating the actions of PEA (after both i.c.v. and s.c. administration) was suggested by the lack of anti‐hyperalgesic effects of PEA in mutant mice lacking PPAR‐α (Lo Verme et al., 2006; D'Agostino et al., 2009; Di Cesare Mannelli et al., 2013). Importantly, it was also demonstrated that the i.c.v. administration of PEA was effective at preventing inhibitory κB‐α degradation and NF‐κB nuclear translocation, in dorsal root ganglia after carrageenan‐induced inflammatory pain, suggesting the involvement of this transcriptional factor in the control of hyperalgesia (D'Agostino et al., 2009). Nevertheless, because the antinociceptive effects occurred within minutes of agonist administration in wild‐type mice, it was considered that they could be mediated via a transcription‐independent mechanism (Lo Verme et al., 2006). Thus, it was demonstrated that blockade of large‐conductance KCa channels (BKCa) and intermediate‐conductance KCa channels (IKCa) prevented the antinociceptive actions of GW7647 and PEA in the formalin test, suggesting that PPAR‐α agonists can also modulate nociception through a nongenomic mechanism (Lo Verme et al., 2006). In addition, it was also shown that blockade of PPAR‐α and large‐conductance Ca2 +‐activated K+ channels, by using selective antagonists, such as GW6471 and charybdotoxin, respectively, also prevented PEA effects, which after intra‐periaqueductal grey microinjection reduced the ongoing activity of ON and OFF cells and produced an increase in the latency of the nociceptive reaction (De Novellis et al., 2012). Finally, it has recently been demonstrated that PEA, by acting at PPAR‐α, may also be responsible, at least in part, for NMDA‐NR2b subunit down‐regulation, contributing to reduce pain‐related behaviours (Guida et al., 2015).
CB1 receptors, PPAR‐γ and TRPV1 channels have also been suggested as potential targets for the analgesic actions of PEA, for example in the CCI model of neuropathic pain (Costa et al., 2008). In fact, antagonists of these receptors blocked the anti‐allodynic and anti‐hyperalgesic effects induced by i.p. administration of PEA, suggesting that the most likely mechanism of action of PEA was through entourage effects (Costa et al., 2008), due to the enhancement of tissue levels of AEA (Di Marzo et al., 2001), which in turn is able to produce analgesia by activating CB1 receptors (Guindon and Hohmann, 2009) or by desensitizing TRPV1 channels (Starowicz et al., 2012). The conclusions put forward by Costa et al. (2008) were supported by previous findings in rats with CCI, where thermal hyperalgesia and mechanical allodynia were accompanied by increased AEA and 2‐AG levels in three brain areas involved in nociception (the dorsal raphe, periaqueductal grey and rostral ventral medulla), while PEA levels were significantly decreased (Petrosino et al., 2007) (Table 2). These two studies, taken together, suggest that, although the endocannabinoids are up‐regulated during pain conditions, their elevation may not be sufficient to exert an analgesic action because of reduced PEA levels (Petrosino et al., 2007) and that exogenously administered PEA could be an effective alternative to potentiate the endogenous anti‐nociceptive mechanism exerted by endocannabinoids (Costa et al., 2008). On the other hand, Jhaveri et al. (2008) demonstrated that the levels of AEA and PEA are decreased in the hind paw after carrageenan‐induced inflammatory pain (Table 2) and that the administration of a FAAH inhibitor, URB597, significantly elevated the levels of the two mediators, while causing anti‐nociceptive effects that were blocked by the PPAR‐α antagonist, GW6471 (Jhaveri et al., 2008). Thus, a synergistic interaction among the several mechanisms of actions of PEA may occur, thereby allowing PEA to exert its analgesic and anti‐nociceptive effects.
The analgesic properties of micronized and ultra‐micronized formulations of PEA, that is, m‐PEA and um‐PEA, respectively, were initially shown in a rat model of carrageenan‐induced inflammatory pain, where carrageenan‐induced paw oedema and thermal hyperalgesia were markedly and significantly reduced by oral treatment with either formulation (Impellizzeri et al., 2014). Later, clinical studies have demonstrated the efficacy of m‐PEA and um‐PEA, both when administered alone and in combination with the antioxidant stilbene, polydatin. In particular, it was shown that the administration of m‐PEA (300 mg p.o. twice daily) reduced pain scores in diabetic patients suffering from peripheral neuropathy (Schifilliti et al., 2014). Importantly, no alterations related to m‐PEA treatment were revealed from haematological and urine analyses, and no adverse events were observed (Schifilliti et al., 2014). Interestingly, in a mouse model of streptozotocin‐induced type 1 diabetes, it has been recently observed that PEA treatment (i.p.) was not only able to relieve mechanical allodynia but also to improve insulin levels and preserve Langerhans islet morphology by reducing the development of insulitis (Donvito et al., 2015). The effectiveness of um‐PEA has instead been evaluated both in patients with neuropathic pain due to lumbosciatalgia and in patients with chronic pain caused by different etiopathogenesis (Dominguez et al., 2012; Gatti et al., 2012). In the former study, the addition of um‐PEA (600 mg·day−1 p.o.) to a standard treatment was well tolerated and showed an improvement in pain relief (Dominguez et al., 2012). In the latter study conducted on 610 patients, um‐PEA (600 mg p.o. twice daily) was administered in addition to standard analgesic therapies or as a single therapy (Gatti et al., 2012). The results demonstrated that the decrease in pain intensity induced by um‐PEA was also present in patients without concomitant analgesic therapy and that PEA produced no adverse effects (Gatti et al., 2012). Furthermore, short‐term efficacy of um‐PEA was also demonstrated in patients with diabetic or traumatic neuropathic pain, where the administration of the formulation (1200 mg·day−1 p.o.) in addition to standard therapies improved both the visual analogue scale and neuropathic pain symptom inventory scores within the first 10 days (Cocito et al., 2014). Importantly, a pooled data meta‐analysis has recently been performed to evaluate the efficacy and safety of m‐PEA and um‐PEA on pain intensity in patients suffering from chronic and/or neuropathic pain (Paladini et al., 2016). This study confirmed that PEA‐induced pain relief is progressive, age‐ and gender‐independent and not related to the aetiopathogenesis of chronic pain (Paladini et al., 2016). Moreover, PEA administration lacked acute and chronic toxicity and was not associated with gastric mucosal lesions (Paladini et al., 2016). In contrast to these promising actions, a more recent report showed that um‐PEA as add‐on therapy was ineffective on neuropathic pain in individuals with SCI (Andresen et al., 2016). This negative outcome is not surprising considering that the study included patients (i) with different causes and severities of SCI and, more importantly, with an average time since injury of 10 years; (ii) receiving concomitant analgesic medication (dosing and length of treatment not specified); and (iii) with high pain scores at entrance (possibly indicating they were refractory to pain treatment) (Andresen et al., 2016). All the aforementioned factors could have synergistically contributed to the lack of effect. Accordingly, the study might suggest that the administration of um‐PEA may be beneficial if administered in early stages of SCI, as observed in experimental studies. Furthermore, it has been reported that the new combination m‐PEA‐polydatin (one tablet twice daily), constituted of m‐PEA (400 mg) and the antioxidant stilbene polydatin (40 mg), is effective at reducing pelvic pain in women with endometriosis (Indraccolo and Barbieri, 2010; Cobellis et al., 2011; Giugliano et al., 2013). Likewise, it has been demonstrated that the oral administration of um‐PEA is effective at reducing viscerovisceral hyperalgesia in a rat model of endometriosis plus ureteral calculosis (Iuvone et al., 2016). These findings are in agreement with the recently reported elevation of the plasma levels of PEA (and AEA) in patients with moderate‐to‐severe dysmenorrhea and dyspareunia compared with those with low‐to‐moderate pain symptoms (Sanchez et al., 2016).
In summary, these findings suggest that PEA, alone or in combination with polydatin, represents a new promising and well‐tolerated therapeutic strategy for the management of chronic pain in different pathological conditions.
Inflammatory diseases
The anti‐inflammatory effects of PEA seem to be mainly related to its ability to modulate mast cell activation and degranulation, and this action is also known as the ALIA (autacoid local inflammation antagonism) mechanism (Aloe et al., 1993; Facci et al., 1995). The first evidence of the anti‐inflammatory effects of PEA in animal models was reported by Mazzari et al. (1996), who demonstrated that orally administered PEA is able to decrease the amount of degranulated mast cells and plasma extravasation induced by substance P injection in the mouse ear pinna (Mazzari et al., 1996). Oral PEA also reduced paw oedema induced by carrageenan, dextran and formalin, suggesting that the compound directly down‐modulates mast cell activation in vivo and suppresses pathological consequences initiated by mast cell activation regardless of the activating stimuli (Mazzari et al., 1996). Later, the involvement of CB2 receptors in these effects was suggested, based on the finding that the CB2 receptor antagonist, SR144528, prevented the anti‐oedema effect produced by PEA (Conti et al., 2002) but did not reverse its curative effect after carrageenan‐induced acute inflammation (Costa et al., 2002).
Indeed, it was later shown that PPAR‐α also mediates the anti‐inflammatory effects of PEA, since both after carrageenan‐induced paw oedema and phorbol ester‐induced ear oedema, the topically applied compound attenuated inflammation in wild‐type mice but had no effect in mice deficient in PPAR‐α, whereas the PPAR‐α agonist, GW7647, mimicked the effects of PEA (Lo Verme et al., 2005a). Importantly, acute i.c.v. administration of PEA reduced carrageenan‐induced paw oedema, restored carrageenan‐induced PPAR‐α reduction, prevented IkB‐α degradation and NF‐kβ nuclear translocation in the spinal cord, suggesting the involvement of this transcriptional factor also in the central control of peripheral inflammation (D'Agostino et al., 2007). These anti‐inflammatory effects of PEA were again mimicked by a PPAR‐α agonist, GW7647, and absent in mutant mice lacking PPAR‐α (D'Agostino et al., 2007).
The anti‐inflammatory effects of PEA have also been investigated in numerous inflammatory diseases. In particular, the efficacy of this lipid compound was demonstrated in several animal models of inflammatory bowel diseases, such as croton oil‐induced chronic intestinal inflammation, dextran sodium sulphate (DSS)‐induced ulcerative colitis, oil of mustard (OM)‐induced accelerated transit and dinitrobenzene sulfonic acid (DNBS)‐induced colitis. Initially, it was observed that endogenous PEA levels were decreased in the small intestine of mice with chronic intestinal inflammation induced by croton oil and were associated with increased intestinal transit (Capasso et al., 2001) (Table 2). Accordingly, exogenous PEA administration (i.p.) decreased intestinal transit and motility, and these effects were not blocked by the CB1 and CB2 receptor antagonists, SR141716A and SR144528, respectively, suggesting that they were independent from cannabinoid receptor activation (Capasso et al., 2001). Therefore, the role of PPAR‐α in inflammatory bowel diseases was also studied, and in a mouse model of DSS‐induced ulcerative colitis as well as in cultured human biopsies deriving from patients with ulcerative colitis, PEA treatment improved the macroscopic signs of ulcerative colitis, decreased the expression and release of pro‐inflammatory cytokines as well as neutrophil infiltration (Esposito et al., 2014), all these anti‐inflammatory effects being abolished by PPAR‐α antagonists (Esposito et al., 2014). More recently, the effects of PEA on inflammation‐associated angiogenesis in mice with DSS‐induced ulcerative colitis and in patients with ulcerative colitis were also studied (Sarnelli et al., 2016). PEA treatment, in a PPAR‐α‐dependent manner, inhibited colitis‐associated angiogenesis by decreasing VEGF release and new vessel formation via the mammalian target of rapamycin/PKB (mTOR/Akt) axis (Sarnelli et al., 2016). These results suggest that PEA may exert its protective effects, in both inflammation and cancer, by reducing mucosal damage, disease progression and the shift towards carcinogenesis (Sarnelli et al., 2016). On the other hand, in a mouse model of delayed accelerated transit – a pathological condition that persists after the resolution of colonic inflammation and is known as post‐inflammatory irritable bowel syndrome – induced by OM, the CB1 receptor antagonist, SR141716A, blocked the inhibitory effect of PEA on accelerated transit, whereas the TRPV1 antagonist, 5′‐iodo‐resiniferatoxin (I‐RTX), increased it (Capasso et al., 2014). Likewise, in a mouse model of DNBS‐induced colitis, the TRPV1 antagonist, capsazepine, increased the anti‐inflammatory effects of PEA, which instead were reversed not only by a CB2 antagonist but also by GPR55 and PPAR‐α antagonists (Borrelli et al., 2015). These studies, taken together, suggest that, although a direct activation of GPR55 or PPAR‐α occurs, PEA can produce its anti‐inflammatory action in the gut also via indirect activation of CB1 and CB2 receptors, probably due to the ability of this compound to potentiate the action of endocannabinoids at these receptors. Indeed, elevated levels of AEA and 2‐AG were found both in the intestine of mice with accelerated transit induced by OM and in the colon of mice with colitis induced by DNBS (Capasso et al., 2014; Borrelli et al., 2015).
The anti‐inflammatory effects of PEA have also been demonstrated in in vitro and in vivo models of contact allergic dermatitis (CAD). Specifically, in human keratinocyte (HaCaT) cells stimulated with polyinosinic polycytidylic‐acid (poly‐(I:C)) and treated with PEA, the TRPV1 antagonist, I‐RTX, reversed PEA inhibitory effects on the expression and release of chemokine monocyte chemoattractant protein 2 (MCP‐2 also known as CCL8) (Petrosino et al., 2010a). Moreover, in dinitrofluorobenzene (DNFB)‐sensitized mice, PEA anti‐inflammatory effects (after i.p. administration) were counteracted by another TRPV1 antagonist, capsazepine (Petrosino et al., 2010a). Also in this case, the TRPV1‐mediated effects of PEA were attributed to the elevated levels of AEA and oleoylethanolamide (another endogenous TRPV1 agonist) detected both in poly‐(I:C)‐HaCaT cells and in the DNFB in vivo model of CAD (Karsak et al., 2007; Petrosino et al., 2010a) (Table 2) and hence to an entourage effect of PEA on the ‘endovanilloid’ desensitizing action over this channel (De Petrocellis et al., 2001; Ho et al., 2008). The ability of PEA to down‐regulate canine mast cell activation by inhibiting the release of histamine, PGD2 and TNF‐α has also been reported (Cerrato et al., 2010), and an increase in endocannabinoid and PEA levels was found in the skin of dogs with atopic dermatitis (Abramo et al., 2014) (Table 2). Therefore, the efficacy of um‐PEA was recently investigated in canine atopic dermatitis (Noli et al., 2015). Oral administration of um‐PEA to 160 dogs with atopic dermatitis and moderate pruritus was effective and safe in reducing pruritus and skin lesions in dogs (Noli et al., 2015). PEA also strongly reduces the cutaneous allergic inflammatory reaction induced by different immunological and non‐immunological stimuli in Ascaris suum hypersensitive Beagle dogs (Cerrato et al., 2012), and this action is likely to be due to an entourage effect on the skin levels of the endocannabinoid 2‐AG, which were dramatically elevated by PEA following oral administration (Petrosino et al., 2016a). Interestingly, an entourage effect on the skin levels of PEA has also been demonstrated (Petrosino et al., 2016b). In fact, a synthetic PEA analogue, adelmidrol, was able to increase the endogenous levels of PEA in human and canine keratinocytes and reduce the skin inflammatory response, that is, chemokine production following allergic stimulation (Petrosino et al., 2016b). These results suggested that the increase in endogenous concentrations of PEA might partially mediate the anti‐inflammatory effect of adelmidrol (Petrosino et al., 2016b).
Recently, the anti‐inflammatory effects of PEA have also been investigated in animal models of uveitis, retinal inflammation and cystitis. In rats subjected to endotoxin‐induced uveitis, PEA treatment (i.p.) was effective at reducing the degree of ocular inflammation, because it decreased inflammatory cell infiltration, TNF‐α and intercellular adhesion molecule 1 (ICAM‐1) levels, and inhibited iNOS expression as well as nuclear NF‐κB translocation (Impellizzeri et al., 2015). Likewise, oral PEA attenuated the degree of ocular inflammation in rats with experimental diabetic retinopathy and preserved the blood‐retinal barrier (Paterniti et al., 2015). Finally, in a female rat model of cyclophosphamide‐induced cystitis, it has been reported that pain behaviour, bladder inflammation and voiding dysfunction were associated with increased bladder levels of PEA, up‐regulation of CB1 receptor expression and down‐regulation of PPAR‐α expression (Pessina et al., 2015) (Table 2). Oral administration of um‐PEA produced both anti‐inflammatory and analgesic effects, which were counteracted both by CB1 receptor and PPAR‐α antagonists (Pessina et al., 2015).
The effectiveness of the combinations co‐ultraPEALut and m‐PEA‐polydatin has also been shown in animal models of inflammation. In mice subjected to collagen‐induced arthritis, treatment with co‐ultraPEALut (i.p.) reduced periarticular erythema and paw oedema, nitrotyrosine and malondialdehyde levels, as well as plasma levels of cytokines and chemokines (Impellizzeri et al., 2013). In rats subjected to carrageenan‐induced acute inflammation, the efficacy of an oral combination of m‐PEA and polydatin was compared with that of a new co‐micronized composite containing PEA and polydatin, given by the same route, with the latter showing stronger anti‐inflammatory and anti‐hyperalgesic effects compared with the simple association of two compounds (Esposito et al., 2016).
Conclusions
Although PPAR‐α is the molecular target that directly mediates some of the neuroprotective, anti‐(neuro)inflammatory and analgesic effects of PEA (Mattace Raso et al., 2014), the existence of indirect mechanisms of action for this compound has also often been demonstrated. In particular, endocannabinoid‐mediated mechanisms of action following the activation of CB1, CB2 receptors or TRPV1 channels, known as the entourage effect (Di Marzo et al., 2001; Petrosino et al., 2016a), and a CB2 receptor‐ TRPV1‐mediated mechanism of action via PPAR‐α (Ambrosino et al., 2013, 2014; Guida F, Maione S and Di Marzo V, personal communication), have been identified. These findings suggest that the existence of the ‘direct or via PPAR‐α mechanisms’ does not exclude the entourage effect, and in fact, a synergistic interaction can occur between the various mechanisms and explain why PEA has multiple effects and the ability to act on different cell types. Indeed, while the ALIA mechanism (Aloe et al., 1993), that is, the ability of PEA to inhibit mast cell degranulation, has been widely confirmed (Facci et al., 1995; Cerrato et al., 2010; De Filippis et al., 2011), the participation of astrocytes, microglia and keratinocytes in PEA anti‐inflammatory actions has also been revealed (Petrosino et al., 2007; Bettoni et al., 2013; Luongo et al., 2013; Guida et al., 2015). These features distinguish PEA from classical steroidal and non‐steroidal anti‐inflammatory drugs that act by inhibiting the cascade of arachidonic acid. Preclinical and human studies indicate that PEA, especially when co‐micronized together with antioxidants, such as luteolin and polydatin, and in micronized or ultra‐micronized forms, is a therapeutic tool with high potential for the effective treatment of different pathologies characterized by neurodegeneration, (neuro)inflammation and pain. Likewise, PEA also shows high safety, as recently reported by Nestmann (2016). While it is intuitive why, for a particularly water insoluble compound such as PEA, any formulation aimed at enhancing its specific surface, such as m‐PEA and um‐PEA, is likely to increase tissue exposure to this compound following oral administration, or why co‐administration of PEA with anti‐oxidants should enhance its efficacy, it must be emphasized that full pharmacokinetic/pharmacodynamic comparisons between normal and m‐PEA or um‐PEA, or between PEA composites and the single components thereof, in the same study, have not been reported yet.
Author contributions
S.P. and V.D. drafted the manuscript. All authors have read and approved the final manuscript.
Conflict of interest
S.P. is an employee of Epitech Group.
Petrosino, S. , and Di Marzo, V. (2017) The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. British Journal of Pharmacology, 174: 1349–1365. doi: 10.1111/bph.13580.
References
- Abramo F, Campora L, Albanese F, della Valle MF, Cristino L, Petrosino S et al. (2014). Increased levels of palmitoylethanolamide and other bioactive lipid mediators and enhanced local mast cell proliferation in canine atopic dermatitis. BMC Vet Res 10: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad A, Genovese T, Impellizzeri D, Crupi R, Velardi E, Marino A et al. (2012a). Reduction of ischemic brain injury by administration of palmitoylethanolamide after transient middle cerebral artery occlusion in rats. Brain Res 1477: 45–58 .13 [DOI] [PubMed] [Google Scholar]
- Ahmad A, Crupi R, Impellizzeri D, Campolo M, Marino A, Esposito E et al. (2012b). Administration of palmitoylethanolamide (PEA) protects the neurovascular unit and reduces secondary injury after traumatic brain injury in mice. Brain Behav Immun 26: 1310–1321. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The concise guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015d). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloe L, Leon A, Levi‐Montalcini R (1993). A proposed autacoid mechanism controlling mastocyte behaviour. Agents Actions 39: 145–147. [DOI] [PubMed] [Google Scholar]
- Ambrosino P, Soldovieri MV, Russo C, Taglialatela M (2013). Activation and desensitization of TRPV1 channels in sensory neurons by the PPARα agonist palmitoylethanolamide. Br J Pharmacol 168: 1430–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosino P, Soldovieri MV, De Maria M, Russo C, Taglialatela M (2014). Functional and biochemical interaction between PPARα receptors and TRPV1 channels: potential role in PPARα agonists‐mediated analgesia. Pharmacol Res 87: 113–122. [DOI] [PubMed] [Google Scholar]
- Andresen SR, Bing J, Hansen RM, Biering‐Sørensen F, Johannesen IL, Hagen EM et al. (2016). Ultramicronized palmitoylethanolamide in spinal cord injury neuropathic pain: a randomized, double‐blind, placebo‐controlled trial. Pain 157: 2097–2103. [DOI] [PubMed] [Google Scholar]
- Artamonov M, Zhukov O, Shuba I, Storozhuk L, Khmel T, Klimashevsky V et al. (2005). Incorporation of labelled N‐acylethanolamine (NAE) into rat brain regions in vivo and adaptive properties of saturated NAE under x‐ray irradiation. Ukr Biokhim Zh (1999) 77: 51–62. [PubMed] [Google Scholar]
- Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Makriyannis A et al. (2001). Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J 15: 300–302. [DOI] [PubMed] [Google Scholar]
- Basu S, Dittel BN (2011). Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol Res 51: 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdyshev EV, Schmid PC, Dong Z, Schmid HH (2000). Stress‐induced generation of N‐acylethanolamines in mouse epidermal JB6 P+ cells. Biochem J 2: 369–374. [PMC free article] [PubMed] [Google Scholar]
- Berger C, Schmid PC, Schabitz WR, Wolf M, Schwab S, Schmid HH (2004). Massive accumulation of N‐acylethanolamines after stroke. Cell signalling in acute cerebral ischemia? J Neurochem 88: 1159–1167. [DOI] [PubMed] [Google Scholar]
- Bettoni I, Comelli F, Colombo A, Bonfanti P, Costa B (2013). Non‐neuronal cell modulation relieves neuropathic pain: efficacy of the endogenous lipid palmitoylethanolamide. CNS Neurol Disord Drug Targets 12: 34–44. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Maurelli S, Melck D, De Petrocellis L, Di Marzo V (1997). Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem 272: 3315–3323. [DOI] [PubMed] [Google Scholar]
- Blancaflor EB, Kilaru A, Keereetaweep J, Khan BR, Faure L, Chapman KD (2014). N‐Acylethanolamines: lipid metabolites with functions in plant growth and development. Plant J 79: 568–583. [DOI] [PubMed] [Google Scholar]
- Borrelli F, Romano B, Petrosino S, Pagano E, Capasso R, Coppola D et al. (2015). Palmitoylethanolamide, a naturally occurring lipid, is an orally effective intestinal anti‐inflammatory agent. Br J Pharmacol 172: 142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braissant O, Foufelle F, Scotto C, Dauça M, Wahli W (1996). Differential expression of peroxisome proliferator‐activated receptors (PPARs): tissue distribution of PPAR‐alpha, −beta, and ‐gamma in the adult rat. Endocrinology 137: 354–366. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D (1998). Control of pain initiation by endogenous cannabinoids. Nature 394: 277–281. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Piomelli D (2001). Antinociceptive activity of the endogenous fatty acid amide, palmitylethanolamide. Eur J Pharmacol 419: 191–198. [DOI] [PubMed] [Google Scholar]
- Caltagirone C, Cisari C, Schievano C, Di Paola R, Cordaro M, Bruschetta G et al. (2016). Co‐ultramicronized palmitoylethanolamide/luteolin in the treatment of cerebral ischemia: from rodent to man. Transl Stroke Res 7: 54–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campora L, Miragliotta V, Ricci E, Cristino L, Di Marzo V, Albanese F et al. (2012). Cannabinoid receptor type 1 and 2 expression in the skin of healthy dogs and dogs with atopic dermatitis. Am J Vet Res 73: 988–995. [DOI] [PubMed] [Google Scholar]
- Capasso R, Izzo AA, Fezza F, Pinto A, Capasso F, Mascolo N et al. (2001). Inhibitory effect of palmitoylethanolamide on gastrointestinal motility in mice. Br J Pharmacol 134: 945–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso R, Orlando P, Pagano E, Aveta T, Buono L, Borrelli F et al. (2014). Palmitoylethanolamide normalizes intestinal motility in a model of post‐inflammatory accelerated transit: involvement of CB₁ receptors and TRPV1 channels. Br J Pharmacol 171: 4026–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraceni P, Viola A, Piscitelli F, Giannone F, Berzigotti A, Cescon M et al. (2010). Circulating and hepatic endocannabinoids and endocannabinoid‐related molecules in patients with cirrhosis. Liver Int 30: 816–825. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997). The capsaicin receptor: a heat‐activated ion channel in the pain pathway. Nature 389: 816–824. [DOI] [PubMed] [Google Scholar]
- Cerrato S, Brazis P, della Valle MF, Miolo A, Puigdemont A (2010). Effects of palmitoylethanolamide on immunologically induced histamine, PGD2 and TNFalpha release from canine skin mast cells. Vet Immunol Immunopathol 133: 9–15. [DOI] [PubMed] [Google Scholar]
- Cerrato S, Brazis P, Della Valle MF, Miolo A, Petrosino S, Di Marzo V et al. (2012). Effects of palmitoylethanolamide on the cutaneous allergic inflammatory response in Ascaris hypersensitive Beagle dogs. Vet J 191: 377–382. [DOI] [PubMed] [Google Scholar]
- Chen J, Matias I, Dinh T, Lu T, Venezia S, Nieves A et al. (2005). Finding of endocannabinoids in human eye tissues: implications for glaucoma. Biochem Biophys Res Commun 330: 1062–1067. [DOI] [PubMed] [Google Scholar]
- Clemente S (2012). Amyotrophic lateral sclerosis treatment with ultramicronized palmitoylethanolamide: a case report. CNS Neurol Disord Drug Targets 11: 933–936. [DOI] [PubMed] [Google Scholar]
- Cobellis L, Castaldi MA, Giordano V, Trabucco E, De Franciscis P, Torella M et al. (2011). Effectiveness of the association micronized N‐Palmitoylethanolamine (PEA)‐transpolydatin in the treatment of chronic pelvic pain related to endometriosis after laparoscopic assessment: a pilot study. Eur J Obstet Gynecol Reprod Biol 158: 82–86. [DOI] [PubMed] [Google Scholar]
- Coburn AF, Graham CE, Haninger J (1954). The effect of egg yolk in diets on anaphylactic arthritis (passive Arthus phenomenon) in the guinea pig. J Exp Med 100: 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocito D, Peci E, Ciaramitaro P, Merola A, Lopiano L (2014). Short‐term efficacy of ultramicronized palmitoylethanolamide in peripheral neuropathic pain. Pain Res Treat 2014: 854560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti S, Costa B, Colleoni M, Parolaro D, Giagnoni G (2002). Antiinflammatory action of endocannabinoid palmitoylethanolamide and the synthetic cannabinoid nabilone in a model of acute inflammation in the rat. Br J Pharmacol 135: 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaro M, Impellizzeri D, Paterniti I, Bruschetta G, Siracusa R, De Stefano D et al. (2016). Neuroprotective effects of co‐UltraPEALut on secondary inflammatory process and autophagy involved in traumatic brain injury. J Neurotrauma 33: 132–146. [DOI] [PubMed] [Google Scholar]
- Cortright DN, Szallasi A (2004). Biochemical pharmacology of the vanilloid receptor TRPV1. An update. Eur J Biochem 271: 1814–1819. [DOI] [PubMed] [Google Scholar]
- Costa B, Conti S, Giagnoni G, Colleoni M (2002). Therapeutic effect of the endogenous fatty acid amide, palmitoylethanolamide, in rat acute inflammation: inhibition of nitric oxide and cyclo‐oxygenase systems. Br J Pharmacol 137: 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa B, Comelli F, Bettoni I, Colleoni M, Giagnoni G (2008). The endogenous fatty acid amide, palmitoylethanolamide, has anti‐allodynic and anti‐hyperalgesic effects in a murine model of neuropathic pain: involvement of CB(1), TRPV1 and PPARgamma receptors and neurotrophic factors. Pain 139: 541–550. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB (1996). Molecular characterization of an enzyme that degrades neuromodulatory fatty‐acid amides. Nature 384: 83–87. [DOI] [PubMed] [Google Scholar]
- Cristino L, de Petrocellis L, Pryce G, Baker D, Guglielmotti V, Di Marzo V (2006). Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 139: 1405–1415. [DOI] [PubMed] [Google Scholar]
- Crupi R, Impellizzeri D, Bruschetta G, Cordaro M, Paterniti I, Siracusa R et al. (2016). Co‐ultramicronized palmitoylethanolamide/luteolin promotes neuronal regeneration after spinal cord injury. Front Pharmacol 7: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Agostino G, La Rana G, Russo R, Sasso O, Iacono A, Esposito E et al. (2007). Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator‐activated receptor‐alpha agonist, modulates carrageenan‐induced paw edema in mice. J Pharmacol Exp Ther 322: 1137–1143. [DOI] [PubMed] [Google Scholar]
- D'Agostino G, La Rana G, Russo R, Sasso O, Iacono A, Esposito E et al. (2009). Central administration of palmitoylethanolamide reduces hyperalgesia in mice via inhibition of NF‐kappaB nuclear signalling in dorsal root ganglia. Eur J Pharmacol 613: 54–59. [DOI] [PubMed] [Google Scholar]
- D'Agostino G, Russo R, Avagliano C, Cristiano C, Meli R, Calignano A (2012). Palmitoylethanolamide protects against the amyloid‐β25‐35‐induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology 37: 1784–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmani NA, Izzo AA, Degenhardt B, Valenti M, Scaglione G, Capasso R et al. (2005). Involvement of the cannabimimetic compound, N‐palmitoyl‐ethanolamine, in inflammatory and neuropathic conditions: review of the available pre‐clinical data, and first human studies. Neuropharmacology 48: 1154–1163. [DOI] [PubMed] [Google Scholar]
- Daynes RA, Jones DC (2002). Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol 2: 748–759. [DOI] [PubMed] [Google Scholar]
- De Filippis D, D'Amico A, Cipriano M, Petrosino S, Orlando P, Di Marzo V et al. (2010). Levels of endocannabinoids and palmitoylethanolamide and their pharmacological manipulation in chronic granulomatous inflammation in rats. Pharmacol Res 61: 321–328. [DOI] [PubMed] [Google Scholar]
- De Filippis D, Luongo L, Cipriano M, Palazzo E, Cinelli MP, de Novellis V et al. (2011). Palmitoylethanolamide reduces granuloma‐induced hyperalgesia by modulation of mast cell activation in rats. Mol Pain 7: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Novellis V, Luongo L, Guida F, Cristino L, Palazzo E, Russo R et al. (2012). Effects of intra‐ventrolateral periaqueductal grey palmitoylethanolamide on thermoceptive threshold and rostral ventromedial medulla cell activity. Eur J Pharmacol 676: 41–50. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Davis JB, Di Marzo V (2001). Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett 506: 253–256. [DOI] [PubMed] [Google Scholar]
- Demuth DG, Molleman A (2006). Cannabinoid signalling. Life Sci 78: 549–563. [DOI] [PubMed] [Google Scholar]
- Di Cesare Mannelli L, D'Agostino G, Pacini A, Russo R, Zanardelli M, Ghelardini C et al. (2013). Palmitoylethanolamide is a disease‐modifying agent in peripheral neuropathy: pain relief and neuroprotection share a PPAR‐alpha‐mediated mechanism. Mediators Inflamm 2013: 328797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, De Petrocellis L, Sepe N, Buono A (1996). Biosynthesis of anandamide and related acylethanolamides in mouse J774 macrophages and N18 neuroblastoma cells. Biochem J 316: 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, De Petrocellis L (2010). Endocannabinoids as regulators of transient receptor potential (TRP) channels: a further opportunity to develop new endocannabinoid‐based therapeutic drugs. Curr Med Chem 17: 1430–1449. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Orlando P, Bisogno T, Zagoory O, Bifulco M et al. (2001). Palmitoylethanolamide inhibits the expression of fatty acid amide hydrolase and enhances the anti‐proliferative effect of anandamide in human breast cancer cells. Biochem J 358: 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez CM, Martín AD, Ferrer FG, Puertas MI, Muro AL, González JM et al. (2012). N‐palmitoylethanolamide in the treatment of neuropathic pain associated with lumbosciatica. Pain Manag 2: 119–124. [DOI] [PubMed] [Google Scholar]
- Donvito G, Bettoni I, Comelli F, Colombo A, Costa B (2015). Palmitoylethanolamide relieves pain and preserves pancreatic islet cells in a murine model of diabetes. CNS Neurol Disord Drug Targets 14: 452–462. [DOI] [PubMed] [Google Scholar]
- Edwards JG (2014). TRPV1 in the central nervous system: synaptic plasticity, function, and pharmacological implications. Prog Drug Res 68: 77–104. [DOI] [PubMed] [Google Scholar]
- Epps DE, Schmid PC, Natarajan V, Schmid HH (1979). N‐Acylethanolamine accumulation in infarcted myocardium. Biochem Biophys Res Commun 90: 628–633. [DOI] [PubMed] [Google Scholar]
- Esposito E, Cuzzocrea S (2013). Palmitoylethanolamide in homeostatic and traumatic central nervous system injuries. CNS Neurol Disord Drug Targets 12: 55–61. [DOI] [PubMed] [Google Scholar]
- Esposito E, Impellizzeri D, Mazzon E, Paterniti I, Cuzzocrea S (2012). Neuroprotective activities of palmitoylethanolamide in an animal model of Parkinson's disease. PLoS One 7 .e41880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Capoccia E, Turco F, Palumbo I, Lu J, Steardo A et al. (2014). Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4‐dependent PPAR‐α activation. Gut 63: 1300–1312. [DOI] [PubMed] [Google Scholar]
- Esposito E, Impellizzeri D, Bruschetta G, Cordaro M, Siracusa R, Gugliandolo E et al. (2016). A new co‐micronized composite containing palmitoylethanolamide and polydatin shows superior oral efficacy compared to their association in a rat paw model of carrageenan‐induced inflammation. Eur J Pharmacol 782: 107–118. [DOI] [PubMed] [Google Scholar]
- Facci L, Dal Toso R, Romanello S, Buriani A, Skaper SD, Leon A (1995). Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc Natl Acad Sci U S A 92: 3376–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar‐Smith WP, Rice AS (2001). Administration of endocannabinoids prevents a referred hyperalgesia associated with inflammation of the urinary bladder. Anesthesiology 94: 507–513. [DOI] [PubMed] [Google Scholar]
- Franklin A, Parmentier‐Batteur S, Walter L, Greenberg DA, Stella N (2003). Palmitoylethanolamide increases after focal cerebral ischemia and potentiates microglial cell motility. J Neurosci 23: 7767–7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielsson L, Mattsson S, Fowler CJ (2016). Palmitoylethanolamide for the treatment of pain: pharmacokinetics, safety and efficacy. Br J Clin Pharmacol 82: 932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley OH, Graessle OE, Robinson HJ (1958). Anti‐inflammatory activity on compounds obtained from egg yolk, peanut oil, and soybean lecithin. J Lab Clin Med 51: 709–714. [PubMed] [Google Scholar]
- Gatti A, Lazzari M, Gianfelice V, Di Paolo A, Sabato E, Sabato AF (2012). Palmitoylethanolamide in the treatment of chronic pain caused by different etiopathogenesis. Pain Med 13: 1121–1130. [DOI] [PubMed] [Google Scholar]
- Genovese T, Esposito E, Mazzon E, Di Paola R, Meli R, Bramanti P et al. (2008). Effects of palmitoylethanolamide on signaling pathways implicated in the development of spinal cord injury. J Pharmacol Exp Ther 326: 12–23. [DOI] [PubMed] [Google Scholar]
- Ghafouri N, Ghafouri B, Larsson B, Turkina MV, Karlsson L, Fowler CJ et al. (2011). High levels of N‐palmitoylethanolamide and N‐stearoylethanolamide in microdialysate samples from myalgic trapezius muscle in women. PLoS One 6 .e27257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghafouri N, Ghafouri B, Larsson B, Stensson N, Fowler CJ, Gerdle B (2013). Palmitoylethanolamide and stearoylethanolamide levels in the interstitium of the trapezius muscle of women with chronic widespread pain and chronic neck‐shoulder pain correlate with pain intensity and sensitivity. Pain 154: 1649–1658. [DOI] [PubMed] [Google Scholar]
- Ghafouri N, Ghafouri B, Fowler CJ, Larsson B, Turkina MV, Karlsson L et al. (2014). Effects of two different specific neck exercise interventions on palmitoylethanolamide and stearoylethanolamide concentrations in the interstitium of the trapezius muscle in women with chronic neck shoulder pain. Pain Med 15: 1379–1389. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Piomelli D (1998). Isotope dilution GC/MS determination of anandamide and other fatty acylethanolamides in rat blood plasma. FEBS Lett 422: 373–376. [DOI] [PubMed] [Google Scholar]
- Giugliano E, Cagnazzo E, Soave I, Lo Monte G, Wenger JM, Marci R (2013). The adjuvant use of N‐palmitoylethanolamine and transpolydatin in the treatment of endometriotic pain. Eur J Obstet Gynecol Reprod Biol 168: 209–213. [DOI] [PubMed] [Google Scholar]
- Gómez del Pulgar T, Velasco G, Guzmán M (2000). The CB1 cannabinoid receptor is coupled to the activation of protein kinase B/Akt. Biochem J 347: 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia‐Figueira S, Nording ML (2014). Development and validation of a sensitive UPLC‐ESI‐MS/MS method for the simultaneous quantification of 15 endocannabinoids and related compounds in milk and other biofluids. Anal Chem 86: 1186–1195. [DOI] [PubMed] [Google Scholar]
- Grillo SL, Keereetaweep J, Grillo MA, Chapman KD, Koulen P (2013). N‐Palmitoylethanolamine depot injection increased its tissue levels and those of other acylethanolamide lipids. Drug Des Devel Ther 7: 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guida F, Luongo L, Marmo F, Romano R, Iannotta M, Napolitano F et al. (2015). Palmitoylethanolamide reduces pain‐related behaviors and restores glutamatergic synapses homeostasis in the medial prefrontal cortex of neuropathic mice. Mol Brain 8: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG (2009). The endocannabinoid system and pain. CNS Neurol Disord Drug Targets 8: 403–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henstridge CM, Balenga NA, Schröder R, Kargl JK, Platzer W, Martini L et al. (2010). GPR55 ligands promote receptor coupling to multiple signalling pathways. Br J Pharmacol 160: 604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WS, Barrett DA, Randall MD (2008). ‘Entourage’ effects of N‐palmitoylethanolamide and N‐oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br J Pharmacol 155: 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannotti FA, Hill CL, Leo A, Alhusaini A, Soubrane C, Mazzarella E et al. (2014). Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: potential for the treatment of neuronal hyperexcitability. ACS Chem Neurosci 5: 1131–1141. [DOI] [PubMed] [Google Scholar]
- Iannotti FA, Di Marzo V, Petrosino S (2016). Endocannabinoids and endocannabinoid‐related mediators: targets, metabolism and role in neurological disorders. Prog Lipid Res 62: 107–128. [DOI] [PubMed] [Google Scholar]
- Impellizzeri D, Esposito E, Di Paola R, Ahmad A, Campolo M, Peli A et al. (2013). Palmitoylethanolamide and luteolin ameliorate development of arthritis caused by injection of collagen type II in mice. Arthritis Res Ther 15: R192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impellizzeri D, Bruschetta G, Cordaro M, Crupi R, Siracusa R, Esposito E et al. (2014). Micronized/ultramicronized palmitoylethanolamide displays superior oral efficacy compared to nonmicronized palmitoylethanolamide in a rat model of inflammatory pain. J Neuroinflammation 11: 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impellizzeri D, Ahmad A, Bruschetta G, Di Paola R, Crupi R, Paterniti I et al. (2015). The anti‐inflammatory effects of palmitoylethanolamide (PEA) on endotoxin‐induced uveitis in rats. Eur J Pharmacol 761: 28–35. [DOI] [PubMed] [Google Scholar]
- Indraccolo U, Barbieri F (2010). Effect of palmitoylethanolamide‐polydatin combination on chronic pelvic pain associated with endometriosis: preliminary observations. Eur J Obstet Gynecol Reprod Biol 150: 76–79. [DOI] [PubMed] [Google Scholar]
- Issemann I, Green S (1990). Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347: 645–650. [DOI] [PubMed] [Google Scholar]
- Iuvone T, Affaitati G, De Filippis D, Lopopolo M, Grassia G, Lapenna D et al. (2016). Ultramicronized palmitoylethanolamide reduces viscerovisceral hyperalgesia in a rat model of endometriosis plus ureteral calculosis: role of mast cells. Pain 157: 80–91. [DOI] [PubMed] [Google Scholar]
- Izzo AA (2004). Cannabinoids and intestinal motility: welcome to CB2 receptors. Br J Pharmacol 142: 1201–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggar S, Hasnie FS, Sellaturay S, Rice AS (1998). The anti‐hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain 76: 189–199. [DOI] [PubMed] [Google Scholar]
- Jean‐Gilles L, Feng S, Tench CR, Chapman V, Kendall DA, Barrett DA et al. (2009). Plasma endocannabinoid levels in multiple sclerosis. J Neurol Sci 287: 212–215. [DOI] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Robinson I, Garle MJ, Patel A, Sun Y et al. (2008). Inhibition of fatty acid amide hydrolase and cyclooxygenase‐2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator‐activated receptor‐alpha in a model of inflammatory pain. Neuropharmacology 55: 85–93. [DOI] [PubMed] [Google Scholar]
- Julius D (2013). TRP channels and pain. Annu Rev Cell Dev Biol 29: 355–384. [DOI] [PubMed] [Google Scholar]
- Karsak M, Gaffal E, Date R, Wang‐Eckhardt L, Rehnelt J, Petrosino S et al. (2007). Attenuation of allergic contact dermatitis through the endocannabinoid system. Science 316: 1494–1497. [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K et al. (1999). Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19: 4544–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilaru A, Blancaflor EB, Venables BJ, Tripathy S, Mysore KS, Chapman KD (2007). The N‐acylethanolamine‐mediated regulatory pathway in plants. Chem Biodivers 4: 1933–1955. [DOI] [PubMed] [Google Scholar]
- Kondo S, Sugiura T, Kodaka T, Kudo N, Waku K, Tokumura A (1998). Accumulation of various N‐acylethanolamines including N‐arachidonoylethanolamine (anandamide) in cadmium chloride‐administered rat testis. Arch Biochem Biophys 354: 303–310. [DOI] [PubMed] [Google Scholar]
- Kuehl FA, Jacob TA, Ganley OH, Ormond RE, Meisinger MAP (1957). The identification of N‐(2‐hydroxyethyl)‐palmitamide as a naturally occurring anti‐inflammatory agent. J Am Chem Soc 79: 5577–5578. [Google Scholar]
- Lam PM, Marczylo TH, Konje JC (2010). Simultaneous measurement of three N‐acylethanolamides in human bio‐matrices using ultra performance liquid chromatography–tandem mass spectrometry. Anal Bioanal Chem 398: 2089–2097. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K (2008). GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A 105: 2699–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi‐Montalcini R, Skaper SD, Dal Toso R, Petrelli L, Leon A (1996). Nerve growth factor: from neurotrophin to neurokine. Trends Neurosci 19: 514–520. [DOI] [PubMed] [Google Scholar]
- Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A et al. (2005a). The nuclear receptor peroxisome proliferator‐activated receptor‐alpha mediates the anti‐inflammatory actions of palmitoylethanolamide. Mol Pharmacol 67: 15–19. [DOI] [PubMed] [Google Scholar]
- Lo Verme J, La Rana G, Russo R, Calignano A, Piomelli D (2005b). The search for the palmitoylethanolamide receptor. Life Sci 77: 1685–1698. [DOI] [PubMed] [Google Scholar]
- Lo Verme J, Russo R, La Rana G, Fu J, Farthing J, Mattace‐Raso G et al. (2006). Rapid broad‐spectrum analgesia through activation of peroxisome proliferator‐activated receptor‐alpha. J Pharmacol Exp Ther 319: 1051–1061. [DOI] [PubMed] [Google Scholar]
- Loría F, Petrosino S, Mestre L, Spagnolo A, Correa F, Hernangómez M et al. (2008). Study of the regulation of the endocannabinoid system in a virus model of multiple sclerosis reveals a therapeutic effect of palmitoylethanolamide. Eur J Neurosci 28: 633–641. [DOI] [PubMed] [Google Scholar]
- Luongo L, Guida F, Boccella S, Bellini G, Gatta L, Rossi F et al. (2013). Palmitoylethanolamide reduces formalin‐induced neuropathic‐like behaviour through spinal glial/microglial phenotypical changes in mice. CNS Neurol Disord Drug Targets 12: 45–54. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990). Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346: 561–564. [DOI] [PubMed] [Google Scholar]
- Mattace Raso G, Russo R, Calignano A, Meli R (2014). Palmitoylethanolamide in CNS health and disease. Pharmacol Res 86: 32–41. [DOI] [PubMed] [Google Scholar]
- Mazzari S, Canella R, Petrelli L, Marcolongo G, Leon A (1996). N‐(2‐hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down‐modulating mast cell activation. Eur J Pharmacol 300: 227–236. [DOI] [PubMed] [Google Scholar]
- Merriam FV, Wang ZY, Hillard CJ, Stuhr KL, Bjorling DE (2011). Inhibition of fatty acid amide hydrolase suppresses referred hyperalgesia induced by bladder inflammation. BJU Int 108: 1145–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muccioli GG, Stella N (2008). Microglia produce and hydrolyze palmitoylethanolamide. Neuropharmacology 54: 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu‐Shaar M (1993). Molecular characterization of a peripheral receptor for cannabinoids. Nature 365: 61–65. [DOI] [PubMed] [Google Scholar]
- Murillo‐Rodriguez E, Désarnaud F, Prospéro‐García O (2006). Diurnal variation of arachidonoylethanolamine, palmitoylethanolamide and oleoylethanolamide in the brain of the rat. Life Sci 79: 30–37. [DOI] [PubMed] [Google Scholar]
- Naccarato M, Pizzuti D, Petrosino S, Simonetto M, Ferigo L, Grandi FC et al. (2010). Possible anandamide and palmitoylethanolamide involvement in human stroke. Lipids Health Dis 9: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy I, Friston D, Valente JS, Torres Perez JV, Andreou AP (2014). Pharmacology of the capsaicin receptor, transient receptor potential vanilloid type‐1 ion channel. Prog Drug Res 68: 39–76. [DOI] [PubMed] [Google Scholar]
- Nestmann ER (2016). Safety of micronized palmitoylethanolamide (microPEA): lack of toxicity and genotoxic potential. Food Sci Nutr . doi:10.1002/fsn3.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noli C, Della Valle MF, Miolo A, Medori C, Schievano C (2015). Efficacy of ultra‐micronized palmitoylethanolamide in canine atopic dermatitis: an open‐label multi‐centre study. Vet Dermatol 26: 432–440. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N (2004). Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem 279: 5298–5305. [DOI] [PubMed] [Google Scholar]
- Paladini A, Fusco M, Cenacchi T, Schievano C, Piroli A, Varrassi G (2016). Palmitoylethanolamide, a Special Food for Medical Purposes, in the Treatment of Chronic Pain: A Pooled Data Meta‐analysis. Pain Physician 19: 11–24. [PubMed] [Google Scholar]
- Palma E, Reyes‐Ruiz JM, Lopergolo D, Roseti C, Bertollini C, Ruffolo G et al. (2016). Acetylcholine receptors from human muscle as pharmacological targets for ALS therapy. Proc Natl Acad Sci U S A 113: 3060–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterniti I, Impellizzeri D, Crupi R, Morabito R, Campolo M, Esposito E et al. (2013a). Molecular evidence for the involvement of PPAR‐δ and PPAR‐γ in anti‐inflammatory and neuroprotective activities of palmitoylethanolamide after spinal cord trauma. J Neuroinflammation 10: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterniti I, Impellizzeri D, Di Paola R, Navarra M, Cuzzocrea S, Esposito E (2013b). A new co‐ultramicronized composite including palmitoylethanolamide and luteolin to prevent neuroinflammation in spinal cord injury. J Neuroinflammation 10: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterniti I, Cordaro M, Campolo M, Siracusa R, Cornelius C, Navarra M et al. (2014). Neuroprotection by association of palmitoylethanolamide with luteolin in experimental Alzheimer's disease models: the control of neuroinflammation. CNS Neurol Disord Drug Targets 13: 1530–1541. [DOI] [PubMed] [Google Scholar]
- Paterniti I, Di Paola R, Campolo M, Siracusa R, Cordaro M, Bruschetta G et al. (2015). Palmitoylethanolamide treatment reduces retinal inflammation in streptozotocin‐induced diabetic rats. Eur J Pharmacol 769: 313–323. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (1997). Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther 74: 129–180. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (2007). GPR55: a new member of the cannabinoid receptor clan? Br J Pharmacol 152: 984–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessina F, Capasso R, Borrelli F, Aveta T, Buono L, Valacchi G et al. (2015). Protective effect of palmitoylethanolamide in a rat model of cystitis. J Urol 193: 1401–1408. [DOI] [PubMed] [Google Scholar]
- Petrosino S, Palazzo E, de Novellis V, Bisogno T, Rossi F, Maione S et al. (2007). Changes in spinal and supraspinal endocannabinoid levels in neuropathic rats. Neuropharmacology 52: 415–422. [DOI] [PubMed] [Google Scholar]
- Petrosino S, Cristino L, Karsak M, Gaffal E, Ueda N, Tüting T et al. (2010a). Protective role of palmitoylethanolamide in contact allergic dermatitis. Allergy 65: 698–711. [DOI] [PubMed] [Google Scholar]
- Petrosino S, Iuvone T, Di Marzo V (2010b). N‐palmitoyl‐ethanolamine: biochemistry and new therapeutic opportunities. Biochimie 92: 724–727. [DOI] [PubMed] [Google Scholar]
- Petrosino S, Schiano Moriello A, Cerrato S, Fusco M, Puigdemont A, De Petrocellis L et al. (2016a). The anti‐inflammatory mediator palmitoylethanolamide enhances the levels of 2‐arachidonoyl‐glycerol and potentiates its actions at transient receptor potential vanilloid type‐1 channels. Br J Pharmacol 173: 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S, Puigdemont A, Della Valle MF, Fusco M, Verde R, Allarà M et al. (2016b). Adelmidrol increases the endogenous concentrations of palmitoylethanolamide in canine keratinocytes and down‐regulates an inflammatory reaction in an in vitro model of contact allergic dermatitis. Vet J 207: 85–91. [DOI] [PubMed] [Google Scholar]
- Rahimi A, Faizi M, Talebi F, Noorbakhsh F, Kahrizi F, Naderi N (2015). Interaction between the protective effects of cannabidiol and palmitoylethanolamide in experimental model of multiple sclerosis in C57BL/6 mice. Neuroscience 290: 279–287. [DOI] [PubMed] [Google Scholar]
- Rasenack N, Müller BW (2004). Micron‐size drug particles: common and novel micronization techniques. Pharm Dev Technol 9: 1–13. [DOI] [PubMed] [Google Scholar]
- Re G, Barbero R, Miolo A, Di Marzo V (2007). Palmitoylethanolamide, endocannabinoids and related cannabimimetic compounds in protection against tissue inflammation and pain: potential use in companion animals. Vet J 173: 21–30. [DOI] [PubMed] [Google Scholar]
- Richardson D, Pearson RG, Kurian N, Latif ML, Garle MJ, Barrett DA et al. (2008). Characterisation of the cannabinoid receptor system in synovial tissue and fluid in patients with osteoarthritis and rheumatoid arthritis. Arthritis Res Ther 10: R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J et al. (2007). The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152: 1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailler S, Schmitz K, Jäger E, Ferreiros N, Wicker S, Zschiebsch K et al. (2014). Regulation of circulating endocannabinoids associated with cancer and metastases in mice and humans. Oncoscience 1: 272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez MG, Ruiz‐Llorente L, Sánchez AM, Díaz‐Laviada I (2003). Activation of phosphoinositide 3‐kinase/PKB pathway by CB(1) and CB(2) cannabinoid receptors expressed in prostate PC‐3 cells. Involvement in Raf‐1 stimulation and NGF induction. Cell Signal 15: 851–859. [DOI] [PubMed] [Google Scholar]
- Sanchez AM, Cioffi R, Viganò P, Candiani M, Verde R, Piscitelli F et al. (2016). Elevated systemic levels of endocannabinoids and related mediators across the menstrual cycle in women with endometriosis. Reprod Sci 23: 1071–1079. [DOI] [PubMed] [Google Scholar]
- Sarnelli G, D'Alessandro A, Iuvone T, Capoccia E, Gigli S, Pesce M et al. (2016). Palmitoylethanolamide modulates inflammation‐associated vascular endothelial growth factor (VEGF) signaling via the Akt/mTOR pathway in a selective peroxisome proliferator‐activated receptor alpha (PPAR‐α)‐dependent manner. PLoS One 11 .e0156198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäbitz WR, Giuffrida A, Berger C, Aschoff A, Schwaninger M, Schwab S et al. (2002). Release of fatty acid amides in a patient with hemispheric stroke: a microdialysis study. Stroke 33: 2112–2114. [DOI] [PubMed] [Google Scholar]
- Schifilliti C, Cucinotta L, Fedele V, Ingegnosi C, Luca S, Leotta C (2014). Micronized palmitoylethanolamide reduces the symptoms of neuropathic pain in diabetic patients. Pain Res Treat 2014: 849623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomacher M, Müller HD, Sommer C, Schwab S, Schäbitz WR (2008). Endocannabinoids mediate neuroprotection after transient focal cerebral ischemia. Brain Res 1240: 213–220. [DOI] [PubMed] [Google Scholar]
- Schreiber D, Harlfinger S, Nolden BM, Gerth CW, Jaehde U, Schömig E et al. (2007). Determination of anandamide and other fatty acyl ethanolamides in human serum by electrospray tandem mass spectrometry. Anal Biochem 361: 162–168. [DOI] [PubMed] [Google Scholar]
- Schuel H, Burkman LJ, Lippes J, Crickard K, Forester E, Piomelli D et al. (2002). N‐Acylethanolamines in human reproductive fluids. Chem Phys Lipids 121: 211–227. [DOI] [PubMed] [Google Scholar]
- Scuderi C, Esposito G, Blasio A, Valenza M, Arietti P, Steardo L Jr et al. (2011). Palmitoylethanolamide counteracts reactive astrogliosis induced by β‐amyloid peptide. J Cell Mol Med 15: 2664–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuderi C, Valenza M, Stecca C, Esposito G, Carratù MR, Steardo L (2012). Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator‐activated receptor‐α. J Neuroinflammation 9: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuderi C, Stecca C, Valenza M, Ratano P, Bronzuoli MR, Bartoli S et al. (2014). Palmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer's disease. Cell Death Dis 5 .e1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharir H, Console‐Bram L, Mundy C, Popoff SN, Kapur A, Abood ME (2012). The endocannabinoids anandamide and virodhamine modulate the activity of the candidate cannabinoid receptor GPR55. J Neuroimmune Pharmacol 7: 856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu MY, Fowler AJ, Kao J, Schmuth M, Schoonjans K, Auwerx J et al. (2002). Topical peroxisome proliferator activated receptor‐alpha activators reduce inflammation in irritant and allergic contact dermatitis models. J Invest Dermatol 118: 94–101. [DOI] [PubMed] [Google Scholar]
- Siracusa R, Paterniti I, Impellizzeri D, Cordaro M, Crupi R, Navarra M et al. (2015a). The association of palmitoylethanolamide with luteolin decreases neuroinflammation and stimulates autophagy in Parkinson's disease model. CNS Neurol Disord Drug Targets 14: 1350–1365. [DOI] [PubMed] [Google Scholar]
- Siracusa R, Paterniti I, Bruschetta G, Cordaro M, Impellizzeri D, Crupi R et al. (2015b). The association of palmitoylethanolamide with luteolin decreases autophagy in spinal cord injury. Mol Neurobiol 53: 3783–3792. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Skaper SD, Facci L, Barbierato M, Zusso M, Bruschetta G, Impellizzeri D et al. (2015). N‐Palmitoylethanolamine and neuroinflammation: a novel therapeutic strategy of resolution. Mol Neurobiol 52: 1034–1042. [DOI] [PubMed] [Google Scholar]
- Solorzano C, Zhu C, Battista N, Astarita G, Lodola A, Rivara S et al. (2009). Selective N‐acylethanolamine‐hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci U S A 106: 20966–20971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaziano G, Luongo L, Guida F, Petrosino S, Matteis M, Palazzo E et al. (2015). Exposure to allergen causes changes in NTS neural activities after intratracheal capsaicin application, in endocannabinoid levels and in the glia morphology of NTS. Biomed Res Int 2015: 980983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence JK, Bhattachar SN, Wesley JA, Martin PJ, Babu SR (2005). Increased dissolution rate and bioavailability through comicronization with microcrystalline cellulose. Pharm Dev Technol 10: 451–460. [DOI] [PubMed] [Google Scholar]
- Starowicz K, Cristino L, Di Marzo V (2008). TRPV1 receptors in the central nervous system: potential for previously unforeseen therapeutic applications. Curr Pharm Des 14: 42–54. [DOI] [PubMed] [Google Scholar]
- Starowicz K, Makuch W, Osikowicz M, Piscitelli F, Petrosino S, Di Marzo V et al. (2012). Spinal anandamide produces analgesia in neuropathic rats: possible CB(1)‐ and TRPV1‐mediated mechanisms. Neuropharmacology 62: 1746–1755. [DOI] [PubMed] [Google Scholar]
- Stella N (2010). Cannabinoid and cannabinoid‐like receptors in microglia, astrocytes, and astrocytomas. Glia 58: 1017–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Piomelli D (2001). Receptor‐dependent formation of endogenous cannabinoids in cortical neurons. Eur J Pharmacol 425: 189–196. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Kishimoto S, Miyashita T, Nakane S, Kodaka T et al. (2000). Evidence that 2‐arachidonoylglycerol but not N‐palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL‐60 cells. J Biol Chem 275: 605–612. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Sun YX, Okamoto Y, Araki N, Tonai T, Ueda N (2005). Molecular characterization of N‐acylethanolamine‐hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J Biol Chem 280: 11082–11092. [DOI] [PubMed] [Google Scholar]
- Turu G, Hunyady L (2010). Signal transduction of the CB1 cannabinoid receptor. J Mol Endocrinol 44: 75–85. [DOI] [PubMed] [Google Scholar]
- Ueda N, Yamanaka K, Yamamoto S (2001). Purification and characterization of an acid amidase selective for N‐palmitoylethanolamine, a putative endogenous anti‐inflammatory substance. J Biol Chem 276: 35552–35557. [DOI] [PubMed] [Google Scholar]
- Vacondio F, Bassi M, Silva C, Castelli R, Carmi C, Scalvini L et al. (2015). Amino acid derivatives as palmitoylethanolamide prodrugs: synthesis, in vitro metabolism and in vivo plasma profile in rats. PLoS One 10 .e0128699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables BJ, Waggoner CA, Chapman KD (2005). N‐Acylethanolamines in seeds of selected legumes. Phytochemistry 66: 1913–1918. [DOI] [PubMed] [Google Scholar]
- Walter L, Franklin A, Witting A, Moller T, Stella N (2002). Astrocytes in culture produce anandamide and other acylethanolamides. J Biol Chem 277: 20869–20876. [DOI] [PubMed] [Google Scholar]
- Whiteside GT, Lee GP, Valenzano KJ (2007). The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists. Curr Med Chem 14: 917–936. [DOI] [PubMed] [Google Scholar]
- Yang H, Zhou J, Lehmann C (2016). GPR55 ‐ a putative "type 3" cannabinoid receptor in inflammation. J Basic Clin Physiol Pharmacol 27: 297–302. [DOI] [PubMed] [Google Scholar]
- Zhukov OD (1999). (1999). [Distribution of N‐([1‐14C]‐palmitoyl)ethanolamine in rat tissues]. Ukr Biokhim Zh 71: 124–125. [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V et al. (1999). Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400: 452–457. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Ermund A, Movahed P, Andersson DA, Simonsen C, Jönsson BA et al. (2013). Monoacylglycerols activate TRPV1‐‐a link between phospholipase C and TRPV1. PLoS One 8 .e81618 [DOI] [PMC free article] [PubMed] [Google Scholar]