

Abstract
RNA-binding proteins are functionally diverse within cells, being involved in RNA-metabolism, translation, DNA damage repair, and gene regulation at both the transcriptional and post-transcriptional levels. Much has been learnt about their interactions with RNAs through structure determination techniques and computational modeling. This review gives an overview of the structural data currently available for protein–RNA complexes, and discusses the technical issues facing structural biologists working to solve their structures. The review focuses on three techniques used to solve the 3-dimensional structure of protein–RNA complexes at atomic resolution, namely X-ray crystallography, solution nuclear magnetic resonance (NMR) and cryo-electron microscopy (cryo-EM). The review then focuses on the main computational modeling techniques that use these atomic resolution data: discussing the prediction of RNA-binding sites on unbound proteins, docking proteins, and RNAs, and modeling the molecular dynamics of the systems. In conclusion, the review looks at the future directions this field of research might take.
Keywords: Protein–RNA complexes, RNA-binding proteins, X-ray crystallography, Nuclear magnetic resonance, Cryo-electron microscopy, Computational modeling, RNA-binding prediction, Structural biology
Introduction
RNA-binding proteins are functionally diverse within cells, and much has been learnt about their interactions with RNAs though structure determination and computational modeling. The 3-dimensional structure of the first protein–RNA complex at atomic resolution was solved using X-ray crystallography (Chen et al. 1989). Since then, nuclear magnetic resonance (NMR) and electronic microscopy (EM) methods have also contributed to the growing number of protein–RNA complexes (1,809 as of 22/07/2016) deposited in the RCSB Protein Data Bank (PDB) (Berman et al. 2000). These structures have been hugely influential in revealing information about protein–RNA interactions. However, the path to protein–RNA complex structure determination has been (and still is) a difficult one, and only now is this field gathering apace and providing the basis for the development of computational modeling techniques.
This review focuses on the challenges faced by those solving protein–RNA structures using X-ray crystallography, NMR, and cryo-EM, and the computational techniques for prediction and analysis of protein–RNA interactions based on this data. It begins with a brief introduction to the functional importance of RNA-binding proteins and summarizes the structural data currently available. The issues using X-ray crystallography and NMR to solve protein–RNA complexes at atomic resolution are then discussed, and the revolution that cryo-EM has brought to the field of ribosomes is presented. The review then outlines the main computational modeling techniques that use this atomic resolution data, including the prediction of interaction sites, docking proteins, and RNAs, and modeling the molecular dynamics of the systems. In conclusion, the review looks at the future focus in this field.
Functional importance of RNA-binding proteins
Using the molecular function annotations in the Gene Ontology (GO) project (Blake et al. 2015), it is possible to estimate that eukaryotic proteomes comprise 4–13 % RNA-binding proteins (RBPs). The fact that RBPs represent such a significant component reflects their diverse function. RBPs are involved in RNA-metabolism, translation, DNA damage repair, and gene regulation at both the transcriptional and post-transcriptional levels. Their diverse functions mean they bind many different types of RNA, including transfer-RNA (tRNA), ribosomal-RNA (rRNA), messenger-RNA (mRNA), and micro-RNA (miRNA). These interactions involve both single-stranded RNA (ssRNA) and double-stranded RNA (dsRNA) binding, with proteins acting as structural scaffolds and enzymes. An understanding of the functional mechanisms of RBPs is only possible if the 3D atomic structures of RBP–RNA complexes are known.
Twenty-seven years of protein–RNA structural data
One of the first structures of a protein–RNA complex deposited in the PDB in 1989 was the bean-pod mottle viral capsid protein bound to ssRNA, determined using X-ray crystallography (Chen et al. 1989). Since then, complexes have been deposited at an increasing rate (Fig. 1), due to a combination of technological developments in structure determination and structural genomics consortia (Grabowski et al. 2016). Currently (22/07/2016), 1,809 protein–RNA complexes are deposited in the PDB (Berman et al. 2000), 80 % of which are X-ray crystal structures, 13 % EM, and 6 % NMR. This compares to 117,098 protein structures in total, with 90 % X-ray, 1 % EM and 9 % NMR.
Fig. 1.

Deposition statistics of protein–RNA complexes in the RCSB PDB (Berman et al. 2000) since the first complex was solved in 1989. The stacked column graph (LH x-axis) shows the number of protein–RNA complexes deposited each year divided by structured determination method. The line graphs (RH x-axis) show the cumulative total of protein–RNA complexes solved by X-ray, NMR, and EM over time
The number of complexes solved is small compared with the number of unbound RBPs deposited. Molecular function GO term annotations (Blake et al. 2015) reveal 5,610 proteins annotated with RNA-binding function in the PDB, with an estimated 69 % having no RNA-ligand bound. The relatively small numbers of annotated RBPs, and the even smaller number with RNAs bound, reflects the unique problems faced by those solving structures using biophysical techniques. It also highlights the need for computational methods to predict the interactions in RBPs, and model the docking and dynamics of bound structures.
X-ray crystallography for protein–RNA complexes
Crystals are solid structures formed by regular arrays of molecules that diffract X-rays in regular and predictable patterns (Shi 2014). To obtain an image of a molecule, the resolution needs to be ∼0.1 nm, and the light wavelengths required for such measurements are in the X-ray range (0.01–10 nm). Hence, X-ray diffraction from crystals is one approach to the structure determination of protein–RNA complexes. X-ray crystallography involves a number of different steps, outlined as: (a) expression or synthesis, and purification of target molecules, (b) screening for optimal crystallization conditions, (c) optimization of crystal quality, (d) diffraction data collection, (e) structure determination, and (f) refinement of 3D model (Krauss et al. 2013). The main bottleneck in this process is obtaining diffraction quality crystals, as there are many parameters that affect crystallization, including temperature, pH, sample concentration, and sample homogeneity (Krauss et al. 2013).
Protein–RNA complexes have proved particularly difficult to crystalize due to conformational flexibility of the protein and the flexibility and negative charge of the RNA. RNAs form diverse structures, from flexible single strands to complex tertiary folds, which contain non-canonical base pairs. Such structures pack loosely in crystals and have a high solvent content, which adversely affects X-ray diffraction. In addition, relatively weak intra-molecular interactions in RNA lead to more flexibility and a higher tendency for the RNA to miss-fold. This can lead to non-homogeneous samples that are also hard to crystallize (Ke and Doudna 2004).
The best approach to the crystallization of protein–RNA complexes has been to engineer the RNA–protein construct, rather than sampling many different crystallization conditions (Obayashi et al. 2007). The length and composition of the RNA included in the construct is critical, and is determined by the size of the minimal binding site necessary for tight complex formation and also for the ability of the RNA to be in its native folded state (Ke and Doudna 2004). Short-chain RNAs (<30 nucleotides) can be synthesized chemically (Oubridge et al. 1995), whilst longer RNAs (>30 nucleotides) can be produced by in-vitro transcription using bacteriophage RNA polymerases (Obayashi et al. 2007). When RNAs are short, it is important for them to be homogenous, as heterogeneous 5′- and 3′-ends can make crystal contacts, and structural heterogeneity can lead to poorly ordered crystals, or can stop crystals forming altogether (Ke and Doudna 2004). RNA-binding proteins comprise multiple domains, and another important step is to identify the minimally folded protein fragment, to include in the construct. This can be done using limited proteolysis on the free or liganded protein, and subsequent analysis of fragments by mass spectroscopy (Krauss et al. 2013). It is also important to avoid the use of phosphate buffers and high salt concentrations to achieve crystallization, as these can bind at or shield recognition sites on the protein and prevent complex formation.
Despite all these technical hurdles, over 1,441 protein–RNA complexes have been successfully solved by X-ray crystallography since 1989. One key advantage of the method for protein–RNA complex determination is that at higher resolutions (<2.5 Å) water molecules are visible, and these play a key role in complex formation. The major disadvantage is that the structures obtained are essentially static images of macromolecules known to have dynamic flexibility.
Solution nuclear magnetic resonance (NMR) spectroscopy for protein–RNA complexes
Solution NMR spectroscopy has the advantage of allowing the collection of dynamic information of flexible macromolecules. NMR is based on the fact that nuclei of isotopes such as 1H, 13C, and 15N carry magnetic dipoles, which take up different orientations with different energies in the magnetic field of an NMR spectrometer (Kwan et al. 2011). Energy state transitions occur according to the rules of quantum mechanics when electromagnetic radiation is applied, resulting in NMR signals. Nuclei in different environments resonate (vibrate) at different frequencies, and plotting intensity against frequency gives a 1-dimensional NMR spectrum (Kwan et al. 2011). By correlating the frequencies of two or more nuclei as they magnetize, multidimensional spectra can be collected (Kwan et al. 2011). Such spectra are combined with constraint information using computer algorithms to calculate a 3D structure, which is an ensemble of different models.
As with X-ray crystallography, NMR of protein–RNA complexes relies on finding optimal RNA and protein constructs at the start of the process. However, an additional problem faced with NMR is the fact that as the size of the macromolecular increases, the NMR signal broadens and decays. Hence, the NMR technique implemented varies dependent upon the size of the complex. RNA–protein complexes < 50 kDa can be solved using standard NMR techniques, using 13C/15N labeling of either the protein and/or the RNA (Carlomagno 2014). The first structure of a human protein–RNA complex solved by NMR was the N-terminal RNA recognition motif (RRM) of the U1A 100 amino acid protein in complex with a 30-nt stem-loop RNA (Allain et al. 1997). As many protein–RNA complexes comprise multiple domain proteins bound to large RNA’s, this molecular weight limitation initially restricted the number of protein–RNA complexes solved by NMR.
However, in recent years an increasing number of high molecular weight protein–RNA complexes (>50 kDa) have been solved by coupling NMR with complementary techniques (Carlomagno 2014). These include small angle X-ray scattering (SAXS) (Hennig and Sattler 2014), and electron paramagnetic resonance (EPR) (Duss et al. 2014). The complementary techniques are used to yield additional restraints for structure calculation and/or validation from sparse NMR data. The 70 kDa complex of RsmZ/RsmE with a noncoding RNA was solved by using NMR for the structures of the individual protein domains, and then using long-range EPR restraints to construct the complete complex (Duss et al. 2014). One of the largest NMR protein–RNA complexes is the 390 kDa archaeal box C/D ribonucleoprotein enzyme bound to RNA (Lapinaite et al. 2013). This structure was solved by coupling NMR with SAXS and small-angle neutron scattering (SANS) (Lapinaite et al. 2013).
The use of complementary techniques alongside solution NMR has led to a total of 109 protein–RNA complexes currently (22/07/2016) being solved. The advantage of NMR for protein–RNA complexes is that the experiments produce ensembles of structures that give a dynamic picture of these flexible complexes (Daubner et al. 2013), but one disadvantage is that the positions of water molecules are not resolved.
Cryo-electron microscopy (cryo-EM) revolutionises knowledge of the ribosome
Whilst crystallography and NMR has given key insights into the structure of protein–RNA complexes, the size of the complex is still a limiting factor. This is a key problem, as many protein–RNA interactions occur within very large macromolecular assemblies such as the ribosome and the spliceosome. The structures of such assemblies were first solved using cryo-EM. The basis of cryo-EM is the imaging of radiation-sensitive molecules in a transmission electron microscope using very low temperature (cryogenic) conditions (Milne et al. 2013), but initially the technique gave structures in excess of 10 Å (Callaway 2015). However, the development of detectors that capture thousands of images (with many frames stored per second), and the advancement of software to build 2D images into a single 3D structure, have recently revolutionized the method (Callaway 2015), leading to the near atomic resolution structure of the human ribosome (Khatter et al. 2015). The 80S human ribosome structure comprises 69 protein chains and five nucleic acid chains, and gave new insights into the tRNA-binding sites (Khatter et al. 2015).
However, one problem with cryo-EM is that the resolution can be highly variable within a single structure (Kucukelbir et al. 2014). The structure of human 80S ribosome varies from 3.6 Å to 2.8 Å resolution, reflecting different local stabilities within the complex (Khatter et al. 2015). In a recent development, the structure of the E.coli ribosome complexed with an elongation factor Tu, an aminoacyl tRNA, and an antibiotic have been solved to a near-uniform 2.9 Å resolution using spherical aberration (Cs)-corrected cryo-EM (Fischer et al. 2015). Spherical aberration is the distortion of an image that occurs when a spherical lens brings rays to a premature focus, leading to a blurred image and reduced resolution (Hawkes 2009). Spherical aberration compensation plates are now being used in cryo-EM systems, and the E.coli ribosome was solved using an aberration corrector (Fischer et al. 2015).
There are currently 249 EM protein–RNA complexes in the PDB (22/07/2016), and of these 210 have been solved using cryo-EM. However, the telling statistic is that only 17 of these complexes have a resolution ≤3.5 Å. A combination of further technological developments in hardware and software for cryo-EM offer the prospects of higher (2.0 Å) resolution macromolecular structures (Glaeser 2016). When this technique becomes mainstream the numbers of atomic resolution protein–RNA complexes solved by cryo-EM will rapidly increase.
Computational methods for protein–RNA interactions
The atomic coordinates of protein–RNA complexes solved by X-ray crystallography, NMR and cryo-EM provide the basis for computational techniques for their analysis and prediction. Computational methods have focused on three key areas: (a) predicting RNA-binding sites on the structures of RNA-BPs solved in the unbound state, (b) docking RNAs with RNA-BPs solved in the unbound state, and (c) modeling of the molecular dynamics of protein–RNA complexes.
Predicting RNA binding sites using structures
There are many methods available to predict the location of RNA-binding sites on the structures of RNA-binding proteins, solved in the unbound state (termed here “target” proteins). In general, methods calculate physical and/or chemical parameters of known RNA-binding residues, and then use this information to search for similar sites on target proteins. The common sequence features used are position-specific scoring matrices (PSSMs) and amino acid propensities, which quantify the propensity for an amino acid to be in a binding site (Murakami et al. 2010). Frequently used structural features include the solvent accessibility of amino acids, surface electrostatic potentials, and geometrical properties such as shape (Sun et al. 2016). The prediction techniques that use these features include machine learning (ML), template methods, and scoring methods.
The most common techniques use supervised ML, in which algorithms learn from the features of known RNA-binding residues (training data) for make predictions for target proteins (Tiwari and Srivastava 2014). Random forests (e.g., Barik et al. 2015), support vector machines (e.g., Maetschke and Yuan 2009) and Naïve Bayes classifiers (e.g., Terribilini et al. 2007) have all been used to predict of RNA-binding sites. One of the most recent methods (RNAProSite) constructs a random forest classifier using electrostatic surface potential and a triplet interface propensity (Sun et al. 2016). Other methods have used ensemble learning, in which multiple ML classifiers are independently trained, and then combined to make predictions for target proteins (e.g., Ren and Shen 2015).
A second prediction technique uses 3D templates to characterize binding sites. This technique extracts and assembles a library of 3D templates of known RNA-binding sites and then structurally aligns them to target proteins. If structural similarity is detected, additional criteria are used to assess the probability RNA binding. One example of this technique is SPOT-Seq-RNA, which uses an energy function to assess the likelihood of binding after template similarity has been established (Zhao et al. 2014).
The ML techniques rely on parameterization of specific training datasets, and template techniques rely on template libraries derived from existing complexes. This means that on novel datasets the accuracy of the predictions can degrade. Scoring methods are designed to overcome this problem to some extent, and they involve the combination of calculated physiochemical and evolutionary features into a scoring function, upon which the probability of RNA-binding is derived (Chen et al. 2014). One technique, RBscore, has a scoring function that integrates electrostatic potential, solvent accessibility, and sequence conservation (Miao and Westhof 2015a).
With so many techniques for RNA-binding prediction available, attempts have been made at benchmarking (Walia et al. 2012; Miao and Westhof 2015b). However, such comparisons are dependent upon the specific datasets used, the definitions of RNA-binding residues, and the statistics used to assess the predictions. Another problem is that some of the false positives result from the misidentification of DNA-binding sites as RNA-binding sites, as they share many characteristics (Shazman et al. 2011). In addition, many binding sites can be identified that fit specific physicochemical criteria, but are not biologically relevant in any system, or only in some specific systems. A further problem with many techniques is their reliance on the existence of close homologs of target proteins to calculate features, such as PSSMs and sequence conservation. When working with model organisms this is not a problem, but when working with non-model species such as agricultural crops or plant pathogens, many techniques have limited prediction potential.
Docking RNAs and proteins
Methods for docking two molecules generally consist of four parts: (a) docking of the structures, usually using fast rigid body docking, (b) scoring of the resulting complexes using a potential based on chemical and/or structural properties derived from known complexes, (c) selection of the ‘best’ models based on the core, and (d) model refinement. There are many methods available for docking proteins with RNAs: GRAMM (Katchalski-Katzir et al. 1992), Haddock (Van Zundert et al. 2016), Hex (Ritchie and Kemp 2000), PatchDock (Schneidman-Duhovny et al. 2005), and FTDock (3D-Dock) (Gabb et al. 1997)). But most were developed for docking protein–protein complexes, and hence many originally lacked specific protein–RNA scoring potentials (Puton et al. 2012).
However, potentials based on reverse Boltzmann statistics [DARS-RNP and QUASI-RNP (Tuszynska and Bujnicki 2011)], propensities [e.g., OPRA (Perez-Cano and Fernandez-Recio 2010)] and statistical mechanics [e.g., ITScore (Huang and Zou 2014)] have since been developed and are available for use with different methods. One recent method, NPDock, exemplifies recent contributions to this field (Tuszynska et al. 2015). Designed for the non-expert user, it combines GRAMM and the protein–RNA specific potentials (DARS-RNP and QUASI-RNP) (Tuszynska and Bujnicki 2011) with tools for clustering, selection, and model optimization. NPDock (Tuszynska et al. 2015), and other similar methods, produce protein–RNA models worthy of further analysis when the RNA is double stranded. But docking ssRNAs to proteins is more difficult due to the huge flexibility of the RNA. However, this has been addressed in a new fragment-based approach that uses the structure of the protein and the sequence of the ssRNA (Chauvot de Beauchene et al. 2016). The method deals with the flexibility of the ssRNA by building potential structures from small fragments.
Simulating the molecular dynamics of protein–RNA interactions
One key issue in both docking and modeling the molecular dynamics (MD) of protein–RNA complexes is the flexibility within the system. RNA molecules undergo conformational changes triggered by a range of signals, and nucleotides contacting amino acids are enriched in unusual and unique conformations (Kligun and Mandel-Gutfreund 2015). Conformational changes, both small and large, have also been observed in RNA-BPs (Ellis and Jones 2008). RNA-complex formation can involve coupled association and folding process, and in some RNA-BPs, regions that are unstructured in the unbound protein become structured upon binding (Qin et al. 2010).
This flexibility and the representation of water molecules presents problems when molecular dynamics is used to assess the energy landscape accessible to protein–RNA complexes (Barik and Bahadur 2014). Modelling the MD of proteins (Dror et al. 2012) and RNA (Šponer et al. 2012) as separate entities is complex, with the selection of an appropriate force field for simulations a key aspect. The frequently used force fields such as AMBER and CHARMM do have variants for simulating dsRNA dynamics, but achieving accurate simulations over a meaningful timeframe for protein–RNA complexes with non-canonical dsRNA structures or ssRNAs is still challenging (Fulle and Gohlke 2010). However, interesting examples have been achieved. An MD simulation of the binding of P22 N-peptide with RNA revealed that the electrostatic field of the RNA influenced transitions in secondary structure (coil to α-helix) outside of the RNA-binding site (Bahadur et al. 2009). In more recent work, explicit solvent MD simulations of Csy4 endoribonuclease complexed with a CRISPR (clustered regularly interspaced short palindromic repeat)-derived RNA gave insight into the potential catalytic mechanisms of the endonuclease, but also highlighted limitations in the use of the AMBER derived force field (Estarellas et al. 2015).
One difficulty in the simulation of the CSY4/RNA complex was the fact that the endonuclease was multi-domain. In such proteins, domains are commonly connected by flexible linkers, and this means that the domains can interact with the RNA in different ways (Mackereth and Sattler 2012). One interesting mechanism proposed for RNA-complex formation is conformational selection. In this mechanism, the protein in the absence of the RNA exists in a dynamic equilibrium of two states, bound and unbound. The bound state preexists before binding, and the binding of the RNA captures the bound conformation, leading to a population shift in the equilibrium. This mechanism has actually been observed from the solution structures of the RBP U2AF65, which is involved in pre-mRNA splicing (Mackereth et al. 2011). Such observations highlights the importance of enhancing techniques for solving RBPs in solution, the necessity for protein–RNA specific software for MD simulations, and the development of more sophisticated RNA-specific force fields.
Lessons learnt from structural data and modeling
The availability of over 5,000 RBP structures and 1,809 protein–RNA complex structures means that much has been learnt about protein–RNA interactions. Initial observations showed that there were a number of common folded domains, including the RNA recognition motif (RRM), K- homology (KH) domain, double-stranded RNA-binding domain (dsRBD), Paz domain, and Piwi domain (Lunde et al. 2007). These have defined secondary structure elements forming the RNA-binding sites (Fig. 2a–e), but others such as the zinc finger motif ZNF-CCCH are predominantly unstructured binding sites (Fig. 2f). Like other classes of proteins, many RBPs have multiple domains; with each domain making contact with 2–10 RNA nucleotides. The domains are commonly connected by flexible linkers which allow the recognition of diverse RNA sequences using different interaction mechanisms (Mackereth and Sattler 2012). Individual domains often bind with low affinity, but cooperative binding of multiple domains (either the same or different ones) leads to increased affinity and in many cases specificity (Cook et al. 2015). The structures of ribosomes from a number of different organisms revealed that these large dynamic assemblies comprise RBPs with many different RNA-binding folds, as well as many disordered regions.
Fig. 2.
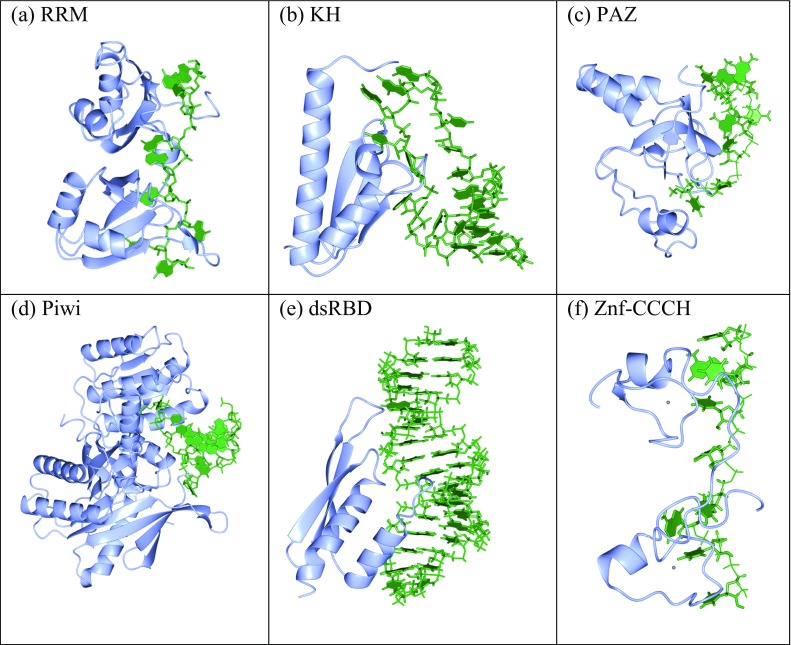
Examples of RNA-binding domains revealed using X-ray crystallography and NMR structure determination techniques. a RNA-recognition motif (RRM): example PDB:1CVJ. b K-homology domain (KH): example PDB:1EC6. c PAZ domain: example PDB:2XFM. d PIWI domain: example PDB:2BGG. e Double-stranded RNA-binding domain (dsRBD): example PDB:2L3C. f Zinc finger CCCH domain: example PDB:1RGO. In each figure, the protein is shown in ribbon format depicting the secondary structure (blue) and the RNA is shown in ball-and-stick format with nucleic acid base blocks (green). The figures have been rendered using the CCP4MG package (McNicholas et al. 2011)
Messenger RNA interactome data has also revealed that one third of RBPs, conserved from yeast to humans, do not feature the classical domains and did not have a previously known RNA-binding function (Beckmann et al. 2015). A significant number of these these potentially new RBPs were glycolytic enzymes (Beckmann et al. 2015). This has led to connections being proposed between RNA-biology and other cellular functions such as metabolism (Beckmann et al. 2016).
Future directions
Structural biology has provided data that has made fundamental contributions to the field of protein–RNA interactions. Computational biology has used this data to increase our understanding of these macromolecules even further. We now have a clearer understanding of the importance of protein and RNA flexibility, and unstructured regions of the protein. Future work needs to focus on how non-classical RBP domains and unstructured regions contribute to binding. In addition, more work is needed to derive the structures of RBPs with ssRNAs bound, and new software for more accurate docking of RNA–protein complexes, and for simulating the dynamics at the molecular level needs to be developed. The insights into the ribosome that have been provided by cryo-EM mean that further technological developments in this field will be key to solving large RNA and protein assemblies at even higher resolutions. The comprehensive array of RNA-binding proteins recently derived from interactome capture methods (Castello et al. 2012; Beckmann et al. 2015) will also provide new targets for structural genomics consortia in the future.
The recent report of an engineered RBP protein PumHD (Pumlio Homology Domain) being used to measure RNA translation within living cells demonstrates the advancing importance of RBPs (Adamala et al. 2016). Such work serves to highlight the need for further understanding of RNA recognition by proteins through structure determination and computational biology. The last 27 years of protein–RNA structural data has clearly paved the way for many more.
Acknowledgements
SJ was funded by the Scottish Government’s Rural and Environment Science and Analytical Services Division (RESAS) research programme. SJ would like to thank the editors for the invitation to contribute to this special issue and to wish a very happy 80th birthday to Professor Don Winzor.
Compliance with ethical standards
Conflict of Interest
Susan Jones declares that she has no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by the authors.
Footnotes
This article is part of a Special Issue on ‘Analytical Quantitative Relations in Biochemistry’ edited by Damien Hall and Stephen Harding
References
- Adamala KP, Martin-Alarcon DA, Boyden ES. Programmable RNA-binding protein composed of repeats of a single modular unit. Proc Natl Acad Sci. 2016;113(19):E2579–E2588. doi: 10.1073/pnas.1519368113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allain FHT, Howe PWA, Neuhaus D, Varani G. Structural basis of the RNA-binding specificity of human U1A protein. EMBO J. 1997;16:5764–5774. doi: 10.1093/emboj/16.18.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahadur RP, Kannan S, Zacharias M. Binding of the bacteriophage P22 N-peptide to the boxB RNA motif studied by molecular dynamics simulations. Biophys J. 2009;97:3139–3149. doi: 10.1016/j.bpj.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik A, Bahadur RPR. Hydration of protein–RNA recognition sites. Nucleic Acids Res. 2014;42:10148–10160. doi: 10.1093/nar/gku679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik A, Nithin C, Karampudi NBR, et al. Probing binding hot spots at protein–RNA recognition sites. Nucleic Acids Res. 2015;44(2):e9. doi: 10.1093/nar/gkv876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann BM, Horos R, Fischer B, et al. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat Commun. 2015;6:10127. doi: 10.1038/ncomms10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann BM, Castello A, Medenbach J. The expanding universe of ribonucleoproteins: of novel RNA-binding proteins and unconventional interactions. Pflügers Arch – Eur J Physiol. 2016;468(6):1029–1040. doi: 10.1007/s00424-016-1819-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake JA, Christie KR, Dolan ME, et al. Gene ontology consortium: going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E. The revolution will not be crystallized. Nature. 2015;525:172–174. doi: 10.1038/525172a. [DOI] [PubMed] [Google Scholar]
- Carlomagno T. Present and future of NMR for RNA–protein complexes: a perspective of integrated structural biology. J Magn Reson. 2014;241:126–136. doi: 10.1016/j.jmr.2013.10.007. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Chauvot de Beauchene I, de Vries SJ, Zacharias M. Binding site identification and flexible docking of single stranded RNA to proteins using a fragment-based approach. PLoS Comput Biol. 2016;12:1–21. doi: 10.1371/journal.pcbi.1004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Stauffacher C, Li Y, et al. Protein–RNA interactions in an icosahedral virus at 3.0 A resolution. Science. 1989;245:154–159. doi: 10.1126/science.2749253. [DOI] [PubMed] [Google Scholar]
- Chen YC, Sargsyan K, Wright JD, et al. Identifying RNA-binding residues based on evolutionary conserved structural and energetic features. Nucleic Acids Res. 2014;42(3):e15. doi: 10.1093/nar/gkt1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KB, Hughes TR, Morris QD. High-throughput characterization of protein–RNA interactions. Brief. Funct. Genomics. 2015;14:74–89. doi: 10.1093/bfgp/elu047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubner GM, Cléry A, Allain FHT. RRM–RNA recognition: NMR or crystallography…and new findings. Curr Opin Struct Biol. 2013;23:100–108. doi: 10.1016/j.sbi.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Dror R, Dirks R, Grossman J, et al. Biomolecular simulation: a computational microscope for molecular biology. Annu Rev Biophys. 2012;41:429–452. doi: 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- Duss O, Michel E, Yulikov M, et al. Structural basis of the non-coding RNA RsmZ acting as a protein sponge. Nature. 2014;509:588–592. doi: 10.1038/nature13271. [DOI] [PubMed] [Google Scholar]
- Ellis JJ, Jones S. Evaluating conformational changes in protein structures binding RNA. Proteins. 2008;70:1518–1526. doi: 10.1002/prot.21647. [DOI] [PubMed] [Google Scholar]
- Estarellas C, Otyepka M, Koča J, et al. Molecular dynamic simulations of protein/RNA complexes: CRISPR/Csy4 endoribonuclease. Biochim Biophys Acta. 2015;1850:1072–1090. doi: 10.1016/j.bbagen.2014.10.021. [DOI] [PubMed] [Google Scholar]
- Fischer N, Neumann P, Konevega AL, et al. Structure of the E. coli ribosome–EF-Tu complex at <3 Å resolution by Cs-corrected cryo-EM. Nature. 2015;520:567–570. doi: 10.1038/nature14275. [DOI] [PubMed] [Google Scholar]
- Fulle S, Gohlke H. Molecular recognition of RNA: challenges for modelling interactions and plasticity. J Mol Recognit. 2010;23:220–231. doi: 10.1002/jmr.1000. [DOI] [PubMed] [Google Scholar]
- Gabb HA, Jackson RM, Sternberg MJ. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol. 1997;272:106–120. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
- Glaeser RM. How good can cryo-EM become? Nat Methods. 2016;13:28–32. doi: 10.1038/nmeth.3695. [DOI] [PubMed] [Google Scholar]
- Grabowski M, Niedzialkowska E, Zimmerman MD, Minor W. The impact of structural genomics: the first quindecennial. J. Struct. Funct. Genom. 2016;17:1–16. doi: 10.1007/s10969-016-9201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes PW. Aberration correction past and present. Philos Trans R Soc A Math Phys Eng Sci. 2009;367:3637–3664. doi: 10.1098/rsta.2009.0004. [DOI] [PubMed] [Google Scholar]
- Hennig J, Sattler M. The dynamic duo: Combining NMR and small angle scattering in structural biology. Protein Sci. 2014;23:669–682. doi: 10.1002/pro.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SY, Zou X. A knowledge-based scoring function for protein–RNA interactions derived from a statistical mechanics-based iterative method. Nucleic Acids Res. 2014;42:1–12. doi: 10.1093/nar/gkt1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katchalski-Katzir E, Shariv I, Eisenstein M, et al. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci U S A. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke A, Doudna JA. Crystallization of RNA and RNA-protein complexes. Methods. 2004;34:408–414. doi: 10.1016/j.ymeth.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Khatter H, Myasnikov AG, Natchiar SK, Klaholz BP. Structure of the human 80S ribosome. Nature. 2015;520:640–645. doi: 10.1038/nature14427. [DOI] [PubMed] [Google Scholar]
- Kligun E, Mandel-Gutfreund Y. The role of RNA conformation in RNA–protein recognition. RNA Biol. 2015;12:720–727. doi: 10.1080/15476286.2015.1040977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss IR, Merlino A, Vergara A, Sica F. An overview of biological macromolecule crystallization. Int J Mol Sci. 2013;14:11643–11691. doi: 10.3390/ijms140611643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan AH, Mobli M, Gooley PR, et al. Macromolecular NMR spectroscopy for the non-spectroscopist. FEBS J. 2011;278:687–703. doi: 10.1111/j.1742-4658.2011.08004.x. [DOI] [PubMed] [Google Scholar]
- Lapinaite A, Simon B, Skjaerven L, et al. The structure of the box C/D enzyme reveals regulation of RNA methylation. Nature. 2013;502:519–523. doi: 10.1038/nature12581. [DOI] [PubMed] [Google Scholar]
- Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackereth CD, Sattler M. Dynamics in multi-domain protein recognition of RNA. Curr Opin Struct Biol. 2012;22:287–296. doi: 10.1016/j.sbi.2012.03.013. [DOI] [PubMed] [Google Scholar]
- Mackereth CD, Madl T, Bonnal S, et al. Multi-domain conformational selection underlies pre-mRNA splicing regulation by U2AF. Nature. 2011;475:408–411. doi: 10.1038/nature10171. [DOI] [PubMed] [Google Scholar]
- Maetschke SR, Yuan Z. Exploiting structural and topological information to improve prediction of RNA-protein binding sites. BMC Biochem. 2009;10:341. doi: 10.1186/1471-2105-10-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas S, Potterton E, Wilson KS, Noble MEM. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr Sect D: Biol Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Z, Westhof E. Prediction of nucleic acid binding probability in proteins: a neighboring residue network based score. Nucleic Acids Res. 2015;43:5340–5351. doi: 10.1093/nar/gkv446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Z, Westhof E. A large-scale assessment of nucleic acids binding site prediction programs. PLoS Comput Biol. 2015;11:1–23. doi: 10.1371/journal.pcbi.1004639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JLS, Borgnia MJ, Bartesaghi A, et al. Cryo-electron microscopy — a primer for the non-microscopist. FEBS J. 2013;280:28–45. doi: 10.1111/febs.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Spriggs RV, Nakamura H, Jones S. PiRaNhA: a server for the computational prediction of RNA-binding residues in protein sequences. Nucleic Acids Res. 2010;38(Suppl):W412–6. doi: 10.1093/nar/gkq474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi E, Oubridge C, Pomeran D, Nagai K. Macromolecular crystallography protocols: Volume 1: Preparation and crystallization of macromolecules. Berlin Heidelberg: Springer; 2007. Crystallization of RNA–protein complexes; pp. 259–276. [Google Scholar]
- Oubridge C, Ito N, Teo CH, et al. Crystallisation of RNA-protein complexes. II. The application of protein engineering for crystallisation of the U1A protein–RNA complex. J Mol Biol. 1995;249:409–423. doi: 10.1006/jmbi.1995.0306. [DOI] [PubMed] [Google Scholar]
- Perez-Cano L, Fernandez-Recio J. Optimal protein–RNA area, OPRA: a propensity-based method to identify RNA-binding sites on proteins. Proteins Struct Funct Bioinf. 2010;78:25–35. doi: 10.1002/prot.22527. [DOI] [PubMed] [Google Scholar]
- Puton T, Kozlowski L, Tuszynska I, et al. Computational methods for prediction of protein–RNA interactions. J Struct Biol. 2012;179:261–268. doi: 10.1016/j.jsb.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Qin F, Chen Y, Wu M, et al. Induced fit or conformational selection for RNA/U1A folding. RNA. 2010;16:1053–1061. doi: 10.1261/rna.2008110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Shen Y. RNA-binding residues prediction using structural features. BMC Bioinf. 2015;16:249. doi: 10.1186/s12859-015-0691-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie DW, Kemp GJL. Protein docking using spherical polar fourier correlations. Proteins Struct Funct Genet. 2000;39:178–194. doi: 10.1002/(SICI)1097-0134(20000501)39:2<178::AID-PROT8>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:363–367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shazman S, Elber G, Mandel-Gutfreund Y. From face to interface recognition: a differential geometric approach to distinguish DNA from RNA binding surfaces. Nucleic Acids Res. 2011;39:7390–7399. doi: 10.1093/nar/gkr395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. A glimpse of structural biology through X-ray crystallography. Cell. 2014;159:995–1014. doi: 10.1016/j.cell.2014.10.051. [DOI] [PubMed] [Google Scholar]
- Šponer J, Otyepka M, Banáš P, et al. Molecular dynamics simulations of RNA molecules. Innov Biomol Model Simulat. 2012;2:129–155. doi: 10.1039/9781849735056-00129. [DOI] [Google Scholar]
- Sun M, Wang X, Zou C, et al. Accurate prediction of RNA-binding protein residues with two discriminative structural descriptors. BMC Bioinf. 2016;17:231. doi: 10.1186/s12859-016-1110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terribilini M, Sander JD, Lee JH, et al. RNABindR: a server for analyzing and predicting RNA-binding sites in proteins. Nucleic Acids Res. 2007;35:1–7. doi: 10.1093/nar/gkm294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari AK, Srivastava R. A survey of computational intelligence techniques in protein function prediction. Int J Proteomics. 2014;2014:845479. doi: 10.1155/2014/845479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynska I, Bujnicki JM. DARS-RNP and QUASI-RNP: New statistical potentials for protein–RNA docking. BMC Bioinf. 2011;12:348. doi: 10.1186/1471-2105-12-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynska I, Magnus M, Jonak K, et al. NPDock: a web server for protein–nucleic acid docking. Nucleic Acids Res. 2015;43:W425–W430. doi: 10.1093/nar/gkv493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zundert GCP, Rodrigues JPGLM, Trellet M, et al. The HADDOCK2.2 Web server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428:720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Walia RR, Caragea C, Lewis BA, et al. Protein–RNA interface residue prediction using machine learning: an assessment of the state of the art. BMC Bioinf. 2012;13:1–20. doi: 10.1186/1471-2105-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Yang Y, Janga SC, et al. Prediction and validation of the unexplored RNA-binding protein atlas of the human proteome. Proteins. 2014;82:640–647. doi: 10.1002/prot.24441. [DOI] [PMC free article] [PubMed] [Google Scholar]