

Abstract
Adoption of Electronic Health Record (EHR) systems has led to collection of massive healthcare data, which creates oppor- tunities and challenges to study them. Computational phenotyping offers a promising way to convert the sparse and complex data into meaningful concepts that are interpretable to healthcare givers to make use of them. We propose a novel su- pervised nonnegative tensor factorization methodology that derives discriminative and distinct phenotypes. We represented co-occurrence of diagnoses and prescriptions in EHRs as a third-order tensor, and decomposed it using the CP algorithm. We evaluated discriminative power of our models with an Intensive Care Unit database (MIMIC-III) and demonstrated superior performance than state-of-the-art ICU mortality calculators (e.g., APACHE II, SAPS II). Example of the resulted phenotypes are sepsis with acute kidney injury, cardiac surgery, anemia, respiratory failure, heart failure, cardiac arrest, metastatic cancer (requiring ICU), end-stage dementia (requiring ICU and transitioned to comfort-care), intraabdominal conditions, and alcohol abuse/withdrawal.
Introduction
A phenotype is an outward physical manifestation of a genotype. Investigating the association between phenotypes and genotypes has been a principal genetic research goal1. Electronic health records (EHRs) are increasingly used to identify phenotypes because EHRs encompass several aspects of patient information such as diagnoses, medication, laboratory results, and narrative reports. Given the importance of these efforts, collaborative groups have been created to develop and share phenotypes obtained from EHRs, such as the Electronic Medical Records and Genomics (eMERGE) Network2 and the Observational Medical Outcomes Partnership3. Two of the main obstacles to generate phenotypes are the needs for substantial time and domain expert knowledge4, 5. Furthermore, phenotypes created using clinical judgement6, 7 or healthcare guidelines5, 8 in one institution often cannot be easily ported to the other institutions, reducing generalizability and leading to unstandardized phenotype definitions9.
Consequently, phenotyping based on machine learning has been proposed to facilitate extraction of meaningful phenotypes automatically from EHRs without human supervision through a process called computational phenotyping. The most widely used approach is unsupervised feature extraction that derives meaningful and interpretable characteristics without supervision on data label. Frequent pattern mining defines phenotypes as a pattern that is frequently observed set of ordered items from sequential numerical data such as laboratory10, 11. A natural language processing technique extracts frequent terms from clinical narrative notes and defines phenotypes as a set of relevant and frequent terms12–14. These frequent set mining methods are useful but unable to learn underlying latent characteristics. Deep learning methods such as autoencoders or skip-grams represent patient as a vector15–17, but it is hard to derive understandable latent concepts due to the nonlinear combinations of multiple layers.
Recently, dimensionality reduction phenotyping methods have been introduced to handle sparse and noisy data from EHRs’ large and heterogeneous features. These methods represent phenotypes as latent medical concepts18. That is, the phenotypes are defined as a probabilistic membership to medical components, and patients also have a probabilistic membership to the phenotypes. For example, Bayesian finite mixture modeling discovers Parkinson’s disease phenotypes as latent subgroups19. Another dimensionality reduction technique, matrix factorization, decomposes time-series matrix data from EHRs into latent medical concepts20–22. Most recently, nonnegative tensor factorization (NTF) is becoming particularly popular due to its ability to capture high dimensional data. It generates latent medical concepts using interaction between components from multiple information source23–27. Ho et al. first introduce NTF for phenotyping23, 24. They define phenotypes as sets of co-occurring diagnoses and prescrptions, and obtain the phenotypes from latent representation of the co-occurrence. They use Kullback-Leibler divergence to decompose the observed co-occurrences that follow Poisson distribution based on CP decomposition. Ho et al. also incorporate sparsity constraints by setting thresholds for negligibly small values. Wang et al. enforce orthogonality constraints on NTF to derive less overlapping phenotypes25. Another NTF based on Tucker decomposition discovers (high-order) feature subgroups as decomposing the tensor into a core tensor multiplied by orthogonal factor matrices for each mode. It uses the core tensor to encode interactions among elements in each mode26, 28.
One of important characteristics that phenotypes should have is to be discriminative to a certain clinical outcome of interest such as mortality, readmission, cost, et al. So far, however, there has been little consideration about discriminative phenotypes associated with certain clinical outcomes. The discriminative phenotypes can be beneficial to clinicians because they can directly apply the phenotypes to their daily practice to improve the clinical outcome of interest. For example, clinicians can use our phenotype to evaluate patients’ risk of hospital death like APACHE II or SAPS score does, and improve resource allocation and quality-of-care in ICUs. Membership to the several different phenotypes can provide an insight on the situation of a patient beyond a single score. In addition, another crucial characteristic for phenotypes is to be distinct from each other, because otherwise clinicians cannot interpret and use the phenotypes easily. For example, let us say a patient suffers from hypertension and diabetes. To represent the patient, we can use a mixture of two phenotypes. We prefer Phenotype 1 = {hypertension, ACE inhibitors}, Phenotype 2 = {diabetes, insulin} to Phenotype 1 = {hypertension, ACE inhibitors, insulin}, Phenotype 2 = {diabetes}, because the former is more distinct and meaningful than the latter. Yet another critical concern about phenotypes is the compactness. Generally speaking, compact representation is more preferable than the lengthy one to end users if both have the same discrimination power and distinctness.
This paper proposes a new tensor factorization methodology for generating discriminative and distinct phenotypes. We defined phenotypes as the sets of co-occurring diagnoses and prescriptions. We used a tensor to represent diagnosis and prescription information from EHRs, and decomposed the tensor into latent medical concepts (i.e., phenotypes). To discriminate a high-risk group (high mortality), we incorporated the estimated probability of mortality from logistic regression during the decomposition process. We also found cluster structures of diagnoses and prescriptions using contextual similarity between the components, and absorbed the cluster structure into the tensor decomposition process.
Methods
We first describe a computational phenotyping method that we developed (Fig. 1) and experiment design.
Figure 1.
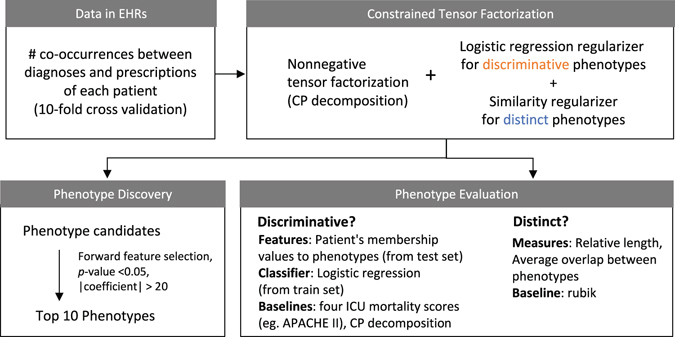
Workflow of our phenotyping method. We constructed a tensor using the number of co-occurrences between diagnoses and prescriptions of each patient in EHRs. We then decomposed the tensor using the proposed constrained tensor factorization that incorporates regularizers for discriminative and distinct phenotypes. We defined phenotype as a set of co-occurring diagnoses and prescriptions, which can be inferred using decomposed tensors, and evaluated their discriminative and distinct power. We also selected top 10 representative phenotypes and presented its meaning and usefulness.
Phenotyping based on tensor factorization
We built a third-order tensor with co-occurrences of patients, diagnoses, and prescriptions from intensive care unit (ICU) EHRs. Detailed tensor construction can be found in Supplementary methods. The co-occurrence is a natural representation of interactions between many diagnoses and prescriptions. We only focused on diagnosis and prescription data as previous phenotyping definition29–31, but we can extend the tensor to a high order (>3) to utilize additional data such as laboratory results and procedures. Specifically, we first built a matrix for individual patient to represent association between prescription and diagnosis. For example, let us say patient 1 is diagnosed with acute respiratory failure and hypertension, and is ordered the medicine phenylephrine during his or her admission. Then, each co-occurrence of acute respiratory failure and phenylephrine, and hypertension and phenylephrine is one, respectively (Fig. 2). Again, let us say patient I is diagnosed with Alzheimer’s disease and is ordered medicine morphine sulfate twice. Then, the co-occurrence of Alzheimer’s disease and morphine sulfate is 2. We collected all the matrices from all the patients and built the third-order observed tensor . Entries at (i, j, k) of the tensor (i.e., i,j,k) is the number of co-occurrence of diagnosis j and prescription k for patient i.
Figure 2.
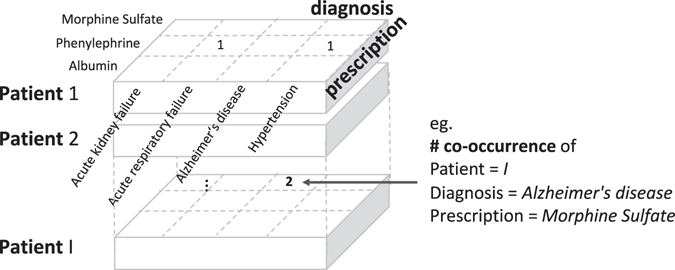
Constructing tensor from EHRs. We built a third-order tensor with co-occurrences of patients, diagnoses, and prescriptions from EHRs. Patient I is diagnosed with Alzheimer’s disease and is ordered morphine sulfate twice.
To decompose the tensor, we used CP algorithm32, 33; detailed description of CP can be found in Supplementary methods. Recently, phenotyping based on Tucker model has been proposed26, 28. It provides a more flexible modeling than does CP by allowing subgroups in each mode, but CP has an advantage in that it is computationally cheap and extendable by imposing regularizers. Using CP model, the third-order tensor was decomposed into three factor matrices: A for patient mode, B for diagnosis mode, and C for prescription mode (Fig. 3). A phenotype consisted of diagnoses and prescriptions, and patients were involved in each phenotype. That is, the r th phenotype consisted of J diagnoses and K prescriptions with membership values that describe how much the diagnoses and prescriptions are involved and contribute to the r th phenotype. The membership values were normalized values between 0 and 1, and stored in the normalized vectors and , respectively. Meanwhile, patients were involved in the R phenotypes with membership values that represent how much the patient has the characteristic of the phenotypes. The membership values of patients were also normalized values between 0 and 1, and stored in the normalized vector . Ability of r th phenotype that can capture and describe the data was stored in , because large values in A :r, B :r, and C :r means that the r th phenotype describes large portion of co-occurrence values in . So, conversely, a phenotype with highly co-occurring diagnosis and prescription may have large λ r.
Figure 3.

Phenotyping by tensor factorization. Dark shade, light shade, and no shade represents high membership, low membership, and zero membership to the phenotype, respectively. Patients who died have high membership to Phenotype 2 and Phenotype R.
For example, ICU survived patients (half of total patients) have Phenotype 1 in Fig. 3, which consists of the first two elements of diagnosis mode and the first one element of prescription mode. The second diagnosis element has higher membership to the Phenotype 1 than the first element does. The patients who died in ICU have Phenotype 2, which consists of the third diagnosis and the second prescription. Similarly, the deceased patients and a few patients who survived have Phenotype R, which consists of the fourth diagnosis and the third prescription. Note that in this example, elements in a phenotype are not overlapped with elements in other phenotypes; thus, we can interpret the phenotype easily. Also, note that phenotypes for the deceased patients and the patients who survived are separated so that we can easily determine which phenotypes are more associated with mortality; consequently, we can further use the phenotypes to evaluate the risk of patients according to the membership to the phenotypes. We introduced two regularizations to make the phenotype discriminative and distinct in the following sections.
Supervised phenotyping for discriminative power
We proposed a supervised approach to encourage the phenotypes separated according to mortality by adding a logistic regression regularization. In the previous section, patients had the membership values to the phenotypes. We used the membership as a feature vector to express patients, and used the feature vector to predict mortality. As a previous work on graph-based phenotyping method21, we added a regularization for supervised term. Let us say y i is a binary indicator of mortality, i.e., y i = 1 if i th patient dies during hospital admission and y i = −1 otherwise. The i th patient in training set L (i ∈ L) was represented as the membership values to the phenotypes, A i:, which is the i th row vector of A. Given logistic regression parameters θ, a probability of i th patient’s mortality to be y i is
| 1 |
where δ i = [A i:, 1] · θ. We then maximized the log-probability, or minimize the negative log-probability,
| 2 |
Thus, the objective function for updating each row A i: is
| 3 |
with a weighting constant ω ( refers to Khatri-Rao product). Note that this objective function is with respect to row A i: not the whole patient factor matrix A. Gradient of f(A i:) is
| 4 |
and hessian of f(A i:) is
| 5 |
Using Newton’s gradient descent method, if i ∈ L, we update A i: as
| 6 |
If i ∉ L, we update A i: as Eq. (6) with ω = 0, which is a traditional CP decomposition without any regularization. Time complexity of Eq. (6) is bounded by O(JKR 2) for i ∈ L; total time complexity to update A is bounded by O(IJKR 2) (Table S1). The supervised term had negligible effects on the total time complexity. This updating rule can be linearly scaled up to the size of A. Updating the logistic regression parameters θ followed a typical logistic regression modeling method. We added a ridge penalty to shrink the size of θ and avoid overfitting (c is a weighting constant)34 as
| 7 |
Similarity-based phenotyping for distinct power
To derive distinct phenotypes with less overlapping with each other, we made phenotypes only consist of similar elements. We first derived components’ similarities from contexts in EHRs, used the similarities to infer cluster structures, and let phenotypes reflect the cluster structures.
Deriving contextual similarity
We derived contextual similarities from EHRs. Farhan et al. generate a vector representation of medical events (or elements in phenotype)17. Based on this work, we generated sequences that consist of diagnoses and prescriptions from EHRs in time order (Table 1). We applied Word2Vec, a two-layer neural network for natural language processing for numerical representation of discrete words35. We input the time-ordered EHRs sequences into Word2Vec and derived a set of vectors for each diagnosis or prescription. After several trials, we set cardinality of the vector as 500 and window size of the sequence (i.e., the number of diagnoses or prescriptions in a sequence to consider them contextually similar) as 30. We found that, as the cardinality increases, distribution of the pairwise similarities spreads widely (i.e., many similarity values are close to −1 or 1 other than 0), but computation time also increases rapidly. We also observed that most of the pairwise similarities become close to 0 as the window size decreases, and close to 1 as the window size increases.
Table 1.
Examples of time-ordered EHRs sequences.
| Lorazepam → Acetaminophen → Piperacillin-Tazobactam → Ventricular fibrillation |
| Diltiazem → Pneumococcal Vac Polyvalent → Anemia → Chronic obst asthma |
| Pantoprazole Sodium → Acetaminophen |
| Oxycodone → Heparin Flush → Severe sepsis |
Each sequence consists of formulary drug codes (prescription) and ICD-9 codes (diagnosis), and is used in Word2Vec to derive pairwise similarities.
We then computed cosine similarities between the vector representation of elements, and derived a pairwise similarity matrix (either J × J matrix S B for diagnosis or K × K matrix S C for prescription). For example, let us say the j 1 th and j 2 th diagnoses in our dataset refer to atrial fibrillation and congestive heart failure, respectively. The vector representation is atrial fibrillation = (0.1, 0.6, 0.2, 0.1) and congestive heart failure = (0.3, 0.7, 0.1, 0.2). The similarity between them is stored at (j 1, j 2)-entry of S B, and the value is .
We made S sparse for efficiency by ignoring trivial values. Many similarities were close to zero, and their small variance did not provide useful information. Similarities less than zero refer to dissimilarity, which was not the focus of this work. Considering all the less useful similarity values can increase computational overhead. We only used the highest l similarities value for each element, and consider the others as 0. We choose for diagnosis and for prescription according to previous works36, 37.
We converted S into a normalized-cut similarity matrix38. Incorporating the normalized cut similarity helped our problem to increase both the total dissimilarity between the different phenotypes and the total similarity within the phenotypes, thus avoid overlapping between the phenotypes. Converting to the normalized cut similarity matrix is
| 8 |
where D is a diagonal matrix of .
Incorporating cluster structure
With the similarity matrix, we inferred a cluster structure from the similarity and incorporated it to our NTF optimization. The cluster structure contained information on which elements should be in the same phenotype together. We introduced a regularization term for the spectral clustering. We increased the sum of pairwise similarity within a phenotype. Because how much the elements are involved in each phenotype is different, the pairwise similarity was weighted by the two elements’ membership values to the phenotype. That is, in terms of diagnosis similarity matrix S B, the sum of weighted pairwise similarity within a phenotype r is
| 9 |
and the sum of all the similarity in Eq. (9) throughout the R phenotypes is
| 10 |
Here, Tr(B T S B B) is the objective of spectral clustering in which B represent the clustering assignment of each element37. Consequently, the phenotypes preserved the spectral clustering structure by incorporating sum of weighted similarity. Meanwhile, Tr(B T S B B) is also equivalent to symmetric nonnegative matrix factorization of similarity matrix S B 36, 39, i.e.,
| 11 |
because
| 12 |
by relaxing a constraint on B T B = I 39. This transformation is beneficial because it helps phenotypes to be more orthogonal (or distinct) by retaining B T orthogonality approximately39. Thus, the objective function with the cluster structure is
| 13 |
with a weighting constant μ. By incorporating , our phenotyping method can absorb the spectral clustering structure and improve the orthogonality at the same time. Although it is a fourth-order non-convex function and it is difficult to find a global optimum, it can converge to a stationary point36. To find an optimum value, we derived the gradient of g(B):
| 14 |
and hessian of g(B):
| 15 |
where a vec(B) of length JR is a vectorization of B by column i.e., , and refers to Kronecker product. Using Newton’s gradient descent method, we updated B as
| 16 |
Time complexity of Eq. (16) is bounded by O(IJKR) + O(J 3 R 3). The similarity term had negligible effects on the total time complexity (Table S2). The updating rule for B contained matrix inversion of , which may not be scaled up well with large J. In this case, we can use a constant learning rate instead of although sacrificing converging rate.
Similarly, the factor matrix C for prescriptions followed the same update procedure. We repeated the updating procedures for the factor matrices A, B and C and logistic regression parameter θ until convergence. We assumed convergence when where fit is defined as , and fit old is the fit of the previous iteration. After normalizing, we removed trivial values less than threshold ε because those values are too small for meaningful membership value and worsen the conciseness. We summarized the entire updating procedures in Algorithm 1.
| Input: |
| 1: Randomly initialize A, B, C. |
| 2: repeat |
| 3: for all i. |
| 4: Update θ for logistic regression |
| 5: . |
| 6: . |
| 7: until Converged |
| 8: , , , |
| 9: , , |
| 10: return . |
Experiment design
Dataset and preprocessing
We used a large publicly available dataset MIMIC-III (Medical Information Mart for Intensive Care III)40. MIMIC-III contains comprehensive de-identified data on around 46,520 patients in critical care units of the Beth Israel Deaconess Medical Center between 2001 and 2012, and it includes information such as demographics, prescription, diagnosis ICD codes, and clinical outcomes such as mortality. We selected 10,028 patients, including all 5,014 patients who died during admission and a random sample of 5,014 of patients who survived. If a patient who survived had multiple admission histories, we used the first admission. We used 202 diagnosis ICD-9 codes that are appeared in the charts of at least 5% of the patients and 316 prescription codes that appeared in at least 10% of the patients. We excluded diagnosis ICD-9 ‘V’ or ‘E’ codes that describe supplementary factors for health status. We excluded trivial base type prescriptions such as 0.9% sodium chloride, 5% dextrose, and sterile water. Most nonzero co-occurrence values are one, and skewed right (Fig. S1). To prevent small-dosage frequent medicines from having high co-occurrences, we truncated the co-occurrence values to 1% percentile, 10 (Fig. S1).
Evaluation measures
We evaluated our proposed method in terms of discrimination and distinction. We measured the discrimination by the area under the receiver operating characteristic curve (AUC), sensitivity, and specificity. We measured distinction by a relative length of phenotype and an average overlap. An absolute length of r th phenotype is the number of nonzero in membership vector B :r and C :r. The relative length of the phenotype is the absolute length divided by the maximum length J + K. We averaged the R relative lengths of phenotype. The average overlap41 measures the degree of overlapping between all phenotype pairs. It is defined as the average of cosine similarities between phenotype pairs:
| 17 |
Setting R = 50, we repeatedly ran our models ten times for 10-fold cross validation. We used the training set to compute the regression parameter θ and the likelihood term in supervised phenotyping, and used the test set to measure the discrimination (Table S3). Because tensor factorization is not deterministic method, the factorized tensors are different in each trial; so, we computed mean and 95% confidence interval.
Baselines
We compared the discrimination and the distinction of our proposed methods with that of several baseline methods. The baselines are:
APACHE II, SAPS II, OASIS, APS III score: Disease severity scores for predicting mortality in intensive care unit (for comparing discrimination only)42–45. These scores assess the severity of disease using variables from pre-existing conditions, physiological measurements, biochemical/hematological indices, and source of admission. The weighted sum of individual values produces the severity scores46.
-
Rubik: A state-of-the-art computational phenotyping method based on CP. Rubik generates a phenotype candidate using count of diagnoses and treatments. It incorporates the orthogonality between phenotypes to derive concise phenotypes41. We assume no existing knowledge term and bias term.
Our proposed methods are:- The supervised phenotyping that incorporates the prediction term for discriminative phenotypes (ω ≠ 0, μ = 0).
- The similarity-based phenotyping that incorporates the cluster structure term for distinct phenotypes (ω = 0, μ ≠ 0).
- The final model that incorporates the both supervised and similarity-based approach (ω ≠ 0, μ ≠ 0).
When evaluating discrimination (AUC, sensitivity, specificity) of NTF-based models, we used the patient’s membership values (i.e., of size 1 × R) as features to fit a binary logistic regression to predict mortality. Particularly, for the supervised model, we fitted a binary logistic regression (after normalization) other than θ that are used during updating procedures. To examine the performance of the supervised and similarity-based phenotyping respectively, we compared the discrimination of CP and the supervised phenotyping (regardless of similarity term), and also compared the distinction of Rubik and similarity-based phenotyping (regardless of supervised term). We then combined the supervised approach and similarity-based approach together to achieve both discrimination and distinction. The weighting constants ω and μ were selected as ω = 1 and μ = 1000 after several trials. Note that ω was comparably small because it sensitively applied to each row of A whereas μ applies to the l 2 norm of the whole matrix B or C. We used a tensor Matlab Tensor Toolbox Version 2.549 from Sandia National Laboratories to represent tensors and compute tensor operations.
Results
We present the experimental evaluation and phenotypes derived from our method.
Discriminative and distinction power comparison
We found that our methods outperformed other baselines in terms of discrimination and distinction. The supervised phenotyping method showed the highest AUC and sensitivity among the other methods including APACHE II and SAPS II (Table 2). The similarity-based phenotyping method showed the lowest relative length and average overlap among the other methods. Particularly when compared with Rubik25 that considers orthogonality for the distinction, the similarity-based method improved the distinction significantly (the relative length of 0.3934 vs 0.0714).
Table 2.
Discriminative and distinction power comparison.
| RMSE | AUC | Sensitivity | Specificity | Rel. Length | Avg. overlap | |
|---|---|---|---|---|---|---|
| APACHE II42 | — | 0.7364 | 0.6712 | 0.6728 | — | — |
| SAPS II43 | — | 0.8129 | 0.7970 | 0.6720 | — | — |
| OASIS44 | — | 0.7227 | 0.6253 | 0.7077 | — | — |
| APS III45 | — | 0.7419 | 0.6861 | 0.6994 | — | — |
| CP32, 33 | 2.2153 (±0.0015) | 0.8469 (±0.0156) | 0.8375 (±0.0391) | 0.7342 (±0.0401) | 0.6807 (±0.0047) | 0.3777 (±0.0064) |
| Supervised | 2.2152 ±(0.0016) | 0.8568 (±0.0106) | 0.8392 (±0.0377) | 0.7518 (±0.0393) | 0.6828 (±0.0019) | 0.3787 (±0.0059) |
| Rubik25 | 2.5025 (±0.0003) | 0.7779 (±0.0247) | 0.7310 (±0.0304) | 0.7242 (±0.0377) | 0.3934 (±0.0102) | 0.2806 (±0.0075) |
| Sim.-based | 2.5069 (±0.0130) | 0.7796 (±0.0204) | 0.7615 (±0.0378) | 0.7097 (±0.0473) | 0.0714 (±0.0406) | 0.0013 (±0.0014) |
| Supervised + Sim.-based | 2.3014 (±0.0060) | 0.8389 (±0.0199) | 0.8223 (±0.0387) | 0.7487 (±0.0409) | 0.3958 (±0.0137) | 0.1267 (±0.0100) |
RMSE, discrimination (AUC, sensitivity, specificity) and distinction (Relative length, Average overlap) with 95% confidence interval of baselines and our proposed models when R = 50. CP = CP decomposition, Supervised = the supervised phenotyping for discriminative power, Sim.-based = the similarity-based phenotyping for distinct power, Supervised + Sim. -based = the final model that incorporates the both supervised and similarity-based phenotyping.
Phenotypes
We presented the phenotypes that are derived from the similarity-based phenotyping method for maximum conciseness. After the tensor decomposition procedures with R = 50, we selected 25 phenotypes by forward feature selection50 to remove phenotypes that are redundant and not statistically significant for predicting mortality (Table 3). Among them, we reported ten representative phenotypes in which coefficients from the feature selection were large enough (absolute value of coefficient >20) to discriminate mortality (Table 4): sepsis with acute kidney injury, cardiac surgery, anemia, respiratory failure, heart failure, cardiac arrest, metastatic cancer (requiring ICU), end-stage dementia (requiring ICU – sepsis, aspiration, trauma – and transitioned to comfort care), intraabdominal conditions, and alcohol abuse/withdrawal.
Table 3.
Logistic regression coefficient from feature selection, p-value, and prevalence.
| Phenotype | Coefficient | p-value | λ | Prevalence |
|---|---|---|---|---|
| Intercept | −0.19 | <0.001 | — | — |
| 1 | 28.47 | <0.001 | 749 | 94.53 |
| 3: Sepsis with acute kidney injury | 44.64 | <0.001 | 96 | 45.24 |
| 4: Cardiac surgery | −138.00 | <0.001 | 95 | 50.43 |
| 5: Anemia | −19.76 | <0.001 | 58 | 36.81 |
| 6: Respiratory failure | 88.87 | <0.001 | 56 | 30.98 |
| 10: Heart failure | 30.79 | <0.001 | 39 | 27.19 |
| 11 | 15.13 | <0.001 | 37 | 16.74 |
| 13 | −15.23 | <0.001 | 31 | 22.48 |
| 15 | −7.74 | 0.02 | 30 | 19.02 |
| 16 | 8.69 | <0.001 | 29 | 42.99 |
| 18: Cardiac arrest | 47.08 | <0.001 | 28 | 9.14 |
| 20 | −11.49 | <0.001 | 23 | 9.70 |
| 21 | −5.54 | 0.02 | 22 | 18.46 |
| 23: Metastatic cancer requiring ICU | 25.10 | <0.001 | 20 | 12.29 |
| 24: End-stage dementia requiring ICU | 34.46 | <0.001 | 20 | 12.72 |
| 25 | 12.81 | <0.001 | 18 | 15.08 |
| 28 | −9.00 | <0.001 | 17 | 10.23 |
| 29 | 10.78 | <0.001 | 16 | 18.06 |
| 31 | 10.42 | 0.01 | 16 | 6.13 |
| 32: Intraabdominal conditions | −19.21 | <0.001 | 15 | 4.84 |
| 33 | −6.41 | 0.04 | 14 | 5.12 |
| 34: Alcohol abuse/withdrawal | −22.82 | <0.001 | 13 | 12.57 |
| 41 | −19.89 | <0.001 | 10 | 16.23 |
| 46 | 13.54 | <0.001 | 8 | 7.20 |
| 47 | −9.78 | <0.001 | 6 | 7.96 |
Ten representative phenotypes are 3: Sepsis with acute kidney injury, 4: Cardiac surgery, 5: Anemia, 6: Respiratory failure, 10: Heart failure, 18: Cardiac arrest, 23: Metastatic cancer requiring ICU, 24: End-stage dementia requiring ICU for comport care, 32: Intraabdominal conditions, 34: Alcohol abuse/withdrawal. λ r = ||A :r||F||B :r||F||C :r||F (for frequency). Prevalence = (the number of patients whose membership to the phenotype is non-zero/the total number of patients) × 100%.
Table 4.
Ten representative phenotypes. Listed in order of frequency.
| Sepsis with acute kidney injury | Cardiac surgery (CABG/valve replacements) | ||
|---|---|---|---|
| Diagnosis | Prescription | Diagnosis | Prescription |
| Acute kidney failure NOS, Acute kidny fail - tubr necr, Acute respiratry failure, Severe sepsis, Septic shock, Septicemia NOS | Vancomycin, Ciprofloxacin, Piperacillin-Tazobactam, CefePIME, Linezolid, Meropenem, Miconazole Powder, Nystatin Oral Suspension, Alteplase, Fluconazole, Loperamide HCl | Hypertension NOS, Crnry athrscl natve vssl, Hyperlipidemia NEC/NOS, Atrial fibrillation, DMII wo cmp nt st uncntr, Pure hypercholesterolem, Surg compl-heart, Aortic valve disorder | Phenylephrine HCl, Neostigmine, Aspirin EC, Ketorolac, Oxycodone-Acetaminophen, Ranitidine, Milk of Magnesia, Furosemide, Ibuprofen, TraMADOL (Ultram) |
| Anemia (variation in other diagnoses) | Respiratory failure | ||
| Anemia NOS, Ac posthemorrhag anemia, Chr blood loss anemia, Iron defic anemia NOS | Insulin, Metformin | Acute respiratry failure, Pulmonary insufficiency following trauma and surgery, Other pulmonary insuff, Acute & chronc resp fail | Albumin, PHENYLEPHrine, Dextrose 50%, Chlorhexidine Gluconate, Milrinone, Epinephrine |
| Heart failure | Cardiac arrest | ||
| CHF NOS | Morphine Sulfate, Nitroprusside Sodium, Nitroglycerin, Aspirin EC, Sucralfate | Ventricular fibrillation, Cardiogenic shock, Parox ventric tachycard, Atriovent block complete, Cardiac arrest, AMI anterior wall - init | Acetaminophen IV, Fentanyl Citrate, Influenza Virus Vaccine, Morphine Sulfate, NORepinephrine, Glucagon, Readi-Cat 2, Midazolam, Omeprazole |
| Metastatic cancer requiring ICU (cord compression, need for bronch, etc) | End-stage dementia requiring ICU (sepsis, aspiration, trauma) and transitioned to comfort care | ||
| Secondary malig neo bone, Secondary malig neo brain/spine, Secondary malig neo lung, Secondary malig neo liver, Neurohypophysis dis NEC | Propofol, Midazolam, Fentanyl Citrate, Dexmedetomidine HCl, Vecuronium Bromide | Alzheimer’s disease, Paralysis agitans, Dementia w/o behav dist, Mental disor NEC oth dis | Morphine Sulfate, Scopolamine Patch |
| Intraabdominal conditions–alcoholic pancreatitis, gallstone pancreatitis, perforated ulcer, etc | Alcohol abuse/withdrawl | ||
| Paralytic ileus, Digestive system complications not elsewhere classified, Acute pancreatitis, Cholangitis | Captopril, Metoprolol Tartrate | Alcohol dep NEC/NOS-unspec, Alcohol withdrawal, Alcohol dep NEC/NOS-contin, Bipolar disorder NOS | Hydromorphone, Diphenhydramine HCl, Morphine Sulfate, Prochlorperazine |
We categorized the phenotypes into four groups according to frequency (common or rare) and risk (high or low). Common phenotypes were the top five with high λ and prevalence (and rare otherwise). High-risk (low-risk) phenotypes were ones with positive (negative) logistic regression coefficients (Table 3). As a result, common and high-risk phenotypes are sepsis with acute kidney injury, respiratory failure, and heart failure; rare but high-risk phenotypes are cardiac arrest, metastatic cancer requiring ICU, and end-stage dementia requiring ICU; common but low-risk phenotypes are anemia and cardiac surgery; and rare and low-risk phenotypes are intraabdominal conditions and alcohol abuse/withdrawal (Fig. 4).
Figure 4.
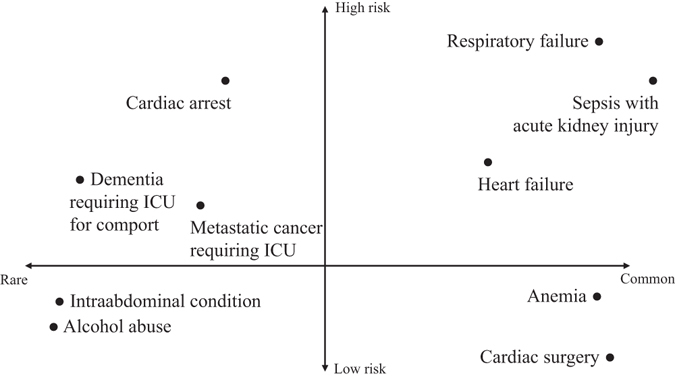
Phenotype maps. Phenotypes are positioned according to frequency and mortality risk.
To examine the risk of each phenotype in detail, we computed mortality of patients who were highly involved to each phenotype (Table 5). We observed that the mortality of patients who have high membership to phenotypes that are denoted as high-risk in Fig. 4 tends to increase to 1.
Table 5.
Patient’s mortality distribution.
| Phenotype | Membership | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| [0, 0.1) | [0.1, 0.2) | [0.2, 0.3) | [0.3, 0.4) | [0.4, 0.5) | [0.5, 0.6) | [0.6, 0.7) | [0.7, 0.8) | [0.8, 0.9) | [0.9, 1) | |
| Sepsis with acute kidney injury | 0.48 | 0.79 | 0.80 | 0.85 | 0.82 | 0.87 | 0.63 | 0.86 | — | — |
| Cardiac surgery | 0.58 | 0.39 | 0.25 | 0.18 | 0.08 | 0.05 | 0.04 | 0.04 | 0.04 | 0.05 |
| Anemia | 0.53 | 0.49 | 0.50 | 0.35 | 0.34 | 0.30 | 0.29 | 0.24 | 0.10 | 0.18 |
| Respiratory failure | 0.48 | 0.84 | 0.85 | 0.91 | 0.86 | 0.88 | 0.80 | 0.77 | 0.92 | 0.73 |
| Heart failure | 0.50 | 0.72 | 0.74 | 0.67 | 0.67 | 0.65 | 0.64 | 0.73 | 0.71 | 0.84 |
| Cardiac arrest | 0.51 | 0.83 | 0.76 | 0.84 | 0.85 | 0.91 | 1.00 | 0.83 | 0.88 | 1.00 |
| Metastatic cancer requiring ICU | 0.51 | 0.80 | 0.71 | 0.81 | 0.65 | 0.78 | 0.87 | 0.80 | 0.75 | 0.74 |
| End-stage dementia requiring ICU | 0.51 | 0.81 | 0.80 | 0.81 | 0.74 | 0.75 | 0.90 | 0.93 | 0.84 | 0.91 |
| Intraabdominal conditions | 0.52 | 0.52 | 0.39 | 0.45 | 0.38 | 0.33 | 0.17 | 0.27 | — | — |
| Alcohol abuse/withdrawal | 0.53 | 0.44 | 0.36 | 0.36 | 0.30 | 0.42 | 0.20 | 0.13 | 0.08 | 0.19 |
The distribution is computed as the number of patients who died/the total number of patients whose membership value is in the range. Empty values when the number of patients <10. Note that our dataset contained half patients who died and half patients who survived.
Discussion
The objective of this study was to develop a phenotyping method that can generate discriminative and distinct phenotypes. As a result, we derived phenotypes that consist of interactions between related diagnoses and prescriptions, and patients had membership to each phenotype. The phenotypes from the supervised model were more discriminative than APACHE II, SAPS II scores and the phenotypes from CP model32, 33; the phenotypes from the similarity-based model were more distinct than the phenotypes from Rubik25. We also observed that the supervised phenotyping and the similarity-based phenotyping have an opposite effect on each other in terms of the discrimination and distinction. The distinct phenotypes from the similarity-based approach lost its discriminative power, and the discriminative phenotypes from the supervised approach lost distinction power. A possible explanation for this trade-off is that the similarity-based model tends to ignore less relevant elements in a phenotype to achieve the best distinction, although the “less relevant elements“ can contribute to increasing the discriminative power overall. However, the combined phenotypes from both approaches achieved the high discrimination and distinction at the same time (Table 2). When combining the supervised and the similarity-based phenotyping, the discrimination increased (with the AUC of 0.8389) compared to the similarity model (with the AUC of 0.7796), and distinction improved (with the relative length of 0.3958 and average overlap of 0.1267) compared to the supervised model (with the relative length of 0.6828 and average overlap of 0.3787).
We also described the most representative phenotypes: sepsis with acute kidney injury, cardiac surgery, anemia, respiratory failure, heart failure, cardiac arrest, metastatic cancer (requiring ICU), end-stage dementia (requiring ICU and transitioned to comfort care), intraabdominal conditions, and alcohol abuse/withdrawal. These conditions are fairly consistent with the list of conditions known to require ICU care in US hospitals51.
Our study also had some limitations. One limitation is that our approach used the entire ICU stay to generate our predictive models. Other predictive models, such as SAPS II, use only the first 24 hours of data as prediction at that point of the hospitalization is more clinically useful. However, our objective was to demonstrate how our approach could be used with a clinically significant outcome. Future work could create additional phenotypes using only the first 24 hours of data to generate models. A second limitation is that some of the phenotypes generated are not obvious to clinicians. For example, the main medications in the “anemia” phenotype are diabetic medications. This is likely because non-pharmacologic therapy is the main treatment for anemia and diabetic patients were highly represented in the “anemia” population.
With refinement, future applications of our proposed computational phenotyping method include clinical decision support to quickly identify subgroups of patients at different levels of important clinical outcomes (e.g., mortality, clinical decompensation, hospital readmission, etc.). It could also be used in cohort identification for quality improvement or research projects to find those who share similar characteristics by representing patients’ heterogeneous medical records into membership of phenotypes. In addition, the phenotypes we derived can provide genomic scientists an insight into genotype-phenotype mapping for precision medicine52, 53. In conclusion, computational phenotyping using non-negative tensor factorization shows promise as an objective method for identification of important cohorts with promise for clinical, quality improvement and research purposes.
Electronic supplementary material
Acknowledgements
This research was partly supported by NHGRI (R00HG008175, R01HG008802), NLM (R00LM011392, R21LM012060), NIPA (NIPA-2014-H0201-14-1001), NRF (2012M3C4A7033344), MSIP/IITP (B0101-15-0307), and MOTIE (10049079).
Author Contributions
Y.K., R.E. and X.J. participated in writing draft; J.S. and X.J. provided important motivations of this study; Y.K. performed the experiments; R.E. and X.J. analyzed data and results; J.S., H.Y. and X.J. provided administrative and supervisory support.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01139-y
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hwanjo Yu, Email: hwanjoyu@postech.ac.kr.
Xiaoqian Jiang, Email: x1jiang@ucsd.edu.
References
- 1.Freimer N, Sabatti C. The human phenome project. Nature genetics. 2003;34:15–21. doi: 10.1038/ng0503-15. [DOI] [PubMed] [Google Scholar]
- 2.McCarty CA, et al. The emerge network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC medical genomics. 2011;4:1. doi: 10.1186/1755-8794-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Overhage JM, Ryan PB, Reich CG, Hartzema AG, Stang PE. Validation of a common data model for active safety surveillance research. Journal of the American Medical Informatics Association. 2012;19:54–60. doi: 10.1136/amiajnl-2011-000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hripcsak G, Albers DJ. Next-generation phenotyping of electronic health records. Journal of the American Medical Informatics Association. 2013;20:117–121. doi: 10.1136/amiajnl-2012-001145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kho AN, et al. Use of diverse electronic medical record systems to identify genetic risk for type 2 diabetes within a genome-wide association study. Journal of the American Medical Informatics Association. 2012;19:212–218. doi: 10.1136/amiajnl-2011-000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen AN, et al. Symbolic rule-based classification of lung cancer stages from free-text pathology reports. Journal of the American Medical Informatics Association. 2010;17:440–445. doi: 10.1136/jamia.2010.003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmiedeskamp M, Harpe S, Polk R, Oinonen M, Pakyz A. Use of international classification of diseases, ninth revision clinical modification codes and medication use data to identify nosocomial clostridium difficile infection. Infection Control & Hospital Epidemiology. 2009;30:1070–1076. doi: 10.1086/606164. [DOI] [PubMed] [Google Scholar]
- 8.Klompas M, et al. Automated identification of acute hepatitis b using electronic medical record data to facilitate public health surveillance. PLOS one. 2008;3:e2626. doi: 10.1371/journal.pone.0002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pathak J, et al. Mapping clinical phenotype data elements to standardized metadata repositories and controlled terminologies: the emerge network experience. Journal of the American Medical Informatics Association. 2011;18:376–386. doi: 10.1136/amiajnl-2010-000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim, Y. et al. Discovery of prostate specific antigen pattern to predict castration resistant prostate cancer of androgen deprivation therapy. BMC Medical Informatics and Decision Making 63, doi:10.1186/s12911-016-0297-0 (2016). [DOI] [PMC free article] [PubMed]
- 11.Moskovitch, R. & Shahar, Y. Medical temporal-knowledge discovery via temporal abstraction. In AMIA (2009). [PMC free article] [PubMed]
- 12.Yu S, et al. Toward high-throughput phenotyping: unbiased automated feature extraction and selection from knowledge sources. Journal of the American Medical Informatics Association. 2015;22:993–1000. doi: 10.1093/jamia/ocv034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Savova GK, et al. Mayo clinical text analysis and knowledge extraction system (ctakes): architecture, component evaluation and applications. Journal of the American Medical Informatics Association. 2010;17:507–513. doi: 10.1136/jamia.2009.001560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman C, Shagina L, Lussier Y, Hripcsak G. Automated encoding of clinical documents based on natural language processing. Journal of the American Medical Informatics Association. 2004;11:392–402. doi: 10.1197/jamia.M1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lasko TA, Denny JC, Levy MA. Computational phenotype discovery using unsupervised feature learning over noisy, sparse, and irregular clinical data. PloS one. 2013;8:e66341. doi: 10.1371/journal.pone.0066341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi, E. et al. Multi-layer representation learning for medical concepts. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 1495–1504 (ACM, 2016).
- 17.Farhan, W. et al. A predictive model for medical events based on contextual embedding of temporal sequences. Journal of medical Interenet Research (2016). [DOI] [PMC free article] [PubMed]
- 18.Winslow RL, Trayanova N, Geman D, Miller MI. Computational medicine: translating models to clinical care. Science translational medicine. 2012;4:158rv11–158rv11. doi: 10.1126/scitranslmed.3003528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White N, et al. Probabilistic subgroup identification using bayesian finite mixture modelling: A case study in parkinson’s disease phenotype identification. Statistical methods in medical research. 2012;21:563–583. doi: 10.1177/0962280210391012. [DOI] [PubMed] [Google Scholar]
- 20.Zhou, J., Wang, F., Hu, J. & Ye, J. From micro to macro: data driven phenotyping by densification of longitudinal electronic medical records. In Proceedings of the 20th ACM SIGKDD international conference on Knowledge discovery and data mining 135–144 (ACM, 2014).
- 21.Liu, C., Wang, F., Hu, J. & Xiong, H. Temporal phenotyping from longitudinal electronic health records: A graph based framework. In Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 705–714 (ACM, 2015).
- 22.Luo, Y., Xin, Y., Joshi, R., Celi, L. & Szolovits, P. Predicting icu mortality risk by grouping temporal trends from a multivariate panel of physiologic measurements. In AAAI, 42–50 (2016).
- 23.Ho JC, et al. Limestone: High-throughput candidate phenotype generation via tensor factorization. Journal of biomedical informatics. 2014;52:199–211. doi: 10.1016/j.jbi.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho, J. C., Ghosh, J. & Sun, J. Marble: high-throughput phenotyping from electronic health records via sparse nonnegative tensor factorization. In Proceedings of the 20th ACM SIGKDD international conference on Knowledge discovery and data mining 115–124 (ACM, 2014).
- 25.Wang, Y. et al. Rubik: Knowledge guided tensor factorization and completion for health data analytics. In Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 1265–1274 (ACM, 2015). [DOI] [PMC free article] [PubMed]
- 26.Luo, Y. et al. Subgraph augmented non-negative tensor factorization (santf) for modeling clinical narrative text. Journal of the American Medical Informatics Association ocv016 (2015). [DOI] [PMC free article] [PubMed]
- 27.Luo, Y., Wang, F. & Szolovits, P. Tensor factorization toward precision medicine. Briefings in bioinformatics bbw026 (2016). [DOI] [PMC free article] [PubMed]
- 28.Perros, I., Chen, R., Vuduc, R. & Sun, J. Sparse hierarchical tucker factorization and its application to healthcare. In Data Mining (ICDM), 2015 IEEE International Conference on 943–948 (IEEE, 2015).
- 29.Ho JC, et al. Limestone: High-throughput candidate phenotype generation via tensor factorization. Journal of biomedical informatics. 2014;52:199–211. doi: 10.1016/j.jbi.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newton KM, et al. Validation of electronic medical record-based phenotyping algorithms: results and lessons learned from the emerge network. Journal of the American Medical Informatics Association. 2013;20:e147–e154. doi: 10.1136/amiajnl-2012-000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richesson RL, et al. A comparison of phenotype definitions for diabetes mellitus. Journal of the American Medical Informatics Association. 2013;20:e319–e326. doi: 10.1136/amiajnl-2013-001952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carroll JD, Chang J-J. Analysis of individual differences in multidimensional scaling via an n-way generalization of “eckart-young” decomposition. Psychometrika. 1970;35:283–319. doi: 10.1007/BF02310791. [DOI] [Google Scholar]
- 33.Harshman, R. A. Foundations of the parafac procedure: Models and conditions for an “explanatory” multi-modal factor analysis (1970).
- 34.Le Cessie S, Van Houwelingen JC. Ridge estimators in logistic regression. Applied statistics. 1992;41:191–201. doi: 10.2307/2347628. [DOI] [Google Scholar]
- 35.Mikolov, T., Sutskever, I., Chen, K., Corrado, G. S. & Dean, J. Distributed representations of words and phrases and their compositionality. In Advances in neural information processing systems 3111–3119 (2013).
- 36.Gegick, M. Symmetric nonnegative matrix factorization for graph clustering. In Proceedings of the 2012 SIAM International Conference on Data Mining (SIAM, 2012).
- 37.Von Luxburg U. A tutorial on spectral clustering. Statistics and computing. 2007;17:395–416. doi: 10.1007/s11222-007-9033-z. [DOI] [Google Scholar]
- 38.Shi, J. & Malik, J. Normalized cuts and image segmentation. In Computer Vision and Pattern Recognition, 1997. Proceedings., 1997 IEEE Computer Society Conference on 731–737 (IEEE, 1997).
- 39.Ding, C. H., He, X. & Simon, H. D. On the equivalence of nonnegative matrix factorization and spectral clustering. In SDM vol. 5, 606–610 (SIAM, 2005).
- 40.Johnson AE, et al. Mimic-iii, a freely accessible critical care database. Scientific data. 2016;3:160035. doi: 10.1038/sdata.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang, Y. et al. Rubik: Knowledge guided tensor factorization and completion for health data analytics. In Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 1265–1274 (ACM, 2015). [DOI] [PMC free article] [PubMed]
- 42.Knaus WA, Draper EA, Wagner DP, Zimmerman JE. Apache ii: a severity of disease classification system. Critical care medicine. 1985;13:818–829. doi: 10.1097/00003246-198510000-00009. [DOI] [PubMed] [Google Scholar]
- 43.Le Gall J-R, Lemeshow S, Saulnier F. A new simplified acute physiology score (saps ii) based on a european/north american multicenter study. Jama. 1993;270:2957–2963. doi: 10.1001/jama.1993.03510240069035. [DOI] [PubMed] [Google Scholar]
- 44.Johnson AE, Kramer AA, Clifford GD. A new severity of illness scale using a subset of acute physiology and chronic health evaluation data elements shows comparable predictive accuracy. Critical care medicine. 2013;41:1711–1718. doi: 10.1097/CCM.0b013e31828a24fe. [DOI] [PubMed] [Google Scholar]
- 45.Pollack MM, Patel KM, Ruttimann UE, et al. The pediatric risk of mortality iii—acute physiology score (prism iii-aps): a method of assessing physiologic instability for pediatric intensive care unit patients. The Journal of pediatrics. 1997;131:575–581. doi: 10.1016/S0022-3476(97)70065-9. [DOI] [PubMed] [Google Scholar]
- 46.Bouch DC, Thompson JP. Severity scoring systems in the critically ill. Continuing Education in Anaesthesia, Critical Care & Pain. 2008;8:181–185. doi: 10.1093/bjaceaccp/mkn033. [DOI] [Google Scholar]
- 47.Carroll JD, Chang J-J. Analysis of individual differences in multidimensional scaling via an n-way generalization of “eckart-young” decomposition. Psychometrika. 1970;35:283–319. doi: 10.1007/BF02310791. [DOI] [Google Scholar]
- 48.Harshman RA. Foundations of the parafac procedure: Models and conditions for an “explanatory” multi-modal factor analysis. UCLA Working Papers in Phonetics. 1970;16:184. [Google Scholar]
- 49.Bader, B. W. & Kolda, T. G. Matlab tensor toolbox version 2.5. Available online, January7 (2012).
- 50.Jain A, Zongker D. Feature selection: Evaluation, application, and small sample performance. IEEE transactions on pattern analysis and machine intelligence. 1997;19:153–158. doi: 10.1109/34.574797. [DOI] [Google Scholar]
- 51.Barrett, M. L., Smith, M. W., Elixhauser, A., Honigman, L. S. & Pines, J. M. Utilization of intensive care services - statistical brief 185. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs (2014).
- 52.Robinson PN. Deep phenotyping for precision medicine. Human mutation. 2012;33:777–780. doi: 10.1002/humu.22080. [DOI] [PubMed] [Google Scholar]
- 53.Zemojtel T, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Science translational medicine. 2014;6:252ra123–252ra123. doi: 10.1126/scitranslmed.3009262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.