

Abstract
Nitrogen (N) is a key macronutrient representing a limiting factor for plant growth and development and affects productivity in wheat. In this study, durum wheat response to N chronic starvation during grain filling was investigated through a transcriptomic approach in roots, leaves/stems, flag leaf and spikes of cv. Svevo. Nitrogen stress negatively influenced plant height, tillering, flag leaf area, spike and seed traits, and total N content. RNA-seq data revealed 4,626 differentially expressed genes (DEGs). Most transcriptomic changes were observed in roots, with 3,270 DEGs, while 963 were found in leaves/stems, 470 in flag leaf, and 355 in spike tissues. A total of 799 gene ontology (GO) terms were identified, 180 and 619 among the upregulated and downregulated genes, respectively. Among the most addressed GO categories, N compound metabolism, carbon metabolism, and photosynthesis were mostly represented. Interesting DEGs, such as N transporters, genes involved in N assimilation, along with transcription factors, protein kinases and other genes related to stress were highlighted. These results provide valuable information about the transcriptomic response to chronic N stress in durum wheat, which could be useful for future improvement of N use efficiency.
Introduction
As a crucial component of amino acids, proteins, nucleic acids, chlorophyll and several plant hormones, nitrogen (N) represents a key macronutrient for crop productivity1. Its availability influences major plant processes such as growth, development, architecture, flowering, senescence, photosynthesis, and allocation of photosynthates in plants2. It is well known that most diffused cereal crop plants, like wheat, rice and maize, use only 30–40% of the applied N fertilizers, while the rest remains unused causing severe environmental pollution3, with eutrophication of water and enrichment of NOx gases in the atmosphere4. The newly formed and released nitrous oxide has 300 times more global warming effect than carbon dioxide5.
There is thus a need to sustain high productivity while decreasing the rate of N application. To this aim, it is important to obtain a better understanding of the molecular regulatory mechanisms underlying morphological and physiological acclimation to N availability in crops6, 7. Although both organic and inorganic N can be used by plants8, 9, inorganic N resources such as nitrate (NO3 −) and ammonium (NH4 +) are the major N forms in soil, the former being more abundant in aerobic soils, the latter the major N compound in flooded wetland or acidic soils7. The primary N metabolism involves several crucial steps. Nitrate is taken up and transported by low and high affinity nitrate transporter genes (NRT1 and NRT2), reduced to nitrite by nitrate reductase (NR), and to ammonium by nitrite reductase (NiR). The ammonium derived from nitrate or directly taken up by ammonium transporters (AMTs) is then incorporated into amino acids mainly through glutamine synthetase (GS) and glutamate synthase (GOGAT) in plastids6. The amino group of glutamate (Glu) can be transferred to amino acids by several aminotransferases10 such as asparagine synthetase (AsnS) which leads to the formation of asparagine (Asn) and Glu from glutamine (Gln) and aspartate. Another enzyme, the NADH–glutamate dehydrogenase (GDH), plays its role in mitochondria, and is able to incorporate ammonium into Glu, in response to high levels of ammonium under stress1. The products of the primary metabolism, can then serve for the biosynthesis of other N-containing compounds.
High-throughput sequencing technologies have gained trust and reliability in the last decade representing the most advantageous and economical tool for deeply exploring genomes, transcriptomes, proteomes and metabolomes11. In particular, the transcriptomic approach has been widely used recently thanks to its high sensitivity and reproducibility in a number of different species (model and non-model) and experimental conditions12–14. Studies concerning cereal responses to nitrogen starvation have been also carried out partially unveiling differential expressed genes (DEGs) involved in these processes for instance in sorghum, rice, barley, and wheat15–18. However, the mechanisms regulating N use efficiency (NUE) in wheat (bread and durum) need to be further investigated. From several studies it is known that at molecular level different biological processes are involved in N stress and the expression of many crucial genes is altered in this particular condition18–22. Durum wheat [Triticum turgidum subsp. durum (Desf.) Husn.] crop is fundamental for the production of pasta and semolina23. It is the most widespread crop in the Mediterranean countries, holding an important role for their economies and traditions. These areas, including southern Europe and northern Africa, are characterized by low-rainfall, with critical problems like drought, salinity and low inorganic matter, causing limitations on growing crops without nitrogen fertilizers or biological nitrogen fixation through legumes24, 25. Farmers tend to increase sustainable agriculture, practicing rotation and limiting fertilization due to the high costs related to an overall poor and highly fertilizer-demanding environment. Being nitrogen directly linked with yield and quality (protein content), it is thus important to investigate how plants cope with N deficiency during plant growth and, specifically, during the process of grain development and senescence26. In this developmental stage, nitrogen plays a particularly important role, in fact low levels of nitrogen supply induce early senescence that is associated with a reduction of protein accumulation in wheat grain27.
From an -omics point of view, the grain filling stage still needs to be deeply investigated and genetic, metabolic and physiological data should be integrated for understanding how plants respond to reduced fertilizer inputs, possibly suggesting new targets for plant breeding.
In this study, using an RNA-sequencing approach, we investigated transcriptomic responses to chronic N-starvation in plants of durum wheat cv. Svevo, at the grain filling stage. To our knowledge, this is the first transcriptome-wide study on response to chronic N starvation in durum wheat. Our results represent a solid resource to clarify and further investigate the mechanisms regulating nitrogen use efficiency in this important crop, and to perform comparative transcriptomics in durum wheat germplasm showing different responses to N stress.
Materials and Methods
Plant growing and phenotypic analysis
Seeds of durum wheat cultivar Svevo were surface sterilized with sodium hypochlorite (0.5%, vol/vol) for 20 min, rinsed thoroughly in sterile water, vernalized at 4 °C for two weeks and then transferred to a hydroponic growth system. Seedlings were grown under 14 h/day illumination at 20 °C in net pots containing three plants each. Hydroponic solution was prepared starting from tap water (containing 13, 2, 26, 14, 2.6, 14 and 63 mgl−1 of Cl−, NO3-N, SO4-S, Na+, K+, Mg2+ and Ca2+, respectively) and using the salts reported in Yin et al.28. Two sets of plants were grown in two different conditions: standard nitrogen nutrition (control) including 2 mM Ca(NO3)2*4H2O, and N stress condition (stressed), with 0 mM Ca(NO3)2*4H2O. The whole root system was maintained submersed in the nutrient solution, which was continuously aerated with air-pumps and replaced every two days. The solution was maintained at pH 6.0 with 0.1N H2SO4.
The following phenotypic traits were collected on plants in the control and stressed conditions at the late milk developmental stage (Z77)29: plant height (PH), flag leaf area (FLA); number of culms per plant (NCPP); number of spikelets per spike (NSPS); number of kernels per spike (KNPS); kernel weight per spike (KWPS); flag leaf (FLDM) and spike (SDM) dry matter. Total nitrogen content in percentage was determined in roots, leaf/stem, flag leaf and spike tissues using a CHNS Analyser.
At the same Z77 stage, root, leaf/stem, flag leaf and spike tissues were sampled from three plants for each condition, and stored at −80 °C for RNA extraction.
Illumina sequencing and quality control
Total RNA was extracted from each sample using the RNeasy Plant Mini Kit (Qiagen, Valencia, USA) and treated with DNaseI (Qiagen) according to the manufacturer’s specification. RNA concentrations were determined using NanoDrop® ND-1000 spectrophotometer (Thermo Scientific, Wilmington, USA) and its integrity was verified on a 1.5% agarose gel in 1X TAE/DEPC water. cDNA was synthesized from one microgram of total RNA using the QuantiTect Reverse Transcription kit (Qiagen), and sent to IGA (Udine, Italy) for processing. Twenty-four cDNA libraries (three biological replicates from each tissue in control and stress conditions) were subsequently prepared for an RNA-seq experiment with the Illumina High-Seq 2000 platform providing 100-bp paired-end reads (IGA). Trimming and clipping were performed with Trimmomatic-0.3330 using the following parameters: HEADCROP:10, LEADING:35 and TRAILING:35 MINLEN:25. The quality assessment was based on the remaining reads using the FASTQC quality control tool version 0.10.0 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc).
Read mapping and differential expression analysis
In order to map each cleaned library to the wheat genome, the bread wheat complete sequence, consisting of 23 pseudomolecules, along with the corresponding annotation file, were downloaded from EnsemblGenomes (ftp://ftp.ensemblgenomes.org/pub/plants/release-26/fasta/triticum_aestivum/dna/). STAR program31 was used for read mapping with outFilterScoreMinOverLread parameter set as 0.3. Gene expression levels were calculated with Cufflinks32 using geometric normalization and per-condition dispersion method by quantifying the Illumina reads according to the FPKM (fragments per kilobase per million mapped fragments). These values were used to perform a principal component analysis (PCA) and to check experimental and control biological replicates. Fold-changes were reported as the log (base 2) of normalized read count abundance for the N-stressed samples divided by the read count abundance of the control samples. Each dataset obtained from each considered tissue was filtered according to fold change values ≥1.5 and ≤−1.5.
Transcript classification and Gene Ontology (GO) term analysis
Filtered data were used to identify and classify transcription factors and protein kinases by family, with iTAK (Plant Transcription factor & Protein Kinase Identifier and Classifier) software (http://bioinfo.bti.cornell.edu/cgi-bin/itak/index.cgi). Gene ontology terms were examined with agriGO33 for GO enrichment with custom annotation. For this analysis, the following parameters were chosen: hypergeometric statistical test method, multi-test adjustment hockberg FDR, significance level: <0.05 and 3 minimum number of mapping entries. Significant values were sorted by enrichment score (Query_item/Query_total)/(Background_item/Background_total) and GO redundancy was removed with REVIGO tool34. Heatmaps and hierarchical clustering were generated with MeV software35.
Quantitative real-time PCR
To validate the reliability of the expression profiles observed in the RNA-seq data, 10 genes were selected for quantitative real-time PCR (qPCR) analyses using iTaq SYBR Green supermix (Bio-Rad, Munich, Germany). The RNase L inhibitor-like protein gene, RLI36 was used as an internal control. RNA material from the same samples used for RNA-seq experiment was used for this validation. Gene specific primers, designed by Primer3, are listed in Supplementary Table S1. The specificity of each primer was monitored by high-resolution melt analysis and by Sanger sequencing. The relative expression value was calculated by the delta-delta CT method and expressed as the fold change referred to the expression in the control (N 2 mM) plants (expression = 1)37. Three biological and three technical replicates per sample were analysed to ensure statistical reliability.
Results
Phenotype observations
Due to its high protein content38, durum wheat cultivar Svevo was selected for an RNA-seq experiment aimed at exploring the molecular response to chronic nitrogen starvation. Plants were grown under N standard or deficiency supply up to Z77 developmental stage. Nitrogen shortage caused serious changes in the phenotype, accelerating plant flowering time and senescence. As awaited, the whole plant growth was severely affected under nitrogen limitation39: all the traits considered sensibly differed between plants grown in the two conditions (Table 1). In particular, stressed plants were shorter than control ones and the flag leaf area was sensibly reduced (68%), affecting also grain filling and yield27, 40. This is consistent with a comparable significant reduction in the flag leaf dry matter. The most striking difference between the two groups of plants was, as expected, tiller number and development. In fact, while in the controls the average number of culms per plant was ≅4, the N starved plants generally did not tiller and showed a single culm. Consequently, also the yield traits were negatively influenced by N starvation. A significant reduction between normal and stressed plants was observed for the number of spikelets per spike (NSPS, over 34%), spike dry matter (SDM over 62%), and number of kernels per spike (KNPS, 75%). The average kernel weight per spike (KWPS) was 0.65 g in the control and 0.16 g in the stressed plants, with a reduction of ≅76% (Table 1). These results strongly support the previously formulated hypothesis that, when nitrogen is limited, a premature leaf senescence occurs affecting the rate and duration of protein accumulation in the seeds41.
Table 1.
Phenotypic analyses on plant groups (control and stressed) of durum wheat grown with different N concentrations.
| Control | Stressed | % RR | |
|---|---|---|---|
| PH (cm) | 54.25 ± 2.67 | 44.09 ± 1.78 | 18.73 |
| FLA (cm2) | 16.73 ± 1.65 | 5.28 ± 0.81 | 68.44 |
| FLDM (g) | 0.061 ± 0.002 | 0.019 ± 0.001 | 68.85 |
| NCPP | 4.14 ± 0.4 | 1 ± 0.01 | 75.86 |
| NSPS | 24.6 ± 1.21 | 16.09 ± 1.09 | 34.65 |
| SDM (g) | 0.37 ± 0.002 | 0.14 ± 0.04 | 62.16 |
| KNPS | 20 ± 1.155 | 5 ± 0.58 | 75 |
| KWPS | 0.65 ± 0.034 | 0.15 ± 0.02 | 76.49 |
For each parameter, mean values (±standard error) and relative reductions between control and stressed conditions are presented.
PH: plant height; FLA: flag leaf area; NCPP: number of culms per plant; NSPS: number of spikelets per spike; SDM: spike dry matter; FLDM: flag leaf dry matter; KNPS: kernel number per spike; KWPS: kernel weight per spike; RR: relative reduction.
In addition, total nitrogen content was determined in the four collected tissues of the two plant groups. The relative N reduction was higher in the roots of N starved plants (≅41%) and decreased towards the top of the plant, with ≅24% reduction in the flag leaf and ≅18% in the spike (Fig. 1), as already observed in bread wheat15.
Figure 1.
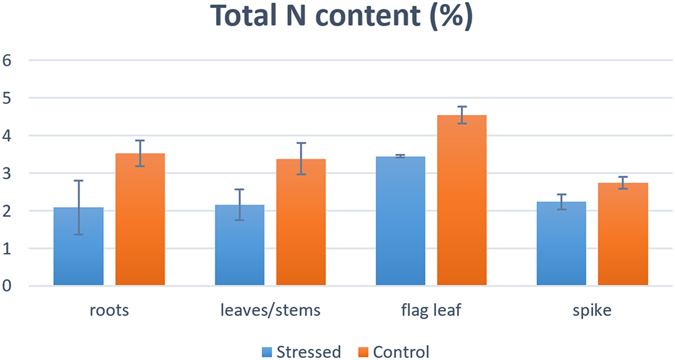
Total nitrogen (N) content detected in four tissues from durum wheat plants grown under standard (control) or N starvation (stressed) conditions. Values represent the N percentage relative to dry matter.
RNA-seq statistics and differentially expressed genes
At the Z77 stage, root, leaf/stem, flag leaf, and spike tissues from control and from N stressed plants were used to prepare 24 cDNA libraries (4 tissues*2 conditions*3 biological replicates). High-throughput sequencing and subsequent read trimming/clipping delivered 25.20 million (M) reads for root, 30.08 M for leaf/stem, 27.85 M for flag leaf, and 26.63 M for spike tissues. The generated reads were then mapped against the bread wheat draft genome (EnsemblGenomes release-26). Varying contents of uniquely mapped reads versus multiple mapped reads were observed in the different tissues. The average of uniquely mapped reads percentage was higher in root (≅53%) with a corresponding low percentage of multiple mapped reads (≅12%). A lower value of uniquely mapped reads was observed in spikes (≅40%), dropping to ≅24% in flag leaf and leaf/stem tissues. Concomitantly, a higher value of multiple mapped reads was observed in spikes (≅30%), flag leaf and leaf/stem tissues (≅48% and ≅51%, respectively). Transcript profiles of the RNA-seq data were analyzed by calculating the read fragments per kilobase per million mapped reads (FPKM). PCA output highlighted the presence of outlier transcripts misleading the analysis for each tissue. These loci were thus removed from the annotation file before performing the mapping analysis. In summary, the number of loci considered for the differential expression analysis was 64991 for root, 64989 for leaf/stem, 64990 for flag leaf and 64989 for spike samples.
Among the total of differentially expressed genes (DEGs) between control and N stressed samples, 3270, 973, 470, and 355 were identified in root, leaf/stem, flag leaf and spike tissues, respectively (p ≤ 0.05 and fold change ≥|1.5|, Table 2 and Fig. 2). All DEGs were annotated according to bread wheat available annotations, and to Arabidopsis thaliana for those lacking annotation (Supplementary Table S2). A Venn diagram was constructed to highlight uniquely and common genes among tissues (Fig. 3). Most of the uniquely DEGs were present in roots (66.10%), followed by leaves/stems (14.70%), flag leaf (6.40%), and spike (5.10%). Most shared genes were between root and leaves/stems (105), followed by leaves/stems and flag leaf (88).
Table 2.
Differentially expressed genes (DEGs) and related enriched categories from GO analysis in response to nitrogen stress.
| Tissue | up/down | DEGs | Filtered DEGs (log2FC) | GO enriched | Filtered GO (FDR) |
|---|---|---|---|---|---|
| leaves/stems | up | 966 | 377 | 275 | 32 |
| down | 904 | 586 | 566 | 186 | |
| flag leaf | up | 206 | 144 | 156 | 9 |
| down | 349 | 326 | 339 | 87 | |
| spike | up | 64 | 60 | 71 | 25 |
| down | 295 | 295 | 462 | 217 | |
| roots | up | 2331 | 1411 | 754 | 114 |
| down | 2340 | 1859 | 750 | 129 |
Number of total DEGs and number of DEGs filtered by log base 2 of the fold change (log2FC ≥ |1.5|) are shown. For each set of up- or downregulated genes, total GO categories identified from the enrichment analysis, and GO filtered by false discovery rate (FDR ≤ 0.05) are presented.
Figure 2.
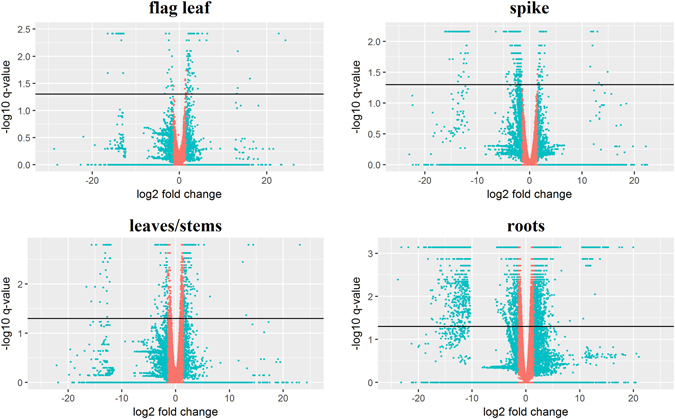
Volcano plot of the differentially expressed genes (DEGs) for each tissue investigated in this work. The y-axis corresponds to the mean expression value of -log10 (q-value), and the x- axis displays the log2 fold change value. The light blue dots represent up and downregulated DEGs, the pink dots denote not DEGs. The black bar indicates the q-value filtering.
Figure 3.
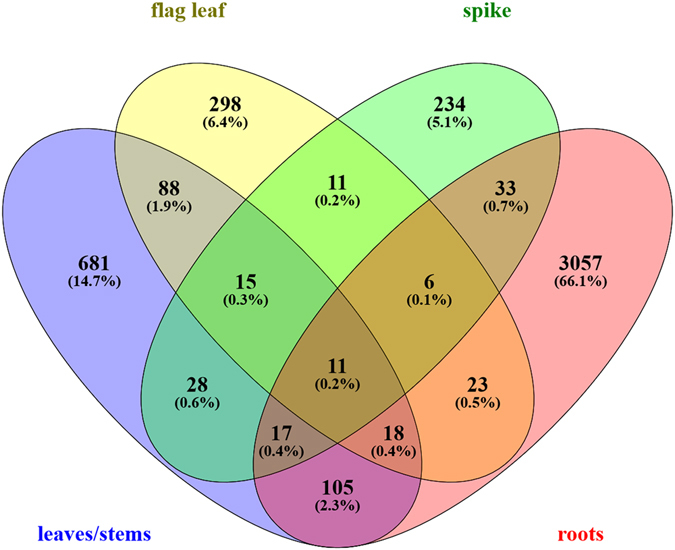
Venn diagram showing overlap of differentially expressed genes between or among tissues. Root, leaves/stems, flag leaf and spike RNA libraries were obtained from durum wheat, Svevo cultivar, grown in nitrogen starvation (0 mM nitrogen), or in standard (2 mM nitrogen) conditions.
In order to validate the results obtained through RNAseq, we performed qPCR expression analyses in specific tissues on ten genes randomly chosen among N transporters, transcription factors, and main genes involved in N metabolism (Table 3), which showed a differential expression in the RNAseq experiment.
Table 3.
RNA-seq results confirmed by quantitative qPCR.
| Locus ID | Tissue | RNA-seq log2 (N−/N+) | RT-PCR log2 (N−/N+) | Putative function |
|---|---|---|---|---|
| S | −12.87 | −1.24 | Probable peptide/nitrate transporter, ZIFL2 | |
| Traes_4AS_86FF10C36 | R | 2.13 | 1.13 | Dof-type zinc finger DNA-binding family protein, DOF1.3 |
| L/S | 2.38 | 3.96 | ||
| FL | 3.02 | 2.14 | ||
| Traes_5BL_CF5A8348D | R | 1.59 | NDE | Chaperone protein dnaJ 2, ATJ2 |
| L/S | 2.54 | 4.87 | ||
| FL | 2.98 | 2.93 | ||
| S | 2.19 | 2.74 | ||
| Traes_4BS_E79CB87B7 | R | −2.77 | −3.79 | ThiaminC, THIC |
| L/S | −3.97 | −1.84 | ||
| FL | −2.77 | −2.13 | ||
| S | −6.51 | −4.49 | ||
| Traes_1BL_241A3B9EF | R | 1.81 | 1.85 | Probable ubiquitin-conjugating enzyme E2 24,UBC24, Phosphate 2 (PHO2) |
| Traes_6DL_0A59B823C | R | 3.21 | 1.09 | Protein NRT1/PTR FAMILY 7.3, NPF7.3 |
| Traes_2BL_309ED7C81 | R | −1.56 | −0.62 | Glutamate dehydrogenase 2,GDH2 |
| Traes_4BL_985CBED5D | R | 2.23 | 1.14 | Protein NRT1/PTR FAMILY 8.4,NPF8.4 |
| Traes_2AS_A8CCC32D3 | R | −1.59 | −1.20 | Protein TIFY 10B,TIFY10B |
| Traes_6AS_AB7CDF374 | R | 1.92 | 0.76 | Nitrate reductase [NADH] 1,NIA1 |
R: root, L/S: leaves/stems, FL: flag leaf, S: spike, NDE: Not Differentially Expressed.
Results from qPCR analyses evidenced that expression trends of these genes were comparable to the ones obtained by means of RNAseq analysis (correlation coefficient R = 0.7086), thus validating the sequencing experiment (Table 3).
GO classification and enrichment analysis of DEGs related to N metabolism
Out of a total of 3373 GO terms identified, 1256 and 2117 were associated to the significantly upregulated and downregulated genes in all tissues, respectively. When the FDR filter was applied to the list of differentially expressed genes, 799 GO terms were identified of which 180 among the upregulated and 619 among the downregulated genes (Table 2).
For summarizing GO categories belonging to each specific tissue, a semantic similarity scatter plot was obtained (Fig. 4). Starting from roots (Fig. 4A), we observed high values for categories involved in cellular response to N levels and cellular N compound metabolism, spermine biosynthesis, cinnamic acid and L-phenylalanine metabolism. Amino acid transmembrane transport and iron transport were also visualized in this graph, as well as trehalose and carotenoid metabolisms.
Figure 4.
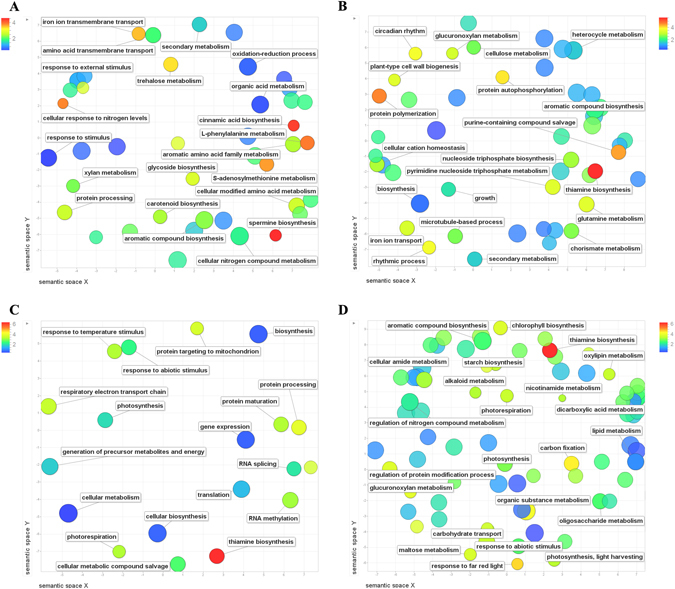
REVIGO semantic diagram summarizing enriched GO terms for roots (A), leaves/stems (B), flag leaf (C), and spike (D). Similar GO terms are placed close together in the plot. Bubble color indicates the log2 of the enrichment value for each category; circle size indicates the frequency of the GO term in GO database.
In leaf/stem tissues (Fig. 4B), thiamine biosynthesis covered the highest values, along with purine-containing compound salvage and protein polymerization. Several categories addressing nucleoside metabolisms were involved in these tissues, including glutamine metabolism along with others already described, such as cell wall biogenesis and cellulose metabolism. In flag leaf (Fig. 4C) we observed high enrichment values addressing photorespiration, respiratory elector transport chain, photosynthesis, response to abiotic stimulus and thiamine biosynthesis. Much more complex and articulate was the situation for spikes (Fig. 4D) in which, along with remarkable values addressing thiamine biosynthesis followed by several categories for carbon metabolism and photosynthesis, we also observed categories addressing regulation of N compound and cellular amide metabolism, aromatic compound biosynthesis, and alkaloid metabolism. Starch biosynthesis category was also remarkably enriched.
DEGs directly involved in nitrogen metabolism
Many genes involved in N absorption and assimilation were differentially expressed under N-stress compared to normal condition. Here, 12 DEGs encoding nitrate transporters were detected (Fig. 5A Supplementary Table S3). Two genes were downregulated while the other ten showed increased expression under N-stress. Two of these transporters are orthologous to the ones differentially expressed in N stress conditions in barley, recently described18. Among the DE transporters responsive to ammonium, two orthologues to AMT3-1 from barley were upregulated in durum wheat stressed roots, while one orthologue to Arabidopsis AMT2 was downregulated in stressed roots and one orthologue to Arabidopsis AMT1-4 was upregulated in both stressed flag leaf and leaf/stem tissues (Fig. 5A, Supplementary Table S3).
Figure 5.

Heatmaps showing the expression patterns of genes involved in nitrogen stress response in durum wheat. Genes are grouped in five categories (A to E). Colours indicate the differential gene expression in nitrogen stressed tissues compared to non-treated plant tissues; green: downregulated, red: upregulated, white: not differentially expressed. R: roots, L: leaves/stems, F: flag leaf, S: spike.
The key genes involved in nitrate assimilation were found differentially expressed mostly in root tissues under N stress conditions. Nitrate reductase [NADH] 1, along with Asparagine synthetase, and the cytosolic Glutamine synthetase genes were found upregulated in roots (Fig. 5A, Supplementary Table S3). On the contrary, the chloroplast/mitochondrial Glutamine synthetase gene appeared downregulated in spikes. Durum wheat genes orthologous to Arabidopsis chloroplast Glutamate synthase 1 [NADH-GOGAT 1] were downregulated in response to N-stress in root and leaf/stem tissues. A downregulation was observed for genes orthologous to three members of the Glutamate dehydrogenase family from Arabidopsis, in response to N-stress (Fig. 5A, Supplementary Table S3).
Other DE transporter genes
The expression of many genes associated with absorption or translocation of other nutrients also changed under N stress. Seven potassium transporters were differentially expressed, including five members of the POT family, which were mostly upregulated in roots and downregulated in flag leaf and spike (Fig. 5B, Supplementary Table S3). Out of five differentially expressed phosphate transporters, two orthologues to PhO1 and one to RHS15 were upregulated and one strongly repressed in stressed roots. A considerable number of DEGs was found for ABC transporters (30), which were mostly upregulated in roots. Iron transporters (21) included orthologues to NRAMP1, VIT1, YSL2,6,8 from Arabidopsis and were generally upregulated in roots. Transporter genes for sugar (15) were mostly downregulated in roots, but also in other tissues. Other DEGs included transporters for peptide (9), aminoacid (7) sulfate (3), zinc (3), and molybdenum (2, Fig. 5B, Supplementary Table S3).
Carbon metabolism
A general alteration was observed for genes participating to carbon metabolism, especially for those involved in glycolysis, tricarboxylic acid (TCA) cycle, oxidative phosphorylation, pentose phosphate pathway, photosynthesis and photorespiration. Most of the genes involved in glycolysis were downregulated under N starvation, including orthologues to phosphoglycerate kinase (PGK) and to biphosphoglycerate mutase (BPGM), which were found strongly downregulated in leaves/stems and roots, respectively (Fig. 5C, Supplementary Table S3). However, among the few upregulated genes, an orthologue to enolase (ENO) was found in roots. Several genes belonging to TCA cycle were upregulated in roots, such as wheat orthologues to malate dehydrogenase (MDH) and succinate dehydrogenase (SDH). A diffused downregulation was observed for genes involved in oxidative phosphorylation, for instance, the orthologues to NADH oxidoreductases (e.g. ndhF, ndhD) and ATP synthases (e.g. atpC and atpI) (Fig. 5C, Supplementary Table S3). Pentose phosphate pathway DEGs identified in spike tissues were downregulated, while a slight majority of those identified in roots were upregulated under N stress. Among these, the genes orthologous to glucose 6-phosphate dehydrogenase (G6PD2) and 6-phosphogluconate dehydrogenase (6PGD) were both downregulated in roots and leaves/stems, whereas the orthologues to ribose-5-phosphate isomerase (RPI) were upregulated in roots. The majority of DEGs involved in photosynthesis was found in spike, while a smaller number was identified in the other tissues. DEGs identified in spike and flag leaf were all downregulated, especially the genes related to photosystem I and II (i.e. psaO, psaK, psbZ, psbE, psbK) (Fig. 5C, Supplementary Table S3). Few genes identified in roots were upregulated, including the 4-hydroxy-3-methylbut-2-enyl-diphosphate synthase (ferredoxin) gene (ISPG). DEGs for photorespiration were all downregulated, for example, the orthologues to NADH dehydrogenase in flag leaf, while the ribulose bisphosphate carboxylase small chain (RBCS) gene in root, flag leaf and spike tissues. Among the other genes involved in carbon and energy metabolism, the orthologues to several genes of the sucrose metabolism were upregulated in roots, including sucrose-phosphate synthase 1 (SPS1), sucrose synthase 3 (SUS3), trehalose-6-phosphate synthase (TPS1), and three starch synthase genes (SS2, SS3, SS4).
DE transcription factors and protein kinases
Due to N starvation, 170 unique genes encoding transcription factors were differentially expressed. Among them, 125 were DE in root, 40 in leaves/stems, 16 in flag leaf, and 18 in spike. The TF genes identified belonged to different families, including MYB (15), bHLH (15), bZIP (11), WRKY (14), mTERF (13), NAC (14), C2C2-Dof (6), NF-Y (10), the auxin-modulated ARF (8) and AUX/IAA (8), and other families (Fig. 5D, Supplementary Table S3). Several TF families were preferentially expressed in specific tissue(s). For instance, most MYB genes were differentially expressed in roots, and NF-Y DE genes were all upregulated in N stressed roots. Additionally, most members (11) of the WRKY family were downregulated in roots, whereas C2C2-Dof TFs were always upregulated in durum wheat root, leaf and stem, and flag leaf tissues following N stress (Fig. 5D, Supplementary Table S3).
In this study, 250 unique protein kinases (PK) were identified in all tissues. In root, 178 genes for protein kinases were found, 88 of which were upregulated and 90 downregulated. In leaves/stems, out of 41 PK genes, 27 were upregulated and 14 were downregulated, while in flag leaf tissues, out of 21 PK genes, 10 were upregulated and 11 were downregulated. In spike tissues, 10 genes for protein kinase were detected, all of which were downregulated. As expected, the largest number of PKs belonged to receptor-like kinase (RLK)/Pelle family, massively expanded in flowering plants and representing more than 60% of the PKs42. We found 208 PKs belonging to this family with a remarkable number (55) of PKs carrying leucine-rich repeat (LRR) domains (Fig. 5E, Supplementary Table S3). Three PKs belonged to AGC (PKA–PKG–PKC), 22 to CAMKs (calcium- and calmodulin-regulated kinases), seven to CMGC (including cyclin-dependent kinases, mitogen-activated protein kinases, glycogen synthase kinase and cyclin-dependent kinases), two to STE (including many kinases functioning in MAP kinase cascades), six to TKL (tyrosine kinase-like kinases), and one plant-specific kinase (belonging to Group-Pl-4) and one WNK_NRBP.
Other DE genes
Nitrogen deficiency causes several plant stress responses. In this study, a number of genes related to detoxification and protection from oxidative stress were upregulated, the majority of which were found in roots. A total of 17 orthologues to Glutathione S-transferases (GSTs) were counted in the DEG dataset, 12 of them were identified in roots, while for leaves/stems and flag leaf only three and two were found, respectively. Particularly, eight GSTs were upregulated, two in leaves/stems (GSTL2) and six in roots (GSTZ1, GSTU24, GSTF13). In addition, 16 orthologues to cytochrome P450 (CYP450) were identified, eleven in roots (nine upregulated), four in leaves/stems (two upregulated) and one in flag leaf (upregulated). Five orthologues to genes involved in the carotenoid/abscisic acid biosynthesis pathway were found upregulated in roots: phytoene synthase (PSY1), phytoene desaturase 3 (PDS), carotenoid isomerase (CRTISO), beta-carotene 3-hydroxylase 2 (BETA-OHASE 2) and zeaxanthin epoxidase (ZEP). Orthologues genes protecting plant cells from oxidative damage were also identified. Catalase 2 (CAT2), superoxide dismutase [Fe] 2 (FSD2), aldehyde dehydrogenase (ALDH) and cinnamyl-alcohol dehydrogenase 1 and 4 (CAD1, CAD4) were upregulated in roots, while methionine sulfoxide reductase B1 (MSRB1) was upregulated in leaves/stems.
The orthologues of homocysteine S-methyltransferase (HMT) and Met S-methyltransferase (MMT), involved in the S-methylmethionine (SMM) cycle, were also upregulated. The former was upregulated in all tissues analyzed (HMT1 in roots and HMT2 in the other tissues), while the latter was upregulated only in roots. Among other DEGs, we also evidenced hormone signaling genes, such as auxin response factor 2 and 18, and the important coenzyme pyridoxal phosphate (PLP)-dependent transferase, all upregulated in roots (Supplementary Table S3).
Discussion
The aim of our study was to investigate the transcriptomic response of durum wheat to N starvation during the grain filling. Instead of analyzing the immediate response to this condition, we focused on genes found differentially expressed after a long-term N stress, and thus representing the cumulative response to N nutrition and availability during the plant cycle until the late milk stage. We used the variety Svevo, which is reported to have a good N use efficiency, and analyzed DEGs in all major organs of the plant, namely roots, leaves/stems, flag leaf and spike in stressed plants compared to plants grown in normal N supply conditions. To our best knowledge, this is the first study on transcriptomic response of durum wheat under N stress, and especially under chronic starvation conditions.
Plants were affected by N shortage primarily on size, showing almost no tillering, decreased height and lower flag leaf area and dry matter. This overall plant depression was also evident in the spikes and in the number of seeds per spike, while the average seed weight was comparable, thus indicating that the stressed plants concentrated all their energy to produce viable seeds.
As observed for the relative reduction of total N content, also the number of DEGs decreased from the roots to the top of the plant. In fact, most DEGs were found in root, which was the main responsive part of the plant to N stress. Gene ontology analysis helped highlight the biological processes mostly involved in the response to this condition. Cellular response to N levels was a highly addressed GO category in roots, comprising several upregulated genes involved in N absorption and assimilation. In above ground tissues, other processes including photosynthesis and photorespiration were highly influenced by N stress. The relation between carbon and nitrogen metabolism has been in fact widely shown, implying alterations in plant growth, physiology and development7, 20.
Two types of transporters, low- and high-affinity nitrate transporters (NRT), carry out nitrate absorption in plant roots. In this study, the N starvation condition led to a marked reduction in total N content in roots, with a parallel upregulation of several genes encoding both low- and high- affinity NRTs. A higher activity of NRTs may be part of the durum wheat adaptation to enhanced N uptake due to N shortage. Among high- affinity NRTs, the gene orthologous to Arabidopsis NRT 2.5 was strongly upregulated in durum wheat. In sorghum, N stress tolerant genotypes showed higher abundance of the transcripts NRT2.2, NRT2.3, NRT2.5, and NRT2.6, related to high affinity NRTs16.
Nitrogen accumulation in wheat occurs in various organs, but predominantly in leaves, since these are more efficient in N remobilization. In particular, N remobilization from flag leaf was positively correlated with N yield per spike and per area43. In N starvation conditions, we observed the upregulation of two genes encoding for oligopeptide transporters in the flag leaf and one in leaves/stems, suggesting that protein degradation might occur in these tissues, with subsequent transportation of N accumulating compounds, such as peptides. Under low N supply, higher N remobilization has been observed compared to high N supply44. This could explain the lower drop in total nitrogen content in the organs mainly involved in N remobilization, that is flag leaf and spike. Considering this basis, our results are in agreement with a metabolic and transcriptional analysis in barley, where it was found that members of amino acid/peptide transporter families are essential components in N remobilization45. Changes in the expression of aminoacid/peptide transporter genes were also observed in roots, suggesting the activation of mechanisms to increase N uptake and accumulation aimed at contrasting N deficiency16. In cereal crops, not only leaves, but also stems, glumes and roots are considered as N sources for grain development, and N remobilization from these organs may also play an important role in grain filling as a buffering leaf senescence mechanism in the post-anthesis period11. This is in line with the observation that a number of proteases were upregulated in most of the tissues here analysed. Among them, the vacuolar processing enzyme gamma (VPE-γ), a possible regulator operating in the early stages of senescence, a putative metacaspase, an asparaginase, and one cysteine protease (papain family) were found upregulated in roots, while another cysteine protease (SAG12) was upregulated in leaves/stems. Interestingly, in common wheat low N plants at the anthesis stage, a cysteine protease of the papain family, involved in bulk degradation of stromal proteins during leaf senescence46, and SAG12, a marker of senescence and remobilization, were upregulated, the latter particularly in leaves and lower stems15.
Glutamine synthetase cytosolic isozyme 1 (GS1) has several metabolic functions, including primary ammonium assimilation in the roots and re-assimilation of catabolism-derived ammonia, for transport and distribution throughout the plant47. During leaf senescence, GS1 is involved in the assimilation and recycling of the ammonia generated from catabolic processes47, 48, remobilizing N to developing grains in cereals49, 50. In this work, an upregulation of a GS1 under N stress conditions was observed in the roots of Svevo. Although it is difficult to establish a global model of cytosolic GS response to variation in nitrogen availability in plant roots and leaves47, in Arabidopsis it has been observed that two cytosolic GS members were upregulated under severe chronic N stress20. The upregulation of GS1 in durum wheat roots under N chronic stress might be related to the re-assimilation of ammonia released from protein degradation occurring under N deficiency20.
One member of the Asparagine synthetase (AsnS) gene family was upregulated in durum wheat roots under N stress. Asparagine contributes to N remobilization in plant tissues, and its synthesis occurs by transferring an amide group from glutamine to aspartate, thanks to asparagine synthetase7. Based on similarity analyses, we realized that the AsnS DEG here detected belongs to class III of AsnS family51. Recent evidence showed that the AsnS rice gene (OsAS1) belonging to class III (according to our phylogenetic analyses, data not shown) is mainly expressed in roots in an NH4 +-dependent manner, similarly to GS1 52. The upregulation of AsnS in durum wheat following N deficiency might be due to a higher availability of ammonium, following protein degradation20.
Many iron transporter genes were activated in durum wheat roots under N stress. Iron plays a crucial role in N metabolism, being a metal cofactor of enzymes of the reductive assimilatory pathway, including nitrate reductase, nitrite reductase, and glutamate synthase53, 54. Moreover, the differential expression of transporter genes for other minerals and nutrients indicates that their uptake in durum wheat plants might be affected by N metabolism under cross-talking regulation.
We detected a high number of DE ABC transporters, mostly upregulated in durum wheat N stressed roots, two in flag leaf, and six downregulated in spike. ABC transporters have been shown to be involved in several processes such as phytate accumulation in seeds, transport of the phytohormones auxin and abscisic acid, peptides, sugars, lipids, etc., thus playing an important role in plant development and nutrition, organ growth, response to abiotic stress, and in plant interaction with the environment55, 56.
Nitrogen stress caused relevant alterations in the expression of genes related to carbon metabolism in durum wheat. It is well known that pathways for carbon and N assimilation are strongly related57, 58; in particular, for many species, N deficiency causes an increase in root growth due to a main redirection of carbon assimilates towards the belowground tissues59. Indeed, total carbon in our samples was higher in stressed tissues compared to control ones, especially in roots and stems (data not shown). An accumulation of C was also observed in low nitrogen common wheat stems, particularly found in the form of fructans15. Under N starvation, several enzymes involved in sucrose and starch metabolism were found upregulated in Svevo roots. This could be linked to a carbon partitioning oriented to root growth, since sucrose plays a role as a stimulant for lateral root formation60; recently it has been hypothesized a role for root starch in root development and in the response to nitrate availability61.
In durum wheat roots, the orthologues to malate dehydrogenase (MDH) and glyceraldehyde 3-phosphodehydrogenase (GAPDH) were upregulated. These genes are particularly relevant for the production of reducing equivalents, thus maintaining the glutathione reduced for the functioning of glutathione S-transferase (GST). GST catalyzes the glutathione-dependent detoxification reactions and the reduction of hydroperoxides. GSTs may act as binding proteins that sequestrate flavonoids in the vacuole for protection against environmental stresses62. On the other hand, we observed a downregulation of the orthologues to genes involved in the oxidative pentose pathway, including those providing reducing power (NADPH). In particular, a downregulation was observed for the orthologues to glucose 6-phosphate dehydrogenase (G6PDH), which is the rate-limiting step in this process and the orthologue to 6-phosphogluconate dehydrogenase (6PGD), which was shown to be nitrate inducible in Arabidopsis 63.
Under prolonged N deficiency, leaf senescence is generally accelerated and ultimately plant growth can be inhibited64. One of the causes of these processes is the decrease in photosynthetic activity of the plant, which is primarily due to the breakdown of Rubisco65. In this work, the majority of DEGs involved in photosynthesis and photorespiration including those coding for proteins related to Photosystem I and II, ATP synthase and Rubisco were downregulated. Among the few upregulated genes, the orthologue to ISPG, was found upregulated in roots. It reduces Ferredoxin (Fd) in plastids and thus it is important for the functioning of Fd-GOGAT and therefore for ammonium assimilation19.
More than 30 TF families were identified to respond to N-starvation in durum wheat, among which MYB and bHLH genes were the most abundant. These two TF families interact to regulate target genes and several R2R3-type MYB and bHLH TFs have been reported to be involved in plant stress responses66. Some R2R3-type MYB proteins have a role in the regulation of phenylpropanoid pathway67, in fact over-expression of specific MYB genes (e.g. PAP1, found upregulated in the present study) enhanced accumulation of lignin and flavonoids20. In the present work, the expression of several Svevo MYB transcription factors was found to be influenced by N conditions; for instance, MYB48 appeared to be upregulated in leaves/stems and root tissues of Svevo plants under N starvation. In rice, the overexpression of OsMYB48-1 enhanced the tolerance toward abiotic stresses probably via the regulation of stress-mediated ABA biosynthesis68. Furthermore, MYB86 and MYB20, which in the present study were downregulated in N stress conditions, were also associated with abiotic stress responses in Arabidopsis 69, 70. These MYB genes could also be regulated by miRNAs as it is the case of Svevo MYB3, which was identified as the target gene of ttu-miR319f71.
Another family of TFs DE in durum wheat under N starvation was the NF-Y TFs. Recent evidence in T. aestivum showed that NF-Y TFs are induced by low nitrogen conditions, increasing nitrogen uptake and grain yield72. In our study, NF-Y factors were mainly upregulated in the roots.
WRKY is one of the largest TF families responsive to N-deficiency in rice, with twelve WRKY members induced in the sheaths versus none in the roots17. In durum wheat, most of the WRKY identified to be responsive to N chronic stress were found in the roots. This difference in WRKY expression profiles between the two Graminaceae species might be due to the large number of WRKY transcription factors and their unknown and diverse roles under complex environmental stimulations73, and to the different plant developmental stage and environmental conditions used in the two species. Thanks to the relevant role they play in plant stress responses, WRKY proteins contribute to the establishment of complex signalling webs and are potential candidates for imparting N stress tolerance.
Moreover, MYB, NAC and WRKY TFs, in combination with hormones (ABA and jasmonic acid), have been shown to be involved in the transition of grain filling and developmental senescence45, 74.
Protein kinases are important for development and adaptation to abiotic stress in plants, by regulating transcription through the phosphorylation of transcription factors75. In the present work, different groups of PK genes were identified and many of them showed to be influenced by N deficiency. Differentially expressed PK genes were more abundant in root and leaves/stems, but were also identified in flag leaf and spike tissues. Most of the protein kinases belong to the RLK/Pelle family, which are involved in many different processes in plants, including signaling networks concerning abiotic environmental stimuli76, 77. Among the numerous RLK/Pelle DEGs, we found some Wall Associated Kinases (WAK) upregulated in roots, which might be involved in root growth under N limitation78.
Kinase genes related to the CIPK23 were downregulated in Svevo roots under N starvation conditions. This observation is in agreement with studies in Arabidopsis, where it has been demonstrated that CIPK23 is a nitrate-inducible protein kinase belonging to the family of CBL-interacting protein kinases being a negative regulator of the high-affinity nitrate transport response79. Therefore, the upregulation of high-affinity nitrate transporters already described here, may be a consequence of the downregulation of its repressors.
This study evidences an upregulation of several important genes for detoxification and protection from oxidative damage under N deficiency. A massive accumulation of reactive oxygen species (ROS) was also observed in Arabidopsis roots under N starvation80. In our study, several root DE orthologues to GST genes along with orthologues to CYP450 and several other genes including catalase 2 (CAT2), superoxide dismutase [Fe] 2 (FSD2) and aldehyde dehydrogenase (ALDH) were observed. This finding might suggest a role of roots in maintaining the redox homeostasis81, in agreement with a number of studies in Arabidopsis 20 and rice19, 56.
As expected, an upregulation of several genes involved in abscisic acid biosynthesis was noticed. This phytohormone, along with auxin and cytokines, has been shown to be involved in N demand and acquisition by bringing changes in plant physiology and morphology82.
An activation of the S-methylmethionine (SMM) cycle has been suggested to be involved in stress adaptation, resulting in the final products of cysteine and several polyamines such as spermine, spermidine and putrescine83. Cysteine plays a critical role in protection against abiotic/biotic stresses thanks to its derivatives, such as GSH and phytochelatin polymers84, 85. Moreover, an upregulation of pyridoxal phosphate (PLP) coenzyme was observed; PLP is important for the functioning of several aminotransferases, thus playing a role in N metabolism, and is notably involved in several stress responses86.
Conclusively, our study provides a landscape of the genes differentially expressed in durum wheat root, leaves/stems, flag leaf and spike following chronic N starvation. The severe phenotypic changes observed in plants under N stress were reflected in an altered transcriptomic activity in all organs of the plant, but especially in roots. To our best knowledge, this is the first comprehensive transcriptomic analysis in durum wheat under N deficiency, and provides valuable resources for a better understanding of durum wheat response to this stress and subsequent improvement of N use efficiency.
Electronic supplementary material
Acknowledgements
We thank Anita Morgese for laboratory assistance. This research was supported by the MIUR projects PON01_01145 ISCOCEM “Sviluppo tecnologico e innovazione per la sostenibilità e competitività della cerealicoltura meridionale”, and PRIN 2010–2011 DD 23/10/2012 no. 719 “Identificazione e caratterizzazione di geni utili ad incrementare la produttività e sostenibilità del frumento duro”. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
Conceived and planned the project: G.S. Designed the experiment: G.S., P.L.C. Plant growth experiment and phenotypic analyses: M.J., P.L.C. and G.S. Performed the bioinformatics analyses: P.L.C., R.A.C. and W.S. Analysed qPCR gene expression: P.L.C. and D.L.Z. Discussed results: P.L.C., R.A.C., D.L.Z. and G.S. Drafted the manuscript: P.L.C. Contributed in writing and revised the paper: G.S., D.L.Z., M.J. and R.A.C. All authors read and approved the final version of manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01377-0
Accession codes:The sequence data generated in this study were deposited in the NCBI Sequence Read Archive (Accession numbers SRR5079759–SRR5079782).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Pasquale L. Curci, Email: luca.curci@ibbr.cnr.it
Gabriella Sonnante, Email: gabriella.sonnante@ibbr.cnr.it.
References
- 1.Masclaux-Daubresse C, et al. Nitrogen uptake, assimilation and remobilization in plants: challenges for sustainable and productive agriculture. Ann. Bot. 2010;105:1141–1157. doi: 10.1093/aob/mcq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prinsi B, Negri AS, Pesaresi P, Cocucci M, Espen L. Evaluation of protein pattern changes in roots and leaves of Zea mays plants in response to nitrate availability by two-dimensional gel electrophoresis analysis. BMC Plant Biol. 2009;9:113. doi: 10.1186/1471-2229-9-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hakeem KR, Ahmad A, Iqbal M, Gucel S, Ozturk M. Nitrogen-efficient rice cultivars can reduce nitrate pollution. Environ. Sci. Pollut. Res. 2011;18:1184–1193. doi: 10.1007/s11356-010-0434-8. [DOI] [PubMed] [Google Scholar]
- 4.Gutierrez RA. Systems biology for enhanced plant nitrogen nutrition. Science. 2012;336:1673–1675. doi: 10.1126/science.1217620. [DOI] [PubMed] [Google Scholar]
- 5.EPA. Methane and Nitrous Oxide Emissions from Natural Sources. U.S. Environmental Protection Agency. Washington, DC: EPA. 430R10001, https://nepis.epa.gov (2010).
- 6.Rennenberg H, Wildhagen H, Ehlting B. Nitrogen nutrition of poplar trees. Plant Biol. 2010;12:275–291. doi: 10.1111/j.1438-8677.2009.00309.x. [DOI] [PubMed] [Google Scholar]
- 7.Xu G, Fan X, Miller AJ. Plant nitrogen assimilation and use efficiency. Ann. Rev. Plant Biol. 2012;63:153–82. doi: 10.1146/annurev-arplant-042811-105532. [DOI] [PubMed] [Google Scholar]
- 8.Näsholm T, Kielland K, Ganeteg U. Uptake of organic nitrogen by plants. New Phytol. 2009;182:31–48. doi: 10.1111/j.1469-8137.2008.02751.x. [DOI] [PubMed] [Google Scholar]
- 9.Gruffman L, Jämtgård S, Näsholm T. Plant nitrogen status and co-occurrence of organic and inorganic nitrogen sources influence root uptake by Scots pine seedlings. Tree Physiol. 2014;34:205–213. doi: 10.1093/treephys/tpt121. [DOI] [PubMed] [Google Scholar]
- 10.Lam HM, Coschigano KT, Oliveira IC, Melo-Oliveira R, Coruzzi GM. The molecular-genetics of nitrogen assimilation into amino acids in higher plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996;47:569–593. doi: 10.1146/annurev.arplant.47.1.569. [DOI] [PubMed] [Google Scholar]
- 11.Kong L, Xie Y, Hu L, Feng B, Li S. Remobilization of vegetative nitrogen to developing grain in wheat (Triticum aestivum L.) Field Crops Res. 2016;196:134–144. doi: 10.1016/j.fcr.2016.06.015. [DOI] [Google Scholar]
- 12.Weeda S, et al. Arabidopsis Transcriptome Analysis Reveals Key Roles of Melatonin in Plant Defense Systems. PLoS One. 2014;9:e93462. doi: 10.1371/journal.pone.0093462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hodgins KA, et al. Genomics of Compositae crops: reference transcriptome assemblies and evidence of hybridization with wild relatives. Mol. Ecol. Resour. 2014;14:166–177. doi: 10.1111/1755-0998.12163. [DOI] [PubMed] [Google Scholar]
- 14.Shankar R, Bhattacharjee A, Jain M. Transcriptome analysis in different rice cultivars provides novel insights into desiccation and salinity stress responses. Sci. Rep. 2016;6:23719. doi: 10.1038/srep23719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruuska SA, et al. Large scale transcriptome analysis of the effects of nitrogen nutrition on accumulation of stem carbohydrate reserves in reproductive stage wheat. Plant Mol. Biol. 2008;66:15–32. doi: 10.1007/s11103-007-9249-5. [DOI] [PubMed] [Google Scholar]
- 16.Gelli M, et al. Identification of differentially expressed genes between sorghum genotypes with contrasting nitrogen stress tolerance by genome-wide transcriptional profiling. BMC Genomics. 2014;15:179. doi: 10.1186/1471-2164-15-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang J, et al. RNA-seq reveals differentially expressed genes of rice (Oryza sativa) spikelet in response to temperature interacting with nitrogen at meiosis stage. BMC Genomics. 2015;16:959–977. doi: 10.1186/s12864-015-2141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quan X, et al. Transcriptome profiling analysis for two Tibetan wild barley genotypes in responses to low nitrogen. BMC Plant Biol. 2016;16:1. doi: 10.1186/s12870-016-0721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lian X, et al. Expression profiles of 10,422 genes at early stage of low nitrogen stress in rice assayed using a cDNA microarray. Plant Mol. Biol. 2006;60:617–631. doi: 10.1007/s11103-005-5441-7. [DOI] [PubMed] [Google Scholar]
- 20.Bi YM, Wang RL, Zhu T, Rothstein SJ. Global transcription profiling reveals differential responses to chronic nitrogen stress and putative nitrogen regulatory components in Arabidopsis. BMC Genomics. 2007;8:281. doi: 10.1186/1471-2164-8-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo TC, et al. Transcription analysis of genes encoding the wheat root transporter NRT1 and NRT2 families during nitrogen starvation. J. Plant Growth Regul. 2014;33:837–848. doi: 10.1007/s00344-014-9435-z. [DOI] [Google Scholar]
- 22.Cormier F, et al. Breeding for increased nitrogen-use efficiency: a review for wheat (T. aestivum L.) Plant Breed. 2016;135:255–278. doi: 10.1111/pbr.12371. [DOI] [Google Scholar]
- 23.Scala V, et al. Climate, soil management, and cultivar affect Fusarium head blight incidence and deoxynivalenol accumulation in durum wheat of Southern Italy. Front. Microbiol. 2016;7:1014. doi: 10.3389/fmicb.2016.01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ercoli L, Lulli L, Mariotti M, Masoni A, Arduini I. Post-anthesis drymatter and nitrogen supply and soil water availability. Europ. J. Agron. 2008;28:138–147. doi: 10.1016/j.eja.2007.06.002. [DOI] [Google Scholar]
- 25.Ryan J, Ibrikci H, Sommer R, McNeill A. Nitrogen in rainfed and irrigated cropping systems in the Mediterranean region. Adv. Agron. 2009;104:53–136. doi: 10.1016/S0065-2113(09)04002-4. [DOI] [Google Scholar]
- 26.Howarth JR, et al. Co-ordinated expression of amino acid metabolism in response to N and S deficiency during wheat grain filling. J. Exp. Bot. 2008;59:3675–3689. doi: 10.1093/jxb/ern218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Distelfeld A, Avni R, Fischer AM. Senescence, nutrient remobilization, and yield in wheat and barley. J. Exp. Bot. 2014;65:3783–3798. doi: 10.1093/jxb/ert477. [DOI] [PubMed] [Google Scholar]
- 28.Yin LP, Li P, Wen B, Taylor D, Berry JO. Characterization and expression of a high-affinity nitrate system transporter gene (TaNRT2.1) from wheat roots, and its evolutionary relationship to other NTR2 genes. Plant Sci. 2007;172:621–631. doi: 10.1016/j.plantsci.2006.11.014. [DOI] [Google Scholar]
- 29.Zadoks JC, Chang TT, Konzak CF. A Decimal Code for the Growth Stages of Cereals. Weed Res. 1974;14:415–421. doi: 10.1111/j.1365-3180.1974.tb01084.x. [DOI] [Google Scholar]
- 30.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trapnell, C. et al. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol.28, 511–515 (2010). [DOI] [PMC free article] [PubMed]
- 33.Du, Z., Zhou, X., Ling, Y., Zhang, Z. & Su, Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70 (2010). [DOI] [PMC free article] [PubMed]
- 34.Supek F, Škunca N, Repar J, Vlahoviček K, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saeed AI, et al. TM4: a free, open-source system from microarray data management and analysis. BioTechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 36.Gimenez MJ, Piston F, Atienza SG. Identification of suitable reference genes for normalization of qPCR data in comparative transcriptomics analyses in the Triticeae. Planta. 2011;233:163–173. doi: 10.1007/s00425-010-1290-y. [DOI] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real- time quantitative PCR and the 2(−delta deltaC(T)) method. Methods. 2001;4:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Blanco A, et al. Relationships between grain protein content and grain yield components through quantitative trait locus analyses in a recombinant inbred line population derived from two elite durum wheat cultivars. Mol. Breed. 2012;30:79–92. doi: 10.1007/s11032-011-9600-z. [DOI] [Google Scholar]
- 39.Walker RL, Burns IG, Moorby J. Response of plant growth rate to nitrogen supply: a comparison of relative addition and N interruption treatments. J. Exp. Bot. 2001;52:309–317. doi: 10.1093/jexbot/52.355.309. [DOI] [PubMed] [Google Scholar]
- 40.Sivasankar S, Oaks A. Regulation of nitrate reductase during early seedling growth. A role for asparagine and glutamine. Plant Physiol. 1995;107:1225–1231. doi: 10.1104/pp.107.4.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martre P, et al. Modelling protein content and composition in relation to crop nitrogen dynamics for wheat. Europ. J. Agron. 2006;25:138–154. doi: 10.1016/j.eja.2006.04.007. [DOI] [Google Scholar]
- 42.Lehti-Shiu MD, Shiu SH. Diversity, classification and function of the plant protein kinase superfamily. Phil. Trans. R. Soc. B. 2012;367:2619–2639. doi: 10.1098/rstb.2012.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H, McCaig TN, DePauw RM, Clarke JM. Flag leaf physiological traits in two high-yielding Canada Western Red Spring wheat cultivars. Can. J. Plant Sci. 2008;88:35–42. doi: 10.4141/CJPS07055. [DOI] [Google Scholar]
- 44.Barbottin A, Lecomte C, Bouchard C, Jeuffroy MH. Nitrogen remobilization during grain filling in wheat: genotypic and environmental effects. Crop Sci. 2005;45:1141–1150. doi: 10.2135/cropsci2003.0361. [DOI] [Google Scholar]
- 45.Kohl S, et al. Metabolic and transcriptional transitions in barley glumes reveal a role as transitory resource buffers during endosperm filling. J. Exp. Bot. 2015;66:1397–1411. doi: 10.1093/jxb/eru492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parrot DL, Martin JM, Fischer AM. Analysis of barley (Hordeum vulgare) lead senescence and protease gene expression: a family C1A cysteine protease is specifically induced under conditions characterized by high carbohydrate, but not low to moderate nitrogen levels. New Phytol. 2010;187:313–331. doi: 10.1111/j.1469-8137.2010.03278.x. [DOI] [PubMed] [Google Scholar]
- 47.Bernard SM, Habash DZ. The importance of cytosolic glutamine synthetase in nitrogen assimilation and recycling. New Phytol. 2009;182:608–620. doi: 10.1111/j.1469-8137.2009.02823.x. [DOI] [PubMed] [Google Scholar]
- 48.Avila-Ospina L, Moison M, Yoshimoto K, Masclaux-Daubresse C. Autophagy, plant senescence, and nutrient recycling. J. Exp. Bot. 2014;65:3799–3811. doi: 10.1093/jxb/eru039. [DOI] [PubMed] [Google Scholar]
- 49.Brestic M, et al. Reduced glutamine synthetase activity plays a role in control of photosynthetic responses to high light in barley leaves. Plant Physiol. Biochem. 2014;81:74–83. doi: 10.1016/j.plaphy.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Martin A, et al. Two cytosolic glutamine synthetase isoforms of maize are specifically involved in the control of grain production. Plant Cell. 2006;18:3252–3274. doi: 10.1105/tpc.106.042689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Todd J, et al. Identification and characterization of four distinct asparagine synthetase (AsnS) genes in maize (Zea mays L.) Plant Sci. 2008;175:799–808. doi: 10.1016/j.plantsci.2008.08.004. [DOI] [Google Scholar]
- 52.Ohashi M, et al. Asparagine Synthetase1, but not Asparagine Synthetase2, is Responsible for the Biosynthesis of Asparagine Following the Supply of Ammonium to Rice Roots. Plant Cell Physiol. 2015;56:769–778. doi: 10.1093/pcp/pcv005. [DOI] [PubMed] [Google Scholar]
- 53.Marschner, H. Mineral nutrition of higher plants. (Academic Press Ltd) p. 1995 (London, 1995).
- 54.Wang R, et al. Microarray analysis of the nitrate response in Arabidopsis roots and shoots reveals over 1,000 rapidly responding genes and new linkages to glucose, trehalose-6-phosphate, iron, and sulfate metabolism. Plant Physiol. 2003;132:556–567. doi: 10.1104/pp.103.021253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang J, et al. Plant ABC transporters. Arabidopsis Book. 2011;9:e0153. doi: 10.1199/tab.0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai H, Lu Y, Xie W, Zhu T, Lian X. Transcriptome response to nitrogen starvation in rice. J. Biosci. 2012;37:731–747. doi: 10.1007/s12038-012-9242-2. [DOI] [PubMed] [Google Scholar]
- 57.Antal T, Mattila H, Hakala-Yatkin M, Tyystjärvi T, Tyystjärvi E. Acclimation of photosynthesis to nitrogen deficiency in Phaseolus vulgaris. Planta. 2010;232:887–898. doi: 10.1007/s00425-010-1227-5. [DOI] [PubMed] [Google Scholar]
- 58.Schluter U, et al. Maize source leaf adaptation to nitrogen deficiency affects not only nitrogen and carbon metabolism but also control of phosphate homeostasis. Plant Physiol. 2012;160:1384–1406. doi: 10.1104/pp.112.204420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smolders E, Merckx R. Growth and shoot:root partitioning of spinach plants as affected by nitrogen supply. Plant, Cell Environ. 1992;15:795–807. doi: 10.1111/j.1365-3040.1992.tb02147.x. [DOI] [Google Scholar]
- 60.Roycewicz P, Malamy JE. Dissecting the effects of nitrate, sucrose and osmotic potential on Arabidopsis root and shoot system growth in laboratory assays. Philos. Trans. R. Soc. B. 2012;367:1489–1500. doi: 10.1098/rstb.2011.0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ikram S, Bedu M, Daniel-Vedele F, Chaillou S, Chardon F. Natural variation of Arabidopsis response to nitrogen availability. J. Exp. Bot. 2012;63:91–105. doi: 10.1093/jxb/err244. [DOI] [PubMed] [Google Scholar]
- 62.Tahkokorpi M, Taulavuori K, Laine K, Taulavuori R. After effects of drought- related winter stress in previous and current year stems of Vaccinium myrtillus L. Environ. Exp. Bot. 2007;61:85–93. doi: 10.1016/j.envexpbot.2007.03.003. [DOI] [Google Scholar]
- 63.Wang R, Guegler K, Labrie ST, Crawford NM. Genomic analysis of a nutrient response in Arabidopsis reveals diverse expression patterns and novel metabolic and potential regulatory genes induced by nitrate. Plant Cell. 2000;12:1491–1509. doi: 10.1105/tpc.12.8.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hortensteiner S, Feller U. Nitrogen metabolism and remobilization during senescence. J. Exp. Bot. 2002;53:927–937. doi: 10.1093/jexbot/53.370.927. [DOI] [PubMed] [Google Scholar]
- 65.Walker RL, Burns IG, Moorby J. Responses of plant growth rate to nitrogen supply: a comparison of relative addition and N interruption treatments. J. Exp. Bot. 2001;52:309–317. doi: 10.1093/jexbot/52.355.309. [DOI] [PubMed] [Google Scholar]
- 66.Pireyre M, Burow M. Regulation of MYB and bHLH transcription factors: A glance at the protein level. Mol. Plant. 2015;8:378–388. doi: 10.1016/j.molp.2014.11.022. [DOI] [PubMed] [Google Scholar]
- 67.Roy S. Function of MYB domain transcription factors in abiotic stress and epigenetic control of stress response in plant genome. Plant Signal. Behav. 2016;11:e1117723. doi: 10.1080/15592324.2015.1117723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xiong H, et al. Overexpression of OsMYB48-1, a novel MYB-related transcription factor, enhances drought and salinity tolerance in rice. PLoS One. 2014;9:e92913. doi: 10.1371/journal.pone.0092913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zeller G, et al. Stress-induced changes in the Arabidopsis thaliana transcriptome analyzed using whole-genome tiling arrays. Plant J. 2009;58:1068–1082. doi: 10.1111/j.1365-313X.2009.03835.x. [DOI] [PubMed] [Google Scholar]
- 70.Rasmussen S, et al. Transcriptome responses to combinations of stresses in Arabidopsis. Plant Physiol. 2013;161:1783–1794. doi: 10.1104/pp.112.210773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.De Paola D, Zuluaga DL, Sonnante G. The miRNAome of durum wheat: isolation and characterisation of conserved and novel microRNAs and their target genes. BMC Genomics. 2016;17:505. doi: 10.1186/s12864-016-2838-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qu B, et al. A wheat CCAAT box-binding transcription factor increases the grain yield of wheat with less fertilizer input. Plant Physiol. 2015;167:411–423. doi: 10.1104/pp.114.246959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen L, et al. The role of WRKY transcription factors in plant abiotic stresses. Biochim. Biophys. Acta. 2011;1819:120–128. doi: 10.1016/j.bbagrm.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 74.Hollmann J, Gregersen PL, Krupinska K. Identification of predominant genes involved in regulation and execution of senescence associated nitrogen remobilisation in flag leaves of field grown barley. J. Exp. Bot. 2014;65:3963–3973. doi: 10.1093/jxb/eru094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hunter T, Karin M. The regulation of transcription by phosphorylation. Cell. 1992;70:375–387. doi: 10.1016/0092-8674(92)90162-6. [DOI] [PubMed] [Google Scholar]
- 76.Sivaguru M, et al. Aluminum-induced gene expression and protein localization of a cell wall-associated receptor kinase in Arabidopsis. Plant Physiol. 2003;132:2256–2266. doi: 10.1104/pp.103.022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hou X, et al. Involvement of a cell wall-associated kinase, WAKL4. Arabidopsis mineral responses. Plant Physiol. 2005;139:1704–1716. doi: 10.1104/pp.105.066910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kiba T, Krapp A. Plant nitrogen acquisition under low availability: regulation of uptake and root architecture. Plant Cell Physiol. 2016;57:707–714. doi: 10.1093/pcp/pcw052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krapp A, et al. Nitrate transport and signalling in. Arabidopsis. J. Exp. Bot. 2014;65:789–798. doi: 10.1093/jxb/eru001. [DOI] [PubMed] [Google Scholar]
- 80.Shin R, Berg RH, Schachtman DP. Reactive oxygen species and root hairs in Arabidopsis root response to nitrogen, phosphorus and potassium deficiency. Plant Cell Physiol. 2005;46:1350–1357. doi: 10.1093/pcp/pci145. [DOI] [PubMed] [Google Scholar]
- 81.Page V, Schwitzguebel JP. The role of cytochromes P450 and peroxidases in the detoxification of sulphonated anthraquinones by rhubarb and common sorrel plants cultivated under hydroponic conditions. Environ. Sci. Pollut. Res. Int. 2009;16:805–816. doi: 10.1007/s11356-009-0197-2. [DOI] [PubMed] [Google Scholar]
- 82.Kiba T, Kudo T, Kojima M, Sakakibara H. Hormonal control of nitrogen acquisition: roles of auxin, abscisic acid, and cytokinin. J. Exp. Bot. 2011;62:1399–1409. doi: 10.1093/jxb/erq410. [DOI] [PubMed] [Google Scholar]
- 83.Lyi SM, Zhou X, Kochian LV, Li L. Biochemical and molecular characterization of the homocysteine S-methyltransferase from broccoli (Brassica oleracea var. italica) Phytochemistry. 2007;68:1112–1119. doi: 10.1016/j.phytochem.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 84.Leustek T, Martin MN, Bick JA, Davies JP. Pathways and regulation of sulphur metabolism revealed through molecular and genetic studies. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2000;51:141–165. doi: 10.1146/annurev.arplant.51.1.141. [DOI] [PubMed] [Google Scholar]
- 85.Noctor G, Gomez L, Vanacker H, Foyer CH. Interactions between biosynthesis, compartmentation and transport in the control of glutathione homeostasis and signalling. J. Exp. Bot. 2002;53:1283–1304. doi: 10.1093/jexbot/53.372.1283. [DOI] [PubMed] [Google Scholar]
- 86.Huang S, Zhang J, Wang L, Huang L. Effect of abiotic stress on the abundance of different vitamin B6 vitamers in tobacco plants. Plant Physiol. Biochem. 2013;66:63–67. doi: 10.1016/j.plaphy.2013.02.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.