

Abstract
MLL-rearranged acute myeloid leukemia (AML) remains a fatal disease with a high rate of relapse and therapeutic failure due to chemotherapy resistance. In analysis of our Affymetrix microarray profiling and chromatin immunoprecipitation (ChIP) assays, we found that ALOX5 is especially down-regulated in MLL-rearranged AML, via transcription repression mediated by Polycomb repressive complex 2 (PRC2). Colony forming/replating and bone marrow transplantation (BMT) assays showed that Alox5 exhibited a moderate anti-tumor effect both in vitro and in vivo. Strikingly, leukemic cells with Alox5 overexpression showed a significantly higher sensitivity to the standard chemotherapeutic agents, i.e., doxorubicin (DOX) and cytarabine (Ara-C). The drug-sensitizing role of Alox5 was further confirmed in human and murine MLL-rearranged AML cell models in vitro, as well as in the in vivo MLL-rearranged AML BMT model coupled with treatment of “5 + 3” (i.e. DOX plus Ara-C) regimen. Stat and K-Ras signaling pathways were negatively correlated with Alox5 overexpression in MLL-AF9-leukemic blast cells; inhibition of the above signaling pathways mimicked the drug-sensitizing effect of ALOX5 in AML cells. Collectively, our work shows that ALOX5 plays a moderate anti-tumor role and functions as a drug sensitizer, with a therapeutic potential, in MLL-rearranged AML.
Introduction
Acute myeloid leukemia (AML), the most common type of acute leukemia attacking adults, is a heterogeneous disease characterized by the malignant expansion of dysfunctional myeloid progenitors and the suppression of normal hematopoiesis1. The mixed lineage leukemia (MLL) gene on chromosome 11q23 is the frequent target of chromosomal translocations and rearrangements in infant and adult leukemias. AML bearing MLL fusion often associates with poor prognosis and is resistant to most of the current clinical chemotherapies2–4. Though immense efforts have been exerted in developing novel therapeutic strategies in the past decades, the standard chemotherapy with intensive administration of chemotherapeutic drugs, e.g. the “7 + 3” regimen (i.e. combined treatment of anthracycline (daunorubicin or doxorubicin) and cytarabine (Ara-C)), remains the main therapeutic approach for treating AML, including MLL-rearranged AML5–8. Resistance to the standard chemotherapy remains the major cause of relapse and therapeutic failure7, 9. Despite intensive chemotherapy, the overall survival of AML patients is still less than 30% for adults and 60% for children, and most of the patients with MLL-rearranged AML fail to survive longer than 5 years4, 9–11. Therefore, it is crucial to understand the mechanisms underlying the pathogenesis and chemotherapy resistance of AML, such as MLL-rearranged AML, and identify potential new therapeutic targets that can suppress AML or improve the response to the currently existing chemotherapies.
The mechanism of AML drug response remains obscure. A number of key factors including transcriptional factors, oncogenes and tumor suppressors, as well as cell metabolism regulators, have been identified to play important roles in determining drug sensitivity or resistance7, 9. Among these factors, the JAK/STAT signaling is known to be closely associated with myeloid malignancies. Activation of JAK/STAT pathway is a hallmark of the myeloproliferative neoplasms, and is found in many AML cases12, 13. JAK/STAT pathway inhibitors, e.g. pacritinib and ruxolitinib, have already been studied in pre-clinical and clinical trials14, 15. The proto-oncogene K-RAS is another factor involved in both leukemogenesis and therapeutic response16. K-RAS mutations have been reported in many AML cases and are associated with prognosis and chemotherapy resistance17, 18. It holds great potential to cure AML by either targeting these factors directly or targeting a common upstream regulator that controls all these factors in AML.
The arachidonate 5-lipoxygenase gene (ALOX5) encodes a non-heme iron-containing enzyme of the lipoxygenase family. It catalyzes the production of leukotrienes (LTs) and reactive oxygen species (ROS) from arachidonic acid19, 20. ALOX5 is known to be involved in various physiological and pathological processes, including oxidative stress response, inflammation and cancer21–25. It was reported previously that loss of Alox5 impairs the function of leukemic stem cells (LSCs) in BCR-ABL-induced chronic myelogenous leukemia (CML)26. To our surprise, our gene profiling reveals that ALOX5 expression is particularly down-regulated in MLL-rearranged AML. Both in vitro and in vivo studies were carried out to investigate the effects and underlying mechanism of ALOX5 in AML pathogenesis and drug response.
Results
ALOX5 is especially down-regulated in MLL-rearranged AML
In analysis of our Affymetrix microarray-based gene expression profiling of 100 human AML samples carrying MLL-rearrangements/t(11q23) (n = 12), inv(16) (n = 27), t(8;21) (n = 30) or t(15;17) (n = 31), along with 9 normal bone marrow (BM) control (3 each for CD34+ hematopoietic stem/ progenitor cell, CD33+ myeloid progenitor cell, and mononuclear cell (MNC)) samples27–29, we found that ALOX5 was expressed at a significantly lower level in MLL-rearranged AML, but not in other AML subtypes, relative to the normal controls (Fig. 1a). Notably, the expression level of ALOX5 in MLL-rearranged AML is also significantly lower than that in other AML subtypes (Fig. 1a); such pattern was also observed in an independent large-scale AML dataset (i.e., GSE1446830, 31) (Fig. 1b). The downregulation of Alox5 in MLL-rearranged AML was verified through both qPCR (Fig. 1c) and Western blotting (Fig. 1d) in MLL-AF9 leukemic blast cells isolated from the relevant leukemic mice, as compared with normal controls.
Figure 1.
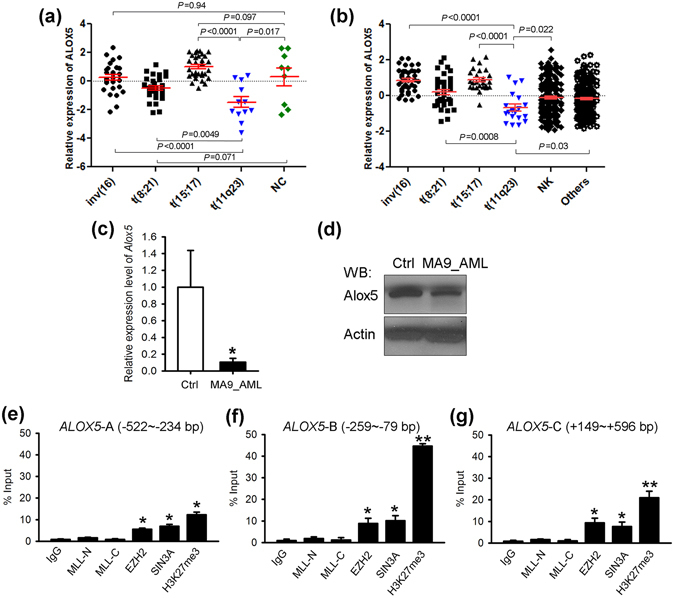
Expression of ALOX5 is significantly down-regulated in MLL-rearranged AML. (a) Comparison of ALOX5 expression between t(11q23)/MLL-rearranged AML and AML cases with inv(16), t(8;21) or t(15;17), or normal controls (NC) in our in-house AML dataset27–29. (b) Comparison of ALOX5 expression between t(11q23) AML and other subtypes of AML cases in GSE1446830. The expression values were log2-transformed and mean centered. The P values were detected by t-test. (c,d) Downregulation of Alox5 in MLL-AF9-AML mouse BM mononuclear cells, as compared with normal controls. Levels of Alox5 were detected through qPCR (c) and Western blotting (d). (e–g) EZH2 and SIN3A bind the promoter of ALOX5. Enrichment of MLL-N terminal (for both wild-type MLL and MLL fusions), MLL-C terminal (for wild-type MLL), EZH2, SIN3A, H3K27me3 or IgG at the ALOX5 promoter region was shown. Relative to transcription start site of ALOX5, locations of the three PCR amplicon sites are: ALOX5-A (e): −522~−234 bp; ALOX5-B (f): −259~−79 bp; ALOX5-C (g): +149~+596 bp32. *P < 0.05; **P < 0.01, two-tailed t-test.
To understand the mechanisms of the repression of ALOX5 in MLL-rearranged AML, we conducted chromatin immunoprecipitation (ChIP) assays. We found no significant enrichment of MLL fusion proteins at the promoter region32 of the ALOX5 locus (Fig. 1e–g). It is known that gene silencing mediated by the Polycomb repressive complex 2 (PRC2) and cofactors, e.g. EZH2 and SIN3A, is essential for MLL-rearranged AML33. Here we show a significant enrichment of EZH2 and SIN3A at the ALOX5 promoter region. Histone H3 lysine 27 tri-methylation (i.e., H3K27me3), a marker for repressive transcription associated with EZH234, 35, also exhibited high enrichement at these genomic locus (Fig. 1e–g). Therefore, it is highly likely that the PRC2 complex mediates the transcriptional repression of ALOX5 in MLL-rearranged leukemia.
Anti-tumor effect of Alox5 in MLL-rearranged AML
To assess the pathological role of Alox5 in MLL-rearranged AML, we cloned Alox5 CDS into MSCV-PIG retroviral vector, and then co-transduced MSCVneo-MLL-AF9 (MA9) with MSCV-PIG-Alox5 (Alox5) or MSCV-PIG (Ctrl) into mouse BM progenitor cells for in vitro colony-forming/replating assays. We showed that along with the increased number of passages, Alox5 overexpression showed a more significant degree of repression on MLL-AF9-induced colony forming (Fig. 2a). We also analyzed cell viability of those retrovirus transduced progenitor cells and showed that forced expression of Alox5 significantly suppressed cell viability (Fig. 2b). Knockdown of ALOX5 with siRNA did not show significant alterations in the viability of MONOMAC-6/t(9;11) cells (Supplementary Figs 1a–c and 3a,b).
Figure 2.

Anti-tumor effect of Alox5 in MLL-AF9-AML. (a) Inhibitory effect of Alox5 on MLL-AF9-induced colony forming/immortalization of normal mouse bone marrow progenitor cells. Normal mouse bone marrow progenitor cells were retrovirally transduced with MSCVneo-MLL-AF9 + MSCV-PIG (MA9 + Ctrl) or MSCVneo-MLL-AF9 + MSCV-PIG-Alox5 (MA9 + Alox5). Colony forming/replating assays (CFAs) were conducted for four passages. 20,000 cells were plated in each dish. (b) Alox5 overexpression represses cell viability of MLL-AF9-transduced mouse bone marrow progenitor cells. After the first passage, 10,000 of the above transduced cells were seeded in liquid culture medium. MTT assays were conducted 72 hours after seeding. (c) Effects of Alox5 on MLL-AF9-induced primary leukemia in vivo. Normal mouse bone marrow progenitor cells (CD45.1) were retrovirally co-transduced with MSCVneo-MLL-AF9 + MSCV-PIG (MA9 + Ctrl) or MSCVneo-MLL-AF9 + MSCV-PIG-Alox5 (MA9 + Alox5). 0.2 × 105 of the transduced cells were used as donor cells and transplanted into each primary recipient mouse (CD45.2). MA9 + Ctrl: n = 14; MA9 + Alox5: n = 16. (d) Inhibitory effects of Alox5 on the maintenance of MLL-AF9-induced AML in secondary BMT recipient mice. The secondary BMT recipients were transplanted with BM blast cells from the primary MLL-AF9 AML mice retrovirally transduced with MSCV-PIG (MA9-AML_Ctrl; n = 7) or MSCV-PIG-Alox5 (MA9-AML_Alox5; n = 8). Kaplan-Meier curves are shown. The P values were determined by log-rank test. (e) Total white blood cell (WBC) counts of the secondary BMT recipients were determined by Hemavet 950 at the mice’ end points. n = 5 for each group. (f) Weights of spleen and liver of each secondary BMT recipient at its end point are shown. n = 5 for each group. (g) Flow cytometry analyses of BM cells of secondary BMT recipients. The “blast” population was gated by FSC/SSC (framed out; upper panel) and the proportions of CD45.1+ (middle panel), Gr1+ and/or Mac1+ cells were analyzed (lower panel). (h) Statistical analysis of the Gr1+Mac1+ population of secondary BMT recipients. n = 5 for each group. *P < 0.05; **P < 0.01, two-tailed t-test. (i) Wright-Giemsa staining of mouse peripheral blood (PB) and BM, or hematoxylin and eosin (H&E) staining of mouse spleen and liver of secondary BMT recipients.
In order to determine the in vivo effect of Alox5 in leukemogenesis, we performed primary BM transplantation (BMT) assay first and found that forced expression of Alox5 showed no significant influence on overall survival (medium overall survival: 108 days for the MA9 + Ctrl group vs. 96 days for MA9 + Alox5; P = 0.556, log-rank test) (Fig. 2c). We then isolated the leukemic blast cells from primary MLL-AF9-leukemic mice and transduced the cells with Alox5 or control retrovirus, and performed secondary BMT assays. Notably, Alox5 overexpression significantly delayed the progression of MLL-AF9 AML in secondary BMT recipients (median overall survival, 42.5 days (MA9_Alox5) vs. 37 days (MA9_Ctrl); P = 0.018, log-rank test) (Fig. 2d). While all the leukemic mice died from AML, overexpression of Alox5 resulted in a significant decrease in peripheral white blood cell count (Fig. 2e) and spleen size (Fig. 2f). Moreover, through flow cytometry analysis, we found that although the Alox5 overexpressing group and the control group have similar degrees of engraftment of MLL-AF9 donor cells in BM at their end points, the former has a significantly lower population of the Mac-1+/Gr-1+ leukemic blast cells than the latter (Fig. 2g,h). Tissue staining showed that, compared with the control group, the BM cells in the Alox5 overexpressing group were more differentiated, consistent with the flow cytometry results, and the infiltration of leukemic cells into spleen, liver, and peripheral blood was less severe (Fig. 2i). Thus, our data indicated that Alox5 exhibited a moderate anti-tumor effect in the maintenance of MLL-rearranged AML and restrains leukemic cell infiltration.
Forced expression of Alox5 sensitizes MLL-rearranged AML cells to standard chemotherapy
The anti-tumor role of Alox5 implies a therapeutic potential in treating AML. Since restoration of Alox5 expression/function alone showed only moderate inhibitory effect on AML progression, we sought to investigate whether Alox5 restoration could facilitate chemotherapeutic response and thus yield a more effective therapeutic effect. We first used lentivirus to overexpress human ALOX5 in MONOMAC-6/t(9;11) AML cells, and treated the transduced cells with doxorubicin (DOX) or cytarabine (Ara-C). Our results showed that overexpression of ALOX5 significantly sensitized MONOMAC-6 cells to DOX and especially Ara-C treatment (Fig. 3a–c; Supplementary Fig. 3c,d).
Figure 3.
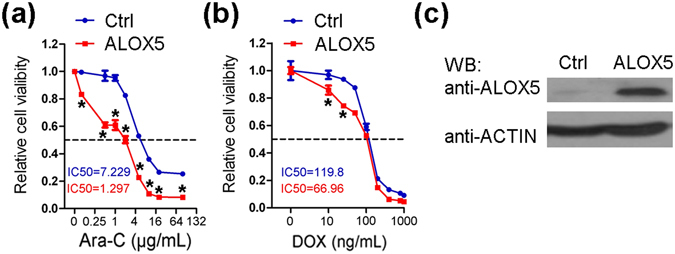
Drug sensitizing effect of ALOX5 in human AML cells. (a,b) MONOMAC-6 cells were transduced with ALOX5 or control lentivirus, and were treated with Ara-C (a) or DOX (b) at indicated doses 7 days after lentiviral infection and selection. MTT assays were conducted 72 hours post-drug treatments. *P < 0.05, two-tailed t-test. (c) Protein level of ALOX5 in the transduced cells was verified through Western blotting.
Similarly, in mouse BM progenitor cells transduced with MLL-AF9, co-expression of Alox5 remarkably enhanced the inhibitory effect of DOX or Ara-C on cell viability and colony forming capacity (Fig. 4a–c). We further tested the drug sensitizing effect of Alox5 in vivo. We transplanted MLL-AF9 AML cells with forced expression of Alox5 (i.e., MA9_Alox5) or without (i.e., MA9) into secondary recipient mice, and after the onset of leukemia, the recipient mice were treated with or without DOX + Ara-C (i.e. “5 + 3” regimen5). As shown in Fig. 4d, while DOX + Ara-C treatment alone only moderately (though statistically significantly) improved survival in mice carrying MLL-AF9 AML, forced expression of Alox5 dramatically improved the response of MLL-AF9 AML to the DOX + Ara-C treatment (Fig. 4d). As a result, amazingly over 70% of mice in the MA9_Alox5 + DOX/Ara-C group survived over 200 days, while all mice in the other groups died from AML within 70 days (Fig. 4d). The infiltration of leukemic cells into the spleen, liver, and peripheral blood was almost completely blocked in the MA9_Alox5 + DOX/Ara-C group of mice (Fig. 4e). Therefore, restoration of Alox5 holds great potential in improving chemotherapeutic response in MLL-rearranged AML patients.
Figure 4.
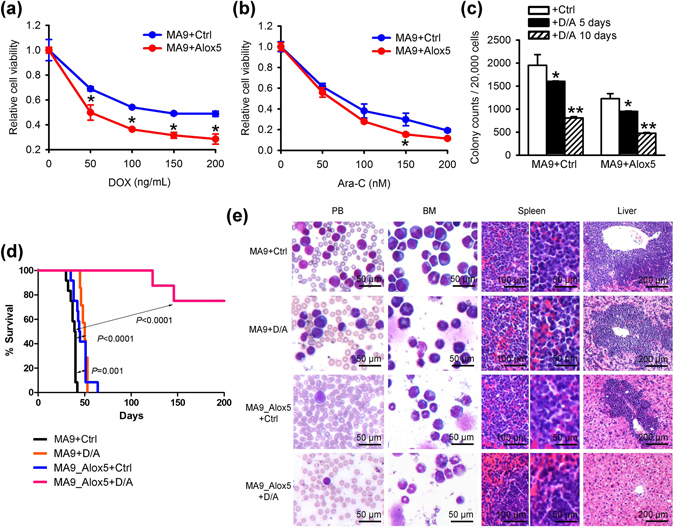
Drug sensitizing effect of Alox5 in mouse MLL-rearranged AML. (a,b) Normal mouse bone marrow progenitor cells were retrovirally transduced with MSCVneo-MLL-AF9 + MSCV-PIG (MA9 + Ctrl) or MSCVneo-MLL-AF9 + MSCV-PIG-Alox5 (MA9 + Alox5). 10,000 of the transduced cells were seeded in liquid culture medium and treated with DOX (a) or Ara-C (b) at indicated doses. MTT assays were conducted 72 hours after drug treatments. (c) Alox5 overexpression enhanced the inhibitory effect of DOX and Ara-C on MLL-AF9-induced colony forming. On the second passage of serial replating, colony cells were treated with control, or 50 ng/mL DOX plus 50 nM Ara-C, for 5 or 10 days. *P < 0.05; **P < 0.01, two-tailed t-test. (d) Alox5 overexpression enhanced the therapeutic effects of DOX and Ara-C on MLL-AF9-AML in vivo. Secondary MLL-AF9-AML recipient mice with Alox5 overexpression (MA9_Alox5) or without (MA9) were treated with PBS (control), or a daily dose of 50 mg/kg Ara-C for five days along with a daily dose of 1.5 mg/kg DOX during the first three days of Ara-C treatment (i.e., ‘5 + 3’ regimen). Kaplan-Meier curves are shown. MA9 + Ctrl: n = 12; MA9 + DOX/Ara-C (D/A): n = 7; MA7_Alox5 + Ctrl: n = 12; MA9_Alox5 + D/A: n = 8. The P values were determined by log-rank test. (e) Wright-Giemsa staining of mouse PB and BM, or H&E staining of mouse spleen and liver of secondary BMT recipients after control or drug treatments.
Signaling pathways correlated with Alox5 overexpression in AML
To delineate the potential molecular mechanism underlying the anti-tumor and drug-sensitizing effects of Alox5, we performed RNA sequencing (RNA-seq) of two pairs of mouse BM leukemic blast cells collected from the MA9_Ctrl and MA9_Alox5 mice in secondary BMT assays shown in Fig. 2d. Through gene set enrichment analysis (GSEA)36, we found that Il-2/Stat5 signaling and K-Ras pathway were significantly suppressed in MA9_Alox5 AML cells as compared with MA9_Ctrl AML cells (Fig. 5a,b). Stat5 and K-Ras function as critical oncogenes in both solid tumors and leukemia, and both STAT5 and K-RAS signaling pathways were known to be closely associated with oncogenesis and drug sensitivity15, 16, 37. c-MYC is a downstream target gene shared by STAT and K-RAS signaling pathways38, 39. We analyzed two independent AML patient databases and found a significant negative correlation between the expression levels of ALOX5 and c-MYC (Fig. 5c,d). In BM leukemic blast cells of MLL-AF9 leukemic mice, overexpression of Alox5 suppressed the expression of c-Myc and Flt3, an upstream key regulator of both Stat5 and K-Ras signaling40 (Fig. 5e,f; Supplementary Fig. 3e–g). In order to determine whether repression of STAT5 and/or K-RAS pathways could mimic the drug sensitizing effect of ALOX5, we treated MONOMAC-6 cells with a combination of DOX or Ara-C, together with control, STAT5 inhibitor sc-35597941, and/or RAS signaling inhibitor Tipifarnib42. Results showed that a combined treatment of both sc-355979 and Tipifarnib significantly sensitized the cells to chemotherapy (Fig. 5g,h). In a data set composed of 82 AML samples (including 26 MLL-rearranged AMLs)43, those bearing K-RAS mutations (n = 5) are associated with a lower ALOX5 level (Supplementary Fig. 2a), indicating a potential positive feedback between K-RAS signaling and ALOX5. Noticeably, the only AML sample with mutated K-RAS that has relatively higher ALOX5 level also bears FLT3 mutation. The remarkable suppression of ALOX5 in MLL-rearranged AML bearing K-RAS mutation was further verified in human CD34+ derived MLL-AF9 cell lines (Supplementary Fig. 2b). Therefore, our results suggest that Alox5 exerts its anti-tumor and drug sensitizing effects in MLL-rearranged AML through suppressing the Stat and K-Ras signaling pathways.
Figure 5.
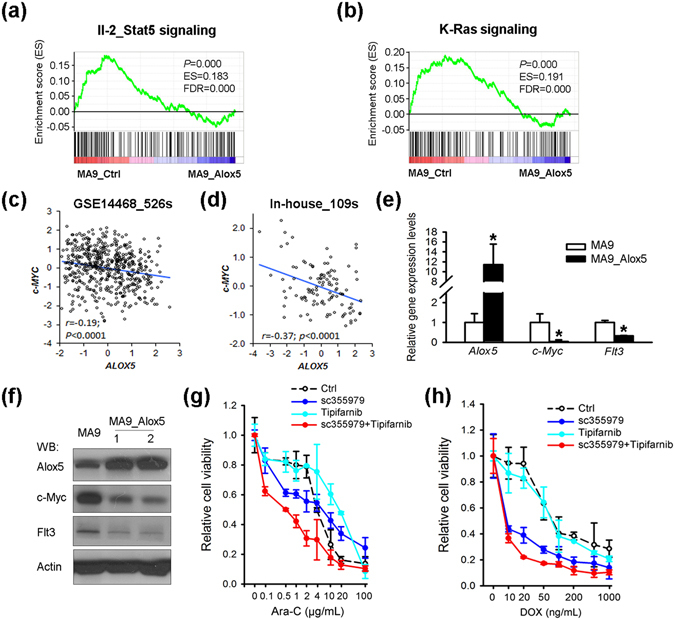
ALOX5 represses both STAT5 and K-RAS signaling pathways in MLL-rearranged AML. (a,b) RNA sequencing and Gene Set Enrichment Analysis (GSEA) of Alox5 overexpressed AML. Shown are gene sets of Il-2/Stat5 pathway (a) and K-Ras pathway (b) that are significantly enriched (P < 0.05) in genes with a lower level in the BM leukemic blast cell samples of secondary MLL-AF9-AML mice with Alox5 overexpression (MA9_Alox5), as compared with secondary MLL-AF9-AML mice without Alox5 overexpression (MA9_Ctrl). ES, enrichment score; FDR, false discovery rate. (c,d) Correlation between the expression levels of ALOX5 and c-MYC, a downstream target genes of shared by STAT and K-RAS signaling pathways, in two independent AML patient datasets. All expression data were log(2) transformed. The correlation coefficient (r) and P values were detected by “Pearson Correlation”, and the correlation regression lines were drawn with the “linear regression” algorithm. (e,f) Levels of Alox5, Myc and Flt3 in BM mononuclear cells of MLL-AF9 leukemic mice and normal controls were detected with qPCR (e) and Western blotting (f). (g,h) Inhibition of STAT5 and K-RAS signaling pathways sensitizes MONOMAC-6 cells to chemotherapy. MONOMAC-6 cells were treated with DMSO control, 1 μM sc-355979, 100 nM Tipifarnib, in combination with control, Ara-C (g) or DOX (h) at indicated doses. Cell viability was tested 48 hours post treatment.
Discussion
In contrast to the oncogenic role of ALOX5 reported previously in CML26, here we show that ALOX5, suppressed by PRC2 at the transcription level, exhibits a moderate anti-tumor effect both in vitro and in vivo in MLL-rearranged AML, in which ALOX5 expression is especially repressed. More interestingly, we show that restoration of Alox5 expression can substantially increase the sensitivity of MLL-rearranged AML cells to standard chemotherapeutic agents such as DOX and Ara-C both in vitro and in vivo, as well as the underlying mechanism through suppressing the STAT and K-RAS oncogenic signaling pathways.
Interestingly, ALOX5 was largely known as an oncogene in solid tumors, e.g. prostate cancer and pancreas cancer24, 25. However, its role remains vague in leukemia. It was reported that Alox5 deficiency resulted in a significant reduction of LSCs in BM, and thus largely prolonged survival of BCR-ABL-induced CML mice26. Nonetheless, there was no significant influence of Alox5 deficiency on normal hematopoietic stem cells (HSCs) or on the induction of lymphoid leukemia by BCR-ABL 26. Another study showed that Alox5 deficiency inhibited in vitro colony-forming capacity of BM progenitor cells induced by some AML oncogenic fusion genes such as AML1-ETO9a, a potent oncogenic isoform of AML1-ETO resulting from t(8;21)44, but had no significant effects on AML1-ETO9a-induced leukemogenesis in vivo 32. Those findings together with our present results suggest that Alox5 likely plays distinct roles at different lineages (myeloid and lymphoid) or different stages (i.e. induction and maintenance) of various hematopoietic malignancies.
One of the major challenges in AML therapy is the high frequency of occurrence of therapeutic resistance and the subsequent relapse7. Thus far, a variety of factors, such as JAK/STAT signaling and RAS pathway, have been identified to be closely related with chemotherapy response9, 13, 16. Abnormal activation of JAK/STAT signaling is found in most de novo AML13. K-RAS is a proto-oncogene essential for many solid tumors and hematopoietic malignancies16. Although RAS (i.e. N-RAS or K-RAS) gene mutations were only found in no more than 10% of AML cases, activation of RAS pathway by mutations in upstream receptors, e.g. FLT3 and c-KIT, or downstream effectors, broadly exist in AML45. A set of inhibitors have been developed to target JAK/STAT signaling (e.g. OPB-31121 and pacritinib) or RAS pathway (e.g., Selumetinib), but their efficacy in clinical trials to treat hematopoietic malignancies was still limited46–48. Our data suggest that restoration of ALOX5 expression/function could suppress both JAK/STAT and RAS signaling pathways simultaneously, and thus represents an alternative strategy, other than individual small-molecule inhibitors of JAK/STAT and RAS signaling pathways, to target these two critical oncogenic pathways in treating AML. Especially, restoration of ALOX5 expression/function in combination with the standard chemotherapy represents a potentially more effective therapeutic strategy for curing AML, at least MLL-rearranged AML. Thus, it would be interesting to identify small-molecule compound(s), such as natural product(s), that can specifically induce endogenous expression of ALOX5 expression in AML cells, and such compound(s) can be applied together with standard chemotherapy to treat MLL-rearranged AML.
Methods
Affymetrix exon array assays of human samples
As described previously27–29, a total of 100 human AML (including 30 t(8;21), 27 inv(16), 31 t(15;17) and 12 MLL-rearranged) and 9 normal BM samples (including 3 each of CD34+ hematopoietic stem/progenitor, CD33+ myeloid, and mononuclear cell (MNC) samples) were analyzed by use of Affymetrix GeneChip Human Exon 1.0 ST arrays (Affymetirx, Santa Clara, CA). Robust Multi-array Average (RMA)49 was used for the data normalization with Partek Genomics Suite (Partek Inc., St. Louis, MI). The complete microarray data set has been deposited into the GEO database under the accession code GSE34184 and GSE3028543.
Plasmid construction
The CDS of ALOX5, encoding the human ALOX5 gene, was amplified through PCR using primers 5′-ATAACCGGTCCACCATGGATTACAAGGATGACGATGACAAGCCCTCCTACACGGTC-3′ and 5′-AGCGAATTCTCAGATGGGCACACTGTTCGGA-3′, and then cloned into pLJM1-EGFP lentiviral vector (Addgene, Cambridge, MA). The CDS of mouse Alox5 gene was amplified through PCR using primers 5′-AATCTCGAGCCACCATGGATTAC AAGGATGACGATGACAAGCCCTCCTACACG and 5′-ATTGAATTCTTAGATGGCTACGCTGTTGGGAAT-3, and then ligated into a retroviral vector, namely MSCV-PIG (i.e., MSCV-puro-IRES-GFP vector; bearing GFP gene), a kind gift from Drs. Gregory Hannon, Scott Hammond, and Lin He (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY).
Cell culture
MONOMAC-6 cells were maintained in RPMI 1640 supplemented with 10% FBS, 1% HEPES, 2 mM L-Glutamine, 100 × Non-Essential Amino Acid, 1 mM sodium pyruvate, 9 μg/ml insulin and 1% penicillin-streptomycin. Mouse leukemic cells were kept in RPMI 1640 supplemented with interleukin 3 (IL-3) and IL-6, 10 ng/ml; SCF 100 ng/ml; 10% FBS and 1% penicillin-streptomycin. siRNAs were transfected with Cell Line Nucleofector Kit V following program T-037, using the Amaxa Nucleofector Technology (Amaxa Biosystems, Berlin, Germany). Experiments were performed 48 h after transfection.
Lentivirus production and infection
pMD2.G, pMDLg/pRRE, pRSV-Rev and pLJM1-EGFP plasmids were purchased from Addgene (Cambridge, MA). 0.5 μg pMD2.G, 0.3 μg pMDLg/pRRE, 0.7 μg pRSV-Rev and 1.5 μg pLJM1-EGFP constructs, i.e. pLJM1-ALOX5 or pLJM1-EGFP control were co-transfected into HEK-293T cells in 60 mm cell culture dish with Effectene Transfection Reagent (QIAGEN, Valencia, CA). Lentiviral particles were harvested at 48 and 72 hours after transfection and concentrated with PEG-itTM Virus Precipitation Solution (SBI). The lentivirus particles were directly added into leukemic cells and the cells were washed with PBS 24 hours after infection.
Cell viability assays
Cells were seeded into 96-well plates at the concentration of 10,000 cells/ well in triplicates and MTT (Promega, Madison, WI) was used to assess cell proliferation and viability following the manufacturer’s instructions.
Chromatin immunoprecipitation (ChIP)
ChIP assay was conducted as described previously50, with SABiosciences Corporation’s ChampionChIP One-Day kit (Qiagen, Frederick, MD) following the manufacturer’s protocol. Chromatin from THP-1 cells were cross-linked, sonicated into an average size of ~500 bp, and then immunoprecipitated with antibodies against the N’-terminal of MLL (MLL-N), the C’-terminal of MLL (MLL-C), EZH2, SIN3A, H3K27Me3 or IgG (Abcam, Cambridge, MA). Purified DNA was amplified by real-time qPCR using primers targeting the promoter of ALOX5 as described before32.
Western blotting
Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and ruptured with RIPA buffer (Pierce, Rockford, IL) containing 5 mM EDTA, PMSF, cocktail inhibitor, and phosphatase inhibitor cocktail. Cell extracts were microcentrifuged for 20 min at 10,000 g and supernatants were collected. Cell lysates (20 μl) were resolved by SDS-PAGE and transferred onto PVDF membranes. Membranes were blocked for 1 hour with 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 and incubated overnight at 4 °C with anti-ALOX5 antibody (Cell Signaling Technology Inc., Danvers, MA) or anti-ACTIN antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Membranes were washed 30 min with Tris-buffered saline containing 0.1% Tween-20, incubated for 1 hour with appropriate secondary antibodies conjugated to horseradish peroxidase, and developed using chemiluminescence substrates.
In vitro colony forming and replating assays (CFAs)
CFAs were conducted as described previously with some modifications29, 50, 51. Briefly, retrovirus vectors were co-transfected with pCL-Eco packaging vector (IMGENEX, San Diego, CA) into HEK293T cells using Effectene Transfection Reagent (Qiagen, Valencia, CA) to produce retrovirus. BM cells were harvested from a cohort of 4- to 6-week-old B6.SJL (CD45.1) donor mice after five days of 5- fluorouracil (5-FU) treatment, and primitive hematopoietic progenitor cells were enriched with Mouse Lineage Cell Depletion Kit (Miltenyi Biotec Inc., Auburn, CA). An aliquot of enriched hematopoietic progenitor cells was added to retroviral supernatant together with polybrene in cell culture plates, which were centrifuged at 2,000 g for 2 hours at 32 °C (i.e., “spinoculation”27–29) and then the medium was replaced with fresh media and incubated for 20 hours at 37 °C. Next day, the same procedure was repeated once.
Then, on the day following the second spinoculation, an equivalent of 2.0 × 104 cells were plated into a 35 mm Petri dish in 1.5 ml of Methocult M3230 methylcellulose medium (Stem Cell Technologies Inc, Vancouver, Canada) containing 10 ng/ml each of murine recombinant IL-3, IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF), and 30 ng/ml of murine recombinant SCF (R&D Systems, Minneapolis, MN), along with 1.0 mg/ml of G418 and 2 μg/ml of puromycin. For each transduction, there were two duplicate dishes. Cultures were incubated at 37 °C in a humidified atmosphere of 5% CO2 in air. The colonies were replated every 7 days under the same conditions. The colony-forming/replating assays were repeated 3 times.
The maintenance, monitoring, and end-point treatment of mice
C57BL/6 (CD45.2), B6.SJL (CD45.1) mice were purchased from the Jackson Lab (Bar Harbor, ME, USA) or Harlan Laboratories, Inc (Indianapolis, IN, USA) and maintained in house. Both male and female mice were used for the experiments. All laboratory mice were maintained in the animal facility at University of Cincinnati and the University of Chicago. All experiments on mice in our research protocol were approved by Institutional Animal Care and Use Committee (IACUC) of University of Cincinnati and the University of Chicago. All methods were performed in accordance with the relevant guidelines and regulations. The maintenance, monitoring, and end-point treatment of mice were conducted as described previously27–29, 50, 52–54.
Bone marrow transplantation assays
For primary bone marrow transplantation (BMT) assays, normal BM cells of B6.SJL (CD45.1) mice were retrovirally co-transduced with MSCVneo-MLL-AF9 + MSCV-PIG (i.e., MA9 + Ctrl) or MSCVneo-MLL-AF9 + MSCV-PIG-Alox5 (i.e., MA9 + Alox5), through two rounds of spinoculation, as described previously27–29, 50, 52–54. Then, the transduced donor cells were injected by tail vein into lethally irradiated (960 rads) 8- to 10-week-old C57BL/6 (CD45.2) recipient mice with 3 × 105 donor cells plus a radioprotective dose of whole BM cells (1 × 106; freshly harvested from a C57BL/6 mouse) per recipient mouse.
For secondary BMT assays, leukemic BM cells (with CD45.1+ percentage >90%) were isolated from primary leukemic mice (CD45.2) that received BMT of wild-type BM progenitor donor cells (CD45.1) infected with MSCVneo-MLL-AF9 retrovirus alone. The leukemic cells from the primary recipients were retrovirally transduced with MSCV-PIG (MA9_Ctrl) or MSCV-PIG-Alox5 (MA9_Alox5), and then transplanted into sublethally irradiated (480 rads) 8- to 10-week-old C57BL/6 (CD45.2) secondary recipient mice via tail vein injection. 1.5 × 105 donor cells were transplanted into each secondary recipient mouse.
Peripheral blood (PB) cells were collected from the transplanted recipient mice monthly via tail bleeding and at any time that the mice showed signs of systemic illness to obtain the complete blood counts with white blood cell (WBC) differentials and the blood smears for the presence of immature or abnormal hematopoietic cells. The engraftment was assessed by flow cytometry analysis of CD45.1 in PB. Leukemic mice were euthanized by CO2 inhalation if they showed signs of systemic illness to collect specimens as controls for further analyses. The spleen, liver and thymus were weighed. Cells were obtained from PB, BM, spleen, and liver for flow cytometric analysis. Blood smear and BM cytospin slides were stained with Wright-Giemsa. For histological analysis, portions of the spleen and liver were fixed in formalin, sectioned, embedded in paraffin, and stained with hematoxylin and eosin (H&E).
Gene set analyses
Gene Set Enrichment Analysis (GSEA)36 was used to analyze the signal pathway enrichment in different groups of samples. “Hallmark gene sets” obtained from MsigDB (The Molecular Signatures Database) were used as the “gene sets database” input. Vertebrate homology resource from the Mouse Genome Database (MGD)55 was extracted to convert between homologous human and mouse gene symbols.
Statistical software
The gene/exon array data analyses and qPCR data analyses were conducted by use of Partek Genomics Suite (Partek Inc, St. Louis, MI), TIGR Mutiple Array Viewer software package (TMeV version 4.6; TIGR, Rockville, MA)56, and/or Bioconductor R packages. The t-test, Kaplan-Meier method, and log-rank test, etc. were performed with WinSTAT (R. Fitch Software), GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA), and/or Partek Genomics Suite (Partek Inc, St. Louis, MI). The P-values less than 0.05 were considered as statistically significant. Significance analysis of microarrays (SAM)57, embedded in the TMeV package (TIGR, Rockville, MA), was used to identify the genes that are significantly (q < 0.05; false discovery rate, FDR < 0.05) dysregulated in human AML samples relative to the normal controls.
Data availability
Data referenced in this study are available in The Gene Expression Omnibus. The Affymetrix exon array data and the microarray data are available under accession codes code GSE34184 and GSE30285. The mouse RNA sequencing data is available under accession code GSE94840. The RNA sequencing results of the AML sample cohort analyzed for the correlation of ALOX5 levels and K-RAS mutations is under accession code GSE62190.
Electronic supplementary material
Acknowledgements
We thank Drs. Gregory Hannon, Scott Hammond, and Lin He for providing us with the retroviral constructs. We thank Dr. James Mulloy for providing us with the CD34+-derived MLL-AF9 cell samples. This work was supported in part by the National Institutes of Health (NIH) R01 Grants CA178454 and CA182528 (J.C.), Leukemia & Lymphoma Society (LLS) Translational Research Grant (J.C.), American Cancer Society (ACS) Research Scholar grant (J.C.), Gabrielle’s Angel Foundation for Cancer Research (J.C., X.J.), The University of Chicago Committee on Cancer Biology (CCB) Fellowship Program (X.J.), Leukemia Research Innovative Team of Zhejiang Province 2011R50015 (Y.W., J.J.), Zhejiang Medical and Health Science and Technology Project 2012KYB080(Y.W).
Author Contributions
Y.W., J.C. and X.J. conceived the project and designed the research. Y.W., J.S., C.H., L.D., K.F., R.S., C.L., H.H., H.W., H.H., X.Q., J.J., J.C. and X.J. performed experiments and/or data analysis. Y.W., J.S., C.H., L.D., K.F., R.S., C.L., H.H., H.W., J.J., J.C. and X.J. contributed reagents/analytic tools and/or grant supports. J.C. and X.J. wrote the paper. All authors discussed the results and commented on the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Yungui Wang, Jennifer R. Skibbe and Chao Hu contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-01913-y
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jianjun Chen, Email: chen3jj@uc.edu.
Xi Jiang, Email: jiangx4@uc.edu.
References
- 1.Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 2.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 3.Behm FG, et al. Rearrangement of the MLL gene confers a poor prognosis in childhood acute lymphoblastic leukemia, regardless of presenting age. Blood. 1996;87:2870–2877. [PubMed] [Google Scholar]
- 4.Rowley JD. Chromosomal translocations: revisited yet again. Blood. 2008;112:2183–2189. doi: 10.1182/blood-2008-04-097931. [DOI] [PubMed] [Google Scholar]
- 5.Zuber J, et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009;23:877–889. doi: 10.1101/gad.1771409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lynch RC, Medeiros BC. Chemotherapy options for previously untreated acute myeloid leukemia. Expert Opin Pharmacother. 2015;16:2149–2162. doi: 10.1517/14656566.2015.1076795. [DOI] [PubMed] [Google Scholar]
- 7.Komanduri KV, Levine RL. Diagnosis and Therapy of Acute Myeloid Leukemia in the Era of Molecular Risk Stratification. Annu Rev Med. 2016;67:59–72. doi: 10.1146/annurev-med-051914-021329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seval GC, Ozcan M. Treatment of Acute Myeloid Leukemia in Adolescent and Young Adult Patients. J Clin Med. 2015;4:441–459. doi: 10.3390/jcm4030441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakamoto KM, et al. Targeting novel signaling pathways for resistant acute myeloid leukemia. Mol Genet Metab. 2015;114:397–402. doi: 10.1016/j.ymgme.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perry AM, Attar EC. New insights in AML biology from genomic analysis. Semin Hematol. 2014;51:282–297. doi: 10.1053/j.seminhematol.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 12.Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–683. doi: 10.1038/nrc2210. [DOI] [PubMed] [Google Scholar]
- 13.Steensma DP, et al. JAK2 V617F is a rare finding in de novo acute myeloid leukemia, but STAT3 activation is common and remains unexplained. Leukemia. 2006;20:971–978. doi: 10.1038/sj.leu.2404206. [DOI] [PubMed] [Google Scholar]
- 14.Hatzimichael E, Tsolas E, Briasoulis E. Profile of pacritinib and its potential in the treatment of hematologic disorders. J Blood Med. 2014;5:143–152. doi: 10.2147/JBM.S51253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott LM, Gandhi MK. Deregulated JAK/STAT signalling in lymphomagenesis, and its implications for the development of new targeted therapies. Blood Rev. 2015;29:405–415. doi: 10.1016/j.blre.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Tsuchida, N., Murugan, A. K. & Grieco, M. Kirsten Ras* oncogene: Significance of its discovery in human cancer research. Oncotarget (2016). [DOI] [PMC free article] [PubMed]
- 17.Greenberger JS. Ras mutations in human leukemia and related disorders. Int J Cell Cloning. 1989;7:343–359. doi: 10.1002/stem.5530070603. [DOI] [PubMed] [Google Scholar]
- 18.Osumi H, et al. Acquired drug resistance conferred by a KRAS gene mutation following the administration of cetuximab: a case report. BMC Res Notes. 2013;6:508. doi: 10.1186/1756-0500-6-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soberman RJ, Christmas P. The organization and consequences of eicosanoid signaling. J Clin Invest. 2003;111:1107–1113. doi: 10.1172/JCI200318338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis RA, Austen KF, Soberman RJ. Leukotrienes and other products of the 5-lipoxygenase pathway. Biochemistry and relation to pathobiology in human diseases. N Engl J Med. 1990;323:645–655. doi: 10.1056/NEJM199009063231006. [DOI] [PubMed] [Google Scholar]
- 21.Kim HA, Lee KH, Lee BH. Neuroprotective effect of melatonin against kainic acid-induced oxidative injury in hippocampal slice culture of rats. Int J Mol Sci. 2014;15:5940–5951. doi: 10.3390/ijms15045940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Enyedi B, Jelcic M, Niethammer P. The Cell Nucleus Serves as a Mechanotransducer of Tissue Damage-Induced Inflammation. Cell. 2016;165:1160–1170. doi: 10.1016/j.cell.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poczobutt JM, et al. Deletion of 5-Lipoxygenase in the Tumor Microenvironment Promotes Lung Cancer Progression and Metastasis through Regulating T Cell Recruitment. J Immunol. 2016;196:891–901. doi: 10.4049/jimmunol.1501648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarveswaran S, Chakraborty D, Chitale D, Sears R, Ghosh J. Inhibition of 5-lipoxygenase selectively triggers disruption of c-Myc signaling in prostate cancer cells. J Biol Chem. 2015;290:4994–5006. doi: 10.1074/jbc.M114.599035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou GX, et al. Inhibition of 5-lipoxygenase triggers apoptosis in pancreatic cancer cells. Oncol Rep. 2015;33:661–668. doi: 10.3892/or.2014.3650. [DOI] [PubMed] [Google Scholar]
- 26.Chen Y, Hu Y, Zhang H, Peng C, Li S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009;41:783–792. doi: 10.1038/ng.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang H, et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci USA. 2013;110:11994–11999. doi: 10.1073/pnas.1310656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, et al. miR-196b directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat Commun. 2012;2:688. doi: 10.1038/ncomms1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang X, et al. Blockade of miR-150 Maturation by MLL-Fusion/MYC/LIN-28 Is Required for MLL-Associated Leukemia. Cancer Cell. 2012;22:524–535. doi: 10.1016/j.ccr.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wouters BJ, et al. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113:3088–3091. doi: 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: an international collaborative study. J Clin Oncol. 2013;31:1172–1181. doi: 10.1200/JCO.2012.44.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeKelver RC, et al. Cooperation between RUNX1-ETO9a and novel transcriptional partner KLF6 in upregulation of Alox5 in acute myeloid leukemia. PLoS Genet. 2013;9:e1003765. doi: 10.1371/journal.pgen.1003765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neff T, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. P Natl Acad Sci USA. 2012;109:5028–5033. doi: 10.1073/pnas.1202258109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krivtsov AV, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernt KM, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagner KU, Rui H. Jak2/Stat5 signaling in mammogenesis, breast cancer initiation and progression. J Mammary Gland Biol Neoplasia. 2008;13:93–103. doi: 10.1007/s10911-008-9062-z. [DOI] [PubMed] [Google Scholar]
- 38.Basham B, et al. In vivo identification of novel STAT5 target genes. Nucleic Acids Res. 2008;36:3802–3818. doi: 10.1093/nar/gkn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu X, Zhao L, Park JW, Willingham MC, Cheng SY. Synergistic signaling of KRAS and thyroid hormone receptor beta mutants promotes undifferentiated thyroid cancer through MYC up-regulation. Neoplasia. 2014;16:757–769. doi: 10.1016/j.neo.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choudhary C, Muller-Tidow C, Berdel WE, Serve H. Signal transduction of oncogenic Flt3. Int J Hematol. 2005;82:93–99. doi: 10.1532/IJH97.05090. [DOI] [PubMed] [Google Scholar]
- 41.Ruppert SM, Falk BA, Long SA, Bollyky PL. Regulatory T Cells Resist Cyclosporine-Induced Cell Death via CD44-Mediated Signaling Pathways. Int J Cell Biol. 2015;2015:614297–10. doi: 10.1155/2015/614297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Braun T, Fenaux P. Farnesyltransferase inhibitors and their potential role in therapy for myelodysplastic syndromes and acute myeloid leukaemia. Brit J Haematol. 2008;141:576–586. doi: 10.1111/j.1365-2141.2008.07099.x. [DOI] [PubMed] [Google Scholar]
- 43.Lavallee VP, et al. The transcriptomic landscape and directed chemical interrogation of MLL-rearranged acute myeloid leukemias. Nat Genet. 2015;47:1030–1037. doi: 10.1038/ng.3371. [DOI] [PubMed] [Google Scholar]
- 44.Yan M, et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med. 2006;12:945–949. doi: 10.1038/nm1443. [DOI] [PubMed] [Google Scholar]
- 45.Patel JP, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koppikar P, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 2012;489:155–159. doi: 10.1038/nature11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jain N, et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: a University of Chicago phase II consortium trial. Clin Cancer Res. 2014;20:490–498. doi: 10.1158/1078-0432.CCR-13-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burgess MR, et al. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood. 2014;124:3947–3955. doi: 10.1182/blood-2014-05-574582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Irizarry RA, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15–15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang X, et al. miR-22 has a potent anti-tumour role with therapeutic potential in acute myeloid leukaemia. Nat Commun. 2016;7:11452. doi: 10.1038/ncomms11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang X, et al. MiR-495 is a tumor-suppressor microRNA down-regulated in MLL-rearranged leukemia. Proc Natl Acad Sci USA. 2012;109:19397–19402. doi: 10.1073/pnas.1217519109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang X, et al. Eradication of Acute Myeloid Leukemia with FLT3 Ligand-Targeted miR-150 Nanoparticles. Cancer Res. 2016;76:4470–4480. doi: 10.1158/0008-5472.CAN-15-2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Z, et al. Overexpression and knockout of miR-126 both promote leukemogenesis. Blood. 2015;126:2005–2015. doi: 10.1182/blood-2015-04-639062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li, Z. et al. PBX3 and MEIS1 cooperate in hematopoietic cells to drive acute myeloid leukemias characterized by a core transcriptome of the MLL-rearranged disease. Cancer Res (2016). [DOI] [PMC free article] [PubMed]
- 55.Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE. The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res. 2015;43:D726–736. doi: 10.1093/nar/gku967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saeed AI, et al. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- 57.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data referenced in this study are available in The Gene Expression Omnibus. The Affymetrix exon array data and the microarray data are available under accession codes code GSE34184 and GSE30285. The mouse RNA sequencing data is available under accession code GSE94840. The RNA sequencing results of the AML sample cohort analyzed for the correlation of ALOX5 levels and K-RAS mutations is under accession code GSE62190.