

ABSTRACT
Locally concentrated nuclear factors ensure efficient binding to DNA templates, facilitating RNA polymerase II recruitment and frequent reutilization of stable preinitiation complexes. We have uncovered a mechanism for effective viral transcription by focal assembly of RNA polymerase II around Kaposi's sarcoma-associated herpesvirus (KSHV) genomes in the host cell nucleus. Using immunofluorescence labeling of latent nuclear antigen (LANA) protein, together with fluorescence in situ RNA hybridization (RNA-FISH) of the intron region of immediate early transcripts, we visualized active transcription of viral genomes in naturally infected cells. At the single-cell level, we found that not all episomes were uniformly transcribed following reactivation stimuli. However, those episomes that were being transcribed would spontaneously aggregate to form transcriptional “factories,” which recruited a significant fraction of cellular RNA polymerase II. Focal assembly of “viral transcriptional factories” decreased the pool of cellular RNA polymerase II available for cellular gene transcription, which consequently impaired cellular gene expression globally, with the exception of selected ones. The viral transcriptional factories localized with replicating viral genomic DNAs. The observed colocalization of viral transcriptional factories with replicating viral genomic DNA suggests that KSHV assembles an “all-in-one” factory for both gene transcription and DNA replication. We propose that the assembly of RNA polymerase II around viral episomes in the nucleus may be a previously unexplored aspect of KSHV gene regulation by confiscation of a limited supply of RNA polymerase II in infected cells.
IMPORTANCE B cells infected with Kaposi's sarcoma-associated herpesvirus (KSHV) harbor multiple copies of the KSHV genome in the form of episomes. Three-dimensional imaging of viral gene expression in the nucleus allows us to study interactions and changes in the physical distribution of these episomes following stimulation. The results showed heterogeneity in the responses of individual KSHV episomes to stimuli within a single reactivating cell; those episomes that did respond to stimulation, aggregated within large domains that appear to function as viral transcription factories. A significant portion of cellular RNA polymerase II was trapped in these factories and served to transcribe viral genomes, which coincided with an overall decrease in cellular gene expression. Our findings uncover a strategy of KSHV gene regulation through focal assembly of KSHV episomes and a molecular mechanism of late gene expression.
KEYWORDS: KSHV, transcription, Kaposi's sarcoma-associated herpesvirus, RNA polymerase II, reactivation, transcriptional factory, regulation of gene expression
INTRODUCTION
Gene expression is regulated by the formation of active chromatin hubs (ACHs) at enhancer regions of the genome, where many tissue-specific gene promoters are brought into proximity (1). The idea of the formation of ACHs is based on the fact that the protein concentration of many nuclear factors is below the dissociation constant of protein-protein or protein-DNA interactions (2). Accordingly, it is necessary to have mechanisms to increase the local concentration of nuclear factors at a given chromatin site for continuous and effective gene transcription. Transcription factors locate their binding sites by three-dimensional scanning of nuclear space and leads to the formation of productive transcription complexes on DNA through an inherently dynamic process (3, 4). Factors that bind transiently to their recognition sites will eventually dissociate if secondary factor(s) do not also bind within a certain cycling time. Thus, the concentration of transcription enzymes and cofactors near transcription initiation sites is a very sensitive limiting factor in the efficiency of transcription (5).
Herpesviruses are known to establish replication compartments (RCs), which are nuclear structures for viral replication. RCs are the result of the assembly of cellular and viral proteins, and they function as factories to facilitate efficient viral genome replication, gene expression, and RNA export (6). Studies have also shown the involvement of RCs in the disruption of cellular pathways that are associated with antiviral defense and biosynthesis by sequestration of key components of the pathways (7, 8). RCs can be established within the nucleus or in the cytoplasm depending on the type of virus. The formation of RCs is mainly studied and characterized in DNA viruses, including the human simplex virus (HSV) and human cytomegaloviruses, as well as nucleocytoplasmic large DNA viruses. Formation of RCs by KSHV has also been studied by transient-transfection analyses, and there is evidence for at least six viral proteins involved in DNA replication that are associated with the RC (9). These include single-stranded DNA binding protein (SSB; open reading frame 6 [ORF6]), polymerase processivity factor (PPF; ORF59), DNA polymerase (Pol; ORF9), primase associated factor (PAF; ORF40/41), primase (PRI; ORF56), and helicase (HEL; ORF44) (9). All three families of herpesvirus are known to establish RCs adjacent to promyelocytic leukemia protein nuclear bodies (PML-NBs) (6). PML-NBs are known to be recruited to incoming viral genomes as an antiviral response, and the virus may take advantage of this cellular reaction for their own use (8, 10).
KSHV is the eighth member of the human herpesvirus family identified in 1994 (11). Infection by KSHV is etiologically linked to the development of Kaposi's sarcoma (KS), an angioproliferative and inflammatory lesion of the endothelium that is the most common neoplasm occurring in untreated AIDS patients (12, 13). KSHV also strongly associates with two human lymphoproliferative diseases: primary effusion lymphoma (PEL) and AIDS-related multicentric Castleman's disease (14–17). Unfortunately, both of these cancers have generally poor outcomes and short median survival times, complicated by their association with AIDS. Understanding how KSHV replicates in infected cells is thus very important to find a strategy to prevent KSHV-associated malignancies.
Like all herpesviruses, the KSHV life cycle consists of two phases, known as latency and lytic replication. In latency, the viral genome persists in the host as nuclear episomes, and its expression is largely silenced except for a few genes (12, 13). As a result, viral particles are not produced, and the latent infected cell presents only a few viral proteins. KSHV lytic replication phase is initiated by the expression of a single viral protein, K-Rta. K-Rta is both necessary and sufficient to induce lytic reactivation of the latent KSHV genome in the BCBL-1 cell line model, as well as in a de novo infection model (18–22). K-Rta is classified as an immediate early gene and its coding sequence is separated into two exons (21, 23). The K-Rta promoter is also activated by K-Rta itself to amplify its own expression (24). Various K-Rta responsive promoters have been identified in vitro and in vivo (21, 25–29), thus expression of K-Rta triggers a cascade of viral gene expression.
Recent genomic studies demonstrated that host cell chromosomes physically redistribute to form genomic hubs in response to external stimuli (30–35). Transcriptionally active genomic sites were marked by RNA in situ hybridization and the association of genomic regions within the nucleus could be examined. These studies showed that inducible gene promoters frequently translocated (in proximity) to regions of active gene transcription and formed genomic hubs (34). It remains unknown whether viral episomes are regulated in a similar manner. Because of the small size of its genome and well-established analytical tools (20, 36, 37), the KSHV episome represents an ideal system to investigate the molecular mechanisms of chromosome movements and the downstream effects on gene expression.
In this study, we developed an approach to fluorescently label transcribing KSHV episomes in infected cells. We also established a technique to visualize ongoing viral DNA replication by targeting single-stranded DNA. By imaging KSHV transcription in situ, we found that KSHV episomes formed “transcriptional factories” during reactivation, toward which the translocation of a significant fraction of cellular RNA polymerase II (RNA Pol II) was observed. Based upon the results of this study, we postulate that the redistribution of viral episomes in the cell nucleus and recruitment of RNA Pol II to generate viral transcriptional factories is a fundamental molecular mechanism for effective viral gene expression.
RESULTS
Establishment of immune-FISH to monitor reactivating episomes in primary effusion lymphoma cells.
Current methods to study viral gene expression rely primarily on quantitative reverse transcription-PCR (qRT-PCR) of total RNA isolated from a mixed population of both latent infected cells and reactivating cells. Using such an approach, it is impossible to know the cell-to-cell variation in response to stimuli—much less the response of individual viral genomes (episomes) in a particular cell. Knowing how and where transcription takes place in the nucleus can provide useful insight into the regulation of viral gene expression. Are only a few episomes actively transcribing RNA, or do all the episomes in the cell transcribe viral genes simultaneously, and if so, how? To answer such questions, we applied an RNA-FISH approach to visualize viral transcripts. This technique was first described in a study of cellular gene regulation, in which target transcripts were marked near the transcribing genomic locus in situ by generating probes specifically to intronic regions (30, 34). Using the same approach, we designed RNA-FISH probes for the K-Rta intron region (Fig. 1A and Table 1), which allowed us to specifically tag pre-mRNAs immediately after transcription from viral DNA (but before splicing and export to the cytoplasm). Transcripts become visible only when a sufficient number of fluorescent oligonucleotide probes are hybridized to the same K-Rta intron region; this mechanism increases the specificity of hybridization signals. KSHV reactivation was induced for 24 h by incubation with 12-O-tetradecanoyl-phorbol-13-acetate (TPA). In combination with LANA immune staining to mark the location of viral episomes in infected cells, we successfully obtained the first images of reactivating KSHV episomes in situ (Fig. 1B). The results show RNA molecules as single dots. The image also reveals that not all of the episomes in a single cell react uniformly to TPA stimulation (Fig. 1C and enlarged figure is shown in Fig. S1 in the supplemental material). Three-dimensional (3D) imaging shows that some KSHV episomes (labeled in green) were adjacent to transcripts (labeled in red), suggesting that these episomes were the likely origin of the RNAs (Fig. 1C). Brighter red signals correspond to higher RNA concentration and could represent points of origin for transcriptional activity. Possibly due to RNA mobility within the nucleus, not all of the RNA molecules were expected to colocalize with LANA signals. Also, as expected, most BCBL-1 cells did not reactivate (as demonstrated by the absence of red signal) with a single TPA treatment. The results also indicated a heterogeneous KSHV reactivation, because a different number of RNA molecules (red dots in cells designated with white arrowheads, in Fig. 1C) were present in the respective cells. Finally, we also noticed that LANA dots tend to aggregate where a larger number of red punctate were present (see below).
FIG 1.

Establishment of immune-FISH approach. (A) Design of RNA-FISH probes. Fluorescence-labeled DNA oligonucleotides were designed to hybridize to the intron region of the K-Rta transcript. Premature and mature mRNA encoding K-Rta are illustrated. ORFs located around ORF50 (K-Rta) are also depicted. (B) Visualization of active episomes in situ. Unsynchronized BCBL-1 cells were treated with TPA (20 ng/ml) for 24 h and stained by immune-FISH. LANA was stained with specific antibody and K-Rta transcripts were visualized by fluorescent labeled oligonucleotides. A phase-contrast image is also shown. (C) Multicolor fluorescence image of BCBL-1 cells viewed in 3D perspective. Reactivation was induced by TPA (20 nM) for 24 h. White arrows indicate reactivating cells. Green, LANA; red, K-Rta RNA.
TABLE 1.
FISH probe sequences
| Probe | Sequence (5′-3′) |
|---|---|
| KSHV Rta_1 | CCCACCTACACCATTGTAAA |
| KSHV Rta_2 | GTAGAGCTTGGCGAACTCTG |
| KSHV Rta_3 | TTAATAAGAGCCCTGACACC |
| KSHV Rta_4 | CTTTGGTCAAGTACACCGAA |
| KSHV Rta_5 | TACAGACCCAACCTAGGTTC |
| KSHV Rta_6 | GATCCTTTTTTGCCTGGTAC |
| KSHV Rta_7 | AACTCTCCTGAAAAGCACCG |
| KSHV Rta_8 | TTGTCCACATAATCAGCACG |
| KSHV Rta_9 | CGGTGCATTTACGAGCAGAA |
| KSHV Rta_10 | CAGCTGTCGTTCAGATGTAC |
| KSHV Rta_11 | ATTTTAACGTGCAGCTGAAC |
| KSHV Rta_12 | TACGAGGACTTTCAGGATAC |
| KSHV Rta_13 | GATGGCCAGCGTGGTAAAAA |
| KSHV Rta_14 | AACAACGCACAACGGGACGT |
| KSHV Rta_15 | GCTCCGAAGTTAGGGATATC |
| KSHV Rta_16 | AGTTTTGACTCTCCAATACC |
| KSHV Rta_17 | AGAGCTCATTAAGGGACTGC |
| KSHV Rta_18 | CTGGCTGCATAGGTTTTGAG |
| KSHV Rta_19 | TATTAACGGCATGCTGCCAC |
| KSHV Rta_20 | GCAACGACAAGATAAGGGGT |
| KSHV Rta_21 | CTGCAGCAGTTGTACAAACT |
| KSHV Rta_22 | TAGGGCGAATTTGGCTTATT |
| KSHV Rta_23 | GACAAAGCGAGCCGTACCTG |
| KSHV Rta_24 | ATTCAGATATTGGTCCAGCA |
| KSHV Rta_25 | GACCCTATACCTTGCAAAGA |
| KSHV Rta_26 | TACCCGCTCTAAACGTTGAG |
| KSHV Rta_27 | TTCAGGAGTTAGATACCCTG |
| KSHV Rta_28 | TTTAGGTATCATTACCCGTC |
| KSHV Rta_29 | AAGATCACCTGTTCAACCAT |
| KSHV Rta_30 | ACAATGACATCGAGAAGGCC |
| KSHV Rta_31 | AGAGTGGCGTGTCATAGTTT |
| KSHV TR_1 | TAAAACAGGGGGGGGGGATG |
| KSHV TR_2 | CACGCCTACTTTTTTTTTCG |
| KSHV TR_3 | CTGGACACTACGTGAACACC |
| KSHV TR_4 | TCAGTGCTTGCTACGTGGAG |
| KSHV TR_5 | CTTGTGTGTGAGCCTGTTTG |
| KSHV TR_6 | TCTACTGTGCGAGGAGTCTG |
| KSHV TR_7 | CCGCGGGAGAAAACGAAAGC |
| KSHV TR_8 | CTCGCACAGTAGAGAGAGGG |
| KSHV TR_9 | CAGGCTCACACACAAGACA |
| KSHV TR_10 | ACGTAGCAAGCACTGAGGAG |
| KSHV TR_11 | AAAAAGTAGGCGTGGCCTAG |
Formation of KSHV transcriptional factories.
Stimulation of cellular transcription has been shown to generate so called “transcriptional factories,” where an increased signal intensity of RNA Pol II was observed by immunofluorescence staining (34, 38). We reason that K-Rta RNA, which has immediately been transcribed from the viral genome, should be in close proximity with RNA Pol II. Accordingly, we stained for RNA Pol II and probed for K-Rta RNA intron-containing transcripts by RNA-FISH. The results showed colocalization of K-Rta RNA with cellular RNA Pol II in BCBL-1 cells (Fig. 2). Accumulated K-Rta RNA signals, presumably at actively transcribing sites, colocalized very well with RNA Pol II, while some distantly spaced K-Rta RNA did not. To ensure that we were not observing a cell line-specific phenomenon, we also repeated the study with two other PEL cell lines, BC2 and HBL-6. KSHV reactivation by TPA and sodium butyrate again induced formation of RNA Pol II foci, which colocalized with K-Rta RNA (Fig. 2). Linear intensity plots confirmed the overlapping distribution of RNA Pol II and K-Rta transcripts (see Fig. S2A in the supplemental material). The results also showed a significantly lower RNA Pol II signal intensity in reactivating cells compared to nonreactivating cells (Fig. 2). The number of cells that had little to no detectable RNA Pol II signal increased at later time points in postreactivation (data not shown). Translocation of cellular RNA Pol II required KSHV reactivation because latent cells (defined by absence of red dots) rarely exhibited punctate staining patterns of RNA Pol II (8.1%, Table 2), while RNA Pol II exhibited punctate staining patterns in approximately 80% of reactivating cells (Fig. 2 and Table 2). As controls, we performed the same set of experiments with slides of BJAB cells (KSHV-negative cells) and RNase-treated BCBL-1. The results showed the absence of K-Rta RNA staining, ensuring the specificity of the RNA FISH signal (see Fig. S2B and C in the supplemental material).
FIG 2.

Hijacking cellular RNA Pol II. Cellular RNA Pol II (green) colocalizes with K-Rta RNA (red). The indicated PEL cells were stimulated by a mixture of TPA (20 nM) with sodium butyrate (1 mM) for 4 h and stained at 24 h after the end of stimulation. DNA was counterstained with DAPI. Areas defined by rectangles in the merge + DAPI images are enlarged at the far right.
TABLE 2.
Relationship between KSHV reactivation and RNA polymerase II translocationa
| PEL cell type and analysis | BCBL-1/Pol II | HBL-6/Pol II | BC2/Pol II | BC3/Pol II | Total |
|---|---|---|---|---|---|
| PEL cells with K-Rta RNA signal | |||||
| K-Rta RNA (+) cells (with red dots) | 142 | 27 | 143 | 180 | 492 |
| RNA Pol II translocation | 119 | 16 | 113 | 155 | 403 |
| RNA Pol II translocation (%) | 83.8 | 59.3 | 79.0 | 86.1 | 81.9 |
| PEL cells without K-Rta RNA signal | |||||
| K-Rta RNA (–) cells (without red dots) | 734 | 279 | 925 | 663 | 2601 |
| RNA Pol II translocation | 37 | 33 | 56 | 98 | 224 |
| RNA Pol II translocation (%) | 5.0 | 11.8 | 6.1 | 14.8 | 8.6 |
Numeric data values are expressed as number of cells, unless noted otherwise in column 1.
Next, we performed immune-FISH with RNA Pol II phospho-specific antibodies. By using the phospho-specific RNA Pol II antibodies, we examined the relationship between K-Rta RNA in situ signal with the RNA Pol II status, in addition to confirming the results with different antibodies targeting the same molecule. C-terminal domain (CTD) of RNA Pol II is known to be phosphorylated and position of CTD phosphorylation sites (p-S2, p-S5) regulates recruitment of different protein complexes and thus regulates the function of RNA Pol II. The results demonstrated that S5-phosphorylated, but not S2-phosphorylated RNA Pol II preferentially colocalized with K-Rta RNA. Interestingly, RNA Pol II S2 signals in proximity to K-Rta RNA signals were significantly lower (Fig. 3).
FIG 3.
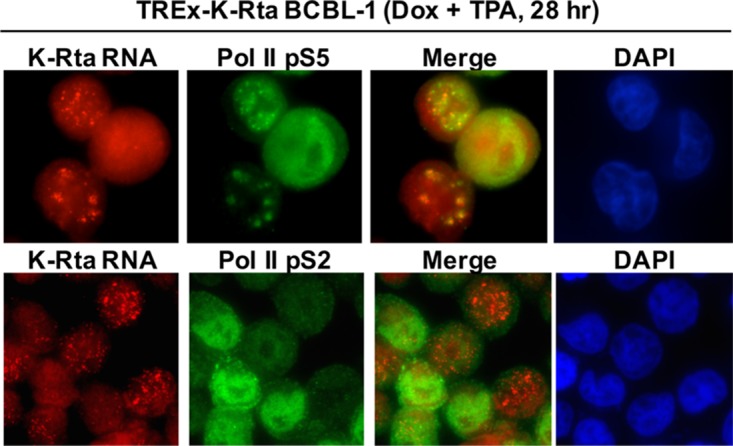
The transcriptional factory predominantly colocalizes with the RNA Pol II p-S5 form. Immune-FISH was performed with phospho-specific RNA Pol II antibodies. KSHV reactivation was induced in TREx-K-Rta BCBL-1 by combination of Dox and TPA for 4 h, and slides were prepared after 24 h after the end of the stimulation. KSHV transcribing sites were marked by RNA-FISH with K-Rta intron probes.
Proteasomes degrade cellular RNA Pol II during KSHV reactivation.
Our immune-FISH studies showed significantly lower RNA Pol II signals in reactivating cells. To examine the possibility that this was mediated by a degradation-associated pathway, we applied a proteasome inhibitor to determine whether we could inhibit the degradation of cellular RNA Pol II. As noted above, nonreactivated cells do not exhibit RNA Pol II degradation; therefore, we needed to increase the number of KSHV reactivated cells in a given tissue culture population to examine this process with a whole population by immunoblotting. Accordingly, we implemented double thymidine blocks to synchronize the cells, followed by KSHV reactivation by the induction of K-Rta expression with doxycycline (Dox) in K-Rta inducible BCBL-1 cells (TREx-K-Rta BCBL-1). TPA was also included during reactivation to further induce reactivation. With this approach, we were able to achieve nearly 100% induction of cells expression of K-Rta RNA (Fig. 4A, bottom left panel). To prevent exogenous K-Rta overexpression, the incubation time with DOX and TPA was limited to 4 h. The K-Rta inducible cassette encodes only exon sequences of K-Rta mRNA and our RNA-FISH probes were designed to hybridize to the intron region of the K-Rta transcript (Fig. 1A), thus we still could probe endogenous K-Rta RNA expression and mark the transcribing episomes in the K-Rta inducible cells (Fig. 4A). By taking time points after reactivation, we monitored cellular and viral protein expression by immunoblotting. As expected, the amount of RNA Pol II began to decrease after approximately 28 h and became nearly undetectable at the 72-h time point (Fig. 4B). Similar results were obtained with two phospho-specific RNA Pol II antibodies, suggesting that there are no differences in RNA Pol II degradation due to the status of RNA Pol II phosphorylation. Although both actin and GAPDH were expressed at significantly higher levels in the cells, both proteins also showed a slight reduction at later time points (Fig. 4B). On the other hand, robust expression of the KSHV early gene product, K-bZIP, was detected at the 28-h time point; this was consistent with the timing of RNA Pol II accumulation. Although a significant fraction of RNA Pol II was degraded by the 72-h time point, the viral late gene product, K8.1, continued to be expressed at 72 h (Fig. 4B). On the other hand, LANA expression was induced at the 12-h time point and gradually decreased thereafter (Fig. 4B). Incubation with a proteasome inhibitor, MG132, slightly recovered the amount of RNA Pol II (Fig. 4C). We added MG132 at the 28-h time point and remained in the cultures for another 20 h. Samples were thus collected at 48 h after reactivation. Incubation of doxycycline and TPA for 4 h induced early lytic gene expression (K-Rta and K-bZIP), and there were no differences by incubation with MG132; however, expression of late gene product, K8.1, was significantly inhibited by the MG132 treatment (Fig. 4C, upper panel). The recovery of RNA Pol II was also confirmed with bortezomib, a U.S. Food and Drug Administration (FDA)-approved proteasome inhibitor. For the bortezomib treatment, K-Rta was induced in the presence or absence of bortezomib at final concentration of 16 nM, which is approximately 3 times above the 50% inhibitory concentration. These results showed that RNA Pol II degradation, at least in part, was mediated by the proteasome (Fig. 4C, lower panel).
FIG 4.

Degradation of RNA Pol II by the proteasome in KSHV-reactivating cells. (A) KSHV reactivation in synchronized TREx-K-Rta BCBL-1 after treatment with a combination of TPA and doxycycline. The cell cycle was synchronized with a double thymidine block, and KSHV reactivation was induced by a combination of Dox (1 μg/ml) and TPA (20 ng/ml). The indicated molecules were stained with immune-FISH at 28 h postinduction. (B) Immunoblotting. TREx-K-Rta BCBL-1 cells were reactivated by TPA and Dox for 4 h after cell cycle synchronization. Cell lysates were prepared at different time points after reactivation, and 50-μg portions of total cell lysates were subjected to immunoblotting. The indicated proteins were probed with specific antibodies. No Dox, no KSHV reactivated cells; Dox & TPA, reactivated by TPA and Dox for 4 h. (C) Proteasome-mediated degradation of RNA Pol II. (a) TREx-K-Rta BCBL-1 cells were reactivated by TPA and Dox for 4 h after cell cycle synchronization. At 28 h after stimulation, MG132 (final concentration, 10 μM) was added to the culture media and remained another 20 h. The indicated proteins were probed with specific antibodies. (b) Bortezomib (final concentration, 16 nM) was added to the culture media after stimulation of KSHV reactivation for 4 h and treatment lasted another 44 h. At 48 h poststimulation, the cells were harvested, and cell lysates were prepared. The indicated proteins were probed with specific antibody.
Biased shifting of RNA Pol II for selected gene expression.
We next examined the impact of viral transcriptional factory formation on cellular and viral gene expression. TREx-K-Rta BCBL-1 cells were reactivated by a combination of Dox and TPA for 4 h, and total RNA was extracted at different time points. Total RNA was also extracted from noninduced cells for comparison. Consistent with there being diminished levels of available RNA Pol II for cellular gene transcription, the expression of GAPDH, β-actin, RNA Pol II (RBP1), and interleukin-10 (IL-10) were all decreased during KSHV reactivation (Fig. 5A, red bars) compared to noninduced cells (Fig. 5A, blue bars). In sharp contrast, KSHV genomes were increasingly transcribed after stimulation (Fig. 5B, red bars). Cellular IL-6, one of the K-Rta cellular target genes (39), also showed increased gene expression with kinetics similar to that of viral early gene expression; the results indicate that not all cellular gene expression was suppressed, despite the degree and extent of RNA Pol II translocation. For these qRT-PCR studies, we used 18S rRNA as an internal control, since it is transcribed by RNA polymerase I, and we found the 18S rRNA expression to be consistent throughout the experiments. Importantly, this observed effect was not due to an artifact of using K-Rta inducible cells, because cellular small ubiquitin-like modifier (SUMO) 2 protein, which is expected to have a short half-life, exhibited drastically decreased signals in KSHV-reactivating cells but not in nonreactivated BCBL-1, when examined at the single-cell level (see Fig. S3 in the supplemental material).
FIG 5.

Biased shifting of RNA Pol II onto viral genomes during reactivation. (A and B) Cellular (A) and viral (B) gene expression. TREx-K-Rta BCBL-1 cells were reacted by TPA and Dox for 4 h after cell cycle synchronization. The indicated cellular or viral gene expression was measured by qRT-PCR. rRNA 18S, which is transcribed by RNA Pol I, was used for normalization. The values of gene-normalized expression at the indicated time points are shown in the bar graph. Blue, No Dox; red, Dox and TPA. (C) Chromatin immunoprecipitation. RNA Pol II occupancies at cellular or viral promoters were determined before and during reactivation (i.e., at 28 h postreactivation). ChIP assays were performed with the indicated genomic regions with specific primers. The relative enrichment over input DNA is shown.
Finally, chromatin immunoprecipitation was performed to examine occupancy of RNA Pol II at cellular and viral genes (Fig. 5C). The results showed that RNA Pol II occupancy on viral genomes was significantly increased during reactivation (Fig. 5C, right graphs). Compared to the cellular genome, RNA Pol II was recruited approximately 10 to 100 times more on viral genomes at 28 h postreactivation. The PAN RNA encoding region recruited the highest levels of RNA Pol II among different viral genomic regions examined (Fig. 5C), which was consistent with significantly higher copies of transcripts (Fig. 5B). Although it was not as clear as we expected from immunostaining images, RNA Pol II occupancies at cellular RNA Pol II (RBP1) and IL-10 coding region (but not actin coding region) were slightly decreased. This is partly due to the presence of nonreactivating cells in the ChIP samples. These results, in conjunction with immune-FISH images, suggest that RNA Pol II formed foci to primarily transcribe viral genomes during KSHV reactivation.
Colocalization of LANA with RNA Pol II in reactivating cells suggests regulation of KSHV gene expression by forming active chromatin hubs with other episomes.
It is known that activated cellular promoters are aggregated at specific enhancer regions to form active chromatin hubs (31–33). The generation of active chromatin hubs facilitates recycling of RNA Pol II, allowing the enzyme to transcribe from assembled tissue specific or transcriptional factor bound promoters without the requirement for distant travel. The observed accumulation of RNA Pol II prompted us to examine if viral episomes are coming together to prepare active chromatin. To do this, we stained both LANA and cellular RNA Pol II along with K-Rta RNA to visualize active transcription sites. K-Rta RNA identifies the location of actively transcribing episomes, while LANA was used to mark the location of viral episomes. In previous work, we found that LANA was dissociated from the unique region of the KSHV genome; however LANA continued to bind to the TR region and remained enriched over 40-fold at the TR region during reactivation (40, 41). The immune-FISH staining clearly demonstrated that LANA dots in reactivating cells were assembling, and RNA Pol II localized very closely to the congregated LANA dots in reactivating cells (Fig. 6A). Similar to Fig. 2, RNA Pol II and K-Rta RNA dots were significantly overlapped, as expected (Fig. 6B [orange dots, K-Rta RNA; red dots, RNA Pol II; green dots, LANA]). Interestingly, the KSHV transcriptional factory appeared to occupy a multilobular space that resembles an “ant's nest” and was largely devoid of DAPI staining (Fig. 2A, and Fig. 6B, right panel with DAPI [4′,6′-diamidino-2-phenylindole]). The size and shape of this nuclear space was different in each reactivating cell. 3D fluorescence deconvolution microscopy further showed that LANA (green) and RNA Pol II (red) frequently formed ring-like shapes, and K-Rta RNAs (light blue) were often seen within the ring structure. LANA was located outside RNA Pol II to form the ring shapes, which is like a cage to trap RNA Pol II (see Fig. S4 in the supplemental material). Taken together, our results strongly suggest that actively transcribing episomes were assembled to create a viral transcriptional factory. Focal concentration of RNA Pol II near the viral episomes then facilitates viral gene expression.
FIG 6.

(A) RNA-FISH showing the assembly of LANA dots with RNA Pol II in BCBL-1. RNA Pol II (red immunofluorescence), LANA (green immunofluorescence), and K-Rta RNA (light blue, RNA-FISH) were visualized. BCBL-1 was stimulated with TPA and sodium butyrate for 4 h. Cells were fixed after 24 h after the end of stimulation. Merge and enlarged merge images overlaid with K-Rta RNA staining are shown. (B) 3D view of KSHV transcriptional factory. Z-stack images were taken, and 3D images without or with DAPI staining were constructed. Green, LANA; red, RNA Pol II; orange, K-Rta transcripts; blue, DAPI.
KSHV DNA replication and transcriptional factory formation.
Next, we examined if viral transcriptional factories are associated with KSHV DNA replication, because both biochemical reactions use the same KSHV genomes as templates. We probed ongoing KSHV DNA replication using a modified DNA-FISH approach. Fluorescence probes were generated to hybridize to the terminal repeat sequence. Targeting terminal repeat sequences increases fluorescence signals due to the presence of multiple tandem copies in a genome. Actively replicating DNA should possess single-stranded DNA (ssDNA), which would then be hybridized by the fluorescence-labeled DNA oligonucleotides without having to perform DNA denaturation. Avoiding denaturation also permitted us to perform multiplex analysis with protein staining, as well as RNA-FISH. Control studies with RNase A treatment did not eliminate the signal with the TR probes, thereby indicating the probes were hybridized to ssDNA (see Fig. S5 in the supplemental material). In fact, staining patterns were significantly different from RNA-FISH staining (see Fig. S5 in the supplemental material). DNA-FISH with RNA Pol II staining showed that RNA Pol II was recruited to the sites of KSHV DNA replication, and colocalized with single-stranded viral DNAs (Fig. 7A). Active DNA replication might be important for the assembly of RNA Pol II foci, because continuous incubation with thymidine inhibited formation of transcriptional factories ∼10-fold (Fig. 7B and C).
FIG 7.

DNA replication and transcriptional factory formation. (A) Colocalization between single-stranded viral DNA and RNA Pol II. The indicated molecules were probed with either antibody or fluorescent oligonucleotide probes. Merged images: green, replicating KSHV genomes; red, RNA Pol II; blue, DAPI. (B) Transcriptional factory formation in presence or absence of thymidine. TREx-K-Rta BCBL-1 cells were reactivated in the presence or absence of thymidine. The aggregation of RNA Pol II is shown. (C) Pictures of five randomly selected fields were taken, and cells with complete colocalization of all three factors (RNA Pol II, LANA, and K-Rta RNA similar to Fig. 6) were counted as transcriptional factories. The numerical results are summarized in panel C.
DISCUSSION
It is known that gene expression is a very complex process that involves multiple biochemical reactions and physical processes, which ultimately leads to significant variability in the abundance of gene products. Collective evidence from single-cell sequencing further highlights the dynamics of transcription (42). In this study, we showed that viral episomes with identical genomic sequence in a single cell are not uniformly transcribed: some episomes appeared to produce K-Rta RNA, while others remained silent (Fig. 1C). We also observed a small number of K-Rta RNA transcripts in latent cells (Fig. 1C); these results suggest that K-Rta expression does not always lead to the initiation of viral replication, and KSHV reactivation may require a threshold amount of K-Rta protein in order to trigger the biochemical reaction to proceed.
KSHV heterogeneity of response to stimulation has also been documented by Darst et al. (43). They used MAPit single molecule foot-printing approach in conjunction with sequencing to analyze the accessibility of M.CviPI DNA methyltransferase. By assessing the accessibility of the enzyme to KSHV episomes, they examined chromatin structure changes in response to TPA (43). Their results showed that only ∼10% of viral episomes can transcribe following a given stimuli. The results also indicated that an open chromatin structure is a prerequisite in order for episomes to respond to an inducer of lytic transcription, although the study does not distinguish whether all episomes in the 10% of cell population possessed open chromatin structure versus whether 10% of the episomes within a cell can be accessed by RNA Pol II. Our studies agreed with these results and showed significant heterogeneity in the response within a cell, at least in the early stage of reactivation (Fig. 1C).
In our experiments, we controlled two possible biological variables within the cell population by synchronizing the cell cycle with thymidine (i.e., S phase) and by using K-Rta inducible cells to further eliminate the variation of effects on the endogenous K-Rta promoter activation. By taking these measures, we were able to achieve nearly 100% uniformity in the activation of endogenous K-Rta expression based on the signals of RNA-FISH in TREx-BCBL-1 K-Rta cells (Fig. 4A). However, we noticed that the translocation of RNA Pol II did not happen in all of the endogenous K-Rta-expressing cells. Although stimulation of reactivation by the combined treatment with TPA and sodium butyrate induced the translocation of cellular RNA Pol II in 80% of K-Rta RNA-expressing cells, the total numbers of K-Rta RNA-positive cells were actually much lower (Fig. 2, ∼20% of the total cell population). This observation suggests that an excess amount of K-Rta protein may inhibit the formation of such structures or that exogenous K-Rta expression might compromise our imaging experiments by generating multiple transcriptional foci, which are too small for us to recognize as viral transcriptional factories.
Given the observed translocation of RNA Pol II to viral genomes, we were interested in studying its impact on cellular gene transcription. As expected, most cellular gene expression, including RNA Pol II (RBP1) itself, was significantly decreased, relative to viral gene expression, during KSHV reactivation. It is clear that the formation of viral transcriptional factories and the activity of lytic viral proteins (44–47) are mechanisms cleverly designed to enhance viral transcript in infected cells. This finding also raises another interesting question as to how RNA Pol II-depleted cells can react and communicate with immune cells. We speculate that, in addition to the action of the lytic viral proteins (48), the depletion/sequestration of RNA Pol II may also contribute to inhibit cellular responses against replicating KSHV by suppressing physiological activity in the host cells.
Interestingly, the expression of some cellular genes, such as cellular IL-6, is induced during KSHV reactivation (Fig. 5A). Cellular IL-6 was previously identified as a K-Rta direct target gene and also escaped from degradation by KSHV ORF37 protein (39, 49). Considering that KSHV captured cellular IL-6 gene into its genome, the cellular IL-6 locus might also be translocated to be neighboring with viral genomes within the transcriptional factory during KSHV reactivation. Physically neighboring cellular IL-6 gene with KSHV genome might then result in genomic recombination between host and viral genomes. A similar scenario has been suggested for cellular genes between MYC and IGH (34). These genomic regions came into close proximity when gene expression was stimulated and increased the chance for recombination (34). It will be important to study which cellular genes were selected by the virus to join their transcriptional factories. Identifying such cellular genomic regions may also provide us an insight into the molecular mechanism(s) of the factory formation. It is important to note that the observation of accumulation of cellular RNA Pol II to viral genomes is also supported by previous studies, in which viral microRNA becomes predominantly associated with the RNA-induced silencing complex (RISC) during KSHV reactivation (50). If the majority of the newly transcribed RNA is derived from viral genomic DNAs, then a large fraction of the RNAs associated with cellular RNA interacting proteins in a reactivating cell would be of viral origin.
It is known that activation of NF-κB translocates NF-κB target promoters to be in proximity to the enhancer region, where highly active RNA Pol II resides (33). Physical movement of one active promoter is important for the activation of another, and active promoters were often aggregated next to each other to form active chromatin hubs (31–35). Similarly, in HSV-1 de novo-infected cells, it has been shown that active transcription of the HSV-1 genome is required to form HSV-1 replication compartments (51). It remains unclear how active promoters are moved to meet each other and which genomic elements or which nuclear factors are required for the regulation. Our studies demonstrated that KSHV episomes were activated in a similar fashion, which provides us an opportunity to answer such fundamental genetic questions by using KSHV reactivation as a unique model system. The combination of a high copy number of viral genomes in a cell, easy readout of viral gene expression by qRT-PCR array, defined viral transcriptional factor necessary for triggering gene expression, and a cloned KSHV genome in a bacterial artificial chromosome to generate mutant episomes (i.e., single nucleotide variants, deletions, etc.) should provide significant advantages for these studies.
Bortezomib, a proteasome inhibitor, is an FDA-approved anti-cancer drug, which has been used to treat lymphoma patients in clinical practice (52, 53). Preclinical mouse studies for PEL showed that administration of bortezomib prolongs survival in a mouse xenograft model (54–57). We found that bortezomib partially prevented RNA Pol II degradation during KSHV reactivation (Fig. 4C); however, bortezomib treatment reduced the production of DNase I-resistant KSHV virions in the culture media by ∼5-fold at 96 h postreactivation (data not shown). We also found that KSHV, in the presence of proteasome inhibitors, failed to form clear transcriptional factories and reduced K8.1 expression (Fig. 4C and see Fig. S6 in the supplemental material). This suggests that degradation of RNA Pol II after onset of viral gene transcription may be important for DNA replication and/or effective DNA encapsidation to produce viral progeny. In addition, like the generation of DNA repair foci (58, 59), protein ubiquitinylation pathways may be important for the assembly of transcriptional factories/replication compartments and hence late gene expression. Further studies are needed to answer these questions. Nonetheless, bortezomib to target the ubiquitinylation/proteasome pathway may have potential benefits as a treatment option for PEL in clinical practice.
Phosphorylation of RNA Pol II at the CTD is associated with the functional status of RNA Pol II, namely, preinitiation complex and elongation in yeast (60). Recent studies from Nojima et al. demonstrated that yeast RNA Pol II and human RNA Pol II are regulated differently (61). The group elegantly demonstrated that RNA Pol II p-S5 is significantly enriched at exon junctions, while p-S2 is predominantly associated with polyadenylation machinery (61). Our immune staining showed that RNA Pol II p-S5 form but not p-S2 form was enriched at transcriptional factories (Fig. 3). Although we tried to confirm the observation by ChIP analyses with phospho-specific RNA Pol II antibodies, we could not differentiate the degree of RNA Pol II subtype recruitment on the KSHV genome. Our results showed that occupancies of both pS2 and pS5 forms were increased significantly during reactivation (data not shown). Another explanation of p-S5 colocalization would be that there may be a responsible kinase such as ORF36 in the viral transcriptional factory (62), which may associate with phosphorylation of RNA Pol II at the serine 5 either directly or indirectly. Given that promoter-proximal splice sites and the process of splicing can enhance transcription significantly (63, 64), it is important to examine how the virus manipulates the cellular transcriptional machinery and what is the meaning of colocalization with p-S5 signals at viral transcriptional factories.
The most important question is how such a significant fraction of cellular RNA Pol II is trapped on the KSHV genome. The trapping of RNA Pol II onto the viral genome is likely associated with the extremely high copy number of viral noncoding RNA, PAN RNA, expression (Fig. 5B) (65). Related to this, a cellular, nuclear noncoding RNA located upstream of the MYC gene, has been shown to play an important role in MYC expression (66). In that study, the authors demonstrated that the physical interaction between MYC and noncoding RNA expression is important for the MYC expression (66). We speculate that one of function of PAN RNA may be to trap cellular RNA Pol II on the KSHV genome, through DNA-RNA, RNA-protein interactions, and/or process of transcription itself (67, 68), forcing RNA Pol II to transcribe viral nuclear noncoding RNA over and over. In accord with this, our ChIP studies showed that significantly higher occupancies (>30-fold) of RNA Pol II at the PAN RNA locus (Fig. 5C). We think that there might be a reason for PAN RNA to be a noncoding nuclear RNA. Being a noncoding nuclear RNA, PAN RNA would not compete with other viral coding RNA for RNA export and translational machinery. We and others have shown that PAN RNA physically interacts with the PAN RNA coding and promoter region of the KSHV genome with chromatin isolation by RNA purification (CHIRP)-seq analyses and that K-Rta protein also directly binds to PAN RNA (69; M. Campbell et al., unpublished data). According to this hypothesis, the PAN RNA transcribing region should function as an enhancer for many of KSHV genes during lytic replication as it maintains RNA Pol II in close proximity to KSHV genomes.
In summary, we established an immune-FISH approach to study KSHV gene expression at the single-episome level. We propose that the physical accumulation of RNA Pol II near viral genomes in the infected cell nucleus represents an additional layer of KSHV gene regulation, which facilitates the reutilization and repurposing of limited quantities of cellular RNA Pol II. Identifying this molecular mechanism of viral transcriptional factory formation should lead to new strategies to inhibit herpesvirus replication.
MATERIALS AND METHODS
Cell culture.
BCBL-1 and BJAB cells were generous gifts from D. Ganem (University of California, San Francisco). BC2 and HBL-6 cells were obtained from Masahiro Fujimuro (Kyoto Pharmaceutical University). TREx-(F3H3)-K-Rta BCBL-1 cells that expresses Flag ×3 and hemagglutinin (HA) ×3 tags at the N-terminal region of K-Rta were generated by using TREx-BCBL-1 cells (a gift from J. Jung, University of Southern California) with Flp-recombination according to the manufacturer's protocol (Invitrogen), and cultured in complete RPMI 1640 containing 50 μg/ml blasticidin and 100 μg/ml hygromycin B. These B cell lines were grown at 37°C in the presence of 5% CO2 in RPMI 1640 medium supplemented with 15% fetal calf serum, 2 mM l-glutamine, and antibiotics. K-Rta expression and KSHV reactivation was induced in TREx K-Rta BCBL-1 cells with 20 ng/ml 12-O-tetradecanoyl-phorbol-13-acetate (TPA) and 100 ng/ml doxycycline. For other PEL cell lines—BCBL-1, BC2, and HBL-6—KSHV reactivation was induced with 20 ng/ml TPA, 1 mM sodium butyrate, or both agents in combination.
Double thymidine block.
When applicable, a double thymidine block was used to synchronize the cell cycle and to synchronize the initiation of reactivation and increase efficacy of viral reactivation (70). TREx K-Rta BCBL-1 and BCBL-1 cells at 30% confluence were incubated with 2 mM thymidine (Sigma-Aldrich) for 18 h. The cells were washed with fresh medium to remove the thymidine and allowed to grow for 9 h. The cells were again treated with 2 mM thymidine for 18 h. After the second thymidine treatment, the cells were washed with medium to remove thymidine, and KSHV reactivation was induced for 4 h with various stimuli described in the figure legend.
RT-qPCR.
Total RNA was purified with the RNeasy Plus minikit (Qiagen). Eluted RNA was converted to cDNA by using Superscript II reverse transcriptase (Thermo Fisher Scientific) and diluted 1:100. Portions (4 μl) of template were used for every 20-μl reaction, and all samples were analyzed in triplicate using SYBR green Supermix (Bio-Rad) on a QuantStudio 3 real-time PCR system using specific primers designed previously by Fakhari and Dittmer (37). Melting curve analysis was then performed to verify product specificity. KSHV gene expression was normalized to the cellular 18S rRNA.
Immunoblotting.
Cells were rinsed in phosphate-buffered saline (PBS) and lysed in radioimmunoprecipitation assay buffer (25 mM Tris-HCl [pH 7.6], 1.0% NP-40, 1% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 0.1% sodium dodecyl sulfate [SDS]). After centrifugation (15,000 × g for 10 min at 4°C), the protein concentration was measured by using a Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific), and equal amounts of total cell lysates were subjected to an SDS-PAGE (6% for Pol II and 10% for the other proteins) and subsequently transferred to a Immobilon-P polyvinylidene difluoride (EMD Millipore) using a semidry transfer apparatus (Bio-Rad). Membranes were blocked for 10 min at room temperature in TBST (20 mM Tris-HCl [pH 7.5], 137 mM NaCl, 0.05% Tween 20)–5% skim milk, followed by incubation with primary antibodies overnight at 4°C. The membranes were subsequently washed with TBST three times for 10 min each time at room temperature and then incubated with horseradish peroxidase-conjugated antibodies for 1 h at room temperature. The membranes were washed three times with TBST and visualized with enhanced with SuperSignal substrate (Thermo Fisher Scientific). Where indicated, the cells were treated with 10 μM MG132 (Sigma) or 16 nM bortezomib to inhibit proteasome. The following antibodies were used for immunoblotting: anti-K-Rta (1:5,000 [62]), anti-K8.1 (1 μg/ml; Santa Cruz), anti-K8α (K-bZIP) (1 μg/ml; Santa Cruz), anti-LANA (1:1,000; Advanced Biotechnologies, Inc.), anti-RNA polymerase II (1 μg/ml; EMD Millipore), anti-GAPDH (0.2 μg/ml; Santa Cruz), anti-β-actin (1 μg/ml; Sigma-Aldrich), anti-RNA polymerase phospho-S2 polyclonal rabbit IgG (Abcam), and anti-RNA polymerase II phospho-S5 (Abcam).
Immuno-FISH.
At 28 h (unless otherwise stated in the legends) after the induction of KSHV reactivation, the cells were harvested and washed three times with diethylpyrocarbonate (DEPC)-treated PBS. The cells were then spotted onto coverslips and subsequently fixed with 4% formaldehyde in DEPC PBS solution for 10 min. After washing three times with DEPC-PBS, the cells were quenched with 100 mM glycine DEPC-PBS solution for 10 min. After a rinsing with DEPC-PBS, the cells were permeabilized with a 1:1 methanol-acetone solution for 15 min and washed with DEPC-PBS. Primary antibodies (anti-RNA polymerase II mouse monoclonal antibody [1:250; EMD Millipore], anti-RNA polymerase phospho-S2 polyclonal rabbit IgG [1:250; Abcam], anti-RNA polymerase II phospho-S5 [1:250; Abcam], anti-LANA rat monoclonal antibody [1:80; Advanced Biotechnologies, Inc.], and anti-Small ubiquitin-like modifier SUMO-2/3 [1:200; Thermo Fisher]) were diluted in dilution buffer (DEPC-PBS, 100 μg/ml yeast tRNA [Sigma-Aldrich]), followed by incubation for 1 h at 37°C. The slides were washed three times with DEPC-PBS and then washed three times with FISH wash buffer (2× SCC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 10% formamide in DEPC–double-distilled water [dH2O]). Coverslips were incubated with a mixture of FISH probes and secondary antibodies in hybridization solution (10% dextran sulfate, 10% formamide, 2× SSC, 100 μg/ml yeast tRNA, 125 nM FISH probe [LGH Science, Inc.], and secondary antibodies) for 18 h at 37°C. The FISH probe sequences are listed in Table 1. The listed Quasar 570-labeled oligonucleotides were combined and used for RNA FISH studies. The slides were then washed with FISH wash buffer three times for 5 min and twice with 2× SSC. DAPI was added in 2× SSC for 5 min at room temperature to stain DNA. For DNA-FISH, probes were designed to hybridize terminal repeat sequence, and RNase A was incubated where indicated as a control. The slides were rinsed with SSC and mounted onto glass slides by using SlowFade Gold antifade reagent (Life Technologies). Fluorescence images were taken in two dimensions using a Keyence BZ-X710 or in three dimensions as follows.
3D fluorescence microscopy and digital image analysis.
Prepared cell specimens were imaged on a computer-automated widefield fluorescence deconvolution microscope (Deltavision personalDV; Applied Precision/GE Healthcare) equipped with a ×60, 1.42NA oil immersion objective lens, xenon arc lamp, and standard filters for DAPI (blue), FITC (green), and TRITC (tetramethyl rhodamine isothiocyanate; red-orange) channels. 3D image stacks were acquired with 0.2-μm spacing in the z-axis; and image deconvolution was performed postacquisition using on-board software (Resolve3D; Applied Precision/GE Healthcare). Image data were transferred to a separate computer for 3D image rendering and subsequent analysis using VoloCITY 3D imaging suite (Improvision; Perkin-Elmer).
ChIP assay.
Chromatin immunoprecipitation (ChIP) assays were performed as described previously (29). The antibodies used were mouse monoclonal anti-RNA polymerase II (Upstate) or control mouse IgG (Santa Cruz). Immunoprecipitated chromatin DNA was analyzed by SYBR green-based quantitative PCR (Bio-Rad) with the primers listed in Table S1 in the supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
We thank Masahiro Fujimuro (Kyoto Pharmaceutical University) for providing the PEL cell lines, HBL-6 and BC2.
This research was supported by a National Institutes of Health grant (R01-DE025985) and by an American Cancer Society Research Scholar Award (RSG-13-383-MPC). This study was also supported by grants from the U.S. Department of Agriculture (2015-67015-23268 and 2014-67015-21787) and by a New Research Initiative grant from the University of California, Davis.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.02491-16.
REFERENCES
- 1.de Laat W, Grosveld F. 2003. Spatial organization of gene expression: the active chromatin hub. Chromosome Res 11:447–459. doi: 10.1023/A:1024922626726. [DOI] [PubMed] [Google Scholar]
- 2.Palstra RJ, de Laat W, Grosveld F. 2008. Beta-globin regulation and long-range interactions. Adv Genet 61:107–142. doi: 10.1016/S0065-2660(07)00004-1. [DOI] [PubMed] [Google Scholar]
- 3.Misteli T. 2001. Protein dynamics: implications for nuclear architecture and gene expression. Science 291:843–847. doi: 10.1126/science.291.5505.843. [DOI] [PubMed] [Google Scholar]
- 4.Hager GL, McNally JG, Misteli T. 2009. Transcription dynamics. Mol Cell 35:741–753. doi: 10.1016/j.molcel.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Droge P, Muller-Hill B. 2001. High local protein concentrations at promoters: strategies in prokaryotic and eukaryotic cells. Bioessays 23:179–183. doi:. [DOI] [PubMed] [Google Scholar]
- 6.Schmid M, Speiseder T, Dobner T, Gonzalez RA. 2014. DNA virus replication compartments. J Virol 88:1404–1420. doi: 10.1128/JVI.02046-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31:145–158. doi: 10.1089/jir.2010.0111. [DOI] [PubMed] [Google Scholar]
- 8.El Asmi F, Maroui MA, Dutrieux J, Blondel D, Nisole S, Chelbi-Alix MK. 2014. Implication of PMLIV in both intrinsic and innate immunity. PLoS Pathog 10:e1003975. doi: 10.1371/journal.ppat.1003975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu FY, Ahn JH, Alcendor DJ, Jang WJ, Xiao J, Hayward SD, Hayward GS. 2001. Origin-independent assembly of Kaposi's sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J Virol 75:1487–1506. doi: 10.1128/JVI.75.3.1487-1506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Everett RD. 2016. Dynamic response of IFI16 and promyelocytic leukemia nuclear body components to herpes simplex virus 1 infection. J Virol 90:167–179. doi: 10.1128/JVI.02249-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 12.Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 120:939–949. doi: 10.1172/JCI40567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer 10:707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schulz TF. 2006. The pleiotropic effects of Kaposi's sarcoma herpesvirus. J Pathol 208:187–198. doi: 10.1002/path.1904. [DOI] [PubMed] [Google Scholar]
- 15.Wong EL, Damania B. 2005. Linking KSHV to human cancer. Curr Oncol Rep 7:349–356. doi: 10.1007/s11912-005-0061-6. [DOI] [PubMed] [Google Scholar]
- 16.Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, Chang Y. 1995. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood 86:2708–2714. [PubMed] [Google Scholar]
- 17.Cesarman E, Knowles DM. 1999. The role of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in lymphoproliferative diseases. Semin Cancer Biol 9:165–174. doi: 10.1006/scbi.1998.0118. [DOI] [PubMed] [Google Scholar]
- 18.Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF50 transactivator, a homolog of the EBV R protein. Virology 252:304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- 19.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 95:10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 77:4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J Virol 73:9348–9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y, AuCoin DP, Huete AR, Cei SA, Hanson LJ, Pari GS. 2005. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J Virol 79:3479–3487. doi: 10.1128/JVI.79.6.3479-3487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J Virol 73:2232–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lan K, Kuppers DA, Robertson ES. 2005. Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J Virol 79:3468–3478. doi: 10.1128/JVI.79.6.3468-3478.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang PJ, Shedd D, Gradoville L, Cho MS, Chen LW, Chang J, Miller G. 2002. Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J Virol 76:3168–3178. doi: 10.1128/JVI.76.7.3168-3178.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng H, Song MJ, Chu JT, Sun R. 2002. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J Virol 76:8252–8264. doi: 10.1128/JVI.76.16.8252-8264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song MJ, Li X, Brown HJ, Sun R. 2002. Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J Virol 76:5000–5013. doi: 10.1128/JVI.76.10.5000-5013.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.West JT, Wood C. 2003. The role of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene 22:5150–5163. doi: 10.1038/sj.onc.1206555. [DOI] [PubMed] [Google Scholar]
- 29.Izumiya Y, Izumiya C, Hsia D, Ellison TJ, Luciw PA, Kung HJ. 2009. NF-κB serves as a cellular sensor of Kaposi's sarcoma-associated herpesvirus latency and negatively regulates K-Rta by antagonizing the RBP-Jκ coactivator. J Virol 83:4435–4446. doi: 10.1128/JVI.01999-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fanucchi S, Shibayama Y, Burd S, Weinberg MS, Mhlanga MM. 2013. Chromosomal contact permits transcription between coregulated genes. Cell 155:606–620. doi: 10.1016/j.cell.2013.09.051. [DOI] [PubMed] [Google Scholar]
- 31.Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, Sim HS, Peh SQ, Mulawadi FH, Ong CT, Orlov YL, Hong S, Zhang Z, Landt S, Raha D, Euskirchen G, Wei CL, Ge W, Wang H, Davis C, Fisher-Aylor KI, Mortazavi A, Gerstein M, Gingeras T, Wold B, Sun Y, Fullwood MJ, Cheung E, Liu E, Sung WK, Snyder M, Ruan Y. 2012. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Razin SV, Gavrilov AA, Ioudinkova ES, Iarovaia OV. 2013. Communication of genome regulatory elements in a folded chromosome. FEBS Lett 587:1840–1847. doi: 10.1016/j.febslet.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 33.Brown JD, Lin CY, Duan Q, Griffin G, Federation AJ, Paranal RM, Bair S, Newton G, Lichtman AH, Kung AL, Yang T, Wang H, Luscinskas FW, Croce KJ, Bradner JE, Plutzky J. 2014. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell 56:219–231. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ, Corcoran AE, Fraser P. 2007. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol 5:e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, Herman B, Happe S, Higgs A, LeProust E, Follows GA, Fraser P, Luscombe NM, Osborne CS. 2015. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet 47:598–606. doi: 10.1038/ng.3286. [DOI] [PubMed] [Google Scholar]
- 36.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fakhari FD, Dittmer DP. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J Virol 76:6213–6223. doi: 10.1128/JVI.76.12.6213-6223.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jackson DA, Hassan AB, Errington RJ, Cook PR. 1993. Visualization of focal sites of transcription within human nuclei. EMBO J 12:1059–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng H, Chu JT, Rettig MB, Martinez-Maza O, Sun R. 2002. Rta of the human herpesvirus 8/Kaposi sarcoma-associated herpesvirus up-regulates human interleukin-6 gene expression. Blood 100:1919–1921. doi: 10.1182/blood-2002-01-0015. [DOI] [PubMed] [Google Scholar]
- 40.Campbell M, Chang PC, Huerta S, Izumiya C, Davis R, Tepper CG, Kim KY, Shevchenko B, Wang DH, Jung JU, Luciw PA, Kung HJ, Izumiya Y. 2012. Protein arginine methyltransferase 1-directed methylation of Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen. J Biol Chem 287:5806–5818. doi: 10.1074/jbc.M111.289496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campbell M, Kim KY, Chang PC, Huerta S, Shevchenko B, Wang DH, Izumiya C, Kung HJ, Izumiya Y. 2014. A lytic viral long noncoding RNA modulates the function of a latent protein. J Virol 88:1843–1848. doi: 10.1128/JVI.03251-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolodziejczyk AA, Kim JK, Tsang JC, Ilicic T, Henriksson J, Natarajan KN, Tuck AC, Gao X, Buhler M, Liu P, Marioni JC, Teichmann SA. 2015. Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 17:471–485. doi: 10.1016/j.stem.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Darst RP, Haecker I, Pardo CE, Renne R, Kladde MP. 2013. Epigenetic diversity of Kaposi's sarcoma-associated herpesvirus. Nucleic Acids Res 41:2993–3009. doi: 10.1093/nar/gkt033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glaunsinger B, Ganem D. 2004. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol Cell 13:713–723. doi: 10.1016/S1097-2765(04)00091-7. [DOI] [PubMed] [Google Scholar]
- 45.Muller M, Hutin S, Marigold O, Li KH, Burlingame A, Glaunsinger BA. 2015. A ribonucleoprotein complex protects the interleukin-6 mRNA from degradation by distinct herpesviral endonucleases. PLoS Pathog 11:e1004899. doi: 10.1371/journal.ppat.1004899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W, Jager S, Shales M, Horner J, Hernandez RD, Krogan NJ, Glaunsinger BA. 2015. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol Cell 57:349–360. doi: 10.1016/j.molcel.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gong D, Kim YH, Xiao Y, Du Y, Xie Y, Lee KK, Feng J, Farhat N, Zhao D, Shu S, Dai X, Chanda SK, Rana TM, Krogan NJ, Sun R, Wu TT. 2016. A herpesvirus protein selectively inhibits cellular mRNA nuclear export. Cell Host Microbe 20:642–653. doi: 10.1016/j.chom.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jung J, Munz C. 2015. Immune control of oncogenic gamma-herpesviruses. Curr Opin Virol 14:79–86. doi: 10.1016/j.coviro.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glaunsinger B, Ganem D. 2004. Highly selective escape from KSHV-mediated host mRNA shutoff and its implications for viral pathogenesis. J Exp Med 200:391–398. doi: 10.1084/jem.20031881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haecker I, Gay LA, Yang Y, Hu J, Morse AM, McIntyre LM, Renne R. 2012. Ago HITS-CLIP expands understanding of Kaposi's sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog 8:e1002884. doi: 10.1371/journal.ppat.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang L, Godinez WJ, Kim IH, Tektonidis M, de Lanerolle P, Eils R, Rohr K, Knipe DM. 2011. Herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late mRNA. Proc Natl Acad Sci U S A 108:E136–E144. doi: 10.1073/pnas.1103411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Camicia R, Winkler HC, Hassa PO. 2015. Novel drug targets for personalized precision medicine in relapsed/refractory diffuse large B-cell lymphoma: a comprehensive review. Mol Cancer 14:207. doi: 10.1186/s12943-015-0474-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siddiqi T, Joyce RM. 2008. A case of HIV-negative primary effusion lymphoma treated with bortezomib, pegylated liposomal doxorubicin, and rituximab. Clin Lymphoma Myeloma 8:300–304. doi: 10.3816/CLM.2008.n.042. [DOI] [PubMed] [Google Scholar]
- 54.Bhatt S, Ashlock BM, Toomey NL, Diaz LA, Mesri EA, Lossos IS, Ramos JC. 2013. Efficacious proteasome/HDAC inhibitor combination therapy for primary effusion lymphoma. J Clin Invest 123:2616–2628. doi: 10.1172/JCI64503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matta H, Chaudhary PM. 2005. The proteasome inhibitor bortezomib (PS-341) inhibits growth and induces apoptosis in primary effusion lymphoma cells. Cancer Biol Ther 4:77–82. doi: 10.4161/cbt.4.1.1378. [DOI] [PubMed] [Google Scholar]
- 56.Abou-Merhi R, Khoriaty R, Arnoult D, El Hajj H, Dbouk H, Munier S, El-Sabban ME, Hermine O, Gessain A, de The H, Mahieux R, Bazarbachi A. 2007. PS-341 or a combination of arsenic trioxide and interferon-alpha inhibit growth and induce caspase-dependent apoptosis in KSHV/HHV-8-infected primary effusion lymphoma cells. Leukemia 21:1792–1801. doi: 10.1038/sj.leu.2404797. [DOI] [PubMed] [Google Scholar]
- 57.Sarosiek KA, Cavallin LE, Bhatt S, Toomey NL, Natkunam Y, Blasini W, Gentles AJ, Ramos JC, Mesri EA, Lossos IS. 2010. Efficacy of bortezomib in a direct xenograft model of primary effusion lymphoma. Proc Natl Acad Sci U S A 107:13069–13074. doi: 10.1073/pnas.1002985107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garcia-Rodriguez N, Wong RP, Ulrich HD. 2016. Functions of ubiquitin and SUMO in DNA replication and replication stress. Front Genet 7:87. doi: 10.3389/fgene.2016.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwertman P, Bekker-Jensen S, Mailand N. 2016. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat Rev Mol Cell Biol 17:379–394. doi: 10.1038/nrm.2016.58. [DOI] [PubMed] [Google Scholar]
- 60.Buratowski S. 2009. Progression through the RNA polymerase II CTD cycle. Mol Cell 36:541–546. doi: 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nojima T, Gomes T, Grosso AR, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, Proudfoot NJ. 2015. Mammalian NET-Seq reveals genome-wide nascent transcription coupled to RNA processing. Cell 161:526–540. doi: 10.1016/j.cell.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Izumiya Y, Izumiya C, Van Geelen A, Wang DH, Lam KS, Luciw PA, Kung HJ. 2007. Kaposi's sarcoma-associated herpesvirus-encoded protein kinase and its interaction with K-bZIP. J Virol 81:1072–1082. doi: 10.1128/JVI.01473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brinster RL, Allen JM, Behringer RR, Gelinas RE, Palmiter RD. 1988. Introns increase transcriptional efficiency in transgenic mice. Proc Natl Acad Sci U S A 85:836–840. doi: 10.1073/pnas.85.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fong YW, Zhou Q. 2001. Stimulatory effect of splicing factors on transcriptional elongation. Nature 414:929–933. doi: 10.1038/414929a. [DOI] [PubMed] [Google Scholar]
- 65.Sun R, Lin SF, Gradoville L, Miller G. 1996. Polyadenylylated nuclear RNA encoded by Kaposi sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 93:11883–11888. doi: 10.1073/pnas.93.21.11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, Ronning P, Reuland B, Guenther K, Beadnell TC, Essig J, Otto GM, O'Sullivan MG, Largaespada DA, Schwertfeger KL, Marahrens Y, Kawakami Y, Bagchi A. 2014. PVT1 dependence in cancer with MYC copy-number increase. Nature 512:82–86. doi: 10.1038/nature13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES. 2016. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539:452–455. doi: 10.1038/nature20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schlackow M, Nojima T, Gomes T, Dhir A, Carmo-Fonseca M, Proudfoot NJ. 2017. Distinctive patterns of transcription and RNA processing for human lincRNAs. Mol Cell 65:25–38. doi: 10.1016/j.molcel.2016.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rossetto CC, Pari G. 2012. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog 8:e1002680. doi: 10.1371/journal.ppat.1002680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McAllister SC, Hansen SG, Messaoudi I, Nikolich-Zugich J, Moses AV. 2005. Increased efficiency of phorbol ester-induced lytic reactivation of Kaposi's sarcoma-associated herpesvirus during S phase. J Virol 79:2626–2630. doi: 10.1128/JVI.79.4.2626-2630.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.