

ABSTRACT
Robust dengue virus (DENV) replication requires lipophagy, a selective autophagy that targets lipid droplets. The autophagic mobilization of lipids leads to increased β-oxidation in DENV-infected cells. The mechanism by which DENV induces lipophagy is unknown. Here, we show that infection with DENV activates the metabolic regulator 5′ adenosine-monophosphate activated kinase (AMPK), and that the silencing or pharmacological inhibition of AMPK activity decreases DENV replication and the induction of lipophagy. The activity of the mechanistic target of rapamycin complex 1 (mTORC1) decreases in DENV-infected cells and is inversely correlated with lipophagy induction. Constitutive activation of mTORC1 by depletion of tuberous sclerosis complex 2 (TSC2) inhibits lipophagy induction in DENV-infected cells and decreases viral replication. While AMPK normally stimulates TSC2-dependent inactivation of mTORC1 signaling, mTORC1 inactivation is independent of AMPK activation during DENV infection. Thus, DENV stimulates and requires AMPK signaling as well as AMPK-independent suppression of mTORC1 activity for proviral lipophagy.
IMPORTANCE Dengue virus alters host cell lipid metabolism to promote its infection. One mechanism for altered metabolism is the induction of a selective autophagy that targets lipid droplets, termed lipophagy. Lipophagy mobilizes lipid stores, resulting in enhanced β-oxidation and viral replication. We show here that DENV infection activates and requires the central metabolic regulator AMPK for its replication and the induction of lipophagy. This is required for the induction of lipophagy, but not basal autophagy, in DENV-infected cells.
KEYWORDS: autophagy, lipid metabolism
INTRODUCTION
Dengue virus (DENV) is an ∼11-kb positive-strand RNA virus of the Flaviviridae family that is transmitted by the Aedes aegypti mosquito, and it is the causative agent of dengue fever. It is a global pathogen that infects ∼390 million people and causes 25,000 deaths annually. There are no therapeutics for DENV infection. Recently, a tetravalent DENV vaccine has been approved in some countries where DENV is endemic that provides protection against ∼2/3 of DENV infections (1).
Macroautophagy (here called autophagy) is a catabolic process essential to cellular and organismal homeostasis (2, 3). During autophagy, de novo-formed double-membrane vesicles, autophagosomes, sequester cytosolic contents and then fuse with the lysosome, where the contents are degraded. The sequestration of cytosolic content into autophagosomes can be random in the case of bulk autophagy. Alternatively, autophagy can be selectively targeted toward cargo, such as aggregated proteins, damaged organelles, or nutrient stores. Autophagy also has a central role in host defense from invading pathogens, both by directly degrading the pathogen or indirectly interfacing with the larger innate and adaptive immune systems (4–6). Many pathogens have elaborate strategies to either evade detection by autophagy or to subvert autophagy for proviral strategies (7, 8).
Multiple studies have shown that DENV induces a proviral autophagy (9–16). Consistent with these findings, studies of various cell types have shown that infection with dengue virus increases the accumulation of autophagosomes, and blockade of autophagy by either genetic approaches or pharmacological inhibitors decreases viral replication. We previously demonstrated that DENV infection elicits the selective targeting of lipid droplets (LDs) by autophagy, termed lipophagy (17). DENV infection increases both the total number of autophagosomes within the cell and the frequency of autophagosomes that localize to LDs. At 24 h postinfection, ∼30% of all autophagosomes localize to LDs. When medium serum concentrations are low, limiting the uptake of extracellular lipids, DENV-induced lipophagy produces a depletion of lipid droplet volume and triglyceride content. This results in the liberation of free fatty acids from triglycerides, which are transported to the mitochondria for β-oxidation (17, 18). Importantly, supplementation of autophagy-deficient cells with exogenous free fatty acids completely complements viral replication. This fatty acid complementation of DENV replication could be prevented by etomoxir, which prevents the transport of fatty acids into the mitochondria for β-oxidation (17). Thus, the major requirement for autophagy in DENV replication is the stimulation of lipid metabolism.
Lipophagy is conserved from yeast to mammals, and much recent work has described conserved transcriptional requirements for lipophagy induction in Caenorhabditis elegans, mice, and humans (19–24). However, the signaling pathways that control lipophagy remain obscure. Likewise, it remains unknown how DENV triggers lipophagy during viral infection.
Integration of signaling is an important upstream regulation of autophagy, often dictating the cellular site and timing of autophagosome biogenesis (2, 25). A central node of signal integration is mammalian target of rapamycin C1 (mTORC1) (2, 26). The activation of mTORC1 suppresses autophagy by antagonizing the activity of the unc51-like kinase 1/2 (ULK1/2) complex through direct inhibitory phosphorylation of ULK1 (27). As a central sensor of nutrient homeostasis, mTORC1 integrates signals from a variety of other nutrient sensors, including AMPK (reviewed in references 28–30). Activation of AMPK is tightly correlated with the cellular energy state of the cell by the differential binding affinity of AMPK to adenylate nucleotides (31–34). Additionally, activation of AMPK requires its phosphorylation by several upstream kinases that integrate cell stresses (35–40).
Activation of AMPK results in the inactivation of mTORC1. This is accomplished by the phosphorylation of tuberous sclerosis complex 2 (TSC2) (41). TSC1 and TSC2 form a heterodimeric complex that functions as a GAP for the small GTPase Rheb, which is essential for mTORC1 activity (42–45). Activation of TSC2 inactivates Rheb, and this leads to suppression of mTORC1 activity (41, 42). AMPK also directly phosphorylates regulatory-associated protein of mTOR (Raptor), an essential adaptor of the mTORC1 complex (46), at Ser 722 and 792 (47). This leads to the recruitment of 14-3-3 and inhibition of mTORC1 activity (47).
In addition to its suppression of mTORC1 activity, AMPK also plays direct roles in the initiation of autophagy through direct phosphorylation of two key complexes in autophagy (27, 48–50). AMPK directly binds and phosphorylates the ULK1/2 complex, which is essential for licensing autophagy under energy-starved conditions (27, 48). Furthermore, AMPK activation also leads to phosphorylation of the vacuolar protein-sorting 34 (Vps34)-Beclin1 complexes to suppress their roles in vesicular trafficking and promote the licensing of autophagosome biogenesis by Vps34-Beclin1–Atg14L complexes (50).
In this paper, we examine the role of AMPK and mTORC1 in DENV-induced lipophagy. We show that DENV infection transiently activates AMPK while inhibiting mTORC1. Inhibition of AMPK and the mTORC1 inhibitor TSC2 decreases autophagy induction, LD depletion, and DENV replication. Thus, DENV requires the activity of AMPK and inactivation of mTORC1 for lipophagy.
RESULTS
DENV induction of proviral lipophagy requires AMPK.
AMPK is a central node in the cellular nutrient stress response to stress and is frequently manipulated during infection with distinct viruses (51). To investigate the role of AMPK in DENV infection, we first examined the requirement of AMPKα1 expression for DENV replication. HepG2 cells were depleted of AMPKα1 by short interfering RNA (siRNA) (Fig. 1A) and then infected with DENV for 24 h. siRNA-mediated depletion of AMPKα1 did not impact cellular viability (Fig. 1B). Compared to cells treated with an irrelevant nontargeting siRNA (IRR), DENV replication was significantly impaired in cells transfected with AMPKα1 siRNA, as measured by reverse transcription-PCR (RT-PCR) quantification of viral RNA (Fig. 1C) and infectious virus production (Fig. 1D).
FIG 1.
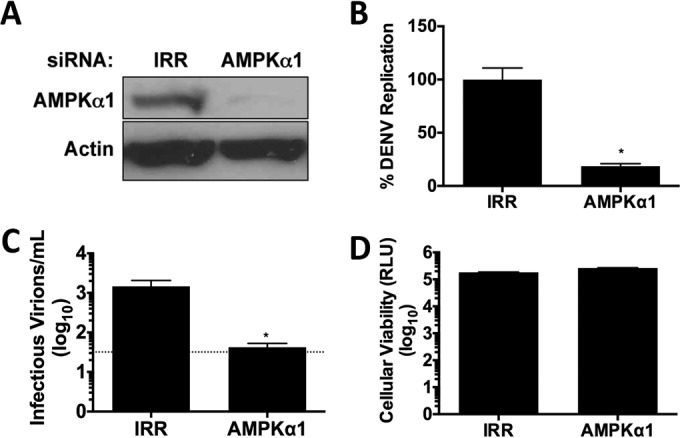
AMPK silencing inhibits DENV replication. (A) AMPKα1 protein levels following treatment with a noncoding (IRR) or AMPKα1 siRNA. (B and C) siRNA-treated cells were infected at an MOI of 1 with DENV. Twenty-four hours postinfection, cellular RNA was harvested to determine viral genome replication (B), and the titers of viral supernatants were determined to delineate the infectious virus produced (C). (D) Cellular viability was assessed 72 h posttransfection of siRNAs.
To determine the role of AMPK in DENV-induced lipophagy, we tested the requirement of AMPKα1 for increased autophagosomes in DENV-infected cells via immunofluorescence analysis of LC3II-positive puncta. HepG2 cells were depleted of AMPKα1 by siRNA or treated with an IRR siRNA and then either mock or DENV infected for 24 h. The infection of IRR-treated HepG2 cells with DENV increased the number of autophagosomes at 24 hpi, consistent with the induction of autophagy by DENV (Fig. 2A and B). While the silencing of AMPKα1 did not alter the number of autophagosomes in mock-infected cells, it prevented the increase in autophagosomes in DENV-infected cells. In contrast, silencing the core autophagy machinery component ATG12 decreased autophagosome number in both mock- and DENV-infected cells (Fig. 2A and B). This suggests that AMPK is required for DENV-induced autophagy, but not basal autophagy, in HepG2 cells.
FIG 2.
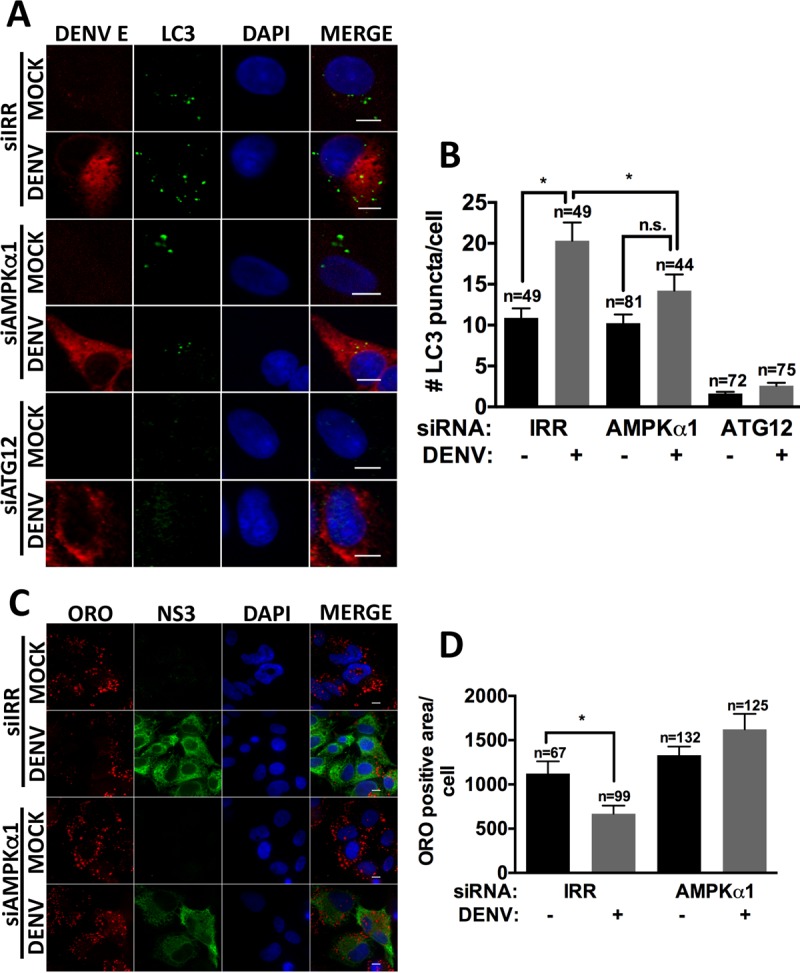
AMPK silencing inhibits DENV-induced lipophagy. Cells were treated with the indicated siRNAs for 48 h, infected with DENV at an MOI of 0.5 for 24 h and 48 h, and probed for DENV E and LC3-II (A) or stained with ORO (C). Nuclei were stained with DAPI. Quantification of LC3-II puncta per cell (B) and LD area (D) was performed using ImageJ. Scale bar, 8 μm; n, numbers of cells counted; n.s., not significant.
We next examined whether AMPK is required for the mobilization of lipids from lipid droplets, as quantified by a decrease in the total area stained by the neutral lipid dye Oil Red O (ORO). HepG2 cells were depleted of AMPKα1 by siRNA or treated with an IRR siRNA and then either mock or DENV infected for 48 h. DENV infection depleted the area of lipid droplets in IRR siRNA-treated cells, consistent with our previous study (17). AMPK silencing prevented the depletion of lipid droplets in DENV-infected cells (Fig. 2C and D). Thus, AMPK is required for both components of DENV-induced lipophagy: the induction of autophagosomes and the mobilization of lipid droplet stores.
DENV-induced lipophagy requires AMPK enzymatic activity.
We tested the requirement of AMPK enzymatic activity for DENV-induced lipophagy and viral replication using the selective AMPK inhibitor compound C (52). HepG2 cells were infected, treated with dimethyl sulfoxide (DMSO) or compound C for 24 h, and assayed for DENV replication (Fig. 3A) or infectious virus production (Fig. 3B). Compound C treatment significantly inhibited DENV replication and infectious virus production without compromising cellular viability (Fig. 3C).
FIG 3.
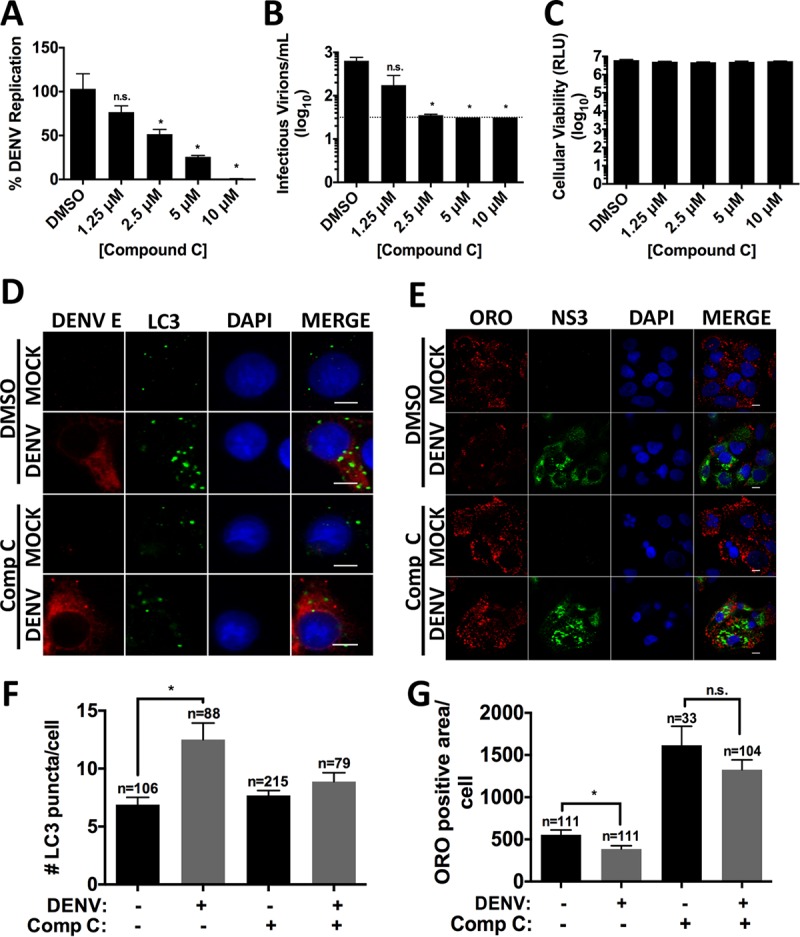
AMPK inhibition diminishes DENV replication and lipophagy induction. Cells were DENV infected at an MOI of 1 for 2 h. After virus adsorption, infected cells were treated with DMSO or various concentrations of compound C. At 24 h postinfection, cellular RNA (A) or supernatants (B) were harvested to assess viral replication and infectious virus release, respectively. (C) Viability was assessed on naive HepG2 cells treated with the indicated concentration of compound C. (D and E) Cells were DENV infected at an MOI of 0.2, and compound C (2.5 μM) was added after virus adsorption. Cells were fixed at 24 and 48 hpi to probe for DENV E and LC3-II (D) or NS3 (E) and stained with ORO. Nuclei were stained with DAPI. Quantification of LC3-II puncta per cell (F) and LD area (G) was performed using ImageJ. Scale bar, 8 μm; n, numbers of cells counted.
Similar to the AMPK-silencing experiments, compound C treatment also prevented DENV-induced lipophagy. In DMSO-treated cells, DENV infection increased autophagosome number and decreased lipid droplet area compared to mock-infected cells. Treatment with compound C blocked autophagy induction and lipid droplet depletion (Fig. 3D to G). Interestingly, treating mock-infected cells with compound C led to an overall increase in lipid droplet area (Fig. 3F and G). This suggests that AMPK regulates aspects of basal lipid metabolism that impact lipid droplet storage, in addition to DENV-induced autophagy.
We confirmed the requirement of AMPK enzymatic activity for DENV replication using an siRNA-resistant trans-complementation approach. Replication-defective lentiviruses expressing siRNA-resistant AMPKα1 that is either wild type (WT) or enzymatically inactive (D156A) were used to infect AMPKα1 siRNA-treated HepG2 cells. We confirmed that AMPKα1 siRNA treatment decreased AMPKα1 expression and that the siRNA-resistant, AMPKα1-expressing lentiviruses restored AMPKα1 expression (Fig. 4A). These cells were then DENV infected in parallel and tested for viral replication. We observed that AMPKα1 silencing decreases DENV replication, as shown in Fig. 1B. The enzymatically active AMPKα1 restores DENV replication, while the enzymatically inactive AMPKα1 fails to rescue DENV replication (Fig. 4B). This confirms the requirement of AMPKα1 enzyme activity for DENV replication, in addition to ruling out off-target effects of the AMPKα1 siRNA.
FIG 4.
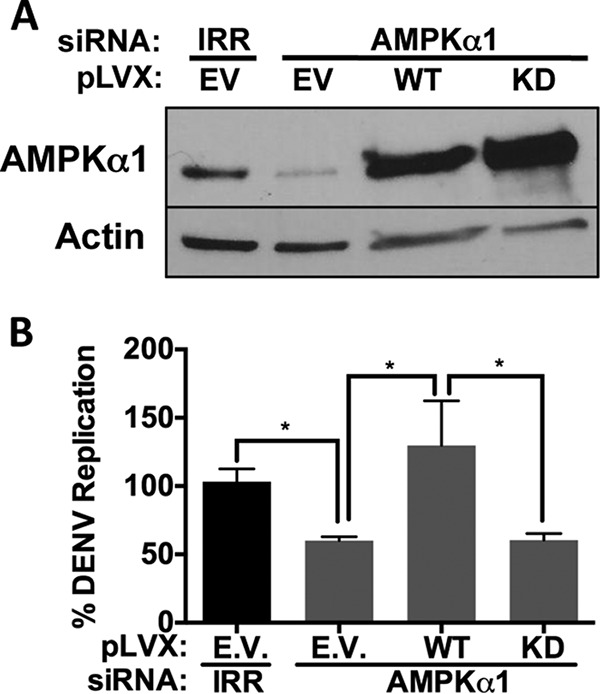
DENV replication requires AMPK kinase activity. HepG2 cells were transduced with lentiviruses derived from either the pLVX empty vector (EV), wild-type (WT) AMPK, or kinase-dead (KD) AMPK and then treated with IRR or AMPK siRNAs. At 72 h after siRNA treatment, cells were infected with DENV, and protein lysates were harvested and subjected to immunoblot analysis for the indicated proteins (A) or RNA was harvested 24 hpi for quantitative RT-PCR analysis (B).
Inhibition of mTOR signaling during DENV infection is required to induce lipophagy.
Activated AMPK phosphorylates TSC2, which then inhibits mTORC1 activity (41). The requirement for AMPKα1 enzymatic activity for DENV-induced lipophagy suggests that its regulation of mTORC1 activity controls this process. This model suggests that the constitutive activation of mTORC1 would prevent DENV-induced AMPK-dependent lipophagy. To test this model, we examined the effect of silencing TSC2 on DENV lipophagy. HepG2 cells were effectively depleted of TSC2 with two different siRNAs to control for off-target effects (Fig. 5A). Infection of HepG2 cells depleted of TSC2 showed a significant decrease in viral replication and infectious virus release, with minimal effects on cellular viability, compared with the infection of IRR siRNA-treated cells (Fig. 5B to D). Thus, depletion of TSC2 and the corresponding constitutive activation of mTORC1 inhibit DENV replication.
FIG 5.
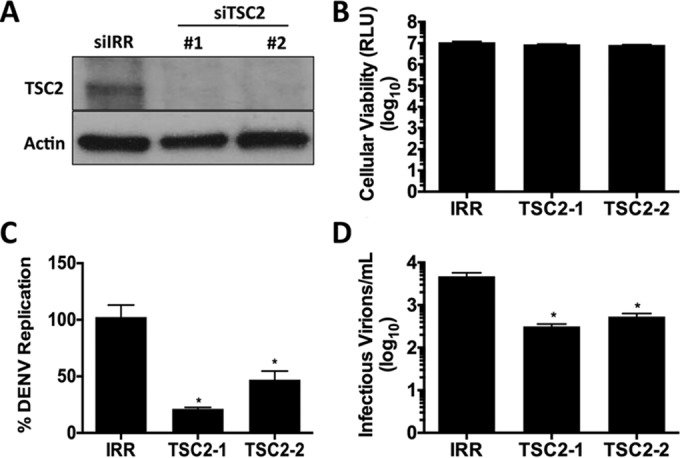
TSC2 silencing inhibits DENV replication. (A) TSC2 protein levels following treatment with IRR or two distinct TSC2 siRNAs. (B) HepG2 cells were assayed for cellular viability 72 h posttransfection with the indicated siRNAs. (C and D) siRNA-treated cells were DENV infected at an MOI of 1. At 24 hpi, RNA was harvested and analyzed for viral replication (C) and titers of supernatants were determined for infectious virus production (D).
In parallel, we probed TSC2-silenced cells that were mock- or DENV-infected cells for markers of lipophagy. Immunofluorescent analysis of DENV-infected cells for endogenous LC3-II showed an increase in autophagosome accumulation in IRR siRNA-treated cells compared to mock-infected cells. This increase in autophagosome number was inhibited in TSC2-depleted DENV-infected cells (Fig. 6A and B). Additionally, TSC2 silencing blocked the significant depletion of lipid droplet area seen in DENV-infected, IRR-treated cells (Fig. 6C and D). Similar to the AMPK silencing phenotype, TSC2 silencing did not alter basal autophagy in mock-infected cells, as opposed to the positive-control ATG12. Thus, TSC2 is required for robust DENV replication and the induction of lipophagy. This suggests that constitutively active mTORC1 blocks DENV-induced lipophagy and thus must be inactivated to initiate lipophagy.
FIG 6.

TSC2 silencing prevents DENV-induced lipophagy. siRNA-treated cells were DENV infected at an MOI of 0.2 and fixed at 24 and 48 hpi to probe for DENV E LC3-II (A) or stain with ORO (C). Nuclei were stained with DAPI. Quantification of LC3-II puncta per cell (B) and LD area (D) was performed using ImageJ. Scale bar, 8 μm; n, numbers of cells counted.
DENV infection activates AMPK signaling.
Our previous experiments demonstrated a requirement of AMPK signaling for DENV-induced lipophagy. We next investigated whether DENV infection activates AMPK signaling. AMPK is a heterotrimeric complex composed of α, β, and γ subunits, where the α subunit, the catalytic core of AMPK, is directly regulated by phosphorylation at threonine 172 (Thr-172) (53, 54). HepG2 cells were infected with DENV, and the levels of AMPK and phospho-AMPK were probed over a time course. We observe that although DENV infection does not alter AMPK protein levels, it results in an increase in phospho-AMPK accumulation at 12 and 24 h postinfection (3.4-fold increase at 12 h and 2-fold increase at 24 h) (Fig. 7A). Increased phospho-AMPK (Thr-172) is a correlate of AMPK activation in DENV-infected cells.
FIG 7.
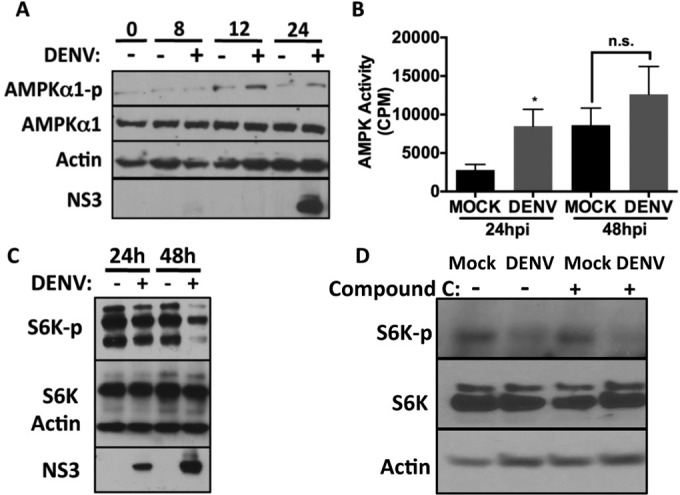
DENV infection activates AMPK and inhibits mTORC1 signaling. (A) HepG2 cells were infected with DENV at an MOI of 5. At the indicated times postinfection, cells were lysed and proteins extracted. Immunoblot analysis was performed to assess the levels of the indicated proteins. (B) HepG2 cells were infected with DENV at an MOI of 10. Lysates (9 μg) were harvested and assayed for AMPK activity. Samples were assayed in triplicate, and the means ± SEM were graphed. (C) HepG2 cells were infected at an MOI of 5 for the indicated times, and lysates were subjected to immunoblot analysis for the indicated proteins. (D) HepG2 cells were infected at an MOI of 5 for 3 h, medium containing DMSO or 10 μM compound C was added to cells for 48 h, and then lysates were harvested and subjected to immunoblot analysis for the indicated proteins.
To more directly test whether DENV infection stimulates AMPK activity, we determined the enzymatic activity of AMPK in crude cellular lysates (55). HepG2 cells were mock or DENV infected for 24 or 48 h, lysed, and assayed for AMPK activation by radiolabeled phosphorylation of a peptide containing the AMPK phosphorylation site from its substrate, acetyl-coenzyme A (CoA) carboxylase (ACC) (55). Infection with DENV for 24 h produced a 3-fold increase in AMPK activity in DENV-infected cell lysates compared to mock-infected cells (Fig. 7B). This activation was transient, as AMPK activity was only moderately elevated at 48 h after DENV infection compared the mock-infected cells. Thus, DENV infection transiently enhances phospho-AMPK accumulation and AMPK enzymatic activity.
AMPK activation should inhibit mTORC1 activity. mTORC1 inhibition can regulate many processes, including the induction of autophagy and the loss of phosphorylation of the mTOR substrate p70 S6 kinase (S6K), which regulates protein translation (56, 57). To test whether the activation of AMPK in DENV infection produced a corresponding decrease in mTORC1 activity, we examined the phosphorylation status of S6K at Thr-389 over a time course of DENV infection. We observed a decrease in phospho-S6K after 24 h of DENV infection that further declined at 48 h (Fig. 7C). Thus, DENV infection increases the accumulation of activated AMPK and produces a corresponding decrease in mTORC1 activity.
We next tested whether AMPK activity is required for the decrease in p70 S6 kinase phosphorylation in DENV infection. Cells were mock- or DENV-infected and treated with compound C or vehicle control for 48 h, and then p70S6K phosphorylation at Thr-389 was assessed. We again observed a decrease in p70S6K phosphorylation in DENV-infected cells (this time treated with DMSO) (Fig. 7D). Surprisingly, this decrease in p70S6K phosphorylation was also observed in DENV-infected compound C-treated cells. Thus, DENV infection stimulates a second, unknown mechanism to alter regulation of p70S6K phosphorylation, in addition to AMPK inactivation of mTORC1.
DISCUSSION
In this study, we investigated the role of AMPK in DENV-induced lipophagy and replication. We observed that AMPK is proviral for DENV replication. Inhibition of AMPK expression or kinase activity suppressed DENV replication and infectious virus production. Similarly, AMPK inhibition prevented the induction of lipophagy, as assayed by autophagosome number (LC3-II puncta) and lipid droplet depletion (ORO area). It is noteworthy that while AMPK inhibition prevented DENV-induced lipophagy, it did not affect basal autophagy under our experimental conditions. LC3-II puncta were unchanged in mock-infected cells by AMPK inhibition, while they were restored to basal levels in DENV-infected cells. In contrast, silencing the core autophagic component ATG12 inhibited autophagy in both mock- and DENV-infected cells.
Given the well-defined role of AMPK in mTORC1 inhibition (and the function of mTORC1 in autophagy suppression), we next tested the impact of constitutively activating mTORC1 on DENV-induced lipophagy. A common approach to studying mTORC1 activation is the silencing of the mTORC1 negative regulator TSC2. We observed that TSC2 silencing mirrored the phenotypes of AMPK inhibition. TSC2 silencing decreased DENV-induced lipophagy and replication. Similarly, TSC2 silencing did not affect basal autophagy. Thus, mTORC1 activation can inhibit DENV-induced lipophagy.
Our initial experiments defined a requirement for AMPK and TSC2 in DENV-induced lipophagy but did not demonstrate that DENV activated this signaling pathway. We next examined the activation of AMPK in DENV infection in two assays. Phospho-AMPK accumulation increased during DENV infection at 12 and 24 h, which is consistent with the kinetics of lipophagy induction by DENV. To directly assess AMPK activity during DENV infection, we measured the AMPK activity in DENV-infected cell lysates and found it was increased during DENV infection compared with that of mock-infected cells. Consistent with these data, DENV infection resulted in a decrease in mTORC1 activity. Accumulation of the mTORC1 product phospho-p70S6K decreased during DENV infection. Although we demonstrate that mTORC1 inhibition is required for DENV-induced lipophagy, it is unknown how this impacts other functions of mTORC1, such as the regulation of protein translation.
Surprisingly, DENV infection can also decrease p70S6K phosphorylation in an AMPK-independent manner; however, the identity of the AMPK-independent pathway is a mystery at this time. The requirement of AMPK for DENV-induced lipophagy, despite its dispensability for mTORC1 regulation, suggests that it has additional targets that regulate lipophagy. Likely candidates include known AMPK substrates that regulate autophagy, including ULK1/2 and Vps34-Beclin complexes (27, 48–50). The requirement of mTORC1 for lipophagy and multiple mechanisms of its inhibition during DENV infection highlight its importance to robust DENV replication.
As a central regulator of the cellular response to energy levels, AMPK has many known interactions with viruses (51). In some viral infections, AMPK exerts antiviral effects. AMPK is activated during Rift Valley fever virus and inhibits its replication by limiting fatty acid synthesis through inactivation of acetyl-CoA carboxylase 1 (ACC1) (58). AMPK also inhibits the replication of Sindbis virus, West Nile virus, and vesicular stomatitis virus in this manner. Hepatitis C virus and human immunodeficiency virus evade AMPK antiviral activity by preventing its activation (59, 60). Alternatively, AMPK can be proviral for many viral infections, including simian virus 40, avian reovirus, and vaccinia virus (61–63). Human cytomegalovirus (HCMV) requires AMPK and activates it in a way that does not inhibit fatty acid synthesis, which is typically the result of AMPK activation (64). This requires the AMPK activator calmodulin-dependent protein kinase kinase beta (CaMKKβ) (64). CaMKKβ-dependent activation of AMPK is also required for rotavirus-induced activation of autophagy (65). Interestingly, activation of AMPK was determined to be a major determinant of cellular permissiveness for rotavirus infection (66).
DENV has a sophisticated modulation of cellular lipid metabolism. Early during DENV infection, lipid droplets are reabsorbed into the endoplasmic reticulum (ER), possibly contributing to the formation of viral replication compartments (67, 68). When DENV infection proceeds under excess serum levels, lipid droplet area increases, which is consistent with an increased uptake and storage of extracellular lipids (67–69). Alternatively, when serum levels are lower, thus limiting lipid uptake, lipid droplet area decreases during DENV infection due to their depletion by lipophagy (17). This results in increased β-oxidation levels and, presumably, an enhanced cellular energetic state (17). Thus, DENV appears to be enhancing lipid metabolic flux both in uptake from serum and subsequent mobilization via lipophagy, resulting in increased β-oxidation levels. In parallel, DENV also induces fatty acid synthesis at sites of viral replication (the ER) (70), which presumably would be incompatible with AMPK activation (58). We envision three possibilities for the activation of both catabolic (AMPK-activated lipophagy) and anabolic (fatty acid synthesis) pathways in DENV infection. There could be mechanistic similarities between HCMV, which also activates AMPK and fatty acid synthesis, and DENV. Alternatively, lipophagy and fatty acid biosynthesis could be either spatially separated into distinct subcellular compartments or kinetically separated at different stages of infection.
The mechanism by which DENV activates AMPK, leading to the induction of autophagy, is unknown. Lipophagy is induced by and required for subgenomic DENV replication, suggesting that the nonstructural proteins 1 to 5 are sufficient for its induction. However, expression of individual DENV NS proteins fails to induce lipophagy (data not shown), suggesting that either multiple viral proteins are involved or that lipophagy is, in part, a cellular response to DENV replication. Future studies will investigate the role of DENV proteins in AMPK activation and the targeting of autophagosomes to the lipid droplet during DENV replication.
MATERIALS AND METHODS
Cells and virus.
HepG2 cells, a human hepatoma cell line (ATCC), and HEK293T cells were maintained in Dulbecco's modified Eagle medium-high glucose and supplemented with 5% fetal bovine serum (FBS), 0.1 mM nonessential amino acids, and 1% penicillin-streptomycin (Life Technologies). Infectious DENV-2 clone 16681 was used, and virus was propagated in C6/36 Aedes albopictus cells (ATCC) as previously described (17). Cellular viability was assayed using Cell Titer-Glo (Promega).
Antibodies and inhibitors.
The antibodies used in this study include AMPK, phospho-AMPK, Thr172, TSC2, S6K, phospho-S6K Thr389 (Cell Signaling), DENV NS3 (70), β-actin (Sigma), LC3B (Novus Biologics), and goat anti-rabbit IgG (Thermo Fischer) for immunoblot analysis. For immunofluorescence analysis, the antibodies used included LC3B (Cell Signaling), DENV NS3, DENV2 E (ATCC), and Alexa Fluor-350, -488, and -594 secondary antibodies (Life Technologies). Compound C was obtained from Cayman Chemicals.
Real-time RT-PCR.
RNA was extracted from cells grown in 96-well plates by an RNeasy 96 kit (Qiagen) per the manufacturer's instructions. Extracts were reverse transcribed and PCR amplified by using the Superscript III Platinum one-step RT-PCR system with Platinum Taq (Life Technologies) as previously described (17). DENV RNAs were amplified using 300 nM forward primer (5′-TCCCAAACGCAGTGATATTACAA-3′), 300 nM reverse primer (5′-TGAGACCTTTGATCGTCAATGC-3′), and 200 nM probe (5′-6-carboxyfluorescein-TGGTGTCCGTTTCCCCACTGCTCTT-IowaBlack-3′) (Integrated DNA Technologies), which recognizes NS2A of DENV 16881. Parallel reaction mixtures used 0.8× the amount of 18S rRNA TaqMan gene expression assay as an internal loading control (Hs01021073_m1; Applied Biosystems). RT-PCR was programmed for 50°C for 30 min, 95°C for 6 min, and then 50 cycles of 95°C for 15 s, 60°C for 30 s, and 72°C for 15 s using an ABI 7300 system (Applied Biosystems). Data were analyzed with SDS v1.4 software (Applied Biosystems) and normalized to 18S controls. Relative quantification was calculated by comparing the cycle threshold (CT) values using 2ΔΔCT.
siRNA transfection.
siRNAs were introduced into cells using Lipofectamine RNAiMax (Life Technologies) per the manufacturer's instructions. At 48 h posttransfection with siRNAs, cells were DENV infected for the indicated times. ATG12 (5′-GCGAACACGAACCAUCCAATT-3′), AMPKα1 (5′-CGGGAUCAGUUAGCAACUATT-3′), and TSC2 (TSC2-1, 5′-GCACCUCUACAGGAACUUUTT-3′; TSC2-2, 5′-CGACGAGUCAAACAAGCCAAUUU-3′) siRNAs were obtained from Life Technologies, and a scrambled negative control (IRR) siRNA was from Dharmacon (71, 72).
Western blot analysis.
Cells were plated in 12-well dishes and, at the indicated times postinfection, were washed 2× in phosphate-buffered saline (PBS) and then harvested in NP-40 lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 2 mM EDTA, 10% glycerol, 10 mM NaF, and protease inhibitors [Roche protease inhibitor cocktail]). Lysate was boiled in 1× SDS sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 12.5 mM EDTA, 0.025% bromphenol blue). Proteins were separated by SDS-PAGE on 4 to 20% gradient gels (Lonza) and transferred to polyvinylidene difluoride (PVDF) membranes (Thermo Fischer). Proteins were detected using SuperSignal West Femto substrate (Thermo Fischer) and exposed to film.
Immunofluorescence.
After infection for the indicated times and multiplicities of infection (MOIs), the indicated cells were fixed on coverslips in either 4% paraformaldehyde or methanol. Coverslips were blocked in PBS containing 30% goat serum and 0.1% saponin and then stained with antibodies in PBS containing 10% goat serum and 0.1% saponin. Lipid droplets were stained with Oil Red O (ORO; MP Biomedical) per the manufacturer's instructions. Stained coverslips were mounted with Prolong Gold with or without 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies). Images were collected with an Olympus DSU confocal microscope with a 100× oil objective. Digital images were taken with Slidebook 5.0 software and processed using ImageJ (National Institutes of Health). Analysis of images was performed with ImageJ and used a set of defined intensity thresholds on all images.
Lentivirus particle production and complementation.
siRNA-resistant AMPKα1 was engineered by silent mutation of the seed sequence sites in the AMPKα1 cDNA (AGCGGAAGCGTTTCAAATT; seed sequence is in boldface, and resistance mutations are underlined) into the lentivirus packaging vector pLVX-IRES-puro (Clontech) between the EcoRI and XbaI sites. HEK293T cells were transfected with empty pLVX-IRES-puro or pLVX-IRES-puro containing siRNA-resistant WT or kinase-dead (D156A) AMPKα1, along with plasmids encoding gag/pol and vesicular stomatitis virus glycoprotein. At 48 h posttransfection, the supernatants were harvested and cell debris spun down by centrifugation at 1,200 × g for 5 min before filtration through a 0.22-μm filter. HepG2 cells were transduced with empty lentiviral pseudoparticles or those encoding an siRNA-resistant wild-type or kinase-dead (D156A) AMPKα1. The following day, the cells were treated with siRNAs against AMPKα1 or an irrelevantly targeted sequence. At 72 h after siRNA treatment, cells were infected and processed at the subsequent indicated time points or, alternatively, protein lysates were harvested in NP-40 lysis buffer and subjected to SDS-PAGE and immunoblot analysis.
AMPK activity assay.
AMPK activity was measured as previously described (55). HepG2 cells were washed 3× with warm Krebs-HEPES buffer (20 mM Na HEPES, pH 7.4, 118 mM NaCl, 3.5 mM KCl, 1.3 mM CaCl2, 1.2 mM MgSO4, 10 mM glucose, 1.2 mM KH2PO4, 0.1% bovine serum albumin) and incubated in Krebs-HEPES buffer for 1 h at 37°C. The buffer was aspirated and dishes were placed on ice with immediate addition of 100 μl ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 50 mM NaF, 5 mM Na pyrophosphate, 1 mM EDTA, 1 mM EGTA, 250 mM mannitol, 1% Triton X-100, 1 mM dithiothreitol [DTT], protease inhibitors). Cells were scraped and lysates transferred to microcentrifuge tubes and incubated on ice for 5 min. Lysates were then centrifuged for 30 min at 14,000 × g and 4°C in preparation for use.
The AMPK activity assay was composed of a total reaction volume of 25 μl that was incubated for 10 min at 30°C. Each reaction mixture consisted of 2.5 μl lysate assay buffer (62.5 mM Na HEPES, pH 7.0, 62.5 mM NaCl, 62.5 mM NaF, 6.25 mM Na pyrophosphate, 1.25 mM EDTA, 1.25 mM EGTA, 1 mM DTT, and protease inhibitor cocktail [Roche]), 2.5 μl of 100 μM [γ-32P]ATP (1 μCi/μl) in 25 mM MgCl2, 2.5 μl of 2 mM AMP in lysate assay buffer, 5 μl of 1 mM SAMS peptide in lysate assay buffer, and 12.5 μl cell lysate. The reaction mixture was spotted on P81 phosphocellulose paper, which was washed twice with 1% phosphoric acid and then once in water and acetone. The radioactivity of the phosphorylated SAMS peptide was quantified by scintillation counting. Assay background was determined by incubating the lysate in the absence of SAMS peptide.
Statistical analysis.
Data are presented as means ± standard errors of the means (SEM). To assess statistical significance, two-tailed, paired Student's t tests were performed.
ACKNOWLEDGMENTS
We thank Yasmine Baktash and Arnold Olali for critically reading the manuscript. We thank Claire Huang for reagents. We thank the University of Chicago Light Microscopy Facility and director Vytas Bindokas.
This work was funded by NIAID (1R01AI080703 and 1R01DK102883). T.X.J. was funded by NIH training grant T32 GM007183 and R01AI080703-S1.
REFERENCES
- 1.Hadinegoro SR, Arredondo-Garcia JL, Capeding MR, Deseda C, Chotpitayasunondh T, Dietze R, Muhammad Ismail HI, Reynales H, Limkittikul K, Rivera-Medina DM, Tran HN, Bouckenooghe A, Chansinghakul D, Cortes M, Fanouillere K, Forrat R, Frago C, Gailhardou S, Jackson N, Noriega F, Plennevaux E, Wartel TA, Zambrano B, Saville M. 2015. Efficacy and long-term safety of a Dengue vaccine in regions of endemic disease. N Engl J Med 373:1195–1206. doi: 10.1056/NEJMoa1506223. [DOI] [PubMed] [Google Scholar]
- 2.Kroemer G, Marino G, Levine B. 2010. Autophagy and the integrated stress response. Mol Cell 40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. 2008. Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levine B, Mizushima N, Virgin HW. 2011. Autophagy in immunity and inflammation. Nature 469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deretic V, Saitoh T, Akira S. 2013. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deretic V. 2011. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev 240:92–104. doi: 10.1111/j.1600-065X.2010.00995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deretic V, Levine B. 2009. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jordan TX, Randall G. 2012. Manipulation or capitulation: virus interactions with autophagy. Microbes Infect 14:126–139. doi: 10.1016/j.micinf.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metz P, Chiramel A, Chatel-Chaix L, Alvisi G, Bankhead P, Mora-Rodriguez R, Long G, Hamacher-Brady A, Brady NR, Bartenschlager R. 2015. Dengue virus inhibition of autophagic flux and dependency of viral replication on proteasomal degradation of the autophagy receptor p62. J Virol 89:8026–8041. doi: 10.1128/JVI.00787-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang YT, Wan SW, Lu YT, Yao JH, Lin CF, Hsu LJ, Brown MG, Marshall JS, Anderson R, Lin YS. 2014. Autophagy facilitates antibody-enhanced dengue virus infection in human pre-basophil/mast cells. PLoS One 9:e110655. doi: 10.1371/journal.pone.0110655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang J, Li Y, Qi Y, Zhang Y, Zhang L, Wang Z, Zhang X, Gui L. 2014. Coordinated regulation of autophagy and apoptosis determines endothelial cell fate during Dengue virus type 2 infection. Mol Cell Biochem 397:157–165. doi: 10.1007/s11010-014-2183-3. [DOI] [PubMed] [Google Scholar]
- 12.Lee YR, Hu HY, Kuo SH, Lei HY, Lin YS, Yeh TM, Liu CC, Liu HS. 2013. Dengue virus infection induces autophagy: an in vivo study. J Biomed Sci 20:65. doi: 10.1186/1423-0127-20-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mateo R, Nagamine CM, Spagnolo J, Mendez E, Rahe M, Gale M Jr, Yuan J, Kirkegaard K. 2013. Inhibition of cellular autophagy deranges dengue virion maturation. J Virol 87:1312–1321. doi: 10.1128/JVI.02177-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khakpoor A, Panyasrivanit M, Wikan N, Smith DR. 2009. A role for autophagolysosomes in Dengue virus 3 production in HepG2 cells. J Gen Virol 90:1093–1103. doi: 10.1099/vir.0.007914-0. [DOI] [PubMed] [Google Scholar]
- 15.Panyasrivanit M, Khakpoor A, Wikan N, Smith DR. 2009. Linking dengue virus entry and translation/replication through amphisomes. Autophagy 5:434–435. doi: 10.4161/auto.5.3.7925. [DOI] [PubMed] [Google Scholar]
- 16.Panyasrivanit M, Khakpoor A, Wikan N, Smith DR. 2009. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J Gen Virol 90:448–456. doi: 10.1099/vir.0.005355-0. [DOI] [PubMed] [Google Scholar]
- 17.Heaton NS, Randall G. 2010. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 8:422–432. doi: 10.1016/j.chom.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. 2009. Autophagy regulates lipid metabolism. Nature 458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. 2014. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 516:112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, Ma J, Kemper B, Kemper JK. 2014. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 516:108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo B, Huang X, Zhang P, Qi L, Liang Q, Zhang X, Huang J, Fang B, Hou W, Han J, Zhang H. 2014. Genome-wide screen identifies signaling pathways that regulate autophagy during Caenorhabditis elegans development. EMBO Rep 15:705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu CC, Visvikis O, Chang JT, Gelino S, Ong B, Davis AE, Irazoqui JE, Dillin A, Hansen M. 2013. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun 4:2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Rourke EJ, Ruvkun G. 2013. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat Cell Biol 15:668–676. doi: 10.1038/ncb2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. 2011. TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell RC, Yuan HX, Guan KL. 2014. Autophagy regulation by nutrient signaling. Cell Res 24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. 2010. mTOR regulation of autophagy. FEBS Lett 584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sengupta S, Peterson TR, Sabatini DM. 2010. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell 40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howell JJ, Manning BD. 2011. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol Metab 22:94–102. doi: 10.1016/j.tem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, Kemp BE. 2011. AMPK is a direct adenylate charge-regulated protein kinase. Science 332:1433–1435. [DOI] [PubMed] [Google Scholar]
- 32.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ. 2011. Structure of mammalian AMPK and its regulation by ADP. Nature 472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, Martin SR, Carling D, Gamblin SJ. 2007. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- 34.Hardie DG, Carling D, Gamblin SJ. 2011. AMP-activated protein kinase: also regulated by ADP? Trends Biochem Sci 36:470–477. doi: 10.1016/j.tibs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 35.Momcilovic M, Hong SP, Carlson M. 2006. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem 281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 36.Hong SP, Momcilovic M, Carlson M. 2005. Function of mammalian LKB1 and Ca2+/calmodulin-dependent protein kinase kinase alpha as Snf1-activating kinases in yeast. J Biol Chem 280:21804–21809. doi: 10.1074/jbc.M501887200. [DOI] [PubMed] [Google Scholar]
- 37.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. 2005. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 38.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. 2005. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. 2005. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 40.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. 2004. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inoki K, Zhu T, Guan KL. 2003. TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 42.Inoki K, Li Y, Xu T, Guan KL. 2003. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Inoki K, Guan KL. 2004. Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol Cell Biol 24:7965–7975. doi: 10.1128/MCB.24.18.7965-7975.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Inoki K, Vikis H, Guan KL. 2006. Measurements of TSC2 GAP activity toward Rheb. Methods Enzymol 407:46–54. doi: 10.1016/S0076-6879(05)07005-9. [DOI] [PubMed] [Google Scholar]
- 45.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. 2005. Rheb binds and regulates the mTOR kinase. Curr Biol 15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 46.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. 2002. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110:163–175. doi: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 47.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. 2011. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JW, Park S, Takahashi Y, Wang HG. 2010. The association of AMPK with ULK1 regulates autophagy. PLoS One 5:e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. 2013. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152:290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chukkapalli V, Heaton NS, Randall G. 2012. Lipids at the interface of virus-host interactions. Curr Opin Microbiol 15:512–518. doi: 10.1016/j.mib.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. 2001. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Investig 108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. 1996. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 54.Weekes J, Hawley SA, Corton J, Shugar D, Hardie DG. 1994. Activation of rat liver AMP-activated protein kinase by kinase kinase in a purified, reconstituted system. Effects of AMP and AMP analogues. Eur J Biochem 219:751–757. [DOI] [PubMed] [Google Scholar]
- 55.Davies SP, Carling D, Hardie DG. 1989. Tissue distribution of the AMP-activated protein kinase, and lack of activation by cyclic-AMP-dependent protein kinase, studied using a specific and sensitive peptide assay. Eur J Biochem 186:123–128. doi: 10.1111/j.1432-1033.1989.tb15185.x. [DOI] [PubMed] [Google Scholar]
- 56.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. 1998. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci U S A 95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Magnuson B, Ekim B, Fingar DC. 2012. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J 441:1–21. doi: 10.1042/BJ20110892. [DOI] [PubMed] [Google Scholar]
- 58.Moser TS, Schieffer D, Cherry S. 2012. AMP-activated kinase restricts Rift Valley fever virus infection by inhibiting fatty acid synthesis. PLoS Pathog 8:e1002661. doi: 10.1371/journal.ppat.1002661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mankouri J, Tedbury PR, Gretton S, Hughes ME, Griffin SD, Dallas ML, Green KA, Hardie DG, Peers C, Harris M. 2010. Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP-activated protein kinase. Proc Natl Acad Sci U S A 107:11549–11554. doi: 10.1073/pnas.0912426107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang HS, Wu MR. 2009. SIRT1 regulates Tat-induced HIV-1 transactivation through activating AMP-activated protein kinase. Virus Res 146:51–57. doi: 10.1016/j.virusres.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 61.Moser TS, Jones RG, Thompson CB, Coyne CB, Cherry S. 2010. A kinome RNAi screen identified AMPK as promoting poxvirus entry through the control of actin dynamics. PLoS Pathog 6:e1000954. doi: 10.1371/journal.ppat.1000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chi PI, Huang WR, Lai IH, Cheng CY, Liu HJ. 2013. The p17 nonstructural protein of avian reovirus triggers autophagy enhancing virus replication via activation of phosphatase and tensin deleted on chromosome 10 (PTEN) and AMP-activated protein kinase (AMPK), as well as dsRNA-dependent protein kinase (PKR)/eIF2alpha signaling pathways. J Biol Chem 288:3571–3584. doi: 10.1074/jbc.M112.390245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar SH, Rangarajan A. 2009. Simian virus 40 small T antigen activates AMPK and triggers autophagy to protect cancer cells from nutrient deprivation. J Virol 83:8565–8574. doi: 10.1128/JVI.00603-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McArdle J, Moorman NJ, Munger J. 2012. HCMV targets the metabolic stress response through activation of AMPK whose activity is important for viral replication. PLoS Pathog 8:e1002502. doi: 10.1371/journal.ppat.1002502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crawford SE, Hyser JM, Utama B, Estes MK. 2012. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-beta signaling is required for rotavirus replication. Proc Natl Acad Sci U S A 109:E3405–E3413. doi: 10.1073/pnas.1216539109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Green VA, Pelkmans L. 2016. A systems survey of progressive host-cell reorganization during rotavirus infection. Cell Host Microbe 20:107–120. doi: 10.1016/j.chom.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 67.Pena J, Harris E. 2012. Early dengue virus protein synthesis induces extensive rearrangement of the endoplasmic reticulum independent of the UPR and SREBP-2 pathway. PLoS One 7:e38202. doi: 10.1371/journal.pone.0038202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang WC, Lin RJ, Liao CL, Lin YL. 2014. Rab18 facilitates dengue virus infection by targeting fatty acid synthase to sites of viral replication. J Virol 88:6793–6804. doi: 10.1128/JVI.00045-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Samsa MM, Mondotte JA, Iglesias NG, Assuncao-Miranda I, Barbosa-Lima G, Da Poian AT, Bozza PT, Gamarnik AV. 2009. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog 5:e1000632. doi: 10.1371/journal.ppat.1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, Randall G. 2010. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci U S A 107:17345–17350. doi: 10.1073/pnas.1010811107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, Chien M, Weir DB, Russo JJ, Ju J, Brownstein MJ, Sheridan R, Sander C, Zavolan M, Tuschl T, Rice CM. 2007. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci U S A 104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Randall G, Grakoui A, Rice CM. 2003. Clearance of replicating hepatitis C virus replicon RNAs in cell culture by small interfering RNAs. Proc Natl Acad Sci U S A 100:235–240. doi: 10.1073/pnas.0235524100. [DOI] [PMC free article] [PubMed] [Google Scholar]