

Abstract
Constructing high-quality libraries of molecular building blocks is essential for successful fragment-based drug discovery. In this communication, we describe eMolFrag, a new open-source software to decompose organic compounds into nonredundant fragments retaining molecular connectivity information. Given a collection of molecules, eMolFrag generates a set of unique fragments comprising larger moieties, bricks, and smaller linkers connecting bricks. These building blocks can subsequently be used to construct virtual screening libraries for targeted drug discovery. The robustness and computational performance of eMolFrag is assessed against the Directory of Useful Decoys, Enhanced database conducted in serial and parallel modes with up to 16 computing cores. Further, the application of eMolFrag in de novo drug design is illustrated using the adenosine receptor. eMolFrag is implemented in Python, and it is available as stand-alone software and a web server at www.brylinski.org/emolfrag and https://github.com/liutairan/eMolFrag.
Introduction
Hit identification, lead generation, and lead optimization are the key steps at the outset of a drug discovery process. Briefly, compounds showing promising activity identified by high-throughput screening as initial hits are filtered and modified to generate lead compounds, which satisfy basic drug-likeliness properties.1 These lead compounds are further optimized to enhance the potency toward the target protein as well as to reduce their nonselectivity and toxicity.2 Conventional hit identification is not only limited to already synthesized compounds often leading to low discovery rates, but it is also expensive and requires time-consuming screening experiments.3 Consequently, virtual screening that can rapidly evaluate millions of compounds has become an integral part of lead identification protocols.4
In order to enhance the chemical diversity of virtual screening libraries, large collections of drug-like compounds can be generated through combinatorial chemistry.5 Since constructing and screening the entire chemical space are not feasible even with the most advanced computers, building extensive yet targeted libraries is critical for the success of virtual screening. A number of fragment- and atom-based techniques have been developed to generate novel chemical compounds for virtual screening, including binding-site point connection methods (LUDI6), fragment connection methods (LEA3D,7 LigBuilder,8 and eSynth9), sequential build-up algorithms (LEGEND10 and SPROUT11), and random connection techniques (CoG12 and Flux13). These de novo methods require an initial set of building blocks or molecular fragments, which ultimately control the properties of the resulting screening compounds and their affinity toward the target protein. Consequently, there is a great interest in efficient fragmentation techniques to generate sets of chemically feasible building blocks for the subsequent molecular synthesis. Retrosynthetic combinatorial analysis procedure (RECAP14) and breaking retrosynthetically interesting chemical substructures (BRICS15) are examples of systematic fragmentation methods. In RECAP, compounds are dissected based on a set of 11 bond types, following simple rules such as leaving cyclic bonds and alkyl groups smaller than five carbons intact. These rules ensure that major structural features of organic compounds, such as ring motifs, are preserved. BRICS expands the bond type criteria used by RECAP from 11 to 16 taking into account the chemical environment of each bond type and the surrounding substructures. Additional filters are also applied in order to prevent generating small and unwanted fragments. Other methods extract and classify chemical scaffolds by pruning side chains and removing peripheral ring moieties.16
In general, the performance of fragment-based chemical synthesis tools such as eSynth,9 CONFIRM,17 and AutoGrow18 could significantly be improved by employing building blocks annotated with empirical connectivity patterns. Although this information could help explore pharmacologically relevant regions of the diverse chemical space,9 many existing fragmentation tools, e.g. Fragmenter19 and molBLOCKS,20 do not consider the chemical context of the fragments. In other words, the connectivity information on a fragment is not stored while extracting building blocks. To address this issue, we developed eMolFrag, a new open-source molecular fragmentation software. eMolFrag decomposes either a single ligand or a library of compounds into two types of chemical building blocks, bricks and the connecting linkers. The resulting complete and nonredundant sets of building blocks are annotated with the comprehensive connectivity information in order to facilitate the construction of novel compounds with combinatorial synthesis software. eMolFrag has been parallelized to decrease the computing time required to analyze large collections of molecules.
Methods
eMolFrag employs a graph-based notation, where molecules are sets of nodes representing atoms connected by edges corresponding to chemical bonds. A fragment is a substructure, which has either all or only some atoms and bonds of a given molecule; fragments are categorized as either bricks or linkers. Given a collection of molecules, the complete set of unique fragments is constructed in two steps shown in Figure 1. The first step, labeled as Part I, involves creating an initial set of fragments, whereas the second step, labeled as Part II, guarantees the uniqueness of the resulting set of fragments.
Figure 1.

Flowchart of eMolFrag. Part I: Input molecules are fragmented with the BRICS algorithm to generate a complete set of building blocks. Part II: Fragment redundancy is removed according to pairwise chemical alignments with the kcombu program. At the end, nonredundant sets of bricks and linkers are reported along with the consolidated connectivity information as well as lists of similar fragments that were removed.
Part I: Fragmentation
In eMolFrag, a set of molecules are first decomposed into constituent fragments with the BRICS algorithm,15 implemented in RDKit.21 Chemical compounds are broken down into larger moieties called bricks connected by linkers based on 16 chemical environments defined by the BRICS model;15 a pseudocode for the fragmentation process is given in the Supporting Information (Algorithm S1). A brick fragment is a molecular construct having at least four non-hydrogen atoms. Subsequently, bricks are removed from a molecule and the remaining fragments are classified as linkers (see Algorithm S2 in the Supporting Information). Broken bonds are replaced by dummy atoms, which are placeholders for those atoms removed from a particular bond. The complete information, including the type of atoms involved in those bonds that were broken, is stored for each brick in order to provide empirical connectivity patterns. Linkers have different auxiliary connectivity information, i.e. these fragments are annotated only with the maximum number of bonds at various positions. Examples of bricks and linkers are provided in the Supporting Information (Examples S1 and S2, respectively). We found that this approach allows to efficiently construct series of new molecules, whose chemistry is similar to that of parent compounds.
Part II: Mitigation of Fragment Redundancy
Since one of the objectives of an effective fragmentation procedure is to employ the resulting fragments in a synthesis procedure, the cardinality of the final set of fragments is critical. On that account, eMolFrag attempts to minimize the size of sets of bricks and linkers by removing redundancy with a partitioning and sieve-based removal scheme presented in the Supporting Information (Algorithm S3). Two fragments are equivalent if the Tanimoto coefficient (TC) calculated for topologically constrained maximum common substructures by the kcombu program22 is equal to 1.0. Information on equivalent atoms provided by kcombu as well as their connectivity information is then used to consolidate identical fragments into a single, unique construct.
Results and Discussion
Benchmarks against the DUD-E Database
We validate the eMolFrag algorithm by conducting a self-reconstruction test as described previously.9 Briefly, given an input molecule m, a set of fragments extracted from m by eMolFrag are passed to a fragment-based construction procedure with eSynth employing its chemical rules.9 A molecule with the highest chemical similarity to m measured by the TC calculated for Daylight fingerprints23 is selected from a series of compounds constructed by eSynth. Here, we employ a fingerprint-based assessment of chemical similarity with OpenBabel24 because this technique is computationally much faster than kcombu. A TC of ≥0.8 indicates that a molecule highly similar to m was generated, whereas a TC of 1.0 indicates that compound m has been reconstructed. As a testing set, we use 20 408 active compounds for 102 protein targets from the Directory of Useful Decoys, Enhanced (DUD-E) database25 covering a diverse chemical space of pharmacologically relevant molecules.
The performance of eMolFrag is compared to molBLOCKS,20 another fragmentation software employing the RECAP algorithm.14Figure 2 shows a two-way box plot of the number of atoms per fragment and the number of fragments per molecule for these two programs. Fragments generated by molBLOCKS typically contain 6–10 (the default protocol) and 6–11 (an extensive mode) atoms, whereas most fragments extracted by eMolFrag consist of 2–7 atoms. The median numbers of fragments per molecule are 3, 8, and 6 for molBLOCKS (default), molBLOCKS (extensive), and eMolFrag, respectively. Finally, molecular synthesis with eSynth9 was conducted employing fragments generated by eMolFrag for active compounds in the DUD-E database. Encouragingly, 82.8% of active compounds were reconstructed with a TC of 1.0 and 92.2% with a TC of ≥0.8. An inspection of the failed cases revealed that the major reason for not generating a relatively small fraction of testing compounds is the fact that the synthesis software does not allow to directly connect two bricks. Overall, the self-reconstruction benchmarking results demonstrate that eMolFrag properly extracts molecular fragments providing sufficient connectivity information to rebuild the majority of parent molecules.
Figure 2.
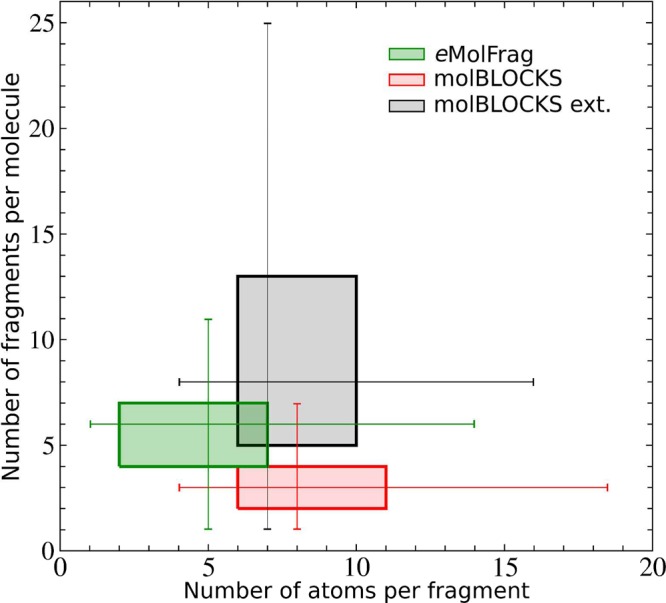
Two-way box plot of the number of fragments per molecule against the number of atoms per fragment. Bioactive compounds from the Directory of Useful Decoys, Enhanced database were fragmented with eMolFrag (green) and molBLOCKS (red: the default protocol, gray: an extensive mode).
Computational Performance
Decomposing large compound libraries can be time-consuming depending on the number of input molecules; therefore, we parallelized the eMolFrag code. The serial and parallel performance of eMolFrag is assessed by fragmenting subsets of DUD-E actives with sizes varying from 100 to 12 800 molecules. All tests were performed on a machine equipped with two 2.6 GHz 8-core Sandy Bridge Xeon 64-bit processors, 32GB 1666 MHz RAM and 500GB HD, running Red Hat Enterprise Linux 6. Figure 3 shows that the wall time for eMolFrag scales linearly with the number of input molecules. The average processing speed of the serial code ranges from 8.7 molecules/s for the smallest data set to 4.8 molecules/s for the largest data set (see Table S1 in Supporting Information). The actual decomposition speed (Part I in Figure 1) is faster for larger sets because the I/O overhead is reduced by efficient data caching. However, removing redundancy (Part II in Figure 1) from large data sets requires significantly longer computing times compared to small data sets, which in turn causes the overall speed to decrease with the increasing number of input molecules. Without removing redundancy, the average processing speed of serial eMolFrag is 9.8 molecules/s for the smallest data set and 23.2 molecules/s for the largest data set. For comparison, a serial version of molBLOCKS, which does not remove redundancy, is capable of processing 6.6 and 12.5 molecules/s for the smallest and the largest data sets, respectively. Thus, eMolFrag is 1.2–1.9× faster than molBLOCKS. Further, algorithms implemented in eMolFrag are polynomial in complexity; the best-fit curves in Figure 3 are y = 0.022x1.238 (R2 = 0.99989) for serial and y = 0.013x1.201 (R2 = 0.99989) for parallel execution. This near-linear scaling gives empirical evidence of the efficient implementation of eMolFrag.
Figure 3.

Serial and parallel performance of eMolFrag. The main graph shows the wall time for the complete fragmentation procedure plotted against the number of input molecules. A serial code is compared to the parallel processing on 16 computing cores. Parallel scaling for a fixed size input data set of 3200 molecules is presented as the inset. Upper and lower bounds for the ideal speedup calculated according to Amdahl’s law are shown as dark and light gray lines, respectively.
The impact of the number of computing cores on parallel processing is assessed by comparing the performance of parallel eMolFrag to the theoretical speedup estimated with Amdahl’s law.26 The inset in Figure 3 shows that executing eMolFrag in parallel for a fixed input data set of 3200 molecules and the number of computing cores varying from 1 to 16 roughly corresponds to a hypothetical code consisting of 47–60% parallel calculations. Note that eMolFrag does not conform exactly to Amdahl’s law because the workload related to removing redundancy (Part II in Figure 1) is unevenly distributed across computing cores. Although the total execution time of eMolFrag diverges from Amdahl’s law, the parallel processing is faster than the serial execution. The average processing speed for the parallel code running on 16 computing cores ranges from 24 molecules per second for the smallest data set to 11.8 molecules per second for the largest data set (see Table S1 in the Supporting Information). This shorter processing time for parallel eMolFrag becomes particularly beneficial for larger data sets. For instance, decomposing 20 408 active compounds from the DUD-E data set for the self-benchmarking test takes 1 h and 18 min on a single core compared to only half an hour on 16 computing cores.
Application to Antagonists of the Adenosine Receptor
To illustrate the application of eMolFrag in de novo drug discovery, we show that bioactive compounds can successfully be constructed from molecular fragments extracted from chemically dissimilar binders of the same target protein. Here, we selected the human adenosine A2a receptor (AA2AR), a member of the G protein-coupled receptor (GPCR) superfamily containing targets for about 27% of all FDA-approved drugs.27Figure 4 presents individual steps of the cross-validation procedure, in which CHEMBL144979, a known bioactive ligand for AA2AR,28 is the target molecule. Four other AA2AR antagonists, called donors, are shown in Figure 4A. Since the chemical similarity of donors to the target, measured by the TC reported by kcombu, is lower than 0.5, CHEMBL144979 can be considered novel with respect to the donor molecules.
Figure 4.

Example of the successful construction of a bioactive of the adenosine receptor by eMolFrag and eSynth. (A) Donor molecules with the chemical similarity to CHEMBL144979 measured by the Tanimoto coefficient (TC). (B) Bricks annotated with the list of atom types that can be attached at various positions. (C) Linkers annotated with the number of the maximum allowed connections. (D) Examples of new molecules synthesized using bricks and linkers. The first molecule shown in a box is a known bioactive of the adenosine receptor. Highlighted in different colors are essential building blocks to generate CHEMBL144979 that are extracted from donor molecules by eMolFrag and used in molecular synthesis by eSynth. Further, the connectivity information inferred from donors that is required to correctly assemble CHEMBL144979 is highlighted in bold in B and C.
Unique sets of 10 bricks and 7 linkers extracted by eMolFrag from 4 donors are shown in Figures 4B and C, respectively. For instance, the triazolo-quinazoline fragment highlighted in pink carrying the chlorine moiety was obtained from CHEMBL95229. This compound is a member of a series of pyrazolo-triazolo-pyrimidines with subnanomolar affinity against ARs created via N5-phenylcarbamoyl substitutions.29 Bricks contain information on atom types that can be attached at various positions (small boxes in Figure 4B), whereas linkers are annotated with the maximum number of allowed bonds (small circles in Figure 4C). The sets of bricks and linkers are complete and nonredundant, i.e. each unique fragment carries the connectivity information extracted from multiple donor compounds. For example, the connectivity information for a benzene ring, which is present in all donors, is consolidated by eMolFrag into a single fragment shown in cyan in Figure 4B.
Subsequently, molecular fragments extracted by eMolFrag were passed to eSynth9 in order to generate a series of compounds. A serial version of eSynth produced 4 492 609 virtual compounds in 12 h. Encouragingly, the first compound in Figure 4D (shown in a box) is CHEMBL144979; therefore, the target molecule has been successfully constructed. Further, the set of virtual molecules comprises 845 compounds, whose TC to CHEMBL144979 is ≥0.7 and as many as 239 656 molecules with a TC of ≥0.5. Three randomly selected virtual molecules are presented in Figure 4D to demonstrate the chemical diversity of compounds generated by eSynth. It is important to note that these retrospective cross-validation benchmarks are designed to mimic real applications by attempting to construct target molecules using building blocks extracted from chemically dissimilar compounds. This case study demonstrates that high-quality fragment sets generated by eMolFrag can be used in fragment-based drug discovery to create targeted screening libraries likely containing novel bioactives.
Conclusions
eMolFrag is a fast and robust tool to extract molecular fragments, classified as bricks and linkers, from small molecule data sets. Subsequently, these fragments can be used to construct targeted libraries for virtual screening. A unique feature of eMolFrag is that it stores the connectivity information for the extracted building blocks to help generate new series of chemically feasible compounds. Although eMolFrag was optimized to work with eSynth, a recently developed molecular synthesis algorithm, it can also be integrated into other cheminformatics toolkits utilizing chemical fragments. eMolFrag is freely available as stand-alone software and a Web server at www.brylinski.org/emolfrag and https://github.com/liutairan/eMolFrag.
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM119524. This work has been partially supported by Army Research Office (ARO) under grant number W911NF-10-1-0495. The authors are grateful to Mr. Dave Marley Dixon for his help with the graphics.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.6b00596.
eMolFrag algorithms, computing speeds, and file formats (PDF)
Author Contributions
T.L. implemented eMolFrag, performed validation simulations and serial and parallel tests, and analyzed results. M.N. prepared validation data sets, analyzed results, and conducted the case study. T.L. and M.N. drafted the manuscript. C.A. and S.M. contributed algorithms. C.A. helped write the manuscript. M.B. coordinated the project, helped analyze results, and prepared the final version of the manuscript.
Author Contributions
○ T.L. and M.N. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Palermo G.; De Vivo M., Computational Chemistry for Drug Discovery. In Encyclopedia of Nanotechnology; Bhushan B., Ed.; Springer: Netherlands, Dordrecht, 2014; pp 1–15. [Google Scholar]
- Leelananda S. P.; Lindert S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. 10.3762/bjoc.12.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestres J. Virtual screening: a real screening complement to high-throughput screening. Biochem. Soc. Trans. 2002, 30, 797–799. 10.1042/bst0300797. [DOI] [PubMed] [Google Scholar]
- Lipinski C.; Hopkins A. Navigating chemical space for biology and medicine. Nature 2004, 432, 855–861. 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- Bohm H. J. The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J. Comput.-Aided Mol. Des. 1992, 6, 61–78. 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- Douguet D.; Munier-Lehmann H.; Labesse G.; Pochet S. LEA3D: a computer-aided ligand design for structure-based drug design. J. Med. Chem. 2005, 48, 2457–2468. 10.1021/jm0492296. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Pei J.; Lai L. LigBuilder 2: a practical de novo drug design approach. J. Chem. Inf. Model. 2011, 51, 1083–1091. 10.1021/ci100350u. [DOI] [PubMed] [Google Scholar]
- Naderi M.; Alvin C.; Ding Y.; Mukhopadhyay S.; Brylinski M. A graph-based approach to construct target-focused libraries for virtual screening. J. Cheminf. 2016, 8, 14. 10.1186/s13321-016-0126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishibata Y.; Itai A. Automatic creation of drug candidate structures based on receptor structure. Starting point for artificial lead generation. Tetrahedron 1991, 47, 8985–8990. 10.1016/S0040-4020(01)86503-0. [DOI] [Google Scholar]
- Gillet V.; Johnson A. P.; Mata P.; Sike S.; Williams P. SPROUT: a program for structure generation. J. Comput.-Aided Mol. Des. 1993, 7, 127–153. 10.1007/BF00126441. [DOI] [PubMed] [Google Scholar]
- Brown N.; McKay B.; Gilardoni F.; Gasteiger J. A graph-based genetic algorithm and its application to the multiobjective evolution of median molecules. J. Chem. Inf. Comput. Sci. 2004, 44, 1079–1087. 10.1021/ci034290p. [DOI] [PubMed] [Google Scholar]
- Fechner U.; Schneider G. Flux (2): comparison of molecular mutation and crossover operators for ligand-based de novo design. J. Chem. Inf. Model. 2007, 47, 656–667. 10.1021/ci6005307. [DOI] [PubMed] [Google Scholar]
- Lewell X. Q.; Judd D. B.; Watson S. P.; Hann M. M. RECAP - retrosynthetic combinatorial analysis procedure: a powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J. Chem. Inf. Comput. Sci. 1998, 38, 511–522. 10.1021/ci970429i. [DOI] [PubMed] [Google Scholar]
- Degen J.; Wegscheid-Gerlach C.; Zaliani A.; Rarey M. On the art of compiling and using ’drug-like’ chemical fragment spaces. ChemMedChem 2008, 3, 1503–1507. 10.1002/cmdc.200800178. [DOI] [PubMed] [Google Scholar]
- Schuffenhauer A.; Ertl P.; Roggo S.; Wetzel S.; Koch M. A.; Waldmann H. The scaffold tree - visualization of the scaffold universe by hierarchical scaffold classification. J. Chem. Inf. Model. 2007, 47, 47–58. 10.1021/ci600338x. [DOI] [PubMed] [Google Scholar]
- Thompson D. C.; Denny R. A.; Nilakantan R.; Humblet C.; Joseph-McCarthy D.; Feyfant E. CONFIRM: connecting fragments found in receptor molecules. J. Comput.-Aided Mol. Des. 2008, 22, 761–772. 10.1007/s10822-008-9221-8. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; Amaro R. E.; McCammon J. A. AutoGrow: a novel algorithm for protein inhibitor design. Chem. Biol. Drug Des. 2009, 73, 168–178. 10.1111/j.1747-0285.2008.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragmenter (ChemAxon). http://www.chemaxon.com/ (accessed November 27, 2015).
- Ghersi D.; Singh M. molBLOCKS: decomposing small molecule sets and uncovering enriched fragments. Bioinformatics 2014, 30, 2081–2083. 10.1093/bioinformatics/btu173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RDKit: Open-source cheminformatics. http://www.rdkit.org/ (accessed November 27, 2015).
- Kawabata T. Build-up algorithm for atomic correspondence between chemical structures. J. Chem. Inf. Model. 2011, 51, 1775–1787. 10.1021/ci2001023. [DOI] [PubMed] [Google Scholar]
- Daylight Chemical Information Systems Inc. http://www.daylight.com/ (accessed November 27, 2015).
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Carchia M.; Irwin J. J.; Shoichet B. K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amdahl G. M. In Validity of the single processor approach to achieving large scale computing capabilities. Proceedings of the AFIPS Spring Joint Computing Conference; ACM: Atlantic City, NJ, 1967; pp 483–485. [Google Scholar]
- Overington J. P.; Al-Lazikani B.; Hopkins A. L. How many drug targets are there?. Nat. Rev. Drug Discovery 2006, 5, 993–996. 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Baraldi P. G.; Cacciari B.; Romagnoli R.; Spalluto G.; Moro S.; Klotz K. N.; Leung E.; Varani K.; Gessi S.; Merighi S.; Borea P. A. Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as highly potent and selective human A(3) adenosine receptor antagonists: influence of the chain at the N(8) pyrazole nitrogen. J. Med. Chem. 2000, 43, 4768–4780. 10.1021/jm001047y. [DOI] [PubMed] [Google Scholar]
- Kim Y. C.; de Zwart M.; Chang L.; Moro S.; von Frijtag Drabbe Kunzel J. K.; Melman N.; IJzerman A. P.; Jacobson K. A. Derivatives of the triazoloquinazoline adenosine antagonist (CGS 15943) having high potency at the human A2B and A3 receptor subtypes. J. Med. Chem. 1998, 41, 2835–2845. 10.1021/jm980094b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.