

Summary
The interplay between transcription factors and chromatin dictates gene regulatory network activity. Germ layer specification is tightly coupled with zygotic gene activation and, in most metazoans, is dependent upon maternal factors. We explore the dynamic genome-wide interactions of Foxh1, a maternal transcription factor that mediates Nodal/TGFβ signaling, with cis-regulatory modules (CRMs) during mesendodermal specification. Foxh1 marks CRMs during cleavage stages and recruits the co-repressor Tle/Groucho in the early blastula. We highlight a population of CRMs that are continuously occupied by Foxh1, and show they are marked by H3K4me1, Ep300, and Fox/Sox/Smad motifs, suggesting interplay between these factors in gene regulation. We also propose a molecular ‘hand-off’ between maternal Foxh1 and zygotic Foxa at these CRMs to maintain enhancer activation. Our findings suggest that Foxh1 functions at the top of a hierarchy of interactions by marking developmental genes for activation beginning with the onset of zygotic gene expression.
Keywords: Foxh1, Tle/Groucho, Smad2/3, Foxa, Sox7, RNA Polymerase II, TGFβ, Nodal, endoderm, mesoderm, ChIP-seq, epigenetics, Ep300, histone modifications, germ layer specification, pioneer factor, zygotic gene activation, Xenopus tropicalis
eTOC Blurb
How maternal transcription factors control the onset of gene regulatory networks in the early embryo is poorly understood. Charney et al. demonstrate dynamic binding of maternal Foxh1 to the embryonic genome well before zygotic gene activation. They elucidate the temporal recruitment of co-factors to cis-regulatory modules controlling mesendoderm specification.
Introduction
How the genome of the early embryo — through a progressive series of modifications controlling zygotic gene expression in ‘time and space’ — ensures proper cellular differentiation programs is a key biological question. Crucial to this process are transcription factors (TF), which regulate gene expression through interactions with cis-regulatory modules (CRM) — combinations of motifs present in the regulatory region of genes. TFs function not linearly, but in a combinatorial fashion to control the network of gene regulation necessary for proper development. TF activities (‘inputs’) are integrated at CRMs, which orchestrate the temporal and spatial output of gene expression. Dissecting this complex regulatory logic in vivo is critical to a deeper understanding of the causal relationship between inputs and cellular phenotypes, and our ability to alter differentiation programs controlling cellular states.
Germ layer specification is one of the earliest developmental events in metazoan organisms, and the study of these events provides a simple in vivo system for the elucidation of gene regulatory network logic. In a majority of animals, including both vertebrates and invertebrates, germ layer specification is dictated by maternally-localized cytoplasmic materials (determinants) that partition into specific blastomeres during cleavage stages (Schier and Talbot, 2005; Heasman, 2006), and then function to specify differentiation of cell types. However, understanding how maternal TFs and signaling pathway effectors are integrated at CRMs in vivo remains challenging in many systems because cell type diversity in embryos introduce difficulty in both experimental design and interpretation of the results. Xenopus is an excellent system to study such integration because we are able to investigate gene regulatory interactions that occur early in development when cellular complexity is minimal. Here, we attempt to tease apart the interplay between the dynamic DNA binding properties of the maternal TF Foxh1 (previously known as Fast1, forkhead activin signal transducer 1), a known mediator of the Nodal signaling pathway, and the CRMs controlling the activation of the mesendoderm gene regulatory program.
In Xenopus, maternal TFs such as the T-box protein Vegt play a pivotal role in zygotic gene activation (ZGA), and the subsequent induction of the mesoderm and endoderm gene regulatory network (Zhang et al., 1998; Kofron et al., 1999). Prior to ZGA, cleavage stage chromatin is relatively free of well-characterized histone modifications such as permissive H3K4me3 and repressive H3K27me3 marks (Akkers et al, 2009; Vastenhouw et al., 2010; reviewed in Bogdanovic et al., 2011). Vegt controls the zygotic transcription of the Xenopus Nodal ligands (Kofron et al., 1999; Xanthos et al., 2001), initiating Nodal signaling and mesendoderm development (reviewed in Whitman, 2001). The Nodal ligands activate the phosphorylation of R-Smad2/3, which complexes with co-Smad4 to activate targets genes (reviewed in Hill, 2001). Foxh1 has been implicated as a critical Smad2/3 co-factor (Chen et al., 1996; Chen et al., 1997), and has an essential function during mesoderm and endoderm development based on studies in Xenopus (Howell et al., 2002; Kofron et al., 2004; Chiu et al., 2014), zebrafish (Pogoda et al., 2000; Kunmar et al., 2003; Sirotkin et al., 2000; Slagle et al., 2011), mouse (Hoodless et al., 2001; Yamamoto et al., 2001) and differentiated human embryonic stem cells (hES cells) (Yoon et al., 2011).
We recently investigated the Nodal/Foxh1 gene network in early gastrula Xenopus tropicalis embryos through a combination of RNA- and ChlP-seq analyses (Chiu et al., 2014). In the current study, we identified Foxh1 binding to enhancers of known mesendodermal target genes as early as the 32-cell cleavage stage, well before ZGA. We then mapped the global occupancy of Foxh1 over a time course from early zygotic transcription through the onset of gastrulation, and compared Foxh1 to the DNA binding properties of other TFs, RNA polymerase II, and histone markings. We find that early blastula Foxh1 binding strongly correlates with the co-repressor Tle (Groucho family), marking the genome before RNA polymerase II recruitment. Finally, we identify a set of CRMs consisting of Fox/Smad/Sox motifs, which are marked by Ep300 and histone modifications. These CRMs are primed by Foxh1, and subsequently recruit TFs such as Smad2/3 and the pioneer factor Foxa to promote mesendodermal programming. This work sheds light on the central role of maternal Foxh1 in laying the groundwork to integrate the TFs controlling the mesendodermal gene regulatory program.
Results
Foxh1 protein is abundant prior to Nodal signaling
Foxh1 functions as a transcriptional mediator for Nodal signaling by interacting with the activated Smad2/4 complex (Chen et al., 1996; Chen et al., 1997; Yoon et al., 2011; Chiu et al., 2014). In Xenopus, Foxh1 mRNA is present in the unfertilized egg (Chen et al., 1996; Howell et al., 2002), and our work in Xenopus tropicalis reveals that Foxh1 mRNA is present equally in animal and vegetal halves of the 8-cell embryo (K.P. and K.W.Y.C, data not shown) and ubiquitously in the early gastrula (Chiu et al., 2014; Blitz et al., in press). Recent high-resolution transcriptome analysis performed using polyA-selected (mRNA-seq) and ribosome-depleted (rdRNA-seq) samples (Owens et al., 2016) reveals that the majority of maternally expressed foxh1 transcripts are non-polyadenylated and undergo rapid polyadenylation shortly after fertilization (Figure 1A, Figure S1A). Foxh1 mRNA levels fall rapidly during late blastula and gastrula stages, and then increase slightly during neurulation.
Figure 1. Foxh1 is maternally expressed prior to Nodal signaling.
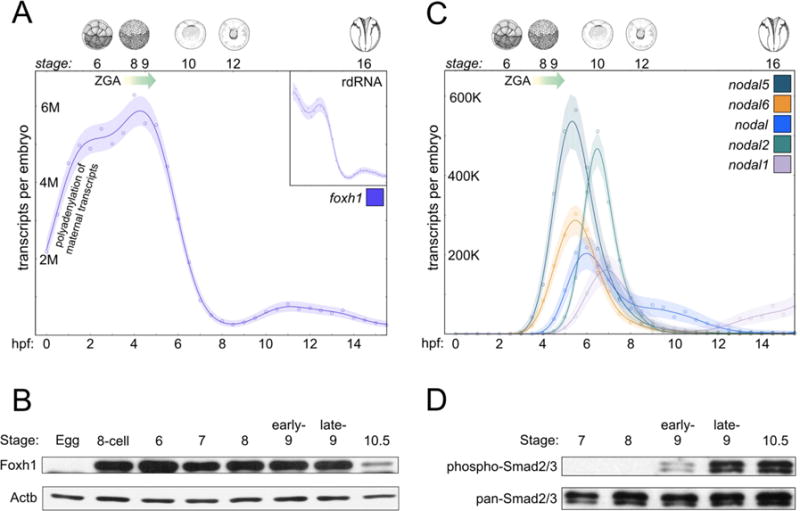
A) mRNA-seq reveals maternally expressed Foxh1 mRNA, which decreases over the course of early Xenopus tropicalis development (Owens et al., 2016). Inset depicts foxh1 in ribo-depleted RNA (rdRNA) samples over the same time period. The major onset of ZGA (Owens et al., 2016) begins at approximately 3.5 hpf, stage 7, and is shown by a graded arrow. B) IP-western blot analysis shows Foxh1 protein over a time course of early development (Foxh1 runs at ~55kDa). Actb ((β-actin) staining used as input control. C) The five Xenopus Nodal-related ligands are zygotically activated, as revealed by mRNA-seq analysis (Owens et al., 2016). D) IP-western blot analysis of phospho-Smad2/3 and pan-Smad2/3 reveal that Nodal signaling is detected beginning in the stage 9 blastula. Digital images of embryos (Nieuwkoop and Faber, 1994) are from Xenbase.
We investigated the temporal expression pattern of endogenous Foxh1 protein (Figure 1B). Foxh1 protein is present at low levels in the unfertilized egg (observed in longer exposure, data not shown), but becomes abundant during cleavage (32-cells, stage 6) and blastula (stages 7, 8, and 9) stages, presumably in response to the increase in polayadenylated Foxh1 transcripts after fertilization. Subsequently, Foxh1 protein expression is drastically reduced in the early gastrula (stage 10.5), which correlates with the rapid drop in transcripts by this stage (Figure 1A). These data suggest that Foxh1 protein levels are dynamically regulated — with a probable half-life of less than a few hours — and its expression peaks between cleavage and late blastula stages, coinciding with the initiation of germ layer specification.
Since Foxh1 is a known transcriptional mediator of Nodal signaling, we investigated the onset of Nodal signaling compared to Foxh1 protein. Transcriptome analysis shows that there are no maternal Nodal transcripts (Figure 1C and Figure S1B). Nodal5 and nodal6 transcripts are the earliest expressed nodal genes detected in Xenopus tropicalis at ~3–3.5 hours post fertilization (hpf), followed by nodal, nodal2, and nodal1, which are transcribed beginning between 4–5 hpf. To determine the onset of Nodal signaling, we investigated the phosphorylation of Smad2/3 over early development (Figure 1D). While Smad2/3 is expressed maternally (Figure S1C and D), the phosphorylated form of Smad2/3 (pSmad2/3) was first detected at the beginning of stage 9 (~1–2,000 cells) in Xenopus tropicalis. This result is consistent with previous reports revealing the detection of pSmad2/3 beginning at stage 9 in Xenopus laevis (Faure et al., 2000; Lee at al., 2001). Using immunoprecipitation, pSmad2/3 was detected in stage 8 Xenopus laevis embryos (Skirkanich et al., 2011). A small difference in embryo staging may have contributed to the difference between our results and those of Skirkanich et al. (2011). Finally, these results demonstrate that while there are other TGFβ ligands that signal through pSmad2/3, such as maternal Gdf1 (also known as Vg1) and zygotic Gdf3 (also known as Derrière) (Figure S1E and F), endogenous Smad2/3 is not activated during cleavage stages and begins in the late blastula (see also Faure et al., 2000). Since Foxh1 protein levels rapidly decline between late blastula stage 9 and early gastrula stage 10.5, these data demonstrate that the overlap of Nodal signaling with abundant Foxh1 expression is restricted to the brief period of approximately stage 9 to 10.5.
Dynamic Foxh1 interactions with the genome during early Xenopus tropicalis development
Since Foxh1 expression is highly dynamic, we investigated Foxh1 binding over a time course of early germ layer development, which coincides with ZGA. Chromatin immunoprecipitation coupled with deep sequencing (ChIP-seq) was performed in the early blastula (stage 8), late blastula (stage 9), and in the early gastrula (stage 10.5), using two biologically independent samples (Figure 2A). We performed pairwise Pearson correlation analyses and found that variance in called peaks between stages is higher than inter-stage variance (Figure S2A), permitting quantitative comparisons between stages. We then used ‘ireproducibility discovery rate’ (IDR) analysis (Li et al., 2011) to identify a set of high confidence peaks at each time point (Figure S2B and see Supplemental Experimental Procedures). A total of 40,884 bound regions were identified over the entire time course (Table S1), some of which are present at a specific stage, and others across multiple stages. Overall, Foxh1’s genomic occupancy is higher in the blastula (stages 8 and 9) than in the early gastrula (stage 10.5), at which point most Foxh1 occupancy is drastically reduced (clusters II and IV) or absent (clusters III, V, and VI) (Figure 2B). The observed reduction of Foxh1 binding by stage 10.5 is in line with the large drop in relative amount of protein by this stage (Figure 1B). While the patterns of Foxh1 enrichment between stage 8 and 9 were similar, we also observed clusters with varying levels of enrichment (cluster V ‘high signal at stage 8’ and cluster IV ‘high signal at stage 9’) between the stages. Thus, binding of Foxh1 to the genome (stage 8; see also below) precedes the activation of Nodal signaling (stage 9).
Figure 2. Foxh1 binding is dynamic across early Xenopus tropicalis development.
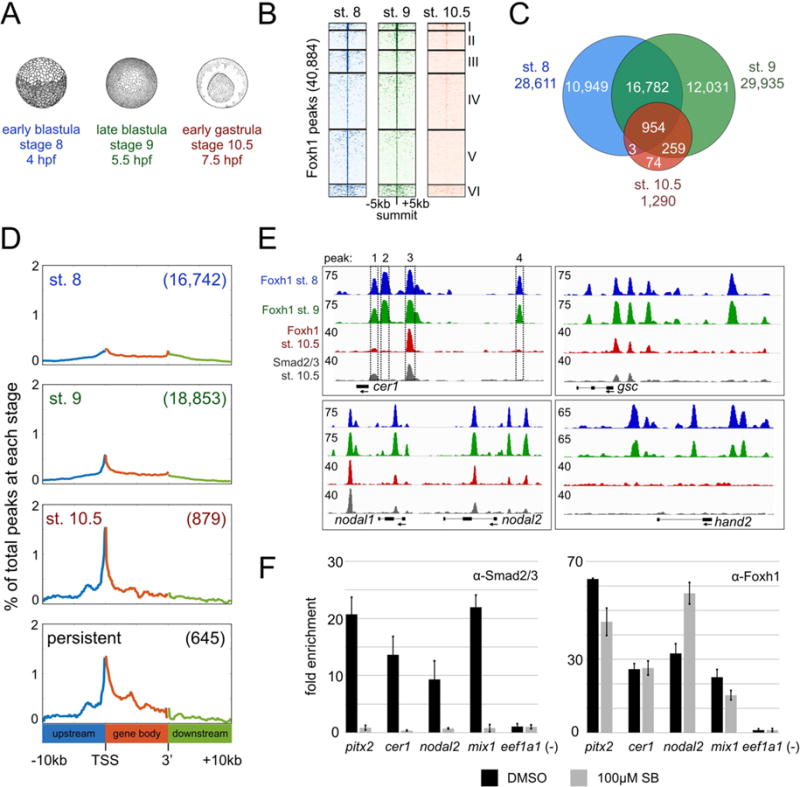
A) Overview of Foxh1 ChIP-seq time course at early blastula (~500 cells), late blastula (~2,000 cells), and early gastrula (~10,000 cells). B) Clustered heatmaps depicting Foxh1 ChIP-seq signal at each stage centered on the summit of all Foxh1 IDR peaks. C) Venn diagram comparing Foxh1 IDR peaks between each stage. There are 77 and 91 fewer total Foxh1 peaks found at stages 8 and 9, respectively, than expected based on the sum total of the peaks in each category of the Venn diagram, due to a small number of ‘tandem peaks’ in one stage overlapping a single peak in a second stage. D) Distribution of Foxh1 peaks within the intervals of 10kb upstream of gene 5’ ends, gene bodies and 10kb downstream of gene 3’ ends. All gene bodies have been scaled to the same length. Numbers in parentheses indicate total number of peaks falling within the interval shown for each analysis. Y-axis values are the percentages of the peaks at coordinates along genomic intervals. E) Genome browser visualization of Foxh1 and Smad2/3 peaks at the target genes cer1 (scaffold_1:102,363,840-102,373,840), gsc (scaffold_8:62,417,448-62,431,448), nodal1 and nodal2 (scaffold_3:5,671,591-5,690,591), and hand2 (scaffold_1:175,354,835-175,366,835). Boxes 1–4 represent 4 Foxh1 peaks identified by IDR peak calls on the cer1 gene. Regions containing peaks in boxes 1 and 3 are also bound by Smad2/3. Y-axis numerical values in each track indicate track height scaling in read depth. F) Anti-Smad2/3 (left) or anti-Foxh1 (right) ChIP-qPCR in DMSO or SB431542 treated embryos cultured until stage 10.5. One representative experiment is shown with mean fold enrichment over the eef1a1 background region +/− SD. While treatment does not significantly affect Foxh1 binding, we note minor differences in Foxh1 enrichment at the pitx2 and nodal2 enhancers, possibly due to tissue-specific impacts of Smad2/3.
IDR analysis identified 28,611 Foxh1 peaks at stage 8 and 29,935 at stage 9, indicating a broad role for Foxh1 (Figure 2C and Table S1). We find that the majority of blastula peaks are not maintained at the beginning of gastrulation (stage 10.5), and only 1,290 regions are bound at this stage (Figure 2C). The vast majority of these (954 peaks) were persistent across all three stages. We refer to these as ‘persistent peaks’ (see also cluster I in Figure 2B). We also identified the genes associated with Foxh1 peaks by assigning peaks to their nearest genes within 10kb of the gene body (Figure S2C and Table S2). This revealed that the persistent peaks associate with 611 genes, many of which are expressed in the mesendoderm (Figure 6E below). Taken together, this analysis reveals that Foxh1 binding is prevalent during blastula stages, and for the first time reveals the highly dynamic nature of the in vivo genome binding patterns of this key TF.
Figure 6. Persistent peaks are enriched for Fox/Smad/Sox cis-regulatory modules.
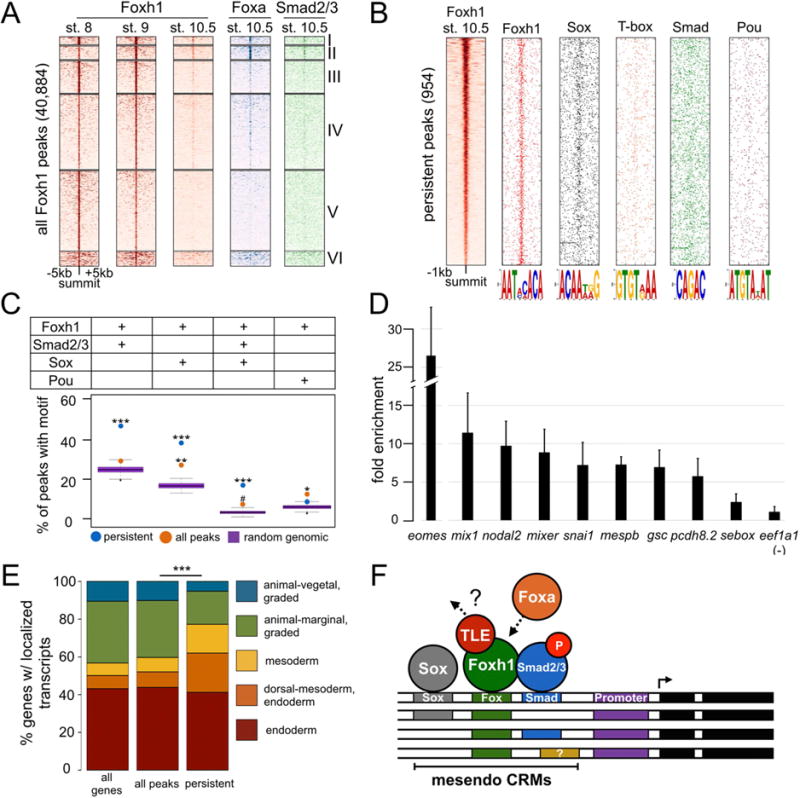
A) Heatmaps of ChIP-seq signal centered on the summit of all Foxh1 peaks, and K-means clustered. B) Positional distribution around the peak summit of select motifs discovered under the Foxh1 persistent peaks. C) Persistent peaks are enriched with the co-occupancy of Foxh1/Smad/Sox motifs. P-values were calculated between Foxh1 peaks and genomic background using the Grubb’s test for outliers (*** p-value < 2.2e-16; ** p-value = 2.08e-09; * p-value = 1.2e-07; # p-value = 0.003). D) Sox7 binding to Foxh1-bound CRMs as detected by ChIP-qPCR. CRMs in the genes shown contain ChIP signal with the exception of sebox, which is not significantly enriched above background. Shown is the mean fold enrichment over the eef1a1 background region +/− SD. E) Genes associated with Foxh1 persistent peaks display a spatial localization profile significantly different from all Foxh1-bound genes. The spatial profile of all Foxh1-bound genes is not significantly different from all genes in the genome. P-value was calculated between all Foxh1 and persistent genes using a chi-squared test (*** p-value < 2.2e-16). F) Model of Foxh1 marking of Fox/Smad/Sox mesendoderm CRMs.
Since our Foxh1 ChIP-seq time course spanned early ZGA, we wanted to investigate whether there were stage-specific differences in the genomic distribution of Foxh1 peaks (Figure S2D). At all stages, the majority of Foxh1 peaks are found in the intergenic region (more than 10kb away from the nearest gene). Interestingly, at the beginning of gastrulation, a higher proportion of Foxh1 peaks were present within 1kb upstream of transcription start sites (TSS) (promoter proximal) (9% at stage 10.5, compared to 3.3% and 4.4% at stages 8 and 9, respectively). In Foxh1 persistent peaks, 8% of the peaks are found proximal to promoters. To gain a more complete picture of this trend, we analyzed the distribution of Foxh1 peaks within 10kb of gene bodies (Figure 2D). At stage 8, the positions of Foxh1 peaks are fairly evenly distributed across this window. We observed a higher proportion of Foxh1 peaks located at the promoters and TSSs of genes as development proceeds, most significantly at stage 10.5. The change in distribution of peaks by stage 10.5 reflects selective retention of Foxh1 at regions corresponding to the persistent peaks. Since these are in closer proximity to gene bodies, this may indicate that persistent peaks are functionally relevant (see more below).
Foxh1 binds independently of Nodal signals
Global analysis indicated that Foxh1 occupancy of putative enhancers is dynamic over the time course. To illustrate this behavior, we highlight Foxh1’s associations with the organizer gene gsc, the nodal ligands nodal1 and nodal2, and the nodal inhibitor cer1, which are regulated via Foxh1-Smad2/3 interactions (Germain et al., 2000; Ring et al., 2002; Chiu et al., 2014) (Figure 2E). We previously identified these genes as Foxh1 direct targets, based on the dual criteria of Foxh1 binding and gene expression changes in response to Foxh1 loss of function (Chiu et al., 2014). We identify four Foxh1 bound regions upstream of cer1 (peaks 1–4, Figure 2E). Two of these regions (peaks 1 and 3) are also bound by Smad2/3 at stage 10.5, indicating that these are Nodal-responsive enhancers. On the other hand, peaks 2 and 4 are only bound by Foxh1 in the blastula, and are not occupied by Smad2/3. Similar dynamics are also observed in Foxh1-Smad2/3 peaks associated with gsc, nodal1, nodal2, and others. Lastly, we observed blastula-specific binding of Foxh1 near the TF hand2 gene (Figure 2E), which is transiently expressed pre-MBT through gastrulation and is negatively regulated by Foxh1 in a Nodal-independent manner (Chiu et al., 2014). These findings indicate that not all Foxh1 binding sites overlap with Smad2/3 binding.
As ChIP-seq analysis suggested that Foxh1 is capable of occupying target regions in a Nodal-independent manner, we examined Foxh1 binding in the presence of a Nodal inhibitor (Inman et al., 2002; Ho et al., 2006; Chiu et al., 2014). Incubation of embryos in medium containing SB431542 until stage 10.5 resulted in a loss of Smad2/3 enrichment at regulatory regions of the Nodal signaling targets pitx2, cer1, nodal2 and mix1 (Chen et al., 1996; Shiratori et al., 2001; Chiu et al., 2014) (Figure 2F, left). We then investigated Foxh1 binding in the absence of Nodal signaling. Inhibition of Nodal signaling did not significantly affect the binding of endogenous Foxh1 to these same regions (Figure 2F, right), suggesting that Foxh1’s interactions with its binding sites are largely independent of its physical interactions with pSmad2/3. In addition, Foxh1 protein is highly expressed shortly after fertilization (Figure 1B), well in advance of Nodal signaling (Figure 1C,D). Overall, these results signify that Foxh1 is able to bind to its regulatory regions in a Nodal-independent fashion.
To further explore the co-occupancy of Foxh1 and Smad2/3, we correlated the set of 40,884 total Foxh1 peaks with our Smad2/3 ChIP-seq performed at stage 10.5 (Chiu et al., 2014). Analysis of Smad2/3 ChIP-seq at early stages revealed very few Smad2/3 peaks until the beginning of gastrulation (Gupta et al., 2014), consistent with rising levels of pSmad2/3 until that stage (Figure 1D). After implementing IDR analysis on two biological replicates, we identified 480 high-confidence Smad2/3 peaks at stage 10.5 (Table S3). Of these, 370 overlapped with Foxh1 peaks (Figure S2E). These 370 co-bound regions associate with 225 genes. Interestingly, 60 additional genes are associated with Smad2/3 peaks, without Foxh1. These findings confirm and expand upon previous reports (Germain et al., 2000; Ring et al., 2002; Cordenonsi et al., 2003; Kunmar et al., 2003; Ku et al., 2005; Slagle et al., 2011; Teo et al., 2011; Yoon et al., 2011; Chiu et al., 2014) that Foxh1 is a major, but not the sole, transcription factor mediating Nodal signaling.
Taken together, our Foxh1 ChIP-seq time course demonstrates for the first time the binding dynamics of a key maternal transcription factor during the onset of ZGA and subsequent germ layer development. The majority (nearly 97%) of Foxh1’s total genomic occupancy during this window of time is blastula-specific. The remaining 3%, representing 954 genomic sites, are persistently bound across stages 8 through 10.5.
Interaction between Foxh1 and Tie in early blastula embryos
Foxh1 contains an EH1 domain (Figure 3A) (Yaklichkin et al., 2007a), which mediates interactions with the Groucho/Tle (transducin-like enhancer of split) family of corepressors (reviewed in Chen and Courey, 2000). EH1 domains of several forkhead family transcription factors have been shown to physically interact with Tle corepressors, including Foxa1/2 and Foxd3 (Wang et al., 2000; Sekiya and Zaret, 2007; Yaklichkin et al., 2007b). First identified in Drosophila, Groucho/Tle is known to participate in developmental processes through the silencing of gene expression. We therefore examined the relationship between endogenous Foxh1 and Tle binding by performing Tle ChIP-seq at stage 8. A heatmap centered on the stage 8 Foxh1 peaks reveals strong Tle enrichment that correlates with Foxh1 (Figure 3B). Sixty percent of stage 8 Foxh1 peaks (16,856 peaks) overlap with Tle peaks (37,919 peaks) (Figure 3C and Table S4). Of the 954 Foxh1 persistent peaks, 686 (72%) overlap with Tle. These data imply a functional interaction between Foxh1 and Tle in the early blastula, perhaps in the repression of target genes.
Figure 3. Foxh1 is required for the early blastula recruitment of the co-repressor Tle.
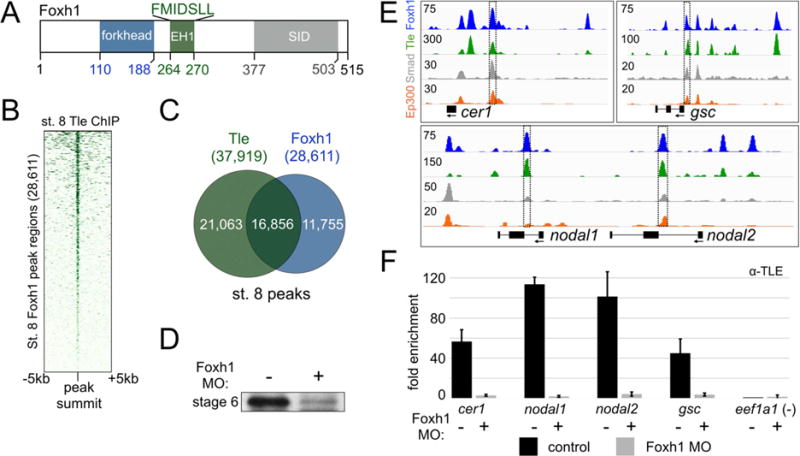
A) A schematic diagram showing domains present in the Foxh1 protein. B) Heatmap of stage 8 Tle ChIP-seq signal centered on the stage 8 Foxh1 peaks. C) Peak calling identified 16,856 overlapping Foxh1 and Tle peaks. D) IP-western blotting reveals the significant reduction of Foxh1 protein at stage 6 (32-cells) after injection of a translation blocking morpholino oligonucleotide (MO) targeting foxh1 mRNA. E) Genome browser visualization of selected Tle/Foxh1 overlapping peaks near target genes cer1 (scaffold_1:102,363,840-102,373,840), gsc (scaffold_8:62,417,448-62,431,448), and nodal1 and nodal2 (scaffold_3:5,671,591-5,690,591). The boxed enhancers were analyzed using ChIP-qPCR in panel (F) below. Ep300 tracks show that many of these Foxh1/Tle-bound regions are active CRMs (see also Figure S5B). F) Stage 8 Tle ChIP-qPCR was performed in control and foxh1 MO-injected embryos. One representative experiment is shown with mean fold enrichment over the eef1a1 background region +/− SD.
Tle lacks a DNA binding domain, and therefore its recruitment to enhancers is due to interactions with other DNA binding transcription factors via the WD domain (Jennings et al., 2006). Since ~60% of stage 8 Foxh1 peaks overlap Tle peaks, we investigated whether Foxh1 binding was required for Tle recruitment. We utilized a previously validated translation blocking morpholino oligonucleotide (MO) targeting foxh1 mRNA to knock down Foxh1 protein synthesis (Chiu et al., 2014). The effectiveness of knocking down Foxh1 in stage 6 (32-cells) embryos after MO injection at the one-cell stage was confirmed (Figure 3D). We next depleted the embryo of Foxh1 protein by foxh1 MO injection, and performed anti-Tle ChIP coupled with quantitative PCR (ChIP-qPCR) in control and MO-injected stage 8 embryos. The bona-fide Foxh1-Smad2/3 binding regions identified in cer1, nodal1, nodal2, and gsc were interrogated (Figure 3E and F). Compared to Tle enrichment in control embryos, Tle binding was abolished in Foxh1 morphants. These data indicate that Foxh1 is required for the early blastula recruitment of Tle.
A subset of Foxh1 bound regions are subsequently marked as active enhancers
Since the binding of Foxh1 and Tle occurs early in development, we correlated Foxh1 occupied regions with the recruitment of the co-activator Ep300 (Yasuoka et al., 2014) and the histone modification H3K4me1 (Gupta et al., 2014). The combination of these marks (Hontelez et al., 2015) allowed us to identify Foxh1-bound enhancers. We correlated all Foxh1 peaks to stage 10.5 Ep300 ChIP-seq signal (Figure 4A), and identified a subset of Foxh1-bound regions that are marked with Ep300 at the beginning of gastrulation. We next investigated whether those Ep300-bound regions were enriched with the histone modification H3K4me1. While little to no H3K4me1 enrichment was observed at stage 8, we observed enrichment at stage 9 and 10.5 at peaks also marked with Ep300 (Figure 4A). This indicates that a subset of Foxh1-bound regions are likely active enhancers during this time period. We also identify a set of Foxh1-bound regions not marked by Ep300 or H3K4me1.
Figure 4. Foxh1 peaks are marked by Ep300 and H3K4me1.
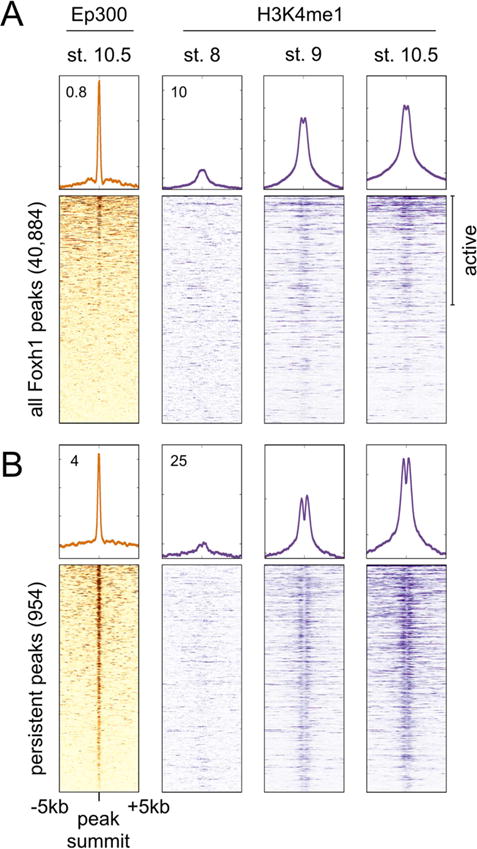
Heatmaps centered on the summits of all Foxh1 peaks (A) and Foxh1 persistent peaks (B) correlate stage 10.5 Ep300 enrichment (Yasuoka et al., 2014) to Foxh1 binding. Peaks marked by Ep300 also show enrichment of the active histone modification H3K4me1 (Gupta et al., 2014), beginning in the late blastula. Heatmaps have been ordered based on the strength of the Ep300 signal. Values 0.8, 4, 12, and 25 indicate read density at the top of windows shown, in units of RPKM.
The Foxh1 ChIP-seq time course identified 954 Foxh1 persistent peaks that are found at all three stages (Figure 2C). We posited that these persistent peaks represent critical Foxh1 occupied enhancers. The vast majority of persistent peaks correlated with Ep300 at stage 10.5, and were also progressively enriched with strong bimodal H3K4me1 enrichment centered on the Foxh1 peak summits (Figure 4B). This supports the notion that the regions containing these persistent Foxh1 peaks are qualitatively different from the total population of Foxh1 peaks, which are predominately found only in the blastula. We also note that no H3K4me1 enrichment is observed surrounding Foxh1 peaks at stage 8, and that Ep300 binding has not been detected prior to stage 9 (Hontelez et al., 2015). Interestingly, the majority of stage 9 Ep300 enrichment is dependent upon zygotic transcription (Hontelez et al., 2015). These findings indicate that Foxh1 binding precedes enrichment of Ep300 and H3K4me1.
Foxh1 marks enhancers in cleavage stage embryos before RNA pol II recruitment
Our finding that Foxh1 binding precedes the enrichment of Ep300 and H3K4me1 suggests that Foxh1 occupies enhancers before they are functionally active. We wondered whether Foxh1 enhancer occupancy precedes the recruitment of RNA polymerase II (RNA pol II). We therefore addressed the relative binding order between Foxh1 and RNA pol II on the Foxh1 direct target genes gsc, cer1, and nodal1. Transcriptome analysis revealed that gsc and nodal1 are zygotically activated between ~4.5 and 5 hpf, while cer1 is activated at ~5.5 hpf (Figure 5A, left). To investigate RNA pol II occupancy at the promoters of these genes, we performed ChIP-qPCR (Figure S3A) and ChIP-seq over a time course of early development (Figure 5A, right), and examined Foxh1 and RNA pol II binding. We observed no RNA pol II enrichment in the stage 7 blastula (~256 cells) (Figure 5A, right). Beginning at stage 8, we observed an accumulation of RNA pol II signal that progressively increases to the end of our timecourse at stage 10.5. Foxh1 binding occurs at nearby enhancers during this period and its interaction with these CRMs decreases between stages 9 and 10.5. This suggests that Foxh1 is critical for the initial activation steps of these direct target genes and that additional TFs are recruited to these enhancers to maintain gene expression after the degradation of maternal Foxh1.
Figure 5. Foxh1 marks enhancers prior to the recruitment of RNA Polymerase II.
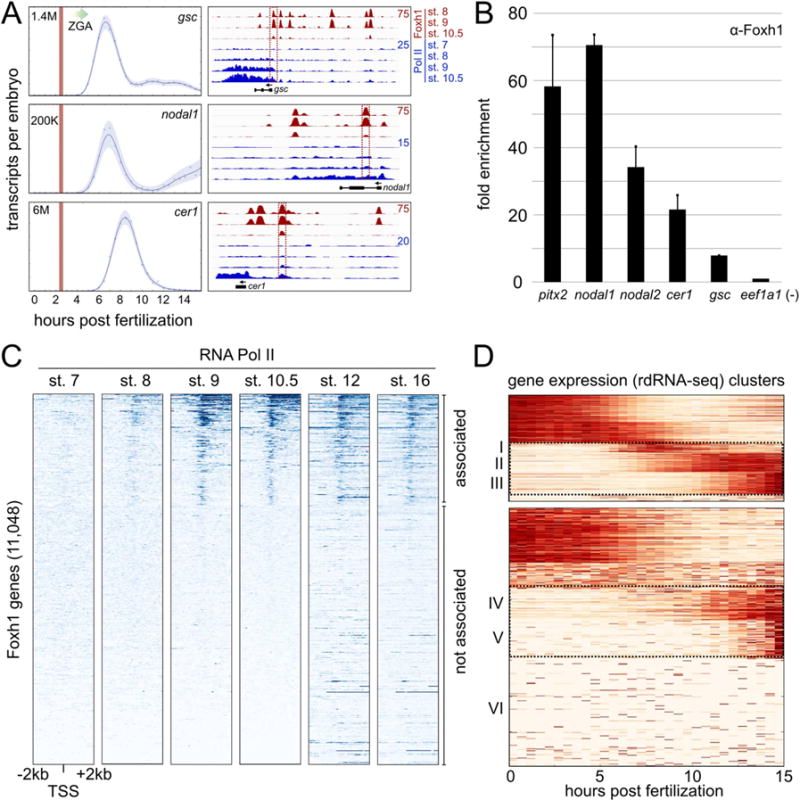
A) Temporal expression patterns of three Foxh1 target genes (left) (Owens et al., 2016). Vertical red lines indicate stage 6 (32-cell) and the major onset of ZGA is indicated by a graded arrow. (Right) Foxh1 and RNA Polymerase II ChIP-seq at Foxh1 target genes gsc (scaffold_8:62,410,054-62,437,258), nodal1 (scaffold_3:5,668,910-5,678,946), and cer1 (scaffold_1:102,363,664-102,374,648). Boxed enhancers were interrogated at stage 6 for Foxh1 binding (panel B), and via qPCR in Figure S3A. B) Foxh1 ChIP-qPCR on stage 6 embryos. Shown is the mean fold enrichment over the eef1a1 background region for two biological replicates +/− SEM C) Heatmaps displaying RNA pol II ChIP-seq signal centered on the transcription start site (TSS) of all Foxh1-associated genes. Gene bodies are oriented to the right. Heatmap gene order is based on K-means clustering at stage 10.5, where clusters 1–3 were collectively called RNA pol II ‘associated’ (cluster boundaries not indicated in figure), and cluster 4 was called ‘not associated.’ Stage 12 and 16 RNA pol II ChIP-seq is from Hontelez et al. (2015). D) Temporal expression patterns of Foxh1 genes ‘associated’ and ‘not associated’ with RNA pol II. The rdRNA-seq (Owens et al., 2016) expression level for each gene was normalized between [0, 1] and clustered using K-means (see also Figure S4).
Given that Foxh1 protein is abundantly expressed in cleavage stage embryos (Figure 1B), and that Foxh1 binds in the absence of Nodal signaling (Figure 2F), we tested the model that Foxh1 is pre-bound to enhancers prior to the onset of ZGA. Foxh1 ChIP-qPCR analysis in 32-cell embryos (stage 6), which occurs at ~2.5 hpf, revealed Foxh1 occupancy at gsc, nodal1, and cer1 enhancers (Figure 5B). The interrogated regions are indicated with red boxes on the genome browser tracks of Figure 5A. We also found Foxh1 pre-binding to pitx2 and nodal2 enhancers (Figure 5B and Figure S3B and C). Interestingly, the nodal1 and pitx2 enhancers have been shown to function during both mesendoderm development and later left-right patterning in a Nodal-dependent fashion (Osada et al., 2000; Shiratori et al., 2001; Faucourt et al., 2001). We therefore conclude that Foxh1 marks enhancers prior to RNA pol II enrichment and the transcription of the target gene.
Foxh1 marked genes display dynamic transcriptional activity
To globally examine the extent of Foxh1 marking, we investigated RNA pol II enrichment over time across the set of Foxh1-bound genes (Figure 5C). We analyzed genes with at least one Foxh1 peak within 10kb upstream of the TSS, resulting in 11,048 total Foxh1-associated genes (Table S5). No significant RNA pol II signal was observed across these genes in the stage 7 blastula. Starting from stage 8 and onwards, RNA pol II signal gradually associates with the TSS and is detected across gene bodies. Finally, we found that not all Foxh1-associated genes display RNA pol II enrichment. We identified ~4,000 Foxh1-bound genes as RNA pol II ‘associated’ and ~7,000 as ‘not associated.’ To investigate the temporal dynamics of the Pol II ‘associated’ and ‘not associated’ genes, we mined the transcriptome data recently published by Owens et al. (2016) across the first 15 hours of development (one-cell to mid-neurula stage 16). We observed that these genes are expressed both maternally and zygotically (Figure 5D and Table S5). Focusing on the zygotic clusters, the RNA pol II ‘associated’ genes are activated at the MBT (clusters I and II) and in the early gastrula (cluster III) (see also Figure S4). ‘Not associated’ genes are activated weakly in the early gastrula (cluster IV) and during neurula stages (cluster V). As expected, approximately half of the ‘not associated’ genes had no RNA expression during this time period (cluster VI) (Figure 5D). Taken together, these data indicate that Foxh1 occupies enhancers during cleavage stages, preceding the recruitment of RNA pol II, and that associated genes are dynamically expressed.
Foxh1 persistent peaks are marked by Foxa, Smad2/3 and Sox TFs
At the onset of zygotic transcription, the mesendoderm regulatory network becomes activated, and a number of zygotic TFs are critical to sustain this program. Having found that Foxh1 binds the genome in the blastula, we investigated whether zygotic TFs are subsequently recruited to these regions. Foxa pioneer TFs are critical for endoderm development across diverse organisms (reviewed in Friedman and Kaestner, 2006; de-Leon, 2011), and are bound to liver-specific enhancers well before they become active (Gualdi et al., 1996). All three Xenopus tropicalis Foxa TFs (foxa1, foxa2, and foxa4) are zygotically transcribed, with foxa4 expression beginning the earliest at ~4.5 hpf (Figure S5A). We therefore performed Foxa ChIP-seq at stage 10.5, and correlated Foxa binding with all Foxh1 peaks (Figure 6A). We find that the strongest Foxa enrichment corresponds predominately with clusters I and II, which is enriched with persistent peaks (see Foxh1 signal). Genome browser visualization of selected endoderm genes (e.g. mix1, hhex) confirms that Foxh1 and Foxa bound regions overlap (Figure S5B). After peak calling, we identified 40,925 Foxa peaks at stage 10.5 (Table S6). These peaks overlap with 878 of the 954 Foxh1 persistent peaks (92%), and are associated with 505 genes within 10kb (Table S6). Thus, Foxh1 persistent peaks significantly correlate with the subsequent binding of the pioneer factor Foxa. This is reminiscent of the binding of Foxd3 to the Alb1 regulatory region in mouse embryonic stem cells, which is replaced by Foxa1 during differentiation to endoderm (Xu et al., 2009).
Interestingly, we also find that the same clusters I and II overlap with Smad2/3 binding at stage 10.5 (Figure 6A), indicating that at least two additional TFs are recruited to Foxh1 marked sites. As the majority of persistent peaks are marked as active enhancers (Figure 4), compared to all peaks, we speculated that these differences might be due to underlying CRM logic at the Foxh1 occupied sites. We therefore performed de novo motif analysis to discover enriched motifs under the stage 8, stage 9, and persistent peaks (Figure 6B, Figure S6A and B, and Table S7). The Foxh1 consensus binding sequence was the most highly enriched motif for all groups. In addition to Foxh1, at each stage, we identified motifs corresponding to Pou, HMG/Sox, Smad2/3 and others (Table S7). Focusing on these motifs, we sought to identify a unique combination of motifs (CRM) that is preferentially associated with the Foxh1 persistent peaks compared to all Foxh1 peaks. To do this, we took a computational approach. We investigated the co-occurrence of the Foxh1 and Smad2/3, Sox, and Pou motifs between persistent and total Foxh1 peaks (Figure 6C, see also Supplemental Experimental Procedures). These co-occurrences were compared to randomly sampled genomic regions containing the Foxh1 motif. We found that the Foxh1/Smad and Foxh1/Sox motif combinations were significantly more prevalent in the persistent peaks compared to all Foxh1 peaks. Among persistent peaks containing the Foxh1 motif, nearly 50% contain Foxh1/Smad motif combinations, while ~40% contain Foxh1/Sox (Figure 6C). Interestingly, the triple occurrence of Foxh1/Smad/Sox motifs was also significantly enriched in persistent peaks, contained in ~20% of Foxh1 motif containing peaks. Unexpectedly, the Pou motif was slightly enriched among all Foxh1 peaks, while not enriched among the persistent peaks. We hypothesize that these combinatory motifs are critical components of Foxh1-bound CRMs functioning in early development.
To determine whether Sox family TFs physically interact with regions identified as Foxh1 persistent peaks, we investigated the in vivo binding of Sox7 to CRMs. Sox7 is the best candidate because it is maternally expressed, vegetally localized, and has been implicated in endodermal specification in both Xenopus and mammals (Futaki et al., 2004; Zhang et al., 2005; Séguin et al., 2008). The only other maternal Sox factor is Sox3, which specifies ectodermal cell fates in the animal pole in opposition to mesendodermal cell fates (Zhang et al., 2003; Zhang et al., 2004), and therefore was not pursued. We determined which persistent peaks contain Sox motifs and selected 9 of these to analyze further. We performed ChIP-qPCR assays using chromatin prepared from stage 8 blastulae and found 8 of these 9 are indeed bound by Sox7 (Figure 6D). These observations support the notion that many Foxh1 CRMs likely are co-regulated by Sox7 to specify mesendodermal gene expression.
Genes associated with Foxh1 persistent peaks are preferentially expressed in the dorsal mesendoderm
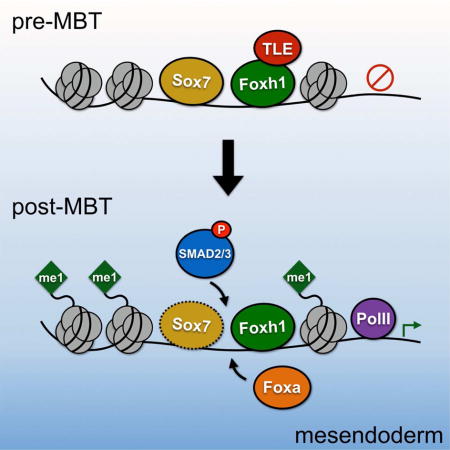
The finding that the Smad2/3 motif was enriched in persistent peaks led us to hypothesize that these peaks are associated with Nodal signaling targets. Through mRNA-seq on dissected Xenopus tropicalis embryos, Blitz et al. (In press) recently examined the tissue-specific gene expression in the early gastrula embryo. Using these data, we investigated whether genes associated with persistent peaks (‘persistent genes’) have a unique spatial profile compared to all Foxh1-associated genes. Focusing on genes with localized expression, we examined the spatial expression domain of all Xenopus tropicalis genes, genes associated with all Foxh1 peaks, and genes associated with persistent peaks. This revealed that the spatial distribution of genes associated with persistent peaks is statistically different from the other two (Figure 6E and Figure S6C). This includes an increase in genes expressed in the dorsal-mesendoderm, and a decrease in genes expressed in the ectoderm. As Nodal signaling is highly active in the dorsal mesendoderm, these findings are consistent with the notion that these persistent genes are enriched with Nodal signaling targets. Based on these findings, we propose a model (Figure 6F) whereby Foxh1 marked CRMs subsequently recruit TFs (e.g. Foxa, Sox), pSmad2/3, and the co-activator Ep300 to initiate the mesendoderm gene regulatory program.
Discussion
We examined the molecular events of early germ layer specification by focusing on the activity of maternal Foxh1. This period of development coincides with zygotic gene activation (ZGA). By examining dynamic Foxh1 binding, we find that Foxh1 sits at the top of the hierarchy of the temporal order of events occurring during early germ layer specification. With these data, together with our bioinformatics analyses, we propose a central role for Foxh1 in regulating the mesendoderm gene regulatory program via CRMs consisting of Foxh1, Smad2/3 and Sox TFs.
Foxh1 protein is abundant during cleavage and blastula stages, and ChIP-seq analyses reveal a broad occupancy at the MBT. This occupancy diminishes at the onset of gastrulation, coinciding with a rapid and large decline in Foxh1 protein level. Strikingly, Foxh1 binds enhancers in 32-cell cleavage stage embryos, which is well before the onset of major zygotic gene expression (Owens et al., 2016), and before Nodal signaling. While current technologies limit our capacity for a genome-wide investigation of Foxh1 binding at very early embryonic stages (e.g. 32-cells), it is tempting to speculate that the observed cleavage stage Foxh1 binding occurs broadly before ZGA.
Foxh1 binds enhancers before Ep300 and transcription of target genes. These early actions of Foxh1 raise the question of whether Foxh1 functions as a pioneer factor. Pioneer factors, considered a special category of TFs that are the first to occupy their target sites in chromatin, can play both ‘passive’ and ‘active’ roles in enhancing transcription (reviewed in Zaret and Carroll, 2011). Passively, a pioneer factor can be an early acting TF that recruits other partner TFs to ‘prime’ the enhancer for activation and increasing the rate in which the target gene is activated. In this report, we demonstrate that endogenous Foxh1 binds before Nodal signaling, independent of pSmad2/3, and subsequently recruits Smad2/3 and Foxa TFs to the Foxh1 marked enhancers. Foxh1 occupies enhancers as early as the 32-cell cleavage stage, when it has been reported that chromatin is nucleosome-dense (Landsberger and Wolffe, 1997; Bogdanovic et al., 2011). These findings suggest that Foxh1, at minimum, functions as a ‘passive’ pioneer factor. Additionally, pioneer factors can play ‘active’ roles by generating local regions of open chromatin (Cirillo et al., 2002). Our finding that Foxh1 binding precedes the enrichment of Ep300 and H3K4me1 suggests that Foxh1 primes enhancers before epigenetic modifications. In the future, it will be important to investigate whether Foxh1 directly functions in affecting the epigenetic landscape.
We discovered that 92% of Foxh1 persistent peaks recruit zygotically-expressed Foxa at early stages of gastrulation. This is intriguing, as the Foxa subfamily of forkhead TFs are well-characterized ‘active’ pioneer factors for hepatic development (reviewed in Zaret and Carroll, 2011), known for their special property of being able to bind their target sites alone in condensed chromatin (Clark et al., 1993; Cirillo et al. 1998), and to coordinate the subsequent binding of other TFs by promoting local chromatin decondensation (Cirillo et al., 2002). Instead, we discovered that the binding of Foxh1 precedes that of Foxa at a subset of sites, reminiscent of Foxd3’s binding preceding interaction of Foxa1 with the mouse Alb1 enhancer (Xu et al., 2009). Since in vivo footprinting analysis revealed Foxa occupancy at an Albumin (Alb) enhancer in mouse endodermal tissue before the expression of the liver-specific Alb mRNA (Gualdi et al., 1996), we examined the orthologous Xenopus alb gene in our ChIP-seq data. Our analysis did not reveal Foxh1 or Foxa binding to alb at these early stages (data not shown). This suggests different roles for Foxa during germ layer development and later liver organogenesis.
In this report, the interaction between Foxh1 and the co-repressor Tle in the early blastula (stage 8) was studied. Using Foxh1 loss of function, we uncovered a Foxh1-dependent recruitment of Tle to Foxh1 enhancers, suggesting that Foxh1 and Tle binding to these CRMs occur in the same cells. Consistent with this model, Reid et al. (2016) recently reported the physical interaction between Foxh1 and Tle4 (also know as Grg4), reminiscent of the interaction between Foxa1 and Tle3/Grg3 (Sekiya and Zaret, 2007). The authors showed that the Foxh1/Tle4 complex is bound at the nodal1 intronic enhancer and negatively regulates the expression of this gene in the absence of pSmad2/3. Thus, Foxh1 is proposed to toggle between interactions with Tle family proteins (repressive state) and pSmad2/3 (active state). Our finding that approximately 50% of early blastula Foxh1 peaks co-localize with Tle is consistent with a model of Foxh1/Tle cooperation. It is possible that this interaction is similar to that of Wnt/β-catenin signaling. In the absence of stabilized β-catenin, the Tcf/Lef TFs interact with Tle and repress transcription of target genes (Cavallo et al., 1998; Roose et al., 1998; Brantjes et al, 2001). Upon Wnt signaling, this repression is relieved by nuclear β-catenin, replacing Tle and switching the bound enhancers to a state of transcriptional activation (Daniels and Weis, 2005). In the case of Foxh1, perhaps under conditions of Nodal signaling, pSmad2/3 replaces Tle, allowing for the activation of mesendodermal genes (Figure 6F). In this scenario, Tle functions as a versatile co-repressor that can impinge upon both Wnt and TGFβ signaling and coordinate the expression of many developmentally regulated genes.
Our analysis identifies thousands of Foxh1 and Tle co-bound sites in the early blastula embryo, suggesting that this complex plays a global role in early Xenopus embryogenesis. We found that the majority of Foxh1’s genomic occupancy occurs in the blastula. While a subset of these regions become enriched with Ep300/H3K4me1 (active enhancers), we note that many do not (Figure 4A and S5B). The strong correlation between Foxh1 and Tle at stage 8 suggests that early Foxh1 binding functions in a largely repressive capacity, perhaps with maternal Pou proteins (see Figure S6). Subsequently, pre-bound Foxh1/Tle may continue to repress target gene expression independently of Nodal signaling in the ectoderm through the recruitment of histone deacetylases. Such a spatial segregation in Foxh1-Tle functions would be similar to a recent report highlighting Tle enrichment in ventral tissue at dorsal (Spemann organizer) CRMs (Yasuoka et al., 2014). Future analysis will focus on Tle activity in a tissue-specific manner, and will require the generation of embryos lacking multiple maternal Tle RNAs.
A number of key TFs have been implicated in Xenopus mesendoderm development, including the maternal T-box TF Vegt, and zygotic factors Foxa, Sox17, and Gata family members (reviewed in Heasman, 2006; Zorn and Wells, 2009). We found that binding of the zygotic, hepatic pioneer factor Foxa is preceded by maternal Foxh1 at the vast majority of persistent peaks. While further confirmation is required, overlapping Foxh1 and Foxa expression in the mesendoderm support the hypothesis that this binding occurs in the same cells. We therefore speculate a mechanism of sequential enhancer binding of maternal Foxh1, followed by zygotic Foxa. This ‘molecular hand-off’ may occur by two different mechanisms. One possibility is that Foxh1 and Foxa TFs bind sequentially but they bind to different sites within these CRMs. The second possibility is a direct competition between Foxh1 and Foxa TFs for binding the same sequences. Foxh1 and Foxa competition would be driven by their binding affinities for DNA and relative Foxh1 and Foxa protein concentrations, which vary dynamically between the blastula and gastrula stages when endodermal cells undergo lineage commitments. We note the inverse expression patterns of foxh1 and foxa around the beginning of gastrulation (compare Figures 1A and S5A), whereby maternal foxh1 is decreasing precipitously while zygotic transcription of foxa ramps up. We also note that both mechanisms for a molecular hand-off could be in play.
Finally, we made a surprising finding that only ~3% of Foxh1 peaks persist into the beginning of gastrulation (954 peaks). Why does Foxh1 remain bound to these regions, and not to others? Our ChIP-seq data revealed that these persistent peaks, in addition to being enriched with Foxa, are strongly associated with Smad2/3 binding. Our bioinformatic analysis also implies the additional involvement of HMG/Sox TFs. We found that the maternal TF Sox7 is associated with these Foxh1-bound regions (Figure 6D), and this Sox family member has been associated with endodermal specification in Xenopus and mammals (Futaki et al., 2004; Zhang et al., 2005; Séguin et al., 2008). Taken together, this suggests an interesting model whereby coordinated mesendodermal gene regulation is under the control of CRMs containing a combination of Foxh1, Sox7, and Smad2/3 motifs (Figure 6F). Possibly, combinatorial TF binding at these CRMs stabilizes the complex, thereby retaining Foxh1 binding despite the drastic decrease in protein level. Finally, our finding that persistent peaks are highly correlated with Ep300 and H3K4me1 enrichment support the notion that these are active CRMs. Our future goals lie in teasing out the interactions between Foxh1 and other TFs critical for mesendoderm formation.
STAR Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ken W. Y. Cho (kwcho@uci.edu)
Experimental model and subject details
Xenopus tropicalis embryos were obtained by in vitro fertilization and staged according to Nieuwkoop and Faber (1994). All embryos were cultured at 25°C in 1/9X Marc’s modified Ringers (MMR) until desired stage. To achieve a knock-down of Foxh1 protein, a total of 22.5ng of foxh1 translation-blocking morpholino antisense oligonucleotide (Chiu et al., 2014) was injected into 4 sites of a 1-cell stage embryo. For Nodal signaling inhibition, 4-cell stage embryos were immersed in 1/9X MMR containing 100μM SB431542 (Tocris Bioscience) or DMSO. Animals were raised and maintained in accordance with the University of California, Irvine Institutional Animal Care Use Committee (IACUC) and guided by husbandry methods developed by the National Xenopus Resource (Marine Biological Laboratory, Woods Hole, MA). In vitro fertilizations were performed according to Ogino et al. (2006) using mature male and female animals raised in the laboratory and/or purchased from NASCO (University of Virginia stock) (https://www.enasco.com/xenopus/page/xen_list#tropicalis).
Method details
Immunoprecipitation and western blotting
Embryos were homogenized in THB (20mM Tris pH 7.5, 150mM NaCl, 2mM MgCl2, 1% Triton X-100) containing protease and phosphatase inhibitors (Roche cOmplete and PhosSTOP) and centrifuged. The supernatant was then re-centrifuged at 140,000rpm, and the lysate was used for immunoprecipitations using anti-Foxh1 (Chiu et al., 2014) or anti-Smad2/3 (BD Bioscience, #610842) antibody coupled to CNBr-activated sepharose beads (GE Healthcare Life Sciences). The immunoprecipitated protein was eluted and subjected to western blotting using anti-Foxh1 or anti-pSmad2/3 (Cell Signaling, #3101). Actb (β-actin) monoclonal antibody was obtained from Sigma Aldrich (A5441). Proteins were visualized using HRP-coupled secondary antibody and ECL Prime reagent (GE Healthcare Life Sciences). For pan-Smad2/3 investigation, Smad2/3 IP-western membranes were re-probed with anti-Smad2/3.
Chromatin immunoprecipitation (ChIP)
ChIP protocol was performed as described (Chiu et al., 2014) with modifications. Embryos were cultured in 1/9X MMR at 25°C until the indicated stage and fixed in 1% formaldehyde at room temperature for 45 minutes with gentle rocking. Crosslinking reactions were neutralized by the removal of the formaldehyde solution and incubation with 1ml 0.125M glycine solution for 10 minutes on ice. Embryos were then washed with cold RIPA buffer (50 mM Tris-HCl pH7.4, 150mM NaCl, 1mM EDTA, 0.25% sodium deoxycholate, 1% NP40, 0.1% SDS, 0.5 mM DTT, and Roche cOmplete protease inhibitor cocktail), flash frozen, and stored at −80°C. The fixed embryos were homogenized in RIPA buffer and incubated on ice for 10 minutes. Samples were then microfuged at 14,000 rpm for 15 minutes at 4°C. Pel lets were resuspended in RIPA buffer and sonicated on ice using a Branson Digital Sonifier 450 resulting in an average fragment size between 200–500bp. The samples were microfuged at 14,000 rpm for 20 minutes at 4°C to remove insoluble cellular debris. The chromatin was “pre-cleared” by incubating with Protein A-coated Dynabeads (Invitrogen) for 2 hour at 4°C with rotation. Antibodies were pre-bound to blocked Protein A Dynabeads by incubating at 4°C for 30 min. A sample of sheared chromatin was frozen for use as an input control. Pre-cleared chromatin was added to antibody-bound Dynabeads, and incubated overnight at 4°C on an end-over-end rotator. The next day, the beads were washed for 20 minutes each with ice-cold ChIP wash solution I (50mM HEPES-KOH pH7.5, 2mM EDTA, 150mM NaCl, 0.1% sodium deoxycholate, 1% Triton X-100, 1mM DTT, and 0.4mM PMSF), ChIP wash solution II (50mM HEPES-KOH pH7.5, 2mM EDTA, 500mM NaCl, 0.1% sodium deoxycholate, 1% Triton X-100, 1mM DTT, and 0.4mM PMSF), ChIP wash solution III (0.25 M LiCl, 1 mM EDTA, 10 mM Tris-HCl pH 8.0, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM DTT, and 0.4 mM PMSF), and TE (10mM Tris, 1mM EDTA, 1 mM DTT, and 0.4 mM PMSF). The DNA was then eluted with TE buffer containing 1% SDS, and reverse-crosslinked at 65°C overnight. The sonicated input control was diluted 3-fold with elution buffer, and also incubated at 65°C. All samples were treated with RNAse A, Proteinase K, phenol/chloroform extracted, and ethanol precipitated overnight. DNA pellets were resuspended in TE (for ChIP-qPCR) or Qiagen EB solution (for ChIP-seq). For stage 6 ChIP-qPCR, 30 embryo equivalents of ChIP DNA were used for each qPCR reaction, while 1-2 embryo equivalents were used for stages 8–10.5. Quantitative PCR was performed on a Roche LightCycler 480 using SYBR Green I master (Roche). Primer information is provided in Table S8.
ChIP-seq library preparation
For Foxh1 (Chiu et al., 2014), RNA pol II (BioLegend, MMS-126R) and Foxa (Santa Cruz Biotechnology, sc-6554) ChIP-seq, 10–30ng of total ChIP DNA was used for library construction using the NEXTflex ChIP-seq kit (Bioo Scientific). Sequencing was performed using the Illumina HiSeq 2500 and 50bp single-end reads were obtained. The stage 8 Tle (Santa Cruz Biotechnology, sc-13373) ChIP-seq library was constructed using the TruSeq ChIP Sample Prep kit (Illumina) and sequenced using the Illumina HiSeq2500 to obtain 100bp single-end reads. A custom X. tropicalis Sox7 antibody (Genscript, Inc.) was designed against a synthetic peptide, QVSQASDIQPSETS, corresponding to amino acids 330–343.
Sequence alignment and visualization
All sequencing data was aligned to Xenopus tropicalis v7.1 genome (Xenbase, http://www.xenbase.org/, RRID:SCR_003280) using Bowtie v1.0.0 (Langmead et al., 2009):
‘bowtie Xentro7 reads.fastq -m 1 -p 32 --sam > $Input_Name1-mapped.sam’
Multi-mapping reads were discarded. ChIP-seq signal was visualized using IGV (Thorvaldsdóttir et al., 2013; Robinson et al., 2011). Duplicate reads were removed from sorted BAM files using the ‘rmdup’ command in Samtools (Li et al., 2009), and biological replicates were concatenated. Bedgraph files were created using HOMER after first making a tag directory (Heinz et al., 2010). Default conditions were applied for the generation of the bedgraph files (http://homer.salk.edu/homer/ngs/ucsc.html).
Irreproducibility Discovery Rate analysis
For Foxh1 ChIP-seq analyses, the irreproducibility discovery rate (IDR) pipeline was used to identify high-confidence peaks between two biological replicates (Li et al., 2011). Uniquely mapped reads from the original replicates were merged into one file to generate one pooled mapped file per stage. Next, the mapped reads for each individual replicate, and the pooled file, were randomly divided to two pseudo-replicates. Using Macs2 v2.0.10 (Zhang et al., 2008), peak calling was performed for all of the original replicates, pseudo replicates, and the pooled data, used a staged-matched control (input) data set and a p-value of 0.001:
‘macs2 callpeak -t treat.bed -c control.bed -n treat-output -f BED -g 1.1e9 -p 1e-3’
IDR scripts (https://sites.google.com/site/anshulkundaje/projects/idr) were used to compare across original and pseudo replicates. The called peaks are evaluated and an IDR score is assigned to them based on their consistency and significance. The recommended IDR score threshold for use in extracting the final set of peaks matched our observations (IDR threshold of 0.01 for consistent peaks between two original replicates, 0.02 for consistent peaks between two pseudo replicates, and 0.0025 for consistent peaks between two pseudo replicates of pooled replicate). For each stage, the final set of peaks used in this manuscript is the optimal set produced by following the IDR pipeline, using only those replicates that were consistent. In this manuscript, we also performed ChIP-seq for RNA pol II, Tle, and Foxa. FASTQ files were uniquely mapped as described above. For Tle and Foxa, Macs2 ‘callpeak’ was used as described above to call peaks against a stage-matched input control.
Heatmaps
deepTools v2.0 (Ramírez et al., 2014) was used to generate heatmaps around peak summits or gene TSS. First, a bigwig file was generated from the BAM files using bamCoverage in deepTools. The expression was normalized to RPKM values and the duplicates were removed.
‘bamCoverage --bam inputBam.bam --binSize 100 -- normalizeUsingRPKM --ignoreDuplicates -p 32 -of bigwig -o bwFILES signalFile.bw’
The ‘computeMatrix’ command with the sub command ‘reference-point’ was used to generate the table underlying the heatmaps, using a bin size of 100bp:
‘computeMatrix reference-point --regionsFileName PeaksFileOrGeneTssFile.bed --scoreFileName SignalFile.bw -- binSize 100 -b 5000 -a 5000 --outFileName OutputFile -- missingDataAsZero --referencePoint center’
‘plotHeatmap’ was used to visualize the table:
‘plotHeatmap --matrixFile OutputFile --outFileName Plot.png -- refPointLabel Summit --kmeans NumberOfClusters --colorMap Colormap --outFileSortedRegions SortedRegion.txt’
To cluster the heatmaps, the inside K-means clustering option in ‘plotHeatmap’ was used.
A heatmap of temporal expressions for Foxh1-associated genes (Figure 5D and Figure S4A) was generated using the absolute abundance measurement data from Owens et al. (2016). The rdRNA-seq data was used to ensure correct identification of maternal transcripts. K-means clustering and visualization was performed in Python v2.7 using Numpy and Scipy library. Five clusters were determined to provide the best representation of the data. For visualization, the expression profile of each gene was normalized to the range of [0,1]. Corresponding line plots (Figure S4B) demonstrating the average transcripts per cluster, were also created using Python.
Motif analysis
Motif analysis was primarily performed using the MEME Suite 4.11.2 (Bailey et al., 2009) (http://meme-suite.org/). De novo analysis was performed using DREME (http://meme-suite.org/tools/dreme) (Bailey et al., 2011) using the 100bp surrounding the summit of the Foxh1 peak. All motifs with an E-value of < 0.05 were identified and shuffled input sequences were used as background control. Motif occurrences within peaks were obtained using HOMER (Heinz et al., 2010) (http://homer.salk.edu/homer/). To identify TFs that recognize the discovered motifs, we used TOMTOM (http://meme-suite.org/tools/tomtom) (Gupta et al., 2007) against the Vertebrates (in vivo and in silico) database, and displayed E-values < 10. All output is available in Table S7. The positional distribution of motifs (Figure 6B and Figure S6A and B) around the Foxh1 peak summits was investigated using CentriMo (http://meme-suite.org/tools/centrimo) (Bailey and Machanick, 2012), as well as visualized through heatmaps. Regions analyzed were 1kb (CentriMo) or 2kb (heatmaps) surrounding the peak summit. CentriMo output used weighted moving average smoothing and a window of 50 (stage 8 and 9) or 80 (persistent peaks). To generate heatmaps (Figure 6B), Foxh1 peak BED files were first annotated for the motif occurrence using ‘annotatePeaks.pl.’
‘annotatepeaks.pl <Foxh1peaks.bed> Xentro7.fa -size 2000 -hist 20 -ghist -m motif.motif -mbed motif.bed > peak.motif.txt’
From this output, the distribution of the motifs in a set of specific genomic regions were extracted and visualized with MATLAB R2015a.
In this report, we investigated the enrichment of various motif combinations under the Foxh1 persistent peaks, all Foxh1 peaks, and within random genomic regions (Figure 6C). To obtain the genomic background, we generated a bed file that contains the location of all Foxh1 motifs (AATHMACA) in the entire genome using the HOMER script ‘findMotifsGenome.pl’ (Heinz et al., 2010) (http://homer.salk.edu/homer/). We sampled this bed file to match the number of persistent peaks 500 times. In addition, the pools of all Foxh1 peaks and persistent peaks were scaled down to include only peaks with a Foxh1 motif. Using a window size of 200bp, we searched for the consensus sequences for Smad (CAGAC), Sox family (ACAAWRG) and Pou family (ATGCAAAT) TFs in the persistent peaks, all foxh1 peaks, and random genomic regions using the HOMER command ‘known.’ The percentage of peaks with each motif combination was calculated. The Grubb’s test for outliers was used to identify the significance of the percentages calculated for the persistent peaks and all Foxh1 peaks, compared to the genomic background.
Spatial localization of Foxh1 genes
To investigate the spatial localization of Foxh1-associated genes (Figure 6E), we utilized expression data from dissected early gastrula (stage 10.5) embryos (Blitz et al., in press; GEO accession number GSE81458). The reads were aligned to Xenopus tropicalis v7.1 genome using RSEM v1.2.12 (Li and Dewey, 2011). Pairwise comparison of differential expression (DE) between the animal cap, dorsal marginal zone, lateral marginal zone, ventral marginal zone, and the vegetal mass was performed using EBseq (Leng et al., 2013). A gene was considered ‘localized’ if the pairwise comparisons identified the gene as differentially expressed in at least one comparison. All localized genes were K-means clustered using R (R Core Team 2014). A Chi-squared test was used to measure the statistical significance of the difference in gene expression between genes with a persistent peak, genes with any Foxh1 peak, and genes in the entire genome (R Core Team 2014).
Additional bioinformatics
When necessary, SAM to BAM conversion was performed using Samtools v0.1.19 (Li et al., 2009), and BAM files were converted to BED files using ‘bamtobed’ from the Bedtools suite v2.19.1 (Quinlan et al., 2010). To find the set of regions (peaks or genes), which were in specific relative position toward another set of genomic regions, we used Bedtools’ ‘closestBed.’ To identify the closest genes to the called peaks, after getting the list of closest genes using ‘closestBed,’ the list was filtered for distance (10kb) between peaks and genes. The intersection of two sets of peaks was generated using Bedtools’ ‘intersectBed,’0 with the default minimum overlap of 1bp.
Quantification and statistical analysis
All statistical tests used in this manuscript can be found in the figure legends.
Data and software availability
All novel high-throughput sequencing data reported in this paper is made publically available using GEO accession number GSE85273. Publically available datasets utilized in this report can be accessed through GEO accession numbers GSE56000 for H3K4me1 ChIP-seq (Gupta et al., 2014), GSE53654 for Foxh1 and Smad2/3 stage 10.5 ChIP-seq data (Chiu et al., 2014), GSE67974 for stage 12.5 and stage 16 RNA pol II ChIP-seq (Hontelez et al., 2015), GSE65785 for absolute transcript temporal profiling (Owens et al., 2016), and GSE81458 for transcriptome profiling of dissected early gastrula (stage 10.5) embryos (Blitz et al., in press). Ep300 ChIP-seq data (Yasuoka et al., 2014) can be accessed at the DDBJ Sequence Read Archive (DRA) under accession number DRA000505.
Additional resources
For Xenopus tropicalis husbandry, please see the National Xenopus Resource, RRID:SCR_013731 (http://www.mbl.edu/xenopus/). For genomic and additional community resources, see Xenbase, RRID:SCR_003280 (www.xenbase.org).
Supplementary Material
Table S1. Foxh1 IDR ChIP-seq peaks, related to Figure 2
Table S2. Foxh1-associated genes, related to Figure 2
Table S3. Smad2/3 IDR ChIP-seq peaks and Foxh1 overlap, related to Figure 2
Table S4. Tle ChIP-seq analysis, related to Figure 3
Table S5. RNA Pol II ChIP-seq analysis, related to Figure 5
Table S6. Foxa ChIP-seq analysis, related to Figure 6
Table S7. Foxh1 motif analysis, related to Figure 6
Highlights.
Foxh1 binds to the genome dynamically and pre-marks target genes at cleavage stages
Foxh1 recruits the co-repressor Tle to cis-regulatory modules in the early blastula
Continuously Foxh1-occupied enhancers are marked by H3K4me1, Ep300, Smad2 and Sox7
Binding of the Foxa pioneer factor to CRMs is preceded by maternal Foxh1 marking
Acknowledgments
This work was made possible, in part, through access to the Genomic High Throughput Facility Shared Resource of the Cancer Center Support Grant (CA-62203) at the University of California, Irvine and NIH shared instrumentation grants 1S10RR025496-01, 1S10OD010794-01 and S10OD021718-01. We thank Xenbase for genomic resources (http://www.xenbase.org/, RRID:SCR_003280), the National BioResource Project (NBRP) in Japan for X. tropicalis embryos, the UC Irvine Office of Information Technology for ongoing support of the High Performance Computing Cluster, and the DNA Sequencing Section at OIST Graduate University. We also thank Gert Veenstra for sharing high-throughput sequencing data. This work was supported by NIH HD073179 to K.W.Y.C., and R.M.C. is a recipient of a US Department of Education GAANN fellowship (P200A120207).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
R.M.C, J.S.C., J.C., K.D.P., Y.Y., S.T., I.L.B. and K.W.Y.C. performed wet bench experiments. R.M.C., E.F. and K.D.P. performed bioinformatic analyses. All authors contributed to experimental design. R.M.C., I.L.B. and K.W.Y.C. wrote the manuscript.
References
- Akkers RC, van Heeringen SJ, Jacobi UG, Janssen-Megens EM, FranCoijs KJ, Stunnenberg HG, Veenstra GJC. A Hierarchy of H3K4me3 and H3K27me3 Acquisition in Spatial Gene Regulation in Xenopus Embryos. Developmental Cell. 2009;17:425–434. doi: 10.1016/j.devcel.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27(12):1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Research. 2009;37:W202–8. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Machanick P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Research. 2012;40(17):e128–e128. doi: 10.1093/nar/gks433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz IL, Paraiso KD, Patrushev I, Chiu WTY, Cho KWY, Gilchrist MJ. A catalog of Xenopus tropicalis transcription factors and their regional expression in the early gastrula stage embryo. Developmental Biology. doi: 10.1016/j.ydbio.2016.07.002. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovic O, van Heeringen SJ, Veenstra GJC. The epigenome in early vertebrate development. Genesis. 2011;50:192–206. doi: 10.1002/dvg.20831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantjes H, Roose J, van de Wetering M, Clevers H. All Tcf HMG box transcription factors interact with Groucho-related co-repressors. Nucleic Acids Research. 2001;29:1410–1419. doi: 10.1093/nar/29.7.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallo RA, Cox RT, Moline MM, Roose J, Polevoy GA, Clevers H, et al. Drosophila Tcf and Groucho interact to repress Wingless signalling activity. Nature. 1998;395:604–608. doi: 10.1038/26982. [DOI] [PubMed] [Google Scholar]
- Chen G, Courey AJ. Groucho/TLE family proteins and transcriptional repression. Gene. 2000;249:1–16. doi: 10.1016/s0378-1119(00)00161-x. [DOI] [PubMed] [Google Scholar]
- Chen X, Rubock MJ, Whitman M. A transcriptional partner for MAD proteins in TGF-β signalling. Nature. 1996;383:691–696. doi: 10.1038/383691a0. [DOI] [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- Chiu WT, Charney Le R, Blitz IL, Fish MB, Li Y, Biesinger J, et al. Genome-wide view of TGF/Foxh1 regulation of the early mesendoderm program. Development. 2014;141:4537–4547. doi: 10.1242/dev.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. The EMBO Journal. 1998;17:244–254. doi: 10.1093/emboj/17.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Molecular Cell. 2002;9:279–289. doi: 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- Clark KL, Halay ED, Lai E, Burley SK. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. 1993;364:412–420. doi: 10.1038/364412a0. [DOI] [PubMed] [Google Scholar]
- Cordenonsi M, Dupont S, Maretto S, Insinga A, Imbriano C, Piccolo S. Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell. 2003;113:301–314. doi: 10.1016/s0092-8674(03)00308-8. [DOI] [PubMed] [Google Scholar]
- Daniels DL, Weis WI. β-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nature Structural & Molecular Biology. 2005;12:364–371. doi: 10.1038/nsmb912. [DOI] [PubMed] [Google Scholar]
- de-Leon SBT. The conserved role and divergent regulation of foxa, a pan-eumetazoan developmental regulatory gene. Developmental Biology. 2011;357:21–26. doi: 10.1016/j.ydbio.2010.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucourt M, Houliston E, Besnardeau L, Kimelman D, Lepage T. The Pitx2 Homeobox Protein Is Required Early for Endoderm Formation and Nodal Signaling. Developmental Biology. 2001;229:287–306. doi: 10.1006/dbio.2000.9950. [DOI] [PubMed] [Google Scholar]
- Faure S, Lee MA, Keller T, ten Dijke P, Whitman M. Endogenous patterns of TGFbeta superfamily signaling during early Xenopus development. Development. 2000;127:2917–2931. doi: 10.1242/dev.127.13.2917. [DOI] [PubMed] [Google Scholar]
- Futaki S, Hayashi Y, Emoto T, Weber CN, Sekiguchi K. Sox7 plays crucial roles in parietal endoderm differentiation in F9 embryonal carcinoma cells through regulating Gata-4 and Gata-6 expression. Mol Cell Biol. 2004;24:10492–503. doi: 10.1128/MCB.24.23.10492-10503.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Kaestner KH. The Foxa family of transcription factors in development and metabolism. Cellular and Molecular Life Sciences. 2006;63:2317–2328. doi: 10.1007/s00018-006-6095-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain S, Howell M, Esslemont GM, Hill CS. Homeodomain and winged-helix transcription factors recruit activated Smads to distinct promoter elements via a common Smad interaction motif. Genes & Development. 2000;14:435–451. [PMC free article] [PubMed] [Google Scholar]
- Gualdi R, Bossard P, Zheng M, Hamada Y, Coleman JR, Zaret KS. Hepatic specification of the gut endoderm in vitro: cell signaling and transcriptional control. Genes & Development. 1996;10:1670–1682. doi: 10.1101/gad.10.13.1670. [DOI] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biology. 2007;8(2):R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Wills A, Ucar D, Baker J. Developmental enhancers are marked independently of zygotic Nodal signals in Xenopus. Developmental Biology. 2014;395:38–49. doi: 10.1016/j.ydbio.2014.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman J. Patterning the early Xenopus embryo. Development. 2006;133:1205–1217. doi: 10.1242/dev.02304. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CS. TGF-β signalling pathways in early Xenopus development. Current Opinion in Genetics & Development. 2001;11:533–540. doi: 10.1016/s0959-437x(00)00229-x. [DOI] [PubMed] [Google Scholar]
- Ho DM, Chan J, Bayliss P, Whitman M. Inhibitor-resistant type I receptors reveal specific requirements for TGF-beta signaling in vivo. Developmental Biology. 2006;295:730–742. doi: 10.1016/j.ydbio.2006.03.050. [DOI] [PubMed] [Google Scholar]
- Hontelez S, van Kruijsbergen I, Georgiou G, van Heeringen SJ, Bogdanovic O, Lister R, Veenstra GJC. Embryonic transcription is controlled by maternally defined chromatin state. Nature Communications. 2016;6:1–11. doi: 10.1038/ncomms10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoodless PA, Pye M, Chazaud C, Labbé E, Attisano L, Rossant J, Wrana JL. FoxH1 (Fast) functions to specify the anterior primitive streak in the mouse. Genes & Development. 2001;15:1257–1271. doi: 10.1101/gad.881501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell M, Inman GJ, Hill CS. A novel Xenopus Smad-interacting forkhead transcription factor (XFast-3) cooperates with XFast-1 in regulating gastrulation movements. Development. 2002;129:2823–2834. doi: 10.1242/dev.129.12.2823. [DOI] [PubMed] [Google Scholar]
- Inman GJ, Nicolás FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Molecular Pharmacology. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- Jennings BH, Pickles LM, Wainwright SM, Roe SM, Pearl LH, Ish-Horowicz D. Molecular Recognition of Transcriptional Repressor Motifs by the WD Domain of the Groucho/TLE Corepressor. Molecular Cell. 2006;22:645–655. doi: 10.1016/j.molcel.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Kofron M, Demel T, Xanthos J, Lohr J, Sun B, Sive H, et al. Mesoderm induction in Xenopus is a zygotic event regulated by maternal VegT via TGFbeta growth factors. Development. 1999;126:5759–5770. doi: 10.1242/dev.126.24.5759. [DOI] [PubMed] [Google Scholar]
- Kofron M, Puck H, Standley H, Wylie C, Old R, Whitman M, Heasman J. New roles for FoxH1 in patterning the early embryo. Development. 2004;131:5065–5078. doi: 10.1242/dev.01396. [DOI] [PubMed] [Google Scholar]
- Ku M, Sokol SY, Wu J, Tussie-Luna MI, Roy AL, Hata A. Positive and negative regulation of the transforming growth factor beta/activin target gene goosecoid by the TFII-I family of transcription factors. Mol Cell Biol. 2005;25:7144–7157. doi: 10.1128/MCB.25.16.7144-7157.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunwar PS, Zimmerman S, Bennett JT, Chen Y, Whitman M, Schier AF. Mixer/Bon and FoxH1/Sur have overlapping and divergent roles in Nodal signaling and mesendoderm induction. Development. 2003;130:5589–99. doi: 10.1242/dev.00803. [DOI] [PubMed] [Google Scholar]
- Landsberger N, Wolffe AP. Remodeling of regulatory nucleoprotein complexes on the Xenopus hsp70 promoter during meiotic maturation of the Xenopus oocyte. The EMBO Journal. 1997;16:4361–4373. doi: 10.1093/emboj/16.14.4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MA, Heasman J, Whitman M. Timing of endogenous activin-like signals and regional specification of the Xenopus embryo. Development. 2001;128:2939–2952. doi: 10.1242/dev.128.15.2939. [DOI] [PubMed] [Google Scholar]
- Leng N, Dawson JA, Thomson JA, Ruotti V. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29(8):1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. The Annals of Applied Statistics. 2011;5:1752–1779. [Google Scholar]
- Nieuwkoop, Faber . Normal table of Xenopus laevis (Daudin) Garland Publishing Inc; New York: 1994. [Google Scholar]
- Ogino H, McConnell WB, Grainger RM. High-throughput transgenesis in Xenopus using I-SceI meganuclease. Nature Protocols. 2006;1(4):1703–1710. doi: 10.1038/nprot.2006.208. [DOI] [PubMed] [Google Scholar]
- Osada SI, Saijoh Y, Frisch A, Yeo CY, Adachi H, Watanabe M, et al. Activin/nodal responsiveness and asymmetric expression of a Xenopus nodal-related gene converge on a FAST-regulated module in intron 1. Development. 2000;127:2503–2514. doi: 10.1242/dev.127.11.2503. [DOI] [PubMed] [Google Scholar]
- Owens NDL, Blitz IL, Lane MA, Patrushev I, Overton JD, Gilchrist MJ, et al. Measuring Absolute RNA Copy Numbers at High Temporal Resolution Reveals Transcriptome Kinetics in Development. Cell Reports. 2016;14:632–647. doi: 10.1016/j.celrep.2015.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogoda HM, Solnica-Krezel L, Driever W, Meyer D. The zebrafish forkhead transcription factor FoxH1/Fast1 is a modulator of Nodal signaling required for organizer formation. Current Biology. 2000;10:1041–1049. doi: 10.1016/s0960-9822(00)00669-2. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Research. 2014;42(Web Server issue):W187–91. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CD, Steiner AB, Yaklichkin S, Lu Q, Wang S, Hennessy M, Kessler DS. FoxH1 mediates a Grg4 and Smad2 dependent transcriptional switch in Nodal signaling during Xenopus mesoderm development. Developmental Biology. 2016;414:34–44. doi: 10.1016/j.ydbio.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring C, Ogata S, Meek L, Song J, Ohta T, Miyazono K, Cho KWY. The role of a Williams-Beuren syndrome-associated helix-loop-helix domain-containing transcription factor in activin/nodal signaling. Genes & Development. 2002;16:820–835. doi: 10.1101/gad.963802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nature Biotechnology. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roose J, Molenaar M, Peterson J, Hurenkamp J, Brantjes H, Moerer P, et al. The Xenopus Wnt effector XTcf-3 interacts with Groucho-related transcriptional repressors. Nature. 1998;395:608–612. doi: 10.1038/26989. [DOI] [PubMed] [Google Scholar]
- Schier AF, Talbot WS. Molecular genetics of axis formation in zebrafish. Annual Review of Genetics. 2005;39:561–613. doi: 10.1146/annurev.genet.37.110801.143752. [DOI] [PubMed] [Google Scholar]
- Séguin CA, Draper JS, Nagy A, Rossant J. Establishment of endoderm progenitors by SOX transcription factor expression in human embryonic stem cells. Cell Stem Cell. 2008;3:182–95. doi: 10.1016/j.stem.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Sekiya T, Zaret KS. Repression by Groucho/TLE/Grg proteins: genomic site recruitment generates compacted chromatin in vitro and impairs activator binding in vivo. Mol Cell. 2007;28:291–303. doi: 10.1016/j.molcel.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiratori H, Sakuma R, Watanabe M, Hashiguchi H. Two-step regulation of left-right asymmetric expression of Pitx2: initiation by nodal signaling and maintenance by Nkx2. Molecular Cell. 2001;7:137–149. doi: 10.1016/s1097-2765(01)00162-9. [DOI] [PubMed] [Google Scholar]
- Sirotkin HI, Gates MA, Kelly PD, Schier AF, Talbot WS. Fast1 is required for the development of dorsal axial structures in zebrafish. Current Biology. 2000;10:1051–1054. doi: 10.1016/s0960-9822(00)00679-5. [DOI] [PubMed] [Google Scholar]
- Skirkanich J, Luxardi G, Yang J, Kodjabachian L, Klein PS. An essential role for transcription before the MBT in Xenopus laevis. Developmental Biology. 2011;357:478–491. doi: 10.1016/j.ydbio.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slagle CE, Aoki T, Burdine RD. Nodal-Dependent Mesendoderm Specification Requires the Combinatorial Activities of FoxH1 and Eomesodermin. PLoS Genetics. 2011;7:e1002072. doi: 10.1371/journal.pgen.1002072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo AKK, Arnold SJ, Trotter MWB, Brown S, Ang LT, Chng Z, Robertson EJ, Dunn NR, Vallier L. Pluripotency factors regulate definitive endoderm specification through eomesodermin. Genes Dev. 2011;25:238–250. doi: 10.1101/gad.607311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw NL, Zhang Y, Woods IG, Imam F, Regev A, Liu XS, et al. Chromatin signature of embryonic pluripotency is established during genome activation. Nature. 2010;464:922–926. doi: 10.1038/nature08866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Waltner-Law M, Yamada K, Osawa H, Stifani S, Granner DK. Transducin-like enhancer of split proteins, the human homologs of Drosophila groucho, interact with hepatic nuclear factor 3beta. J Biol Chem. 2000;275:18418–18423. doi: 10.1074/jbc.M910211199. [DOI] [PubMed] [Google Scholar]
- Whitman M. Nodal signaling in early vertebrate embryos: themes and variations. Developmental Cell. 2001;1:605–617. doi: 10.1016/s1534-5807(01)00076-4. [DOI] [PubMed] [Google Scholar]
- Xanthos JB, Kofron M, Wylie C, Heasman J. Maternal VegT is the initiator of a molecular network specifying endoderm in Xenopus laevis. Development. 2001;128:167–180. doi: 10.1242/dev.128.2.167. [DOI] [PubMed] [Google Scholar]
- Xu J, Watts JA, Pope SD, Gadue P, Kamps M, Plath K, Zaret KS, Smale ST. Transcriptional competence and the active marking of tissue-specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes Dev. 2009;23:2824–38. doi: 10.1101/gad.1861209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaklichkin S, Vekker A, Stayrook S, Lewis M, Kessler DS. Prevalence of the EH1 Groucho interaction motif in the metazoan Fox family of transcriptional regulators. BMC Genomics. 2007a;8:201. doi: 10.1186/1471-2164-8-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaklichkin S, Steiner AB, Lu Q, Kessler DS. FoxD3 and Grg4 physically interact to repress transcription and induce mesoderm in Xenopus. J Biol Chem. 2007b;282:2548–2557. doi: 10.1074/jbc.M607412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Meno C, Sakai Y, Shiratori H. The transcription factor FoxH1 (FAST) mediates Nodal signaling during anterior-posterior patterning and node formation in the mouse. Genes & Development. 2001;15:1242–1256. doi: 10.1101/gad.883901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuoka Y, Suzuki Y, Takahashi S, Someya H, Sudou N, Haramoto Y, et al. Occupancy of tissue-specific cis-regulatory modules by Otx2 and TLE/Groucho for embryonic head specification. Nature Communications. 2014;5:4322. doi: 10.1038/ncomms5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SJ, Wills AE, Chuong E, Gupta R, Baker JC. HEB and E2A function as SMAD/FOXH1 cofactors. Genes & Development. 2011;25:1654–1661. doi: 10.1101/gad.16800511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes & Development. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Basta T, Jensen ED, Klymkowsky MW. The beta-catenin/VegT-regulated early zygotic gene Xnr5 is a direct target of SOX3 regulation. Development. 2003;130:5609–24. doi: 10.1242/dev.00798. [DOI] [PubMed] [Google Scholar]
- Zhang C, Basta T, Hernandez-Lagunas L, Simpson P, Stemple DL, Artinger KB, Klymkowsky MW. Repression of nodal expression by maternal B1-type SOXs regulates germ layer formation in Xenopus and zebrafish. Dev Biol. 2004;273:23–37. doi: 10.1016/j.ydbio.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Zhang C, Basta T, Fawcett SR, Klymkowsky MW. SOX7 is an immediate-early target of VegT and regulates Nodal-related gene expression in Xenopus. Dev Biol. 2005;278:526–41. doi: 10.1016/j.ydbio.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Zhang J, Houston DW, King ML, Payne C, Wylie C, Heasman J. The role of maternal VegT in establishing the primary germ layers in Xenopus embryos. Cell. 1998;94:515–524. doi: 10.1016/s0092-8674(00)81592-5. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biology. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn AM, Wells JM. Vertebrate endoderm development and organ formation. Annual Review of Cell and Developmental Biology. 2009;25:221–251. doi: 10.1146/annurev.cellbio.042308.113344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Foxh1 IDR ChIP-seq peaks, related to Figure 2
Table S2. Foxh1-associated genes, related to Figure 2
Table S3. Smad2/3 IDR ChIP-seq peaks and Foxh1 overlap, related to Figure 2
Table S4. Tle ChIP-seq analysis, related to Figure 3
Table S5. RNA Pol II ChIP-seq analysis, related to Figure 5
Table S6. Foxa ChIP-seq analysis, related to Figure 6
Table S7. Foxh1 motif analysis, related to Figure 6