

Abstract
Chronic pain is a distressing condition, which is experienced even when the painful stimulus, whether surgery or disease related, has subsided. Current treatments for chronic pain show limited efficacy and come with a host of undesirable side‐effects, and thus there is a need for new, more effective therapies to be developed. The mechanisms underlying chronic pain are not fully understood at present, although pre‐clinical models have facilitated the progress of this understanding considerably in the last decade. The mechanisms underlying chronic pain were initially thought to be neurocentric. However, we now appreciate that non‐neuronal cells play a significant role in nociceptive signalling through their communication with neurons. One of the major signalling pathways, which mediates neuron/non‐neuronal communication, is chemokine signalling. In this review, we discuss selected chemokines that have been reported to play a pivotal role in the mechanisms underlying chronic pain in a variety of pre‐clinical models. Approaches that target each of the chemokines discussed in this review come with their advantages and disadvantages; however, the inhibition of chemokine actions is emerging as an innovative therapeutic strategy, which is now reaching the clinic, with the chemokine Fractalkine and its CX 3 CR 1 receptor leading the way.
Keywords: chemokines, chronic pain, proteases, therapy
Abbreviations used
- CCL
chemokine (C–C motif) ligand
- CIPN
chemotherapy‐induced painful neuropathy
- DRG
dorsal root ganglion
- IL
interleukin
- MCP‐1
monocyte chemoattractant protein 1
- PSNL
partial sciatic nerve ligation
Chronic pain is a distressing, debilitating condition, which is poorly managed by clinically available drugs at present. It manifests in a variety of ways, with patients experiencing spontaneous pain, pain in response to innocuous stimuli (allodynia), heightened sensitivity to noxious stimuli (hyperalgesia) or irregular, unpleasant sensations (dysesthesia). Chronic pain can accompany a variety of injuries and conditions that result in lesions or dysfunction of the somatosensory nervous system and can continue for months or years after the initial tissue damage has healed. Such injuries include peripheral nerve injury (post‐surgical); damage to the central nervous system as a result of conditions such as multiple sclerosis (Watson and Sandroni 2016); injury resulting from viral infections (Uebelacker et al. 2015); metabolic disorders, such as diabetes (Davies et al. 2006); autoimmune disorders, such as rheumatoid arthritis (Wolfe et al. 2011) and injury as a result of drug treatment, such as chemotherapy (Dougherty et al. 2007).
Current treatments for chronic pain come with a host of undesirable side‐effects and provide only limited relief to patients. Because of the fact that the underlying mechanisms are not fully understood, effective therapies have yet to be developed. Substantial advances in our understanding of the mechanisms underlying chronic pain have been facilitated by murine pre‐clinical studies, which model chronic pain through surgical, pharmacological and immunization methods.
Chronic pain was historically attributed to a purely neuronal response to injury, which resulted in the development of ‘neurocentric’ strategies, in other words, therapies that focused on targeting neurons. However, extensive pre‐clinical evidence has now implicated significant contributions of non‐neuronal cells in the regulation of chronic pain and, in particular, has identified a pivotal role of neuron/non‐neuronal cell cross‐talk in the modulation of nociceptive signalling. The development of alternative therapeutic strategies has therefore now shifted focus towards targeting the activation of non‐neuronal cells, as well as the multitude of factors released by them, in response to nerve and tissue injury. Although the ultimate aim of developing new therapies is to dampen the neuronal signalling associated with pain, the novelty now comes with the target, which is no longer neurocentric.
One of the major ligand/receptor partnerships that facilitate communication between neurons and neighbouring non‐neuronal cells in the nervous system is chemokine signalling (Ramesh et al. 2013). Chemokines, or ‘chemotactic cytokines’, which were first identified over two decades ago as mediators of leucocyte migration (Oppenheim et al. 1991), are a family of small proteins, typically ranging from 8 to 17 kDa in size. Chemokines are classified according to the organization of cysteine residues on their N‐terminal region and are thus divided into four subfamilies: C, CC, CXC and CX3C (Luster 1998; Bajetto et al. 2002; Laing and Secombes 2004). Chemokines can be released by a variety of cell types in the central nervous system such as microglia, astrocytes and neurons (Bajetto et al. 2002). Various cell types in the peripheral nervous system also have the capacity to express chemokines, for instance, nociceptive sensory neurons in the dorsal root ganglia (Miller et al. 2008) as well as infiltrating monocytes/macrophages and Schwann cells, which constitute the myelin sheath surrounding axons (Kopydlowski et al. 1999; Saika et al. 2012).
While expressed in multiple cell types, in some cases the expression of a specific chemokine is restricted to a particular cell type, for instance, CX3CL1 is principally expressed by neurons (Clark et al. 2009). In most cases, signalling between chemokines and their receptors is promiscuous with one chemokine having the capacity to activate multiple chemokine receptors. For example, chemokine (C–C motif) ligand (CCL)3 is able to activate chemokine receptor (CCR)1, 5 and 9 (Kunkel 1999). In addition, one chemokine receptor can be activated by multiple chemokines. For instance, the CCR5, is activated by multiple chemokines (Combadiere et al. 1996; Raport et al. 1996; Samson et al. 1996; Nibbs et al. 1999).
The majority of chemokines are not constitutively expressed in the nervous system but are instead induced during adverse conditions (Bajetto et al. 2002). The latter feature makes chemokines particularly useful as therapeutic targets, because of the fact that their inhibition would be less likely to interfere with homeostatic processes, thus reducing adverse side‐effects. In addition, chemokine receptors are G protein‐coupled receptors, which are the most commonly targeted receptor class in modern medicine (Wells et al. 2006).
Chemokines have recently been associated with a variety chronic pain conditions clinically. For instance, the homeostatic chemokine CX3CL1, has been reported to be elevated in the bone marrow of rheumatoid arthritis patients (Kuca‐Warnawin et al. 2016). In addition, chemokines such as CCL2 and CCL17 have been observed in blood samples taken from temporomandibular disorder and fibromyalgia patients respectively (Slade et al. 2011; García et al. 2013). This highlights the importance and validity of investigating the role in chemokines in chronic pain disorders. Here, we discuss pre‐clinical evidence for the role of a selected number of chemokines in neuron‐non‐neuronal cell communication in the context of chronic pain and consider the therapeutic potential of disrupting their signalling.
CX3CL1/fractalkine
Expression and distribution
CX3CL1, otherwise known as fractalkine (FKN), is the only member of the CX3C subfamily of chemokines. In the CNS, FKN is expressed predominantly by neurons and unlike the majority of chemokines, is expressed constitutively. Indeed, in the spinal cord, expression of FKN is restricted only to neurons under basal conditions (Lindia et al. 2005; Clark et al. 2009; Yang et al. 2012). In the peripheral nervous system, FKN expression has been observed in the cell bodies of sensory neurons in the dorsal root ganglia (Verge et al. 2004). The expression of FKN at the central terminals of these neurons in the spinal cord, however, is debated with studies reporting both a presence (Verge et al. 2004) and absence (Clark et al. 2009) of FKN at this location. When the FKN expression profile was examined in a FKN reporter mouse (Kim et al. 2011), however, it was confirmed that neurons in the CNS did indeed express FKN, but sensory neurons in the dorsal root ganglion (DRG) as well as their terminals in the spinal cord, did not (Fig. 1). In the periphery, FKN is constitutively expressed in endothelial cells in the skin and intestine (Papadopoulos et al. 1999; Muehlhoefer et al. 2000) as well as in the heart (Harrison et al. 1999) and lungs (Foussat et al. 2000).
Figure 1.
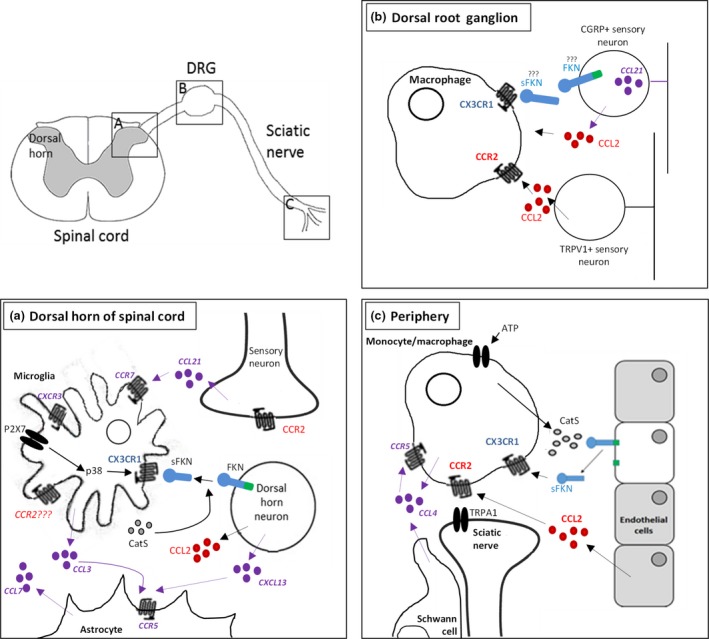
Schematic representation of chemokine/chemokine receptor expression in the dorsal horn of the spinal cord and the peripheral nervous system. Chemokines shown in purple have been less extensively studied. (a) The first pain synapse in the dorsal horn. A major signalling partnership that regulates neuron‐microglia communication in nociception is fractalkine (FKN)/CX 3 CR 1 signalling. FKN is expressed exclusively on neurons in the dorsal horn and is cleaved by Cathepsin S (CatS), which is released by neighbouring microglia, to produce soluble FKN (sFKN). sFKN activates CX 3 CR 1 receptors expressed by microglia. Dorsal horn neurons have also been shown to express chemokine (C–C motif) ligand 2 (CCL 2), however, the expression of the chemokine receptor (CCR)2 receptor, is unclear. Dorsal horn neurons also express CXCL 13, which activates CCR 5 receptors expressed by astrocytes. CCR 5 receptors are also activated by CCL 3, which is expressed by microglia. Afferent nerve terminals in the dorsal horn also express chemokines associated with chronic pain, specifically CCL 21, which has the capacity to activate both CXCR3 and CCR7 receptors expressed by microglia, the latter of which is induced in chronic pain. (b) In the dorsal root ganglion, FKN has been reported to be expressed by sensory neurons (however, this is controversial, see Kim et al. 2011). sFKN activates CX 3 CR 1 receptors, which are expressed by macrophages. Macrophages in the dorsal root ganglion (DRG) also express the CCR 2 receptor, which is activated by CCL 2 released from both CGRP and TRPV 1‐positive sensory neurons. CGRP‐positive sensory neurons also express CCL 21, although CCL 21 actions in the DRG are not established. (c) In the periphery, FKN is expressed by endothelial cells, and is cleaved by CatS, which is found in monocytes/macrophages. sFKN activates CX 3 CR 1 receptors, also expressed by monocytes/macrophages. Both macrophages and Schwann cells also express CCL 4, which can activate CCR 5 receptors expressed by macrophages. Endothelial cells also express CCL 2, which activates CCR 2 receptors in monocytes/macrophages.
FKN exists as both a full length, membrane‐bound form of approximately 100 kDa in size, and a cleaved, soluble form (sFKN) of 80 kDa in size, making this chemokine an anomaly compared to other chemokines in terms of size, with other chemokines typically not exceeding 17 kDa. The cleavage of FKN is both constitutive and inducible. The membrane‐bound form serves an adhesion function in the context of the vascular immune system, and regulates the firm adhesion of leucocytes without the activation of integrins (Fong et al. 1998). sFKN, however, functions as a chemoattractant for T cells, B cells, Natural Killer (NK) cells and monocytes (Imai et al. 1997; Corcione et al. 2009) and is essential for the trans‐endothelial migration of monocytes that express the FKN receptor (Auffray et al. 2007). The cleavage of FKN and thus its shedding from the cell membrane is dependent on two classes of proteases, namely, metalloproteases (ADAM 10 and ADAM 17), which are expressed by endothelial cells (Hundhausen et al. 2007; Hurst et al. 2009) and the cysteine protease cathepsin S (CatS), which is released by microglia in the spinal cord (Clark et al. 2009). While ADAM 10 regulates the constitutive cleavage of FKN (Hundhausen et al. 2003), cleavage by ADAM 17 and CatS are induced in adverse conditions (Clark et al. 2009; Hurst et al. 2009). The ADAMs and CatS target membrane‐bound FKN at different cleavage sites and therefore give rise to different forms of sFKN, which could serve subtly different functions.
Unlike other, typically promiscuous, chemokines, FKN exclusively activates the CX3CR1 receptor. CX3CR1 was initially identified in humans and rats around 20 years ago (Harrison et al. 1994; Imai et al. 1997) and is a seven transmembrane domain G protein‐coupled receptor, specifically of the Gi and Gz subtypes (Al‐Aoukaty et al. 1998). The expression profile of CX3CR1 has been most extensively characterized using the CX3CR1‐GFP reporter mouse line, with the receptor present in murine monocytes (CD11b+Gr1low), NK cells, myeloid and lymphoid dendritic cells and in a subset of cutaneous Langerhans cells (Jung et al. 2000). In the nervous system specifically, CX3CR1 appears to be exclusively expressed in microglia in the spinal cord and brain (Lindia et al. 2005; Yang et al. 2012; Clark et al. 2013).
One caveat to our current understanding of FKN/CX3CR1 expression is that initial studies in which expression has been characterized have relied entirely on antibodies. In some cases, such antibodies have since been shown to be unreliable in the light of the development of transgenic mice, often giving false‐positives. It is therefore prudent to assume that expression patterns revealed by antibodies are only bona fide expression if confirmed in transgenic mice. Inconsistent findings regarding expression must be interpreted with caution and cannot form the basis of therapy development. Here, we will consider selected, critical studies that have used both antibodies and transgenic lines, in which both approaches have been in agreement with each other. The distribution of FKN and CX3CR1 in the spinal cord, which has indeed been confirmed by both antibodies and the use of transgenic mice, is predominantly neuronal and microglial respectively. This strongly indicates that this signalling pair has the potential to mediate neuron‐microglial communication, both homeostatic and pathological.
CX3CL1/R1 signalling in pre‐clinical models of chronic pain
Central mechanisms
The first synapse in the nociceptive pathway – between the terminals of the primary afferents and dorsal horn neurons in the spinal cord – is a key site at which the modulation of nociceptive signalling occurs. It is well established that damage to peripheral nerves, such as the sciatic nerve, disrupts homoeostasis and consequentially results in heightened response states of microglia and astrocytes in the spinal cord (McMahon and Malcangio 2009). This heightened microglial activity results in an increase in neuron‐microglial communication, which has the capacity to amplify nociceptive transmission, resulting in a chronic pain state. In the dorsal horn of the spinal cord, the distribution of FKN and CX3CR1, neuronal and microglial, respectively, makes this signalling pair an ideal candidate for the mediation of this increase in communication. Indeed, in the last decade, pre‐clinical studies have advanced our understanding of, and established the role of FKN/CX3CR1 signalling in chronic pain; thus, bringing the therapeutic potential of targeting this signalling pathway into prominence.
FKN, sFKN in particular, has been shown to be pronociceptive, with intrathecal administration of the FKN domain, but not the full length FKN, resulting in both thermal and mechanical hypersensitivity (Clark and Malcangio 2012; Clark et al. 2007; Zhuang et al. 2007). This effect is mediated by CX3CR1 activation (Clark et al. 2007; Staniland et al. 2010), which in turn induces intracellular phosphorylation of microglial p38 MAPK (Clark et al. 2007; Zhuang et al. 2007) resulting in the release of proinflammatory mediators such as Interleukin 1β (IL‐1β), Interleukin 6 (IL‐6) and nitric oxide (Milligan et al. 2005).
Because of the pronociceptive nature of sFKN/CX3CR1 signalling, the inhibition of this pathway is intuitively a potential therapeutic strategy for the treatment of chronic pain. Indeed, changes in FKN/CX3CR1 signalling in pre‐clinical models of chronic pain as well as the therapeutic potential of disrupting such signalling, has been extensively investigated. Following injury to peripheral nerves, the expression of CX3CR1 in spinal microglia increases significantly (Zhuang et al. 2007; Staniland et al. 2010). It is important to keep in mind, however, that the elevated levels reported could be because of proliferation of CX3CR1‐expressing microglia and therefore increased expression of CX3CR1‐expressing cells as opposed to elevated CX3CR1 expression per se. Although the general consensus is that the expression of total (membrane‐bound and cleaved) FKN remains unchanged following peripheral nerve injury (Clark et al. 2009; Old et al. 2014), levels of sFKN specifically in CSF do appear to increase (Clark et al. 2009; Nieto et al. 2016). Chronic pain models are therefore accompanied by an increase in both sFKN specifically and CX3CR1.
In various pre‐clinical models of peripheral nerve injury, the intrathecal administration of neutralizing antibodies against either FKN or CX3CR1 have been found to delay or attenuate chronic pain‐associated behaviours (Milligan et al. 2004; Clark et al. 2007; Zhuang et al. 2007) through a reduction in p38 MAPK phosphorylation in microglia and, in turn, a reduction in the release of proinflammatory cytokines and dampening of neuron–glia communication (Zhuang et al. 2007). The therapeutic benefits of CX3CR1 inhibition and thus reduced phosphorylation of microglial p38 MAPK, also extend to other models of chronic pain, such as bone cancer, which is also accompanied by microgliosis and elevation of microglial CX3CR1 and p38 MAPK (Yin et al. 2010; Hu et al. 2012). Indeed, intrathecal delivery of a CX3CR1 neutralizing antibody delayed chronic pain‐associated behaviour in this model as well as reduced microgliosis and the associated increases in CX3CR1 and p38 phosphorylation.
The role of FKN/CX3CR1 modulation of neuron‐microglial signalling in chronic pain has also been investigated using CX3CR1 knock‐out mice, which consistently display deficits in chronic pain in various pre‐clinical models, whether they are traumatic or non‐traumatic. For example, CX3CR1 knock‐out mice that are subjected to partial sciatic nerve ligation (PSNL) display reduced mechanical and thermal hypersensitivity compared to wild‐type mice that undergo the same procedure (Staniland et al. 2010). This deficit in chronic pain‐associated behaviour observed in CX3CR1 knock‐out mice is correlated with a reduction in microglial activity. In the case of PSNL, the infiltration of macrophages in the sciatic nerve, the site of injury, was not altered in CX3CR1 knock‐out mice (Staniland et al. 2010), and so it appears that the reduction in pain behaviour observed in these animals is mediated specifically by CX3CR1‐dependent microglial activity in the spinal cord.
The role of CX3CL1/FKN cleavage by CatS in chronic pain
Because of the fact that sFKN specifically has been found to increase following peripheral nerve injury and is pronociceptive, changes in CatS, which regulates cleavage and hence shedding of FKN to form sFKN, has also received attention in recent years in the context of chronic pain pre‐clinical models. Following injury to peripheral nerves, CatS expression increases in microglia that are located in areas of the spinal cord that are innervated by the injured peripheral afferents (Clark et al. 2007) and it is subsequently released from microglia downstream of P2X7 receptor signalling (Clark et al. 2010). This P2X7‐dependent release of CatS results in the shedding of sFKN from neurons in the dorsal horn, which in turn, signals to CX3CR1‐expressing microglia (Clark et al. 2007).
The pronociceptive effects of CatS have also been established ex vivo. For example, in spinal cord slices, which have been prepared from neuropathic animals and still have the damaged dorsal root attached, electrical stimulation of the injured dorsal roots results in the liberation of FKN, indicative of activated or elevated CatS (Clark et al. 2009). This liberation of sFKN appears to only be apparent under conditions in which the microglia are reactive, for example, injury or lipopolysaccharide (a macrophage activator) exposure (Clark et al. 2009). In addition, the pronociceptive effects of intrathecal delivery of CatS in vivo are prevented by neutralization of spinal FKN and in CX3CR1 knock‐out mice (Clark et al. 2007).
The therapeutic potential of inhibiting CatS appears to be most promising for the maintenance phase of chronic pain as opposed to the initial induction. Both systemic (Irie et al. 2008; Zhang et al. 2014) and intrathecal (Clark et al. 2007) delivery of a CatS inhibitor reversed established injury‐induced neuropathic pain, but was not effective if administered during the initial phase of pain (e.g. day 3 post‐surgery), at which time, CatS expression was relatively low in the spinal cord (Clark et al. 2007; Zhang et al. 2014) and peripherally (Barclay et al. 2007). Furthermore, CatS knock‐out mice demonstrate similar levels of chronic pain to wild‐type controls during initial phases of peripheral injury models but display lower levels of pain relative to wild‐type controls from day 3, at which point, pain is considered to have entered the maintenance phase (Zhang et al. 2014).
The inhibition of CatS has also been shown to have therapeutic potential in non‐surgical pre‐clinical models of chronic pain, for example, murine Rheumatoid Arthritis (RA). Rats with collagen‐induced arthritis displayed mechanical hypersensitivity as well as microglial activation and an elevation of IL‐1β in the CSF, all of which was prevented by intrathecal administration of N‐morpholinurea‐homophenylalanyl‐leucyl‐vinylsulfonemethyl (LHVS), a selective CatS inhibitor (Nieto et al. 2016). CatS‐mediated release of sFKN therefore appears to mediate chronic pain maintenance and could provide a therapeutic target that has potential where other current therapies have shown limited efficacy, acting as an alternative or complementary addition to currently available drugs. Indeed, in a recent study, the use of a CatS inhibitor has been shown to enhance the effects of gabapentin and pregabalin in a PSNL model in rats, as well as resulting in a dose‐dependent attenuation of injury‐induced allodynia when used alone (Hewitt et al. 2016).
Peripheral mechanisms
FKN/CX3CR1 signalling in the spinal cord is a promising therapeutic target; however, it is now thought to not be the only location at which this signalling partnership can mediate chronic pain, with FKN/CX3CR1 signalling in the periphery also being implicated in models of chemotherapy‐induced painful neuropathy (CIPN). In paclitaxel‐treated rats for instance, the expression of FKN has been reported to increase in primary sensory neurons in the DRG in vivo and in vitro and macrophage infiltration into the DRG is reported to increase alongside the development of allodynia (Huang et al. 2014). Intrathecal administration of a FKN‐neutralizing antibody was shown to prevent macrophage recruitment to the DRG and reduced activation of p38 MAPK in macrophages in addition to attenuating paclitaxel‐induced allodynia (Huang et al. 2014). The role of FKN/CX3CR1 signalling in sensory neurons in the DRG, however, should be interpreted with caution because of the fact that FKN expression in the DRG is controversial, with contradictory expression patterns being reported, as discussed above. Indeed, the lack of evidence for sensory neuron expression in the FKN reporter transgenic line (Kim et al. 2011) suggests that FKN is unlikely to be expressed in neurons in the DRG and any analgesics effects of its inhibition could be because of CX3CR1 inhibition in macrophages in the DRG, (Old et al. 2014).
In an alternative model of CIPN, Vincristine‐induced neuropathic pain, FKN/CX3CR1 signalling in macrophages that have been recruited to the sciatic nerve following vincristine treatment has provided an additional therapeutic target (Old et al. 2014). Vincristine treatment triggers the recruitment of CX3CR1‐expressing monocytes/macrophages to the sciatic nerve, with CX3CR1 in this location being activated by endothelial FKN (Fig. 1). This results in the downstream activation of Transient receptor potential cation channel, subfamily A, member 1 (TRPA1) receptors in sensory neurons as well as the generation of reactive oxygen species (Old et al. 2014). In this model of CIPN, CX3CR1 knock‐out mice display a delayed onset in mechanical allodynia, which resembles the delay accompanying depletion of macrophages using low‐dose clodronate liposomes, and a decrease in infiltrating macrophages. Critically, no neuronal damage is observed in this model and there are no changes to microglial activity in the dorsal horn of the spinal cord, indicating that in this particular model, FKN/CX3CR1 signalling in the periphery as opposed to the spinal cord, plays a role in chronic pain (Old et al. 2014).
FKN/CX3CR1 signalling therefore provides a promising prophylactic target for the development of pain. Indeed the high fidelity of the partnership ensures that therapies targeted towards this signalling pair are highly specific relative to other therapeutic approaches. One caveat, however, is that unlike the majority of chemokines, FKN is constitutively expressed in neurons as well as a multitude of cells of the immune system suggesting that it also plays an essential role in homeostatic processes, which could be disrupted if signalling is inhibited and could therefore lead to side‐effects. This highlights the importance of targeting sFKN specifically, which could be achieved by targeting CatS, as opposed to full length FKN or CX3CR1. Nonetheless, the FKN/CX3CR1 signalling partnership provides unique therapeutic potential, which is already being appreciated in chronic pain conditions and indeed in other pathological conditions beyond pain, such as pancreatic disease (D'Haese et al. 2010).
CCL2/MCP‐1
Expression and distribution
Another chemokine, which has received much attention in the context of chronic pain is CCL2. CCL2 or monocyte chemoattractant protein 1 (MCP‐1) recruits monocytes to the site of infection, ischaemia or inflammation. While CCL2 is able to recognize the receptors CCR1, 2 and 4, it shows preference for CCR2 (Jung et al. 2009). The expression of CCL2 in primary sensory neurons has been well studied. Under basal conditions, low levels of CCL2 are constitutively expressed in more than 40% of small and medium DRG neurons, with co‐expression being observed with substance P, Calcitonin gene‐related peptide (CGRP) and the Transient receptor potential cation channel, subfamily V, member 1 (TRPV1) (Tanaka et al. 2004; Dansereau et al. 2008). The expression of CCL2 has been found to increase in several pathological conditions. For instance, following PSNL, the expression of CCL2 increased rapidly in DRG neurons, within 4 h of the injury occurring (Tanaka et al. 2004). Likewise, following nerve constriction, CCL2 expression was found to be induced in both small and large diameter neurons in the DRG that also expressed ATF3, an indicator of neuronal injury (Zhang and De Koninck 2006). The expression of the CCR2 receptor, for which CCL2 has the highest affinity, has also been well documented in several models of chronic pain. For example, in situ hybridization shows that chronic constriction of the DRG results in an increase in the expression of CCR2 mRNA in both neuronal and satellite cells in the compressed DRG as well as the adjacent, non‐compressed DRG (White et al. 2005). An increase in CCR2 expression has also been observed in the DRG following sciatic nerve demyelination (Jung et al. 2009).
Expression of CCL2/R2 in the spinal cord, however, is controversial (Fig. 1). CCL2 has been reported to be constitutively expressed in a variety of cell types, having been observed in primary afferent neurons (Zhang and De Koninck 2006) as well as co‐localizing with the astrocytic marker, Glial fibrillary acidic protein (GFAP) (Gao et al. 2009). Expression in astrocytes appears to increase following the induction of different injury models, for example, spinal nerve ligation (Gao et al. 2009), spinal cord contusion injury (Knerlich‐Lukoschus et al. 2008) and demyelinating lesions (Van Der Voorn et al. 1999). Thus, CCL2 expression in astrocytes appears to be elevated in the spinal cord following neuronal injury, regardless of the nature. The expression profile of CCR2 remains debated, with different detection methods producing conflicting results. In CCR2‐GFP reporter mice, for example, a weak but positive GFP signal is apparent in dorsal horn neurons. In situ hybridization, however, suggests an absence of CCR2 mRNA in the spinal cord under basal conditions (Jung and Miller 2008). What has been more consistently reported, however, is the elevation of CCR2 in the spinal cord following nerve injury. For instance, an increase in CCR2 mRNA in the deep dorsal horn and motor neurons has been reported 3 days after spinal nerve injury (Gao et al. 2009). Furthermore, an elevation in CCR2 has been reported in spinal microglia following sciatic nerve injury (Abbadie et al. 2003; Thacker et al. 2009) as well as in astrocytes following spinal cord contusion (Knerlich‐Lukoschus et al. 2008). However, the reliability of antibodies used to detect the presence of CCR2 has been recently doubted and the precise expression profile of CCR2 in the spinal cord has yet to be firmly established.
The fact that both CCL2 and CCR2 have previously been suggested to increase in pre‐clinical models of pain and were initially thought to be located in neurons and microglia, respectively (although expression is debatable and not exclusive to one cell type), makes this signalling pair another potential player in the mechanisms underlying chronic pain, which has received considerable attention over the last decade.
CCL2/R2 signalling in pre‐clinical models of chronic pain
Initial studies investigating CCL2/CCR2 signalling in the spinal cord suggested that this signalling pair played a considerable role in neuron‐microglial communication in chronic pain animal models. For instance, in ex vivo preparations of the dorsal horn of the spinal cord, CCL2 levels obtained from spinal cord superfusates, while similar in unstimulated naive and neuropathic animals, were only elevated as a result of supramaximal electrical stimulation of the dorsal root in neuropathic animals, which also displayed heightened mechanical hypersensitivity (Thacker et al. 2009). In addition, application of intraspinal CCL2 in naïve rats resulted in chronic pain‐like behaviour, while intrathecal delivery of a CCL2‐neutralizing antibody inhibited injury‐associated pain behaviour (Thacker et al. 2009). This suggests that CCL2 is released in an activity‐dependent manner and in turn is able to regulate nociceptive signalling.
CCL2 has also been suggested to play a role in non‐traumatic pre‐clinical models of chronic pain, such as CIPN. In the paclitaxel model of CIPN, for instance, CCL2 is reported to be elevated in the spinal cord of paclitaxel‐treated mice, which display paclitaxel‐induced cold hyperalgesia. Intrathecal administration of an antibody against CCL2 not only completely suppressed the hyperalgesia, but also inhibited paclitaxel‐induced microglial activation (Pevida et al. 2013). This suggests that cold hyperalgesia associated with paclitaxel therapy could be treated by targeting CCL2‐mediated activation of microglia (either directly or indirectly). CCL2 signals through both CCR2 and CCR1 and while CCR2 inhibition appears to alleviate paclitaxel‐induced allodynia, CCR1 inhibition does not (Pevida et al. 2013). This suggests that the pronociceptive effects of CCL2 are mediated via the CCR2 receptor. A caveat to this finding, however, is that the CCR2 inhibitor in the paclitaxel mouse model has been shown to have additional effects on NMDA receptor subunit expression and neuronal nitric oxide synthase (Ren et al. 2015). Both neuronal nitric oxide synthase and the NMDA receptor subunit; NR2B have been found to increase and be associated with pain‐like behaviour in a model of bone cancer chronic pain and they are down‐regulated in parallel to the alleviation of allodynia when the CCR2 inhibitor is administered (Ren et al. 2015). It is therefore possible that the analgesic effects of CCR2 inhibitors in some pre‐clinical models are not because of the inhibition of CCL2/R2 signalling, but additional, non‐specific effects, which could weaken the prominence of CCL2 signalling in the spinal cord in the underlying mechanisms of pain.
CCL2 expressed elsewhere, however, for example, the DRG, could still be involved in mechanisms underlying other forms of pain that are induced by paclitaxel. Paclitaxel has been shown to induce the expression of CCL2 and CCR2 in the primary sensory neurons of the DRG, specifically small, nociceptive neurons and medium‐sized neurons respectively (Zhang et al. 2013). Local blockade of CCL2/R2 signalling using either a neutralizing antibody against CCL2 or a CCR2 antisense oligodeoxynucleotide significantly reduced paclitaxel‐ target for CIPN.
While there is strong associated allodynia (Zhang et al. 2013), suggesting that neuronal CCL2/R2 signalling in the DRG could provide a therapeutic evidence to suggest that CCL2/R2 signalling in the DRG, and potentially the spinal cord, could play a role in chronic pain, the debatable and non‐exclusive expression profiles of both CCL2 and CCR2 currently limits their potential as therapeutic targets and this signalling partnership is in need of further investigation and clarification in order to be translated to the clinic.
Other chemokines in neuropathic pain
Although CX3CL1/FKN and CCL2 are the most extensively studied chemokines in the context of chronic pain, other chemokines are now emerging as potential regulators and thus provide the opportunity for more novel therapies to be developed.
CCL7/MCP‐3
CCL7, otherwise known as MCP‐3, has been shown in the last few years to play a role in the development of chronic pain in some pre‐clinical models. Following partial sciatic nerve ligation, the expression of CCL7 is elevated in astrocytes located in the dorsal horn of the spinal cord up to 2 weeks following the day of surgery (Imai et al. 2013). This elevation appears to be dependent on the proinflammatory cytokine; IL‐6, with IL‐6 knock‐out mice showing no changes in CCL7 expression after PSNL. Indeed, intrathecal delivery of recombinant IL‐6 into IL‐6 knock‐out mice results in an increase in CCL7 mRNA. Both in vitro and in vivo CCL7 treatment results in microglial activation, which is suppressed in vivo by intrathecal administration of a neutralizing antibody against CCL7. In addition, to the suppression of microglial activation, pain behaviours are also reduced (Imai et al. 2013). Because of the expression pattern of CCL7, it is therefore possible that it mediates an astrocyte‐microglia interaction in the context of chronic pain. Indeed, this has recently been shown to be the case in a spinal nerve ligation model of chronic pain (Ke et al. 2016). Following spinal nerve ligation, CCL7 expression in astrocytes in the dorsal horn of the spinal cord increased in correlation with the development of pain. Silencing CCL7 reduced surgery‐induced pain and in vitro, CCL7 has been shown to be sufficient for the proliferation of astroglial cells (Ke et al. 2016). CCL 7 is therefore suggested to be essential for the development of chronic pain via the promotion of astrocyte proliferation.
CCL3 and CCL4/MIP‐1b
CCL3 has also been recently suggested to play a role in the development of chronic pain in both traumatic (surgical) and non‐traumatic (non‐surgical) pre‐clinical models. In a model utilizing chronic constriction injury of the sciatic nerve, both CCL3 and its receptor CCR5 were found to increase alongside microglial activation and the development of both thermal and mechanical hypersensitivity (Sun et al. 2016). Intrathecal delivery of a CCL3‐neutralizing antibody suppressed the up‐regulation of proinflammatory cytokines such as Tumor necrosis factor alpha (TNFα) and IL‐1β as well as the activation of microglial p38 MAPK, indicating that CCL3 regulates, at least in part, the release of proinflammatory cytokines and microglial activation. Furthermore, in CCR5 knock‐out mice, pain behaviour was suppressed (Sun et al. 2016). It is important to keep in mind, however, that CCR5 is not exclusively activated by CCL3 and the effects of the CCR5 knockout could be a result of disrupting the signalling of other chemokines such as CCL4 and CXCL13.
CCL3 has also been implicated in non‐surgical models of chronic pain, for instance, CIPN. Following the treatment of rats with paclitaxel, and the consequential development of mechanical allodynia, microglial activation was reported to increase in the dorsal horn of the spinal cord along with mRNA for both CCL3 and CCR5 (Fig. 1; Ochi‐ishi et al. 2014). The increase in CCL3 is potentially a result of paclitaxel‐induced increase in P2X7 receptors, which are suggested to trigger the release of CCL3 from microglia (Kataoka et al. 2009). Intrathecal administration of a CCL3‐neutralizing antibody not only appeared to attenuate paclitaxel‐induced allodynia, but reverse allodynia during the maintenance phase, indicating that CCL3 could play a role in both induction and maintenance of chronic pain potentially making CCL3 a versatile therapeutic target.
CCL4, otherwise known as macrophage inflammatory protein 1b shares the CCR5 receptor with CCL3 and has also been implicated in the regulation of chronic pain in recent years. Following the induction of a partial sciatic nerve ligation, CCL4 mRNA increases in both macrophages and Schwann cells in the periphery as early as day 1 post‐surgery (Fig. 1; Saika et al. 2012). Following local administration of a CCL4‐neutralizing antibody to the region surrounding the sciatic nerve, both mechanical and thermal allodynia that occurred as a result of surgery, were prevented. In addition, PSNL‐induced up‐regulation of inflammatory chemo/cytokines was also prevented, suggesting that CCL4 could potentially play a peripheral role in the regulation of chronic pain via regulation of proinflammatory chemo/cytokine release. In addition, administration of the CCR5 antagonist also prevented allodynia in this model (Saika et al. 2012). As discussed, however, CCR5 is activated by multiple chemokines and therefore targeting CCR5 will lack the specificity that targeting CCL3 or CCL4, specifically would have.
CCL21
CCL21, another member of the CC subfamily, is another chemokine that has been suggested to play a role in the mechanisms underlying chronic pain. Following peripheral nerve injury, CCL21 expression is rapidly induced in small‐diameter primary sensory neurons and is transported to central terminals in the dorsal horn, where it is suggested to increase the expression of the P2X4 receptor in microglia (Biber et al. 2011). Intrathecal administration of a CCL21‐blocking antibody reduced injury‐induced allodynia suggesting that it, at least in part, mediates nociceptive signalling in this pre‐clinical model. Indeed, CCL21 knock‐out mice do not develop allodynia and in these mice, P2X4 receptor expression in microglia does not significantly increase following injury compared to control mice. Critically, CCL21 injection into CCL21 knock‐out mice, results in the same development of injury‐induced allodynia as that seen in wild‐type controls (Biber et al. 2011). In addition, CCL21 administration in vivo as well as application to microglia in vitro, is sufficient to increase microglial P2X4 receptor expression. CCL21 has been found to activate both the CXCR3 and CCR7 receptors, which are expressed by microglia constitutively and following microglial activation respectively (Biber et al. 2001; Rappert et al. 2002; Dijkstra et al. 2004). While knocking out CXCR3 appears to have no effect on injury‐induced mechanical allodynia, CCR7 knock‐outs display a delayed onset of allodynia (Biber et al. 2011), suggesting that pronociceptive actions of CCL21 are more likely to be elicited via activation of the CCR7 receptor.
CXCL13
The most recent chemokine to receive attention in the context of chronic pain is CXCL13. In the last year, CXCL13 has been shown to regulate chronic pain in both partial infraorbital nerve ligation, resulting in trigeminal neuropathic pain (Zhang et al. 2016) and spinal nerve ligation (Jiang et al. 2016). In the former, both CXCL13 and its receptor CCR5 were found to increase, with the inhibition of either using a shRNA lentivirus, attenuating allodynia. In the latter, CXCL13 expression was found to increase in the spinal cord, and again, suppression of expression using a shRNA lentivirus reduced the injury‐associated allodynia. The potential regulation of pain by CXCL13 has been tentatively suggested to occur via the activation of astrocytes in the dorsal horn, with intrathecal injection of CXCL13 resulting in the activation of astrocytes in naive mice (Jiang et al. 2016).
Conclusion
Chronic pain is a debilitating condition, which is experienced even when the painful stimulus has been resolved. At present, clinically available treatments for chronic pain have limited efficacy and undesirable side‐effects. More effective, novel therapeutic approaches are therefore needed. Although the mechanisms underlying chronic pain were initially thought to be neurocentric, it is now accepted that non‐neuronal cells also play a role in nociceptive signalling and the communication between neurons and non‐neuronal cells in the context of chronic pain has received more attention in the last decade. One of the signalling pathways known to mediate the communication between neurons and neighbouring non‐neuronal cells is chemokine signalling. Here, we considered a selection of chemokines (Fig. 1), which, along with their receptors, have been reported to increase in a variety of traumatic and non‐traumatic pre‐clinical models of chronic pain. Their therapeutic potential has been realized in the light of the reversal of pain that accompanies their inhibition. Each chemokine that has been suggested to play a role in chronic pain comes with its promises and controversies in terms of the development of therapies. In the case of CX3CL1, for example, the high fidelity of its signalling and exclusive expression profile means that drugs that are targeted to this signalling partnership will have highly specific effects. While the debatable and variable expression profile of CCL2/R2 means that currently, targeted therapies will be difficult to develop.
At present, although chemokine signalling inhibitors are in their infancy in the clinic, interest has been rapidly expanding and chemokines, particularly CX3CL1 are becoming more recognized as innovative therapeutic targets for a number of pathological conditions. The CX3CL1 inhibitor, AZD8797, for example, which was originally developed for use in a pre‐clinical model of multiple sclerosis (Ridderstad Wollberg et al. 2014), has shown promise in terms of safety and toxicology in humans and its use in cancer pain may be being considered, signifying that a new chapter of drug development has already begun, with the CX3CL1/CX3CR1 signalling partnership leading the way.
Acknowledgments and conflict of interest disclosure
This work was funded by Medical Research Council (MR/M023893/1). The authors have no conflict of interest to declare.
References
- Abbadie C., Lindia J. A., Cumiskey A. M., Peterson L. B., Mudgett J. S., Bayne E. K., DeMartino J. A., MacIntyre D. E. and Forrest M. J. (2003) Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2 . Proc Natl Acad Sci U S A. 100, 7947–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Aoukaty A., Rolstad B., Giaid A. and Maghazachi A. A. (1998) MIP‐3alpha, MIP‐3beta and fractalkine induce the locomotion and the mobilization of intracellular calcium, and activate the heterotrimeric G proteins in human natural killer cells. Immunology 95, 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffray C., Fogg D., Garfa M., Elain G., Join‐Lambert O., Kayal S., Sarnacki S., Cumano A., Lauvau G. and Geissmann F. (2007) Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317, 666–670. [DOI] [PubMed] [Google Scholar]
- Bajetto A., Bonavia R., Barbero S. and Schettini G. (2002) Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J. Neurochem. 82, 1311–1329. [DOI] [PubMed] [Google Scholar]
- Barclay J., Clark A. K., Ganju P. et al (2007) Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain 130, 225–234. [DOI] [PubMed] [Google Scholar]
- Biber K., Sauter A., Brouwer N., Copray S. C. and Boddeke H. W. (2001) Ischemia‐induced neuronal expression of the microglia attracting chemokine Secondary Lymphoid‐tissue Chemokine (SLC). Glia 121–133. [DOI] [PubMed] [Google Scholar]
- Biber K., Tsuda M., Tozaki‐Saitoh H., Tsukamoto K., Toyomitsu E., Masuda T., Boddeke H. and Inoue K. (2011) Neuronal CCL21 up‐regulates microglia P2X4 expression and initiates neuropathic pain development. EMBO J. 30, 1864–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. K., Yip P. K., Grist J. et al (2007) Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 104, 10655–10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. K. and Malcangio M. (2012) Microglial signalling mechanisms: cathepsin S and fractalkine. Exp. Neurol. 234, 283–292. [DOI] [PubMed] [Google Scholar]
- Clark A. K., Yip P. K. and Malcangio M. (2009) The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J. Neurosci. 29, 6945–6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. K., Old E. A. and Malcangio M. (2013) Neuropathic pain and cytokines: current perspectives. J Pain Res. 6, 803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combadiere C., Ahuja S. K., Tiffany H. L. and Murphy P. M. (1996) Cloning and functional expression of CC CKR5, a human monocyte CC chemokine receptor selective for MIP‐1(alpha), MIP‐1(beta), and RANTES. J. Leukoc. Biol. 147–152. [DOI] [PubMed] [Google Scholar]
- Corcione A., Ferretti E., Bertolotto M. et al (2009) CX3CR1 is expressed by human B lymphocytes and mediates [corrected] CX3CL1 driven chemotaxis of tonsil centrocytes. PLoS ONE 4, e8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansereau M. A., Gosselin R. D., Pohl M. et al (2008) Spinal CCL2 pronociceptive action is no longer effective in CCR2 receptor antagonist‐treated rats. J. Neurochem. 106, 757–769. [DOI] [PubMed] [Google Scholar]
- Davies M., Brophy S., Williams R. and Taylor A. (2006) Painful diabetic polyneuropathy: epidemiology, pain description, and quality of life. Diabetes Res. Clin. Pract. 47, 123–128. [DOI] [PubMed] [Google Scholar]
- D'Haese J. G., Demir I. E., Friess H. and Ceyhan G. O. (2010) Fractalkine/CX3CR1: why a single chemokine‐receptor duo bears a major and unique therapeutic potential. Expert Opin Ther Targets 14, 207–219. [DOI] [PubMed] [Google Scholar]
- Dijkstra I. M., Hulshof S., van der Valk P., Boddeke H. W. and Biber K. (2004) Cutting edge: activity of human adult microglia in response to CC chemokine ligand 21. J Immunol. 172, 2744–2747. [DOI] [PubMed] [Google Scholar]
- Dougherty P. M., Cata J. P., Burton A. W., Vu K. and Weng H. R. (2007) Dysfunction in multiple primary afferent fiber subtypes revealed by quantitative sensory testing in patients with chronic vincristine‐induced pain. J. Pain Symptom Manage. 33, 166–179. [DOI] [PubMed] [Google Scholar]
- Fong A. M., Robinson L. A., Steeber D. A., Tedder T. F., Yoshie O., Imai T. and Patel D. D. (1998) Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J. Exp. Med. 188, 1413–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foussat A., Coulomb‐L'Hermine A., Gosling J., Krzysiek R., Durand‐Gasselin I., Schall T., Balian A., Richard Y., Galanaud P. and Emilie D. (2000) Fractalkine receptor expression by T lymphocyte subpopulations and in vivo production of fractalkine in human. Eur J Immunol. 30, 87–97. [DOI] [PubMed] [Google Scholar]
- Gao Y. J., Zhang L., Samad O. A., Suter M. R., Yasuhiko K., Xu Z. Z., Park J. Y., Lind A. L., Ma Q. and Ji R. R. (2009) JNK‐induced MCP‐1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J. Neurosci. 29, 4096–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García J. J., Cidoncha A., Bote M. E., Hinchado M. D. and Ortega E. (2013) Altered profile of chemokines in fibromyalgia patients. Ann. Clin. Biochem. 51(Pt 5), 576–581. [DOI] [PubMed] [Google Scholar]
- Harrison J. K., Barber C. M. and Lynch K. R. (1994) cDNA cloning of a G‐protein‐coupled receptor expressed in rat spinal cord and brain related to chemokine receptors. Neurosci. Lett. 169, 85–89. [DOI] [PubMed] [Google Scholar]
- Harrison J. K., Jiang Y., Wees E. A., Salafranca M. N., Liang H. X., Feng L. and Belardinelli L. (1999) Inflammatory agents regulate in vivo expression of fractalkine in endothelial cells of the rat heart. J Leukoc Biol. 66, 937–44. [DOI] [PubMed] [Google Scholar]
- Hewitt E., Pitcher T., Rizoska B. et al (2016) Selective cathepsin S inhibition with MIV‐247 attenuates mechanical allodynia and enhances the antiallodynic effects of gabapentin and pregabalin in a mouse model of neuropathic pain. J. Pharmacol. Exp. Ther. 387–396. [DOI] [PubMed] [Google Scholar]
- Hu J. H., Yang J. P., Liu L., Li C. F., Wang L. N., Ji F. H. and Cheng H. (2012) Involvement of CX3CR1 in bone cancer pain through the activation of microglia p38 MAPK pathway in the spinal cord. Brain Res. 1465, 1–9. [DOI] [PubMed] [Google Scholar]
- Huang Z. Z., Li D., Liu C. C., Cui Y., Zhu H. Q., Zhang W. W., Li Y. Y. and Xin W. J. (2014) CX3CL1‐mediated macrophage activation contributed to paclitaxel‐induced DRG neuronal apoptosis and painful peripheral neuropathy. Brain Behav. Immun. 40, 155–165. [DOI] [PubMed] [Google Scholar]
- Hundhausen C., Misztela D., Berkhout T. A. et al (2003) The disintegrin‐like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1‐mediated cell‐cell adhesion. Blood 102, 1186–1195. [DOI] [PubMed] [Google Scholar]
- Hundhausen C., Schulte A., Schulz B. et al (2007) Regulated shedding of transmembrane chemokines by the disintegrin and metalloproteinase 10 facilitates detachment of adherent leukocytes. J Immunol. 178, 8064–8072. [DOI] [PubMed] [Google Scholar]
- Hurst L. A., Bunning R. A., Couraud P. O., Romero I. A., Weksler B. B., Sharrack B. and Woodroofe M. N. (2009) Expression of ADAM‐17, TIMP‐3 and fractalkine in the human adult brain endothelial cell line, hCMEC/D3, following pro‐inflammatory cytokine treatment. J. Neuroimmunol. 210, 108–112. [DOI] [PubMed] [Google Scholar]
- Imai T., Hieshima K., Haskell C. et al (1997) Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 91, 521–530. [DOI] [PubMed] [Google Scholar]
- Imai S. et al (2013) Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long‐lasting neuropathic pain. Brain 136, 828–843. [DOI] [PubMed] [Google Scholar]
- Irie O. et al (2008) Discovery of orally bioavailable cathepsin S inhibitors for the reversal of neuropathic pain. J. Med. Chem. 51, 5502–5505. [DOI] [PubMed] [Google Scholar]
- Jiang B. C., Cao D. L., Zhang X. et al (2016) CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest. 126, 745–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H. and Miller R. J. (2008) Activation of the nuclear factor of activated T‐cells (NFAT) mediates upregulation of CCR2 chemokine receptors in dorsal root ganglion (DRG) neurons: a possible mechanism for activity‐dependent transcription in DRG neurons in association with neuropathic pain. Mol. Cell Neurosci. 37, 170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S., Aliberti J., Graemmel P., Sunshine M. J., Kreutzberg G. W., Sher A. and Littman D. R. (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H., Bhangoo S., Banisadr G., Freitag C., Ren D., White F. A. and Miller R. J. (2009) Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. J. Neurosci. 29, 8051–8062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka A., Tozaki‐Saitoh H., Koga Y., Tsuda M. and Inoue K. (2009) Activation of P2X7 receptors induces CCL3 production in microglial cells through transcription factor NFAT. J. Neurochem. 108, 115–125. [DOI] [PubMed] [Google Scholar]
- Ke B. C., Huang X. X., Li Y., Li L. Y., Xu Q. X., Gao Y., Liu Y. and Sun Luo J. (2016) Neuronal‐derived Ccl7 drives neuropathic pain by promoting astrocyte proliferation. NeuroReport 27, 849–857. [DOI] [PubMed] [Google Scholar]
- Kim K. W., Vallon‐Eberhard A., Zigmond E., Farache J., Shezen E., Shakhar G., Ludwig A., Lira S. A. and Jung S. (2011) In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood 118, e156–e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knerlich‐Lukoschus F., Juraschek M., Blömer U., Lucius R., Mehdorn H. M. and Held‐Feindt J. (2008) Force‐dependent development of neuropathic central pain and time‐related CCL2/CCR2 expression after graded spinal cord contusion injuries of the rat. J. Neurotrauma 25, 427–448. [DOI] [PubMed] [Google Scholar]
- Kopydlowski K. M., Salkowski C. A., Cody M. J., van Rooijen N., Major J., Hamilton T. A. and Vogel S. N. (1999) Regulation of macrophage chemokine expression by lipopolysaccharide in vitro and in vivo. J Immunol. 1537–1544. [PubMed] [Google Scholar]
- Kuca‐Warnawin E. H., Kurowska W. J., Radzikowska A., Massalska M. A., Burakowski T., Kontny E., Słowińska I., Gasik R. and Maśliński W. (2016) Different expression of chemokines in rheumatoid arthritis and osteoarthritis bone marrow. Reumatologia. 54, 51–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel S. L. (1999) Promiscuous chemokine receptors and their redundant ligands play an enigmatic role during HIV‐1 infection. Am. J. Respir. Cell Mol. Biol. 20, 859–860. [DOI] [PubMed] [Google Scholar]
- Laing K. J. and Secombes C. J. (2004) Chemokines. Mol. Immunol. 28, 793–808. [DOI] [PubMed] [Google Scholar]
- Lindia J. A., McGowan E., Jochnowitz N. and Abbadie C. (2005) Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain. 6, 434–438. [DOI] [PubMed] [Google Scholar]
- Luster A. D. (1998) Chemokines–chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 338, 436–445. [DOI] [PubMed] [Google Scholar]
- McMahon S. B. and Malcangio M. (2009) Current challenges in glia‐pain biology. Neuron 64, 46–54. [DOI] [PubMed] [Google Scholar]
- Miller R. J., Rostene W., Apartis E., Banisadr G., Biber K., Milligan E. D., White F. A. and Zhang J. (2008) Chemokine action in the nervous system. J. Neurosci. 11792–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan E. D., Zapata V., Chacur M. et al (2004) Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci. 20, 2294–2302. [DOI] [PubMed] [Google Scholar]
- Milligan E., Zapata V., Schoeniger D., Chacur M., Green P., Poole S., Martin D., Maier S. F. and Watkins L. R. (2005) An initial investigation of spinal mechanisms underlying pain enhancement induced by fractalkine, a neuronally released chemokine. Eur. J. Neurosci. 22, 2775–2782. [DOI] [PubMed] [Google Scholar]
- Muehlhoefer A., Saubermann L. J., Gu X., Luedtke‐Heckenkamp K., Xavier R., Blumberg R. S., Podolsky D. K., MacDermott R. P. and Reinecker H. C. (2000) Fractalkine is an epithelial and endothelial cell‐derived chemoattractant for intraepithelial lymphocytes in the small intestinal mucosa. J Immunol. 164, 3368–3376. [DOI] [PubMed] [Google Scholar]
- Nibbs R. J., Yang J., Landau N. R., Mao J. H. and Graham G. J. (1999) LD78beta, a non‐allelic variant of human MIP‐1alpha (LD78alpha), has enhanced receptor interactions and potent HIV suppressive activity. J. Biol. Chem. 274, 17478–17483. [DOI] [PubMed] [Google Scholar]
- Nieto F. R., Clark A. K., Grist J., Hathway G. J., Chapman V. and Malcangio M. (2016) Neuron‐immune mechanisms contribute to pain in early stages of arthritis. J. Neuroinflammation 13, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi‐ishi R., Nagata K., Inoue T., Tozaki‐Saitoh H., Tsuda M. and Inoue K. (2014) Involvement of the chemokine CCL3 and the purinoceptor P2X7 in the spinal cord in paclitaxel‐induced mechanical allodynia. Mol. Pain. 10, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Old E. A., Nadkarni S., Grist J., Gentry C., Bevan S., Kim K. W., Mogg A. J., Perretti M. and Malcangio M. (2014) Monocytes expressing CX3CR1 orchestrate the development of vincristine‐induced pain. J Clin Invest. 124, 2023–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim J. J., Zachariae C. O., Mukaida N. and Matsushima K. (1991) Properties of the novel proinflammatory supergene “intercrine” cytokine family. Annu. Rev. Immunol. 617–648. [DOI] [PubMed] [Google Scholar]
- Papadopoulos E. J., Sassetti C., Saeki H., Yamada N., Kawamura T., Fitzhugh D. J., Saraf M. A., Schall T., Blauvelt A., Rosen S. D. and Hwang S. T. (1999) Fractalkine, a CX3C chemokine, is expressed by dendritic cells and is up‐regulated upon dendritic cell maturation. Eur J Immunol. 29, 2551–2559. [DOI] [PubMed] [Google Scholar]
- Pevida M., Lastra A., Hidalgo A., Baamonde A. and Menéndez L. (2013) Spinal CCL2 and microglial activation are involved in paclitaxel‐evoked cold hyperalgesia. Brain Res. Bull. 95, 21–27. [DOI] [PubMed] [Google Scholar]
- Ramesh G., MacLean A. G. and Philipp M. T. (2013) Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. 2013, 480739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raport C. J., Gosling J., Schweickart V. L., Gray P. W. and Charo I. F. (1996) Molecular cloning and functional characterization of a novel human CC chemokine receptor (CCR5) for RANTES, MIP‐1beta, and MIP‐1alpha. J. Biol. Chem. 271, 17161–17166. [DOI] [PubMed] [Google Scholar]
- Rappert A., Biber K., Nolte C., Lipp M., Schubel A., Lu B., Gerard N. P., Gerard C., Boddeke H. W. and Kettenmann H. (2002) Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl‐ current and chemotaxis in murine microglia. J Immunol. 168, 3221–3226. [DOI] [PubMed] [Google Scholar]
- Ren F., Jiao H. and Cai H. (2015) Analgesic effect of intrathecal administration of chemokine receptor CCR2 antagonist is related to change in spinal NR2B, nNOS, and SIGIRR expression in rat with bone cancer pain. Cell Biochem. Biophys. 611–616. [DOI] [PubMed] [Google Scholar]
- Ridderstad Wollberg A., Ericsson‐Dahlstrand A., Juréus A. et al (2014) Pharmacological inhibition of the chemokine receptor CX3CR1 attenuates disease in a chronic‐relapsing rat model for multiple sclerosis. Proc Natl Acad Sci U S A. 5409–5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saika F., Kiguchi N., Kobayashi Y., Fukazawa Y. and Kishioka S. (2012) CC‐chemokine ligand 4/macrophage inflammatory protein‐1β participates in the induction of neuropathic pain after peripheral nerve injury. Eur. J. Pain. 1209–1210. [DOI] [PubMed] [Google Scholar]
- Samson M., Labbe O., Mollereau C., Vassart G. and Parmentier M. (1996) Molecular cloning and functional expression of a new human CC‐chemokine receptor gene. Biochemistry 35, 3362–3367. [DOI] [PubMed] [Google Scholar]
- Slade G. D., Conrad M. S., Diatchenko L. et al (2011) Cytokine biomarkers and chronic pain: association of genes, transcription, and circulating proteins with temporomandibular disorders and widespread palpation tenderness. Pain 152, 2802–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staniland A. A., Clark A. K., Wodarski R., Sasso O., Maione F., D'Acquisto F. and Malcangio M. (2010) Reduced inflammatory and neuropathic pain and decreased spinal microglial response in fractalkine receptor (CX3CR1) knockout mice. J. Neurochem. 114, 1143–1157. [DOI] [PubMed] [Google Scholar]
- Sun S., Chen D., Lin F., Chen M., Yu H., Hou L. and Li C. (2016) Role of interleukin‐4, the chemokine CCL3 and its receptor CCR5 in neuropathic pain. Mol Immunol. 77, 184–192. [DOI] [PubMed] [Google Scholar]
- Tanaka T., Minami M., Nakagawa T. and Satoh M. (2004) Enhanced production of monocyte chemoattractant protein‐1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci. Res. 48, 463–469. [DOI] [PubMed] [Google Scholar]
- Thacker M. A., Clark A. K., Bishop T., Grist J., Yip P. K., Moon L. D., Thompson S. W., Marchand F. and McMahon S. B. (2009) CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur. J. Pain 13, 263–272. [DOI] [PubMed] [Google Scholar]
- Uebelacker L. A., Weisberg R. B., Herman D. S., Bailey G. L., Pinkston‐Camp M. M. and Stein M. D. (2015) Chronic pain in HIV‐infected patients: relationship to depression, substance use, and mental health and pain treatment. Pain Med. 16, 1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Voorn P., Tekstra J., Beelen R. H., Tensen C. P., Van Der Valk P. and De Groot C. J. (1999) Expression of MCP‐1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am. J. Pathol. 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verge G. M., Milligan E. D., Maier S. F., Watkins L. R., Naeve G. S. and Foster A. C. (2004) Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 20, 1150–1160. [DOI] [PubMed] [Google Scholar]
- Watson J. C. and Sandroni P. (2016) Central neuropathic pain syndromes. Mayo Clin. Proc. 372–385. [DOI] [PubMed] [Google Scholar]
- Wells T. N., Power C. A., Shaw J. P. and Proudfoot A. E. (2006) Chemokine blockers–therapeutics in the making? Trends Pharmacol. Sci. 27, 41–47. [DOI] [PubMed] [Google Scholar]
- White F. A., Sun J., Waters S. M., Ma C., Ren D., Ripsch M., Steflik J., Cortright D. N., Lamotte R. H. and Miller R. J. (2005) Excitatory monocyte chemoattractant protein‐1 signaling is up‐regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. 14092–14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe F., Häuser W., Hassett A. L., Katz R. S. and Walitt B. T. (2011) The development of fibromyalgia–I: examination of rates and predictors in patients with rheumatoid arthritis (RA). Pain 152, 291–299. [DOI] [PubMed] [Google Scholar]
- Yang J. L., Xu B., Li S. S., Zhang W. S., Xu H., Deng X. M. and Zhang Y. Q. (2012) Gabapentin reduces CX3CL1 signaling and blocks spinal microglial activation in monoarthritic rats. Mol Brain. 5, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q., Cheng W., Cheng M. Y., Fan S. Z. and Shen W. (2010) Intrathecal injection of anti‐CX3CR1 neutralizing antibody delayed and attenuated pain facilitation in rat tibial bone cancer pain model. Behav. Pharmacol. 21, 595–601. [DOI] [PubMed] [Google Scholar]
- Zhang J. and De Koninck Y. (2006) Spatial and temporal relationship between monocyte chemoattractant protein‐1 expression and spinal glial activation following peripheral nerve injury. J. Neurochem. 97, 772–783. [DOI] [PubMed] [Google Scholar]
- Zhang H., Boyette‐Davis J. A., Kosturakis A. K., Li Y., Yoon S. Y., Walters E. T. and Dougherty P. M. (2013) Induction of monocyte chemoattractant protein‐1 (MCP‐1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel‐induced peripheral neuropathy. J Pain. 14, 1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Wu Z., Hayashi Y., Okada R. and Nakanishi H. (2014) Peripheral role of cathepsin S in Th1 cell‐dependent transition of nerve injury‐induced acute pain to a chronic pain state. J. Neurosci. 34, 3013–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Cao D. L., Zhang Z. J., Jiang B. C. and Gao Y. J. (2016) Chemokine CXCL13 mediates orofacial neuropathic pain via CXCR5/ERK pathway in the trigeminal ganglion of mice. J. Neuroinflammation 13, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Z. Y., Kawasaki Y., Tan P. H., Wen Y. R., Huang J. and Ji R. R. (2007) Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury‐induced cleavage of fractalkine. Brain Behav. Immun. 21, 642–651. [DOI] [PMC free article] [PubMed] [Google Scholar]