

Abstract
Polymorphism in cis-regulatory sequences can lead to different levels of expression for the two alleles of a gene, providing a starting point for the evolution of gene expression. Little is known about the genome-wide abundance of genetic variation in gene regulation in natural populations but analysis of allele-specific expression (ASE) provides a means for investigating such variation. We performed RNA-seq of multiple tissues from population samples of two closely related flycatcher species and developed a Bayesian algorithm that maximizes data usage by borrowing information from the whole data set and combines several SNPs per transcript to detect ASE. Of 2,576 transcripts analyzed in collared flycatcher, ASE was detected in 185 (7.2%) and a similar frequency was seen in the pied flycatcher. Transcripts with statistically significant ASE commonly showed the major allele in >90% of the reads, reflecting that power was highest when expression was heavily biased toward one of the alleles. This would suggest that the observed frequencies of ASE likely are underestimates. The proportion of ASE transcripts varied among tissues, being lowest in testis and highest in muscle. Individuals often showed ASE of particular transcripts in more than one tissue (73.4%), consistent with a genetic basis for regulation of gene expression. The results suggest that genetic variation in regulatory sequences commonly affects gene expression in natural populations and that it provides a seedbed for phenotypic evolution via divergence in gene expression.
Keywords: ASE, gene expression evolution, regulatory sequences, RNA-seq
Introduction
The evolution of gene expression is likely to be key to phenotypic evolution (King and Wilson 1975; Stern and Orgogozo 2008). However, variation in gene expression and the evolutionary forces behind this variation are much less well understood than the evolution of diversity and divergence in gene sequences (Wray 2007). One obvious reason for this difference is that gene expression is itself a phenotype that varies both because of underlying genetic diversity and environmental factors (Pastinen 2010; Buil et al. 2015). Moreover, defining a neutral null hypothesis for the evolution of gene expression is not straightforward and, as a consequence, makes inference of selection on gene expression a challenging task (Gilad et al. 2006; Fay and Wittkopp 2008).
A starting point for addressing gene expression evolution is to understand the character of variation within species. This includes quantifying the amount of variation and distinguishing between the genetic and environmental components. It can potentially be approached in a quantitative genetic framework by dissection of the variance components of gene expression and their interaction (Whitehead and Crawford 2006). However, to be able to do this on genome-wide scales, extensive amounts of gene expression data from pedigrees would preferentially be required. This may be difficult in studies of natural populations, especially in light of the need for sampling individuals from different generations in the same developmental stage and under similar environmental conditions.
Cis-acting or trans-acting effects may mediate the genetic basis for variation in gene expression. A trans-factor asserts its effect on the expression of genes elsewhere in the genome while a cis-factor regulates the expression of a nearby gene. A transcription factor regulating the expression of a gene on another chromosome is an example of the former category of regulatory sequences, while the transcription factor binding region upstream of the start site for expression of the gene is an example of the latter. Disentangling the relative roles of these two types of regulatory sequences for gene expression is important for understanding the genetic architecture behind gene expression evolution (Lemos et al. 2008; Goncalves et al. 2012; He et al. 2012; Metzger et al. 2016).
It is well documented that gene expression is a plastic character readily facilitating phenotypic responses to environmental variation (Morris et al. 2014). Coupled with an unknown component of experimental error in measuring gene expression (Whitehead and Crawford 2006; Robinson et al. 2010), observed variation among individuals from a natural population does not easily translate into a picture of the underlying genetic variation (Oleksiak et al. 2002; Tung et al. 2011). In essence, the problem boils down to that we are interested in analyzing genetic diversity and the evolution of sequences that regulate gene expression, yet in the absence of detailed information about such sequence, we often have to draw conclusions from the phenotype that they underlie.
Gene expression is typically measured from the total amount of RNA, that is, the combined transcription of RNA from the two chromosomes (alleles) in diploid organisms. This means that regulatory variation in heterozygous individuals may be concealed and hence not easily quantifiable. However, if expression from the two alleles can be distinguished, cis-regulatory variation is directly revealed by the two alleles being expressed at different levels in heterozygous individuals (Yan et al. 2002; Chen et al. 2016). This forms the basis for analyses of allele-specific expression (ASE). ASE analyses were initially performed to distinguish between cis- and trans-regulatory effects in F1 hybrids. This suggested that cis-regulatory mutations tend to have additive effects on gene expression and are readily visible by selection (Gibson et al. 2004; Wittkopp et al. 2004; Prud'homme et al. 2007; Wray 2007; Fay and Wittkopp 2008; Graze et al. 2012; Bell et al. 2013). Further studies have detected causal relationships between cis-regulatory variation and many types of phenotypic characters including, for example, morphology and mating behaviour (Stern and Orgogozo 2008; Linnen et al. 2009; Arnoult et al. 2013; Santos et al. 2014; Arunkumar et al. 2016).
Studies of cis-regulatory variation by ASE bear the intrinsic power of utilizing within-sample control; both alleles are exposed to the same environment and trans-acting factors (Pastinen 2010). Unfortunately, only a limited number of genome-wide ASE studies have so far focused on natural populations of nonmodel organisms (Tung et al. 2011), in part owing to a lack of necessary genomic resources. In this paper, we report the outcome of an RNA-seq scan for ASE in wild populations of two flycatcher species of the genus Ficedula, the collared flycatcher F. albicollis and the pied flycatcher F. hypoleuca. They are small, trans-Saharan migrant songbirds that breed in deciduous and mixed coniferous forests in Europe, have a generation time of about 2 years and an estimated effective population size (Ne) of about 200,000 (Nadachowska-Brzyska et al. 2016). Four particular aspects make this avian study system and the experimental design applied well suited for assessing cis-regulatory variation via ASE analysis in natural populations. First, a high-quality genome assembly is available for collared flycatcher, with Ensembl annotations of coding sequences (Ellegren et al. 2012; Kawakami et al.2014). Second, the very same individuals that we used for measuring gene expression were subject to whole-genome re-sequencing, meaning that RNA-seq reads could be directly compared with known coding sequence genotypes, which greatly facilitated SNP detection and genotype calling. Third, we introduce a novel statistical method, a Bayesian negative binomial (NB) approach, for modeling variation in ASE. Fourth, in contrast to many previous studies of ASE, we make use of multiple single nucleotide polymorphisms (SNPs) per transcript to distinguish alleles and increase power. The main conclusion of this work is that there is extensive genetic variation for cis-regulation of gene expression in these natural bird populations.
Materials and Methods
Polymorphism Data
Whole-genome re-sequencing and polymorphism data for the same collared flycatcher and pied flycatcher individuals used for transcriptome analyses were taken from Ellegren et al. (2012). SNPs were called separately per species against the collared flycatcher genome assembly version FicAlb_1.4 using GATK v. 2.2 (DePristo et al. 2011) with standard options. Only SNP calls that passed the GATK standard quality criteria were used. SNPs were phased using Beagle v. 3.0.4 with standard options (Browning and Browning 2007) using genome re-sequencing data from all 20 flycatcher individuals from Ellegren et al. (2012), and phased coding SNPs were then extracted.
Transcriptome Data
Five adult male collared flycatchers were collected on the Swedish island of Öland and five pied flycatchers were collected in Uppsala, Sweden. RNA was prepared from brain, kidney, liver, lung, muscle, skin, and testis, and sequenced for 100 cycles on an Illumina Genome Analyzer IIx. Sampling, RNA preparation, library construction, and sequencing have been described in detail in Ellegren et al. (2012). RNA-seq reads were mapped to the SNP-masked genome sequence using TopHat v. 2.0.5 (Kim et al. 2013). The majority of SNPs in collared flycatcher and pied flycatcher is shared between species so mapping efficiency is not biased against pied flycatchers.
Because differential isoform usage could confound our ability to discover ASE, and because Ensembl gene annotations for flycatchers largely lack isoform annotations, we annotated transcripts de novo. Gene models were created using Cufflinks v. 2.0.2 (Trapnell et al. 2012) per species and merged into one annotation in order to retrieve unified gene identifiers using Cuffmerge.
To ensure independence between SNPs required for modeling, we filtered out closely adjacent SNPs and only considered SNP pairs separated by more than 200 bp (i.e., the insert size in our RNA sequencing library). SNPs falling into exonic regions were extracted and read counts mapping to exonic SNPs were summarized per SNP, gene, tissue, and individual. To match our transcript annotations with information from additional databases, we retrieved Ensembl gene IDs for Cufflinks gene models by mapping sequences to the Ensembl-annotated genome assembly.
Statistical Analysis
Variation in coverage among tissues and individuals, and variation in number of SNPs among individuals for a given gene meant that while we had sufficient information and statistical power to detect ASE for some tissue/individual combinations, this was not the case for others. Due to the resulting low overlap of information for ASE detection in different samples (i.e., samples from the same individual or the same tissue) (supplementary fig. S1, Supplementary Material online), data from each individual and tissue were treated independently and, accordingly, no normalization method was applied.
Using phased data we estimated the mean allelic coverage per transcript of individual birds. Mean (μ) and variance (σ2) were incorporated into a statistical model to test whether the observed difference between the frequencies of the haplotypes was greater than what would be expected by chance. Modeling of read count data in ASE analyses has so far mainly employed the Poisson distribution, which is based on an equal mean-variance assumption. However, it predicts less variation than what is usually seen in the data (Mortazavi et al. 2008; Anders and Huber 2010; Robinson et al. 2010; Wang et al. 2010; Anders et al. 2012). To alleviate the assumption and address the extra variation (overdispersion) issue, quasi-Poisson and NB distributions can be applied. The quasi-Poisson distribution assumes that the variance increases as a linear function of the mean, that is, σ2 = ϕμ, whereas the NB distribution assumes that the mean and variance are related via σ2 = μ + ϕμ2. The overdispersion parameter ϕ controls the magnitude of the variance in quasi-Poisson and NB distributions. These models have been applied to analyses of differential expression (DE) of genes among individuals and populations (Anders and Huber 2010; Robinson et al. 2010; Soneson and Delorenzi 2013). Here we extend their use to the detection of ASE, as ASE could be seen as DE between two alleles.
We defined the model structure to include transcript i, SNP j (j = 1, …, J), and allele p (p = 1, 2) (supplementary fig. S2, Supplementary Material online), with the aim to test whether the expression level from all J SNPs were different between alleles p, for each transcript i. We required J > 1 since this is needed to estimate the variance in a generalized linear mixed model (GLMM) framework. The read counts from each transcript, SNP and allele Yijp was formulated as Yijp ∼ h(μijp, σ2ijp), where h() was either a Poisson, quasi-Poisson or NB distribution, decided by the relationship between μijp and σ2ijp.Supplementary figure S3, Supplementary Material online shows the relationship between mean and variance estimates. The quadratic variance function (i.e., the NB distribution) appeared to describe the mean-variance relationship better than the other assumptions. We thus chose to use the conservative NB distribution to minimize the rate of false discoveries.
When the same NB model was fitted for each transcript and a large number of such models were built, it formed the parallel structure of an overall hierarchical model. Taking advantage of such a hierarchical structure, the overdispersion parameter could be estimated precisely and flexibly. Although it is generally agreed upon that overdispersion needs to be addressed in studies of differential gene expression, no consensus on how this should be done has emerged (Pritchard et al. 2001; Robinson and Smyth 2008; Anders and Huber 2010; Robinson et al. 2010; Turro et al. 2011; Soneson and Delorenzi 2013). Basically, three ways have been proposed to estimate overdispersion: local, common and moderate estimation, respectively (supplementary fig. S2, Supplementary Material online). In the first method, local ϕ is estimated from each transcript. In this case, the number of local estimates equals the number of transcripts in the overall hierarchical model and they are likely to differ considerably due to the extent of variance in RNA-seq data. The common estimation utilizes all transcripts to fit one ϕ and then uses this common ϕ for testing each single transcript. Although the common estimate makes use of all information in the data and stabilizes ϕ, it restricts the variability of the dispersion. The moderate dispersion is an approximate empirical Bayesian approach that shrinks the local estimates toward the common estimate using weighted likelihood. The Bayesian prior corresponds to the weight between local and common dispersion, and thereby controlling the extent of dispersion shrinkage. By choosing this prior weight, a Bayesian posterior mean estimator of overdispersion can be calculated using an approximate empirical Bayesian solution. Instead of using a direct estimate of overdispersion, this approximation method is necessary as the NB distribution falls outside of the exponential family and therefore lacks a conjugate prior for overdispersion (Robinson and Smyth 2007, 2008; McCarthy et al. 2012; Zhou et al. 2014).
Instead of a constant weight for all transcripts, we designed a more flexible and pertinent weight () for each transcript depending on how reliably local dispersion could be estimated and approximated by the total number of SNPs (J) within one transcript. Transcripts for which J was comparatively large provided more statistical information and local dispersion was therefore prioritized (overweighted) over common estimation (smaller). Along with this data-dependent prior weight and posterior estimate using the approximate empirical Bayes rule, we employed a weighted quantile-adjusted conditional maximum likelihood to estimate parameters for the NB distribution. Finally, the null hypothesis that the two alleles were balanced in expression level was tested for each transcript.
In practice, we adopted the model of Robinson and Smyth (2007, 2008), as implemented in the edgeR program (Robinson et al. 2010), but adjusted the weight as described above. This adjustment was achieved mathematically by relaxing the fixed residual degree of freedom (d.f.) according to J, that is, d.f. = 2 × J − 2 (as we had two groups under comparison, i.e., two alleles) (supplementary methods, Supplementary Material online). We employed Goodness-of-fit (GOF) as an evaluation criterion for assessing which dispersion would best capture the characteristics of the data. GOF was assessed using Pearson’s χ2 test on a per transcript basis with the null hypothesis that the empirical distribution fits the theoretical distribution (Lee et al. 2008; Oberg et al. 2012). The number of times that the null hypothesis got rejected was summed, meaning that lower GOF values indicate a better fit. Bayesian dispersion performed the best (GOF = 1,954), while common dispersion had limited flexibility (4,626), and local dispersion (2,858) underestimated the overdispersion parameter (supplementary fig. S4, Supplementary Material online).
Simulation
To study the performance of the Bayesian NB approach, we simulated multiple data sets under a variety of scenarios including different allelic imbalance, gene expression levels, and numbers of SNPs (ranges corresponding to real data). For each simulation we randomly selected 8% of genes to be true positive ASE (the same percentage of ASE as observed in real flycatcher data), and the rest to be true negative genes. We chose the level of overdispersion to be 0.38, which was the mean overdispersion in real flycatcher data. One thousand genes were used in each simulation, which was repeated 100 times. We then applied the Bayesian NB approach on the data sets and considered genes with an adjusted P < 0.1 (false discovery rate, FDR, multiple testing correction) to be significant. The average true positive rate and FDR was calculated. Similarly, we simulated a set where we randomly sampled overdispersion, the number of SNPs, coverage, and the allelic imbalance from distributions of flycatcher data and built the receiver operating characteristic curve accordingly (supplementary fig. S5, Supplementary Material online).
Cut-Off Detection
We initially included all data for evaluation of the methods. However, for transcripts with low coverage there is essentially no power to detect ASE, adding noise only. We therefore investigated different coverage cut-offs (3–16 reads per SNP site) in terms of the number of transcripts with significant support for ASE (table 1). The number was highest at a cut-off of 11, and decreased both at lower and higher cut-offs. This pattern can probably be seen as a balance between removing noise in the data and not eliminating significant transcripts. The overlap among significant transcripts at cut-offs of 9–13 was 90% so the choice of cut-off seemed robust.
Table 1.
The Effect of Different Coverage Cut-Off Levels for Detection of Significant ASE Transcripts, Identified by the Bayesian NB Approach
| Cut-Off | Collared Flycatcher |
Pied Flycatcher |
||
|---|---|---|---|---|
| ASE | Total | ASE | Total | |
| 3 | 145 | 31,438 | 62 | 8,987 |
| 4 | 185 | 23,055 | 73 | 6,688 |
| 5 | 221 | 18,668 | 76 | 5,347 |
| 6 | 253 | 15,109 | 90 | 4,346 |
| 7 | 280 | 12,805 | 106 | 3,626 |
| 8 | 283 | 10,861 | 110 | 3,122 |
| 9 | 294 | 9,520 | 117 | 2,734 |
| 10 | 300 | 8,324 | 122 | 2,413 |
| 11 | 324 | 7,481 | 128 | 2,157 |
| 12 | 305 | 6,700 | 116 | 1,937 |
| 13 | 304 | 6,074 | 106 | 1,754 |
| 14 | 302 | 5,472 | 110 | 1,599 |
| 15 | 296 | 5,021 | 103 | 1,482 |
| 16 | 283 | 4,581 | 104 | 1,362 |
Note.—Bold indicates the chosen cut-off level.
Sharing of ASE across Individuals and Tissues
We constructed a Spearman’s rank correlation matrix between all pairs of samples to analyze to what extent ASE was shared among tissues and individuals. For each case of shared significant ASE between pairs of samples, correlation was calculated as the percentage of allelic difference (i.e., the difference between read counts of the two alleles divided by their sum). The correlation matrix was then partitioned into correlations (1) between different tissues within an individual, (2) the same tissue in different individuals, and (3) between different tissues and different individuals, respectively.
Expression breadth was estimated as the inversion of tissue specificity index τ (Yanai et al. 2005; Liao et al. 2006; Park et al. 2011), defined as:
where
and was the expression level of transcript i in the tth tissue, and was the maximum expression level across the total number of k tissues under study for the ith transcript. A low value of τ means low tissue specificity or high expression breadth.
Gene Ontology Enrichment Analysis
Gene ontology (GO) annotations were retrieved from Ensembl (www.ensembl.org). Over-representation of GO categories of ASE genes compared to all other genes were examined using Fisher’s exact test with classic and elim algorithms, provided in the topGO package (Alexa et al. 2006; Rivals et al. 2007). In order to get robust significant GO terms, P-values from both algorithms were considered, and two different thresholds were used in determining significance of the test, which were either FDR < 0.1 in both algorithms or P < 0.05 in both.
Total Expression Levels
For transcripts in which at least one sample showed evidence of ASE, we compared the total expression level per tissue between individuals with evidence of ASE and individuals without. Expression levels were first normalized by the total number of reads per sample and the number of SNPs per transcript. If data for more than one individual was available for a particular tissue-transcript combination, mean values were used. Expression levels were compared by using the ratio of expression levels in ASE and nonASE individuals.
Results
Using RNA-seq to Investigate ASE
By combining SNP data from whole-genome re-sequencing and transcriptome data from RNA-seq of the same five male collared flycatchers we detected 14,858 transcripts with at least two SNPs (a prerequisite for our method). After filtering for adequate RNA-seq coverage at SNP positions (see Materials and Methods), 2,576 transcripts remained, corresponding to 2,418 genes (only 16 transcripts contained overlapping SNPs). The number of filtered transcripts per tissue varied between 328 (muscle) and 994 (skin), and per individual between 713 and 796 (table 2). The mean read coverage per SNP was 41.9 and the mean informative read coverage per transcript was 119.4, with a mean of 3.0 SNPs per transcript. There was no systematic difference in read coverage between the allele corresponding to the genome sequence (reference allele) and the nonreference allele (t-test P = 0.80; supplementary fig. S6, Supplementary Material online).
Table 2.
Number of Analyzed Transcripts for Each Sample (Tissue/Individual Combination)
| ID #1 | ID #2 | ID #3 | ID #4 | ID #5 | Total | |
|---|---|---|---|---|---|---|
| Brain | 85 | 277 | 109 | 99 | 69 | 394 |
| Kidney | 154 | 391 | 308 | 215 | 171 | 741 |
| Liver | 100 | 229 | 62 | 91 | 189 | 403 |
| Lung | 136 | 421 | 148 | 257 | 152 | 656 |
| Muscle | 86 | 231 | 67 | 75 | 74 | 328 |
| Skin | 258 | 597 | 281 | 299 | 243 | 994 |
| Testis | 267 | 624 | 277 | 187 | 252 | 940 |
| Total | 713 | 1,700 | 796 | 740 | 723 |
Note.—In total, there were 7,481 analyzed samples. Totals depict the number of unique transcript per tissue and individual.
A Bayesian NB Approach for the Detection of ASE
To increase the power of detection of ASE, we developed a novel Bayesian NB approach that alleviates the equal mean-variance assumption of the Poisson distribution previously used for modeling read count data in studies of ASE. This new approach shrinks the local (per transcript) estimates of overdispersion toward the common (all transcripts) estimate using weighted likelihood. To evaluate the accuracy of ASE detection with this approach, we noted that for a transcript where the two alleles are expressed at different levels, reads covering different SNPs should be biased toward the same allele. That is, two or more SNPs should agree on the direction of imbalance and thus be concordant. In principle, in the absence of sampling variance and phasing errors, and with accurate detection of ASE, disagreement should be 0%. In contrast, for a transcript where both alleles are expressed at equal levels, disagreement should approach 50% due to sampling variance (fig. 1A).
Fig. 1.
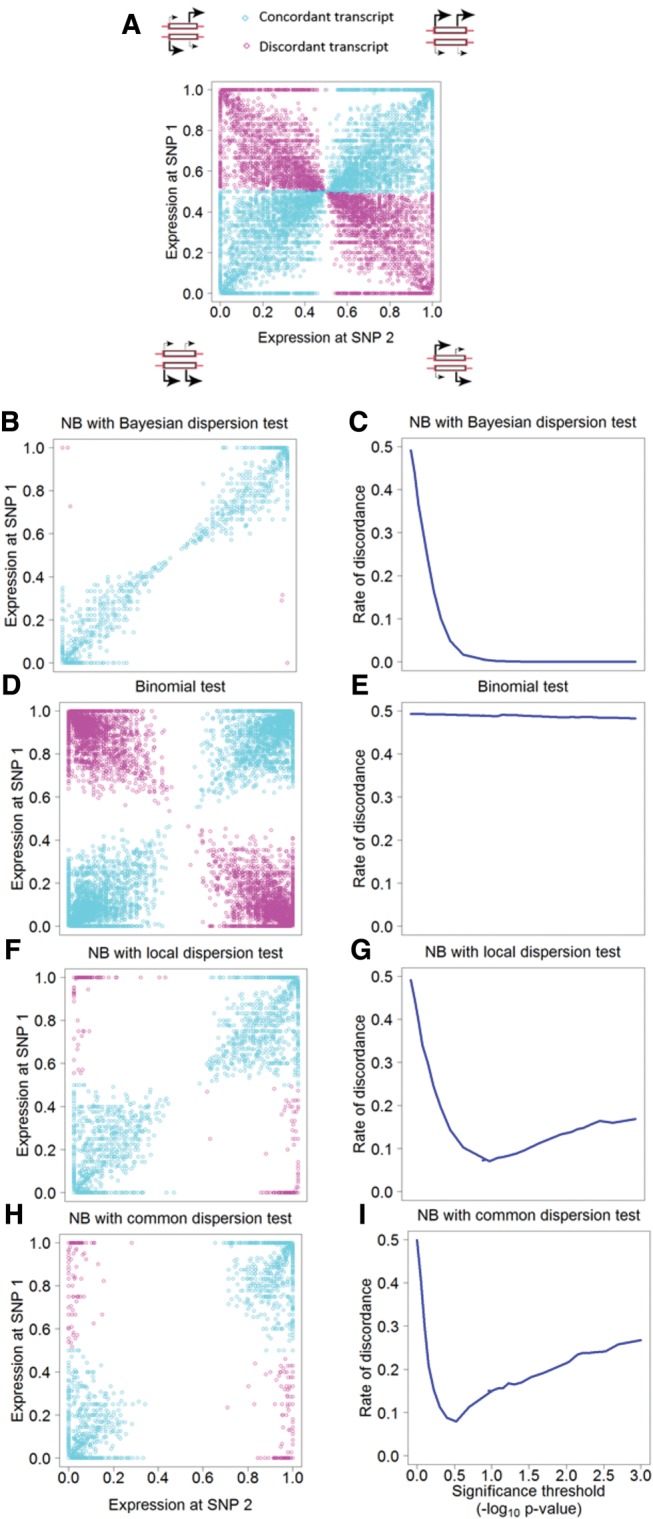
—Concordance analysis of ASE transcripts. A large arrow represents higher and a small arrow indicates lower expression level, respectively. In concordant transcripts (blue) all SNPs have the same direction of imbalance, while in discordant transcripts (red) SNPs differ in the direction of imbalance. (A) Raw data, (B and C) Bayesian NB approach, (D and E) binomial test, (F and G) NB approach with local dispersion, (H and I) NB approach with common dispersion. (C), (E), (G), and (I) show the rate of discordance in relation to different thresholds of statistical significance for the respective models. For the NB approach with local and common dispersion, the rate of discordance was 8.21% and 11.09%, and for the binomial test it was 47.85% at a significance threshold of P = 0.05.
To test the accuracy of the Bayesian NB approach, we constructed pairwise concordance plots for SNPs within each collared flycatcher gene following DeVeale et al. (2012). The rate of discordance was 0.16% (fig. 1B) and decreased asymptotically toward 0 with increased threshold of statistical significance (lowered P) (fig. 1C). This indicates that the Bayesian NB approach is a valid model and has powerful control over noise and variation. The approach outperformed the binomial test and other NB approaches based on local and common dispersion by having much lower discordance rate. More specifically, the binomial test was essentially incapable of removing false positives, as illustrated by close to 50% discordance rate independent of the significance level (fig. 1D and E). Approaches using NB with local (fig. 1F and G) or common dispersion (fig. 1H and I) performed better than the binomial test but showed more disagreement (8.21% and 11.09%, respectively) in the data compared to the Bayesian NB approach. Moreover, in these cases discordance rate increased with increasing significance threshold because highly significant yet discordant ASE transcripts were less efficiently removed from the data.
To further assess the performance of the Bayesian NB approach we applied it to simulated data sets with varying allelic imbalance, read coverage, and number of SNPs per gene, and considered genes with an adjusted (multiple testing correction) P < 0.1 to exhibit ASE (fig. 2). The true positive rate increased with the number of SNPs, read coverage, and, in particular, allelic imbalance (fig. 2A–C). Under strong imbalance (frequency of the major allele, FAM, of 0.9), the average rate of true positives was 0.98 and with a FAM of 0.8 the average rate was 0.84. We found that the FDR was below 0.015 across all tested scenarios (fig. 2D–F), indicating that the model is robust under different settings and that p-value adjustment is effective. Importantly, the average FDR was much lower than the testing threshold of 0.1 implying that the model is conservative. In a simulation of randomly sampled parameters instead of fixed ones, the true positive rate was 0.61 and the FDR was 0.01 (supplementary fig. S5, Supplementary Material online).
FIG. 2.

—The performance of the Bayesian NB model on simulated data. The simulations are based on the number of SNPs in a gene, the coverage per SNP site, and the allelic ratio. The average true positive rate and false positive rate were calculated from 100 simulations with each having 1000 genes.
As an independent validation of the Bayesian NB approach we applied it to a human lymphoblastoid cell line RNA-seq data set (Rozowsky et al. 2011), and compared it to the MBASED (meta-analysis based allele-specific expression detection) method (Mayba et al. 2014). After filtering for minimum coverage and number of heterozygous SNPs, there were 1,014 genes that could be tested. Among them we found 31 ASE genes by the Bayesian NB approach and 45 ASE genes by MBASED, of which 27 were common to both methods (supplementary table S1, Supplementary Material online). The four genes uniquely identified by the Bayesian NB approach failed to pass the MBASED cut-off, which required a FAM > 0.7, although three of them were significant. The 18 genes only detected by MBASED had higher estimated overdispersion compared to other genes (Welch t-test, P = 0.012), implying inconsistent expression level within these genes. We conclude that the two methods produced similar results on this data set. However, since the Bayesian NB approach focuses on the estimation of ASE on the transcript level, it is more sensitive to expression variation between isoforms of a gene.
ASE Is Prevalent in Wild Ficedula Flycatchers
Using the Bayesian NB approach on collared flycatcher expression data we found 185 transcripts (7.2%) that showed evidence of ASE in at least one individual and tissue at an FDR of 0.1 (approximately P < 0.001) (supplementary table S2, Supplementary Material online). The allelic imbalance of ASE transcripts was highly biased toward extensive skews (fig. 3A), with 89.5% of all statistically significant samples showing the major allele in more than 90% of the reads. Relaxed FDR gave almost the same distribution of the FAM but with a slightly inflated tail (fig. 3B). To explore the variation in ASE among tissues we combined data from all individuals. The proportion of transcripts with ASE per tissue varied between 2.4% (testis) and 6.9% (muscle; fig. 4), with testis showing a significantly lower (Fisher’s exact test, P = 6.9 × 10−5 after Bonferroni correction) and muscle a significantly higher (P = 0.018) proportion of ASE genes than the other tissues combined. We found no evidence for overall expression differences of ASE transcripts among tissues (supplementary fig. S7A, Supplementary Material online, Bartlett test P = 0.12, ANOVA P = 0.34, Kruskal–Wallis rank sum test P = 0.11) or differences in the number of detected SNPs per transcript among tissues (supplementary fig. S7B, Supplementary Material online, Bartlett test P = 0.48, ANOVA P = 0.75, Kruskal–Wallis rank sum test P = 0.43). This argues against the possibility that differences in the proportion of ASE genes per tissue would be related to power issues.
FIG. 3.

—The allelic imbalance toward the major allele in significant ASE transcripts. (A) is under FDR < 0.1 and (B) is under a relaxed threshold of FDR < 0.16 which is the optimal cut-off suggested by the simulations.
FIG. 4.

—Proportion of significant ASE transcripts using the Bayesian NB model at an FDR < 0.1 in each tissue. Data points represent the different individuals and medians are denoted with a line.
ASE was detected in more than one individual-tissue combination for 64 transcripts. The most common type of multiple occurrence was in the form of the same individual showing ASE in two or more tissues (73.4%), followed by transcripts showing ASE in the same tissue in multiple individuals (50.0%, with 23.4% of transcripts showing both) (fig. 5). This was also evident from a significantly stronger correlation between the degree of allelic imbalance in different tissues for the same individual and transcript (mean Spearman’s ρ = 0.73) than between the degree of imbalance in different individuals for the same tissue and transcript (mean ρ = 0.18; fig. 6A). This strengthens the interpretation of a genetic basis for ASE with heterozygous cis-regulatory sequences leading to unbalanced expression of the two alleles in multiple tissues of the same individual. However, expression breadth of ASE transcripts was significantly lower than that of nonASE transcripts (Welch t-test P = 1.98 × 10−7, fig. 6B), potentially reducing the incidence of ASE across tissues for the same individual and transcript. We also note that for many of the transcripts in which ASE was detected in only one tissue, we had limited power of detection in other tissues due to low expression levels (fig. 5).
FIG. 5.

—Examples of ASE patterns across individuals and tissues. Green indicates that significant ASE was detected and white indicates that no significant ASE was detected (even though the minimum requirements for number of SNPs and read coverage were met). Gray indicates that ASE could not be investigated because of lack of variable SNPs or too low read coverage.
FIG. 6.

—(A) Box plot of Spearman’s rank correlations between ASE between all pairs of significant samples in different sample groups (i.e., between different tissues within an individual, between individuals within the same tissues, and between different tissues and different individuals). (B) Box plot of expression breadth between ASE transcripts and nonASE transcripts. Whiskers extend to 1.5 times the interquartile range with data points extremer than that are plotted as outliers.
We then analyzed a closely related species, the pied flycatcher, again based on whole-genome re-sequencing for SNP detection and deep transcriptome sequencing of seven tissues in five males. Of 908 transcripts for which we had sufficient coverage and SNP information, 77 (8.5%) showed evidence of ASE in at least one individual and tissue (supplementary table S3, Supplementary Material online; see supplementary fig. S8, Supplementary Material online for performance of the method). The proportion of genes with ASE did not differ between the two species (Fisher’s exact test P = 0.18). Only one gene was common to the sets of ASE genes in the two species—multiple coagulation factor deficiency protein 2 (MCFD2)—providing no evidence for enrichment of shared ASE genes.
There was no clear indication that ASE would be particularly common among certain types of functionally annotated genes. There were 21 significant GO terms in collared flycatcher at P < 0.05 (supplementary table S4, Supplementary Material online) and 18 in pied flycatcher (supplementary table S5, Supplementary Material online). These represented a variety of functions, for example including nervous, sensory and immune systems, metabolism, and transcriptional regulation. No GO term was significantly over-represented among ASE genes in collared flycatcher at FDR < 0.1 and only one term was significant in pied flycatchers, polysaccharide binding.
ASE can potentially imply a change in the total amount of expression from a gene due to segregating polymorphisms in cis-regulatory sequences. For example, an individual showing ASE of a gene may carry an allelic variant of a regulatory sequence that leads to up- or down-regulation of gene expression compared to other alleles. On the other hand, feedback mechanisms may counteract any such effects to maintain similar expression levels across individuals despite the presence of ASE. To gain some preliminary insight into these processes we compared the total amount of expression of ASE genes between individuals that showed evidence of ASE and individuals that did not (fig. 7). There was no general trend for ASE genes to have lower or higher total expression levels compared to the same gene in individuals without ASE. Moreover, there were examples of both increased and decreased total expression following from ASE.
FIG. 7.

—Distribution of the ratio of total expression of ASE genes in individuals showing evidence of ASE and total expression in individuals not showing evidence of ASE of such genes.
Discussion
We quantified ASE in multiple tissues of population samples of two closely related flycatcher species using a novel Bayesian approach. The state of ASE of a gene can be seen as a marker for polymorphism in cis-regulatory sequences influencing the expression level of the gene. The overall context of this study was thus the investigation of within-species diversity of gene expression, which constitutes the raw material for evolution of gene expression.
In line or species crosses, the haplotypes of cis-regulatory and coding sequences are generally known and can easily be inferred in F1 individuals. This is not the case in population samples from species with low levels of linkage disequilibrium (LD), like in flycatchers (Backström, et al. 2006). We could thus not treat individuals as biological replicates even if they carried the same alleles at a particular SNP. To overcome this limitation and to increase the ability of identifying ASE, we developed a new computational method, which aggregates dispersion across multiple SNPs into one measurement and estimates ASE on a per-individual basis. By modeling several SNPs per transcript, the method differs from statistical models that have been used for ASE detection at the level of single SNPs or that have used summed reads from phased SNPs at the gene level (Dimas et al. 2009; Zhang et al. 2009; Pickrell et al. 2010; Rozowsky et al. 2011; Romanel et al. 2015; Van de Geijn et al. 2015; Edsgärd et al. 2016).
Using information from several SNPs within a transcript helps shrinking the variance and thus increases the statistical power. This way we could efficiently use data from transcripts with a coverage as low as 11 reads per SNP. Previous studies that have tested for ASE at the level of single SNPs, or from summed reads across SNPs, using binomial or χ2 distributions have typically required a read coverage of 20 or higher (Fontanillas et al. 2010; McManus et al. 2010; Li et al. 2012), limiting the number of genes that have been possible to include in the analyses. Reducing the minimal coverage required for statistical testing of ASE is crucial in RNA-seq experiments since expression profiles are often highly biased toward a few genes with very high expression levels, whereas most genes are represented by relatively few reads. If we had used a coverage cut-off of 20, the number of tested transcripts would have been reduced by approximately a factor of three, from 2,576 to less than 900. To be fair, given that we required at least two SNPs per transcript, at least 22 reads per transcript were needed. Using our Bayesian NB model, we arrived at a data-driven cut-off that reduced the noise while keeping informative data.
When comparing the Bayesian NB approach to binomial test and other NB approaches for use on the flycatcher data, it outperformed all others in having the lowest rate of discordance. When validating the Bayesian NB approach using a human data set, there was a considerable amount of overlap of detected ASE genes with the method of MBASED. Simulations showed that the Bayesian NB approach can maintain a low level of false positives while keeping a reasonably high level of true positives under most simulated scenarios. Taken together, this suggests that the Bayesian NB model can reliably identify ASE. It should be emphasized that studying ASE in natural populations without pedigree information lacks many of the convenient features when studying ASE using hybrids of inbred lines (Cowles et al. 2002; Emerson et al. 2010; Salinas et al. 2016), such as small within- and large between-population genetic variation, and complete knowledge of the phase in the F1 generation. To compensate for this, we devised a framework that reliably estimates variance between the alleles within an individual.
Confounding factors in a study like this include experimental and technical noise in sequencing, allelic mapping, and phasing of SNPs. Consequently, strictly controlling for the false discovery rate is necessary when determining statistical significance. The Bayesian NB approach accomplishes this by using information from the whole data set to tune overdispersion estimates to an appropriate minimum per transcript, thereby increasing statistical power. However, there is a trade-off between the false positive rate and the false negative rate where a lower false positive rate inevitably comes at the cost of a higher false negative rate. In that respect, the Bayesian NB approach, just like other tests with a low false discovery rate, has the statistical limitation of a potentially high false negative rate. In our study, this appears to be the case especially for genes with less pronounced allelic imbalance (i.e., small effect size), leading to a conservative estimate of the incidence of ASE. Other than by sacrificing a low false positive rate, the false negative rate can be improved by using biological replicates but this is not feasible when studying a natural population. Moreover, the approach we used to statistically infer haplotypes based on population allele frequencies (Beagle) has limitations when sample size is small (Browning and Browning 2007). Incorrect haplotype phasing may lead to inflated variances and thereby less power, which would result in an even more conservative estimate of the incidence of ASE.
The fact that the Bayesian NB approach can only be implemented for transcripts with multiple SNPs excludes testing for ASE in single-SNP transcripts in which calculation of Bayesian dispersion is precluded. A common dispersion could instead be used for single-SNP transcripts. However, it results in very few significant transcripts (<1% detected ASE), likely reflecting an inflated false negative rate and would hinder concordant verification. We therefore only focused on transcripts with multiple SNPs to achieve reliable and confident estimation.
Note that allele-specific splice isoform expression is potentially detected as ASE by our approach. Using shot-gun RNA-seq, it is difficult to disentangle such cases from other forms of ASE, although it would certainly be interesting to do so. The expression of different splice isoforms between individuals does not affect our analyses as we do not compare ASE directly between individuals. Within any individual, allele-specific isoform expression can be regarded as one of several forms of ASE.
Positional biases affecting SNP identification and quantification within a transcript, such as transcript edge effects (impaired read mapping in transcript) should not affect our analyses since we only compare the expression of alleles at the same position within the same transcript. The edge effect will reduce our power to identify ASE when SNPs reside near the edge of transcripts, but should not introduce a bias.
The Biology of ASE
We found that 7.2% (collared flycatcher) and 8.5% (pied flycatcher) of the analyzed transcripts showed ASE in at least one tissue. Tissues differed in their amount of ASE, with muscle having the highest and testis the lowest prevalence, respectively. Similar observations of differences among tissues have been made in humans (Ardlie et al. 2015). When transcripts showed ASE in more than one sample, they more often did so in different tissues within the same individual than within the same tissue in different individuals. This is consistent with a strong genetic basis of ASE with some, but not all, individuals being heterozygous in regulatory sequences for particular genes. The fact that there was hardly any overlap in ASE transcripts between the two investigated species speaks in favor of that ASE is the result of transient polymorphism in regulatory sequences where segregating variants will eventually get fixed or lost. Furthermore, ASE transcripts were generally less broadly expressed than nonASE transcripts. This could potentially be related to that broadly expressed genes are on average more pleiotropic than tissue-specific genes; mis-regulation resulting from ASE may thus more often lead to detrimental effects (Aguet et al. 2016; Shen et al. 2012; Uebbing et al. 2016).
The estimated proportions of ASE transcripts in these bird populations are likely to be underestimated, for several reasons. Most importantly, we lacked power to detect ASE for many individual/tissue combinations of the 2,576 transcripts included in the study. This was primarily a combined effect of too low read coverage (preventing ASE detection in lowly expressed genes) and that SNPs were homozygous in some individuals. Moreover, when we had power, the detection was biased toward pronounced differences in expression level of the two alleles (fig. 3A). Many cases of more subtle differences in expression level are therefore likely to have remained undetected by not reaching statistical significance. Furthermore, with only five individuals per species analyzed, many regulatory variants segregating at rare to moderate frequencies in the studied populations are likely to have been absent from these relatively small population samples. We therefore suggest that the frequency of ASE in these species is higher, and perhaps significantly higher, than the observed frequency of 7–8%. A larger number of sampled individuals can help to detect rarer alleles and greater sequencing depth can help to detect instances of ASE with smaller effect size. Decreasing sequencing costs and an increasing sequencing throughput will facilitate both in future studies. This study nevertheless yields interesting insight in that transcripts showing strongly allele-biased expression patterns, are more likely to have strong phenotypic effects than less biased transcripts. As such, they will be most interesting to explore in more detail using larger sample sizes and more powerful techniques. It had been interesting to find certain types of genes to be more prone to ASE than others, indicating that such genes would be more amenable to variation in gene expression level, potentially indicating the absence of selection for a specified expression level. Instead, the absence of an enrichment of particular gene ontology categories among ASE transcripts may indicate that ASE occurs widely across the genome and across genes categories. It will be interesting to revisit this question with more powerful sampling schemes that are able to detect smaller differences in ASE which may uncover traits or categories of traits among which small-scale natural variation in gene expression levels is more prevalent than in others.
Studies like this in other natural population are rare. Most previous analyses of ASE have focused on F1 crosses of closely related species or inbred lines to estimate the degree of cis-regulatory divergence (Cowles et al. 2002; Emerson et al. 2010; Gaur et al. 2013; Salinas et al. 2016). Naturally, these latter estimates will depend on the overall divergence of the investigated taxa as well as the precise statistical models used, and observed frequencies of ASE genes have varied widely in studies of Arabidopsis (Zhang and Borevitz 2009; He et al. 2012), Capsella (Josephs et al. 2015; Steige et al. 2015), Drosophila (Graze et al. 2012; Coolon et al. 2014), mice (Lagarrigue et al. 2014; Crowley et al. 2015), and cows (Chamberlain et al. 2015). For population-based studies like ours, ASE frequencies between 6% (Ardlie et al. 2015) and 20% (Serre et al. 2008; Zhang et al. 2009) have been reported in humans and Tung et al. (2015) found a frequency of 23% in wild baboons. Taking into account that we likely underestimated the incidence of ASE in flycatchers, our results are roughly comparable to those obtained in primate studies. More generally, they indicate that polymorphisms in regulatory sequences commonly affect gene expression in natural populations. Our study also demonstrates that RNA-seq analysis of population samples provides a simple and cost-effective means to analyze regulatory variation in a population by observing the phenotype resulting from genetic variation in the ignorance of the genotypic basis itself. While sequencing and assembly of whole genomes still represents a prohibiting cost factor in many vertebrate species (Majewski and Pastinen 2011; Sun and Hu 2013; Verta et al. 2016), RNA-seq can readily be used to make inference about the presence of natural gene regulatory variation.
The demonstration of pervasive ASE in natural populations has implications for the evolution of gene expression. ASE can be regarded as a first step in the divergence of expression levels as it results from genetic variation in regulatory sequences upon which selection can act. Given the observed rich source of regulatory diversity, adaptation via changes in the regulation of gene expression may thus play an important role to phenotypic evolution in this and other systems.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the Swedish Research Council (grant numbers 2010-5650 and 2013-8271); the European Research Council (AdG 249976); and the Knut and Alice Wallenberg Foundation. Computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX). We thank Yudi Pawitan for statistical advice, and Paulina Bolivar, Homa Papoli Yazdi, and Carina F. Mugal for valuable discussions. We thank two anonymous reviewers for helpful comments.
Literature Cited
- Aguet F, et al. 2016. Local genetic effects on gene expression across 44 human tissues. bioRxiv. doi: http://dx.doi.org/10.1101/074450.
- Alexa A, Rahnenfuehrer J, Lengauer T.. 2006. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22:1600–1607. [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W.. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, Huber W. 2012. Detecting differential usage of exons from RNA-seq data. Genome Res. 22:2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardlie KG, et al. 2015. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult L, et al. 2013. Emergence and diversification of fly pigmentation through evolution of a gene regulatory module. Science 339:1423–1426. [DOI] [PubMed] [Google Scholar]
- Arunkumar R, Maddison TI, Barrett SC, Wright SI.. 2016. Recent mating‐system evolution in Eichhornia is accompanied by cis‐regulatory divergence. New Phytol. 10:nph.13918.. [DOI] [PubMed] [Google Scholar]
- Backström N, Qvarnström A, Gustafsson L, Ellegren H.. 2006. Levels of linkage disequilibrium in a wild bird population. Biol Lett. 2:435–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell GDM, Kane NC, Rieseberg LH, Adams KL.. 2013. RNA-Seq analysis of allele-specific expression, hybrid effects, and regulatory divergence in hybrids compared with their parents from natural populations. Genome Biol Evol. 5:1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Browning BL.. 2007. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 81:1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buil A, et al. 2015. Gene-gene and gene-environment interactions detected by transcriptome sequence analysis in twins. Nat Genet. 47:88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain AJ, et al. 2015. Extensive variation between tissues in allele specific expression in an outbred mammal. BMC Genomics 16:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, et al. 2016. A uniform survey of allele-specific binding and expression over 1000-Genomes-Project individuals. Nat Commun. 7:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolon JD, McManus CJ, Stevenson KR, Graveley BR, Wittkopp PJ.. 2014. Tempo and mode of regulatory evolution in Drosophila. Genome Res. 24:797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles CR, Hirschhorn JN, Altshuler D, Lander ES.. 2002. Detection of regulatory variation in mouse genes. Nat Genet. 32:432–437. [DOI] [PubMed] [Google Scholar]
- Crowley JJ, et al. 2015. Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance. Nat Genet. 47:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVeale B, van der Kooy D, Babak T.. 2012. Critical evaluation of imprinted gene expression by RNA-Seq: a new perspective. PLoS Genet. 8:e1002600.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, et al. 2009. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 325:1246–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edsgärd D, et al. 2016. GeneiASE: detection of condition-dependent and static allele-specific expression from RNA-seq data without haplotype information. Sci Rep. 6:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, et al. 2012. The genomic landscape of species divergence in Ficedula flycatchers. Nature 491:756–760. [DOI] [PubMed] [Google Scholar]
- Emerson J, et al. 2010. Natural selection on cis and trans regulation in yeasts. Genome Res. 20:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wittkopp PJ.. 2008. Evaluating the role of natural selection in the evolution of gene regulation. Heredity 100:191–199. [DOI] [PubMed] [Google Scholar]
- Fontanillas P, et al. 2010. Key considerations for measuring allelic expression on a genomic scale using high-throughput sequencing. Mol Ecol. 19:212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur U, Li K, Mei S, Liu G.. 2013. Research progress in allele-specific expression and its regulatory mechanisms. J Appl Genet. 54:271–283. [DOI] [PubMed] [Google Scholar]
- Gibson G, et al. 2004. Extensive sex-specific nonadditivity of gene expression in Drosophila melanogaster. Genetics 167:1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Oshlack A, Rifkin SA.. 2006. Natural selection on gene expression. Trends Genet. 22:456–461. [DOI] [PubMed] [Google Scholar]
- Goncalves A, et al. 2012. Extensive compensatory cis-trans regulation in the evolution of mouse gene expression. Genome Res. 22:2376–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graze RM, et al. 2012. Allelic imbalance in Drosophila hybrid heads: exons, isoforms, and evolution. Mol Biol Evol. 29:1521–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, et al. 2012. Genome-wide analysis of cis-regulatory divergence between species in the Arabidopsis genus. Mol Biol Evol. 29:3385–3395. [DOI] [PubMed] [Google Scholar]
- Josephs EB, Lee YW, Stinchcombe JR, Wright SI.. 2015. Association mapping reveals the role of purifying selection in the maintenance of genomic variation in gene expression. Proc Natl Acad Sci USA. 112:15390–15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T, et al. 2014. A high-density linkage map enables a second-generation collared flycatcher genome assembly and reveals the patterns of avian recombination rate variation and chromosomal evolution. Mol Ecol. 23:4035–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, et al. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, Wilson AC.. 1975. Evolution at two levels in humans and chimpanzees. Science 188:107–116. [DOI] [PubMed] [Google Scholar]
- Lagarrigue S, Martin L, Hormozdiari F, Roux P-F, Pan C.. 2014. Analysis of allele-specific expression in mouse liver by RNA-Seq: a comparison with cis-eQTL identified using genetic linkage. Genetics 196:1359–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Hansen KD, Bullard J, Dudoit S, Sherlock G.. 2008. Novel low abundance and transient RNAs in yeast revealed by tiling microarrays and ultra high-throughput sequencing are not conserved across closely related yeast species. PLoS Genet. 4:e1000299.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos B, Araripe LO, Fontanillas P, Hartl DL.. 2008. Dominance and the evolutionary accumulation of cis- and trans-effects on gene expression. Proc Natl Acad Sci USA. 105:14471–14476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, et al. 2012. Identification of allele-specific alternative mRNA processing via transcriptome sequencing. Nucleic Acids Res. 40:e104.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B-Y, Scott NM, Zhang J.. 2006. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol. 23:2072–2080. [DOI] [PubMed] [Google Scholar]
- Linnen CR, Kingsley EP, Jensen JD, Hoekstra HE.. 2009. On the origin and spread of an adaptive allele in deer mice. Science 325:1095–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski J, Pastinen T.. 2011. The study of eQTL variations by RNA-seq: from SNPs to phenotypes. Trends Genet. 27:72–79. [DOI] [PubMed] [Google Scholar]
- Mayba O, et al. 2014. MBASED: allele-specific expression detection in cancer tissues and cell lines. Genome Biol. 15:405.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK.. 2012. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40:4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CJ, et al. 2010. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res. 20:816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger BP, et al. 2016. Contrasting frequencies and effects of cis-and trans-regulatory mutations affecting gene expression. Mol Biol Evol. 33:1131–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MRJ, et al. 2014. Gene expression plasticity evolves in response to colonization of freshwater lakes in threespine stickleback. Mol Ecol. 23:3226–3240. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B.. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628. [DOI] [PubMed] [Google Scholar]
- Nadachowska-Brzyska K, Burri R, Smeds L, Ellegren H.. 2016. PSMC analysis of effective population sizes in molecular ecology and its application to black-and-white Ficedula flycatchers. Mol Ecol. 25:1058–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberg AL, Bot BM, Grill DE, Poland GA, Therneau TM.. 2012. Technical and biological variance structure in mRNA-Seq data: life in the real world. BMC Genomics 13:304.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiak MF, Churchill GA, Crawford DL.. 2002. Variation in gene expression within and among natural populations. Nat Genet. 32:261–266. [DOI] [PubMed] [Google Scholar]
- Park J, Xu K, Park T, Soojin VY.. 2011. What are the determinants of gene expression levels and breadths in the human genome? Hum Mol Genet. 21:46–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastinen T. 2010. Genome-wide allele-specific analysis: insights into regulatory variation. Nat Rev Genet. 11:533–538. [DOI] [PubMed] [Google Scholar]
- Pickrell JK, et al. 2010. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464:768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard CC, Hsu L, Delrow J, Nelson PS.. 2001. Project normal: defining normal variance in mouse gene expression. Proc Natl Acad Sci USA. 98:13266–13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prud'homme B, Gompel N, Carroll SB.. 2007. Emerging principles of regulatory evolution. Proc Natl Acad Sci USA. 104:8605–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivals I, Personnaz L, Taing L, Potier MC.. 2007. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics 23:401–407. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK.. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Smyth GK.. 2007. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 23:2881–2887. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Smyth GK.. 2008. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9:321–332. [DOI] [PubMed] [Google Scholar]
- Romanel A, Lago S, Prandi D, Sboner A, Demichelis F.. 2015. ASEQ: fast allele-specific studies from next-generation sequencing data. BMC Med Genomics 8:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozowsky J, et al. 2011. AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol Syst Biol. 7:522.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas F, et al. 2016. Natural variation in non-coding regions underlying phenotypic diversity in budding yeast. Sci Rep. 6:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos ME, et al. 2014. The evolution of cichlid fish egg-spots is linked with a cis-regulatory change. Nat Commun 5:5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre D, et al. 2008. Differential allelic expression in the human genome: a robust approach to identify genetic and epigenetic cis-acting mechanisms regulating gene expression. PLoS Genet. 4:e1000006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, et al. 2012. A map of the cis-regulatory sequences in the mouse genome. Nature 488:116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Delorenzi M.. 2013. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics 14:91.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steige KA, Reimegård J, Koenig D, Scofield DG, Slotte T.. 2015. Cis-regulatory changes associated with a recent mating system shift and floral adaptation in Capsella. Mol Biol Evol. 32:2501–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DL, Orgogozo V.. 2008. The loci of evolution: how predictable is genetic evolution? Evolution 62:2155–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Hu Y.. 2013. eQTL mapping using RNA-seq data. Stat Biosci. 5:198–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung J, et al. 2011. Allele-specific gene expression in a wild nonhuman primate population. Mol Ecol. 20:725–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung J, Zhou X, Alberts SC, Stephens M, Gilad Y.. 2015. The genetic architecture of gene expression levels in wild baboons. Elife 4:e04729.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turro E, et al. 2011. Haplotype and isoform specific expression estimation using multi-mapping RNA-seq reads. Genome Biol. 12:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uebbing S, et al. 2016. Divergence in gene expression within and between two closely related flycatcher species. Mol Ecol. 25:2015–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Geijn B, McVicker G, Gila Y, Pritchard JK.. 2015. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat Methods 12:1061–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verta JP, Landry CR, MacKay J.. 2016. Dissection of expression‐quantitative trait locus and allele specificity using a haploid/diploid plant system–insights into compensatory evolution of transcriptional regulation within populations. New Phytol. 211:159–171. [DOI] [PubMed] [Google Scholar]
- Wang L, Feng Z, Wang X, Wang X, Zhang X.. 2010. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138. [DOI] [PubMed] [Google Scholar]
- Whitehead A, Crawford DL.. 2006. Variation within and among species in gene expression: raw material for evolution. Mol Ecol. 15:1197–1211. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG.. 2004. Evolutionary changes in cis and trans gene regulation. Nature 430:85–88. [DOI] [PubMed] [Google Scholar]
- Wray GA. 2007. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 8:206–216. [DOI] [PubMed] [Google Scholar]
- Yan H, Yuan WS, Velculescu VE, Vogelstein B, Kinzler KW.. 2002. Allelic variation in human gene expression. Science 297:1143.. [DOI] [PubMed] [Google Scholar]
- Yanai I, et al. 2005. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21:650–659. [DOI] [PubMed] [Google Scholar]
- Zhang K, et al. 2009. Digital RNA allelotyping reveals tissue-specific and allele-specific gene expression in human. Nat Methods 6:613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Borevitz JO.. 2009. Global analysis of allele-specific expression in Arabidopsis thaliana. Genetics 182:943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Lindsay H, Robinson MD.. 2014. Robustly detecting differential expression in RNA sequencing data using observation weights. Nucleic Acids Res. 42:e91.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.