

Abstract

Two new 1,3-oxazin-6-one derivatives (1 and 2) and six new bohemamine-type pyrrolizidine alkaloids (3–8) were isolated from the marine-derived Streptomyces spinoverrucosus strain SNB-048. Their structures including the absolute configurations were fully elucidated on the basis of spectroscopic analysis, ECD spectra, quantum chemical calculations, and chemical methods. Compounds 1 and 2 possess a γ-lactam moiety and a 1,3-oxazin-6-one system.
Pyrrolizidine alkaloids, a group of natural products produced by numerous plant families, are an interesting class of molecules due to their potent biological activities and diverse chemical structures.1 More than 650 pyrrolizidine alkaloids have been isolated from over 6000 plants,2 and many of them have been demonstrated to be hepatotoxic, genotoxic, and mutagenic.3 Secondary metabolites from microbes possessing the pyrrolizidine nucleus, such as pyrrolams,4 jenamidines,5 and bohemamines,6 have also been identified. These structures have attracted the attention of synthetic chemists,7 especially bohemamines, which represented a rare pyrrolizidine subclass due to the methylation pattern and presence of the amide nitrogen on the pyrrolizidine skeleton. To date, only five bohemamine-type pyrrolizidine alkaloids, bohemamine (12), bohemamines B (13) and C (14), 5-chlorobohemamine C (15), and NP25302 (9), have been reported.6
As part of our studies to search for unusual structural features from marine-derived actinomycetes for further investigation, our attention was drawn to a series of metabolites contained in the extract of Streptomyces spinoverrucosus strain SNB-048. These compounds showed UV absorptions similar to those of bohemamines in an LC-UV-MS analysis.6 Chemical investigation on this strain has resulted in the isolation of two novel bohemamine-type pyrrolizidine alkaloids, spithioneines A and B (10 and 11), which possess an unusual ergothioneine moiety.8 Herein, we report another six new bohemamine analogues, bohemamines D–I (3–8), and two new 1,3-oxazin-6-one derivatives, spinoxazines A and B (1 and 2), together with a known bohemamine analogue, NP25302 (9). Structurally, compounds 1 and 2 possess a 1,3-oxazin-6-one system, which is rare in natural products.21
Chart 1.

Results and Discussion
Spinoxazine A (1) was obtained as a white powder. The molecular formula was determined as C13H16N2O4 according to its HRESIMS peak at m/z 265.1183 [M + H]+. There were multiple IR stretches at 1740 cm–1 indicative of a lactam or ester moiety (five-membered-ring lactams absorb in the 1750–1700 cm–1 region; four-membered-ring lactams absorb at 1760–1730 cm–1; esters absorb at 1750–1735 cm–1; α,β-unsaturated esters appears at 1730–1715 cm–1). The 13C NMR spectrum showed 13 carbon signals that were classified by HSQC as three methyl carbons, one sp3 methylene carbon, four methine groups (two olefinic carbons, one carbon connected with oxygen, and one carbon connected with nitrogen), and five nonprotonated carbons (three carbonyls and two olefinic carbons) (Table 1). The 1H NMR spectrum showed five coupled signals at δH 1.23 (H-8, d, J = 6.5), 4.55 (H-4, qd, J = 6.7, 6.6), 4.43 (H-5, ddd, J = 8.8, 8.0, 6.7), 2.67 (H-6β, dd, J = 16.9, 8.0), and 2.63 (H-6α, dd, J = 16.9, 8.8). These data combined with COSY correlations of H-8/H-4/H-5/H-6 and the key HMBC correlations of H-8 to C-4/C-5 and H-4/H-5 to C-6/C-7 (Figure 1) constituted the 4-hydroxy-5-methyl-γ-lactam moiety. Two vinyl methyl groups at δH 2.25 (H-5′, d, J = 1.2 Hz) and 2.03 (H-4′, d, J = 1.2 Hz) and one olefinic proton signal at δH 5.98 (m), along with the HMBC correlations of H-2′ to C-4′/C-5′ and H-4′ to C-5′ (Figure 1), indicated the presence of an isobutenyl fragment. The isobutenyl fragment should be connected with C-1′ (δC 163.7), which was determined by the HMBC correlation of H-2′ to C-1′ (Figure 1). The remaining 1H and 13C NMR signals and the HMBC correlations of H-2 (δH/C 6.73/89.0) to C-1 (δC 159.8) and C-3 (δC 155.3) (Figure 1), along with the molecular formula, provided two possible structures that could not be easily distinguished: a 1,3-oxazin-6-one (1) or an unsaturated β-lactam unit (1a) (Figure 2). The proposed unsaturated β-lactam in 1a would be unprecedented in a natural product. Only one natural product possessing an unsaturated β-lactam moiety was reported,9 but it was later revised to be a six-membered-ring metabolite.10 Synthetic efforts to produce similar unsaturated β-lactams have been reported; however, literature reports suggest that such β-lactams rearrange to form the corresponding 1,3-oxazin-6-ones (Scheme 1).11 In general, the 1H and 13C NMR data for the corresponding β-lactam and 1,3-oxazin-6-one structures for this natural product would be nearly identical. Predicted 13C NMR shifts of both 1,3-oxazin-6-one (1) and β-lactam (1a), which were computed at the B3LYP/6-311++G(2d,p)//B3LYP/6-31G(d) level (full details on the calculation can be found in the Supporting Information),12 strongly support that the chemical shift values of 1 are consistent with a 1,3-oxazin-6-one skeleton (Table S1), particularly the 13C shifts of C-1, C-2, and C-3. The linkage between the γ-lactam and the 1,3-oxazin-6-one could be confirmed by the HMBC correlation of H-4 to C-3.
Table 1. 1H (600 MHz) and 13C (100 MHz) NMR Data for Compounds 1 and 2 in DMSO-d6.
|
1 |
2 |
|||
|---|---|---|---|---|
| no. | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) |
| 1 | 159.8, C | 159.9, C | ||
| 2 | 89.0, CH | 6.73, s | 88.3, CH | 6.79, s |
| 3 | 155.3, C | 155.5, C | ||
| 4 | 57.7, CH | 4.55, qd (6.7, 6.6) | 54.3, CH | 4.65, qd (6.9, 6.6) |
| 5 | 64.1, CH | 4.43, ddd (8.8, 8.0, 6.7) | 24.7, CH2 | 2.21, m; 1.71, m |
| 6 | 39.3, CH2 | 2.67, dd (16.9, 8.0); | 31.4, CH2 | 2.84, m; 2.45, m |
| 2.63, dd (16.9, 8.8) | ||||
| 7 | 173.4, C | 176.0, C | ||
| 8 | 13.3, CH3 | 1.23, d (6.5) | 20.0, CH3 | 1.31, d (6.4) |
| 1′ | 163.7, C | 163.7, C | ||
| 2′ | 116.2, CH | 5.98, m | 116.2, CH | 5.98, m |
| 3′ | 158.7, C | 158.7, C | ||
| 4′ | 28.0, CH3 | 2.03, d (1.2) | 28.1, CH3 | 2.03, s |
| 5′ | 21.0, CH3 | 2.25, d (1.2) | 21.1, CH3 | 2.25, s |
Figure 1.
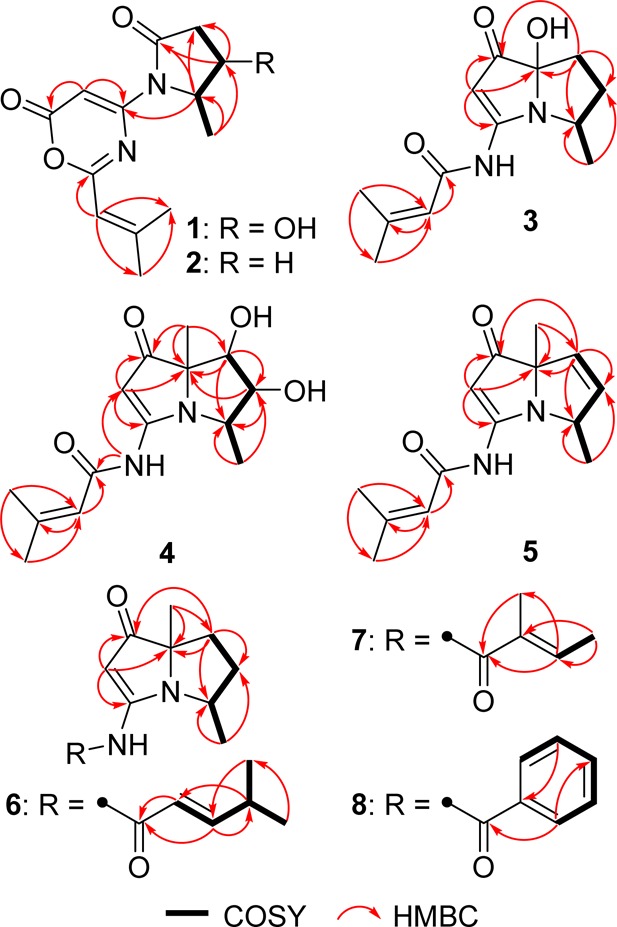
Key correlations for the structural assignment of 1–8.
Figure 2.
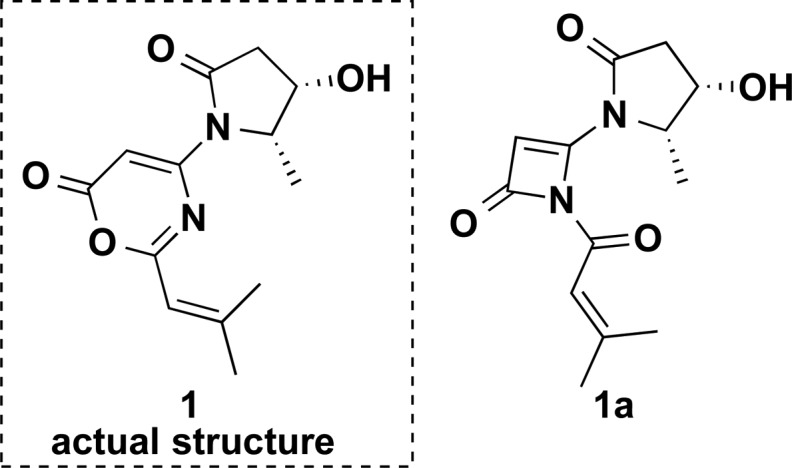
Two structures, 1 and 1a, in general agreement with the NMR data.
Scheme 1. Mechanism for the Conversion of N-Acyl-β-lactams into 1,3-Oxazin-6-ones.

NOE correlations between H-8 and H-5/H-6β, between H-4 and H-5, and between H-5 and H-6α/H-6β were observed in the 2D NOESY spectrum, suggesting the relative configuration of 1 as shown (Figure 3). NOE correlations combined with the coupling constants between H-4 and H-5 (J = 6.7 Hz), between H-5 and H-6α (J = 8.8 Hz), and between H-5 and H-6β (J = 8.0 Hz) also indicated the conformation of 1 as shown in Figure 4. We utilized the empirical electronic circular dichrosim (ECD) rule of γ-lactams13 to determine the absolute configuration of 1. The ECD sign of γ-lactams can be determined by addition of two effects: configuration of Cα and ring chirality. If the γ-lactams have no lone pair electrons on the Cα substituent, the ECD signs are determined by the ring chirality (conformation A gives a negative sign, and B gives a positive sign) (Figure 4).13 The conformation of (4S, 5S)-1, suggested by NOE correlations and chemical calculation (Supporting Information), is consistent with A, which was responsible for the negative Cotton effect (π–π* transition) in accordance with the measured negative ECD Cotton effect at λmax 254 nm of 1 (Figure 4). As the 1,3-oxazin-6-one group connected with the nitrogen nearly lay on the same plane of the amide, it made a very small contribution to the lactam Cotton effect. Moreover, the predicted ECD spectrum was obtained by the TDDFT [B3LYP/6-31G(d)] method (Supporting Information),14 which was subsequently compared with the experimental data. The measured ECD curve of 1 showed a Cotton effect at λmax (Δε) 254 (−4.7) nm, matching with the calculated ECD curve of (4S,5S)-1 (Figure 5).
Figure 3.
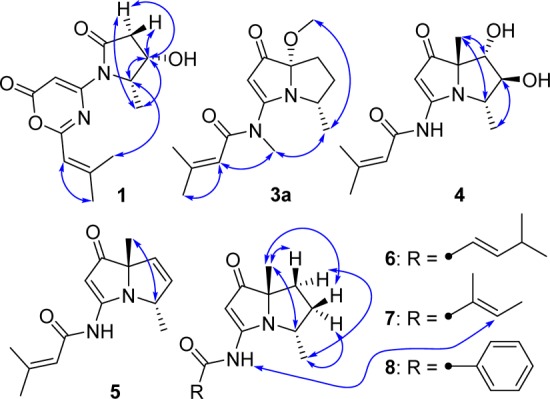
Selected NOESY correlations of 1, 3a, and 4–8.
Figure 4.
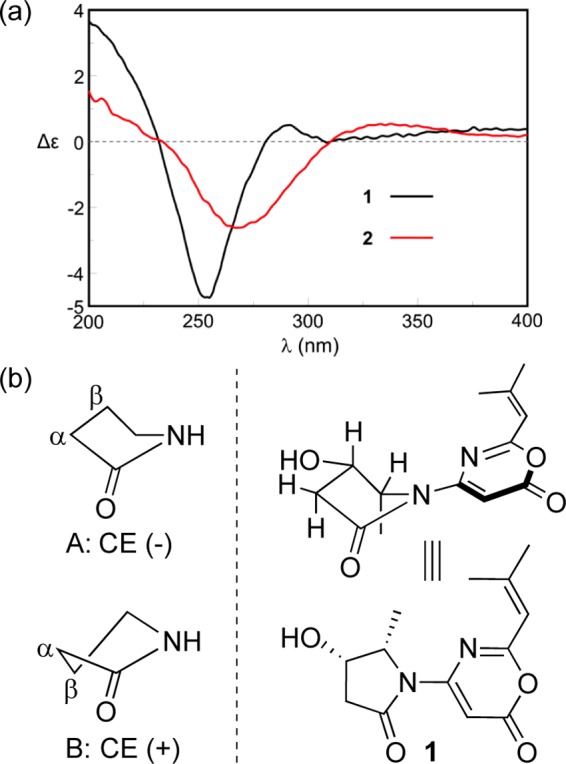
(a) ECD spectra of 1 and 2. (b) Ring chirality and ECD sign of γ-lactams and the conformation of 1.
Figure 5.
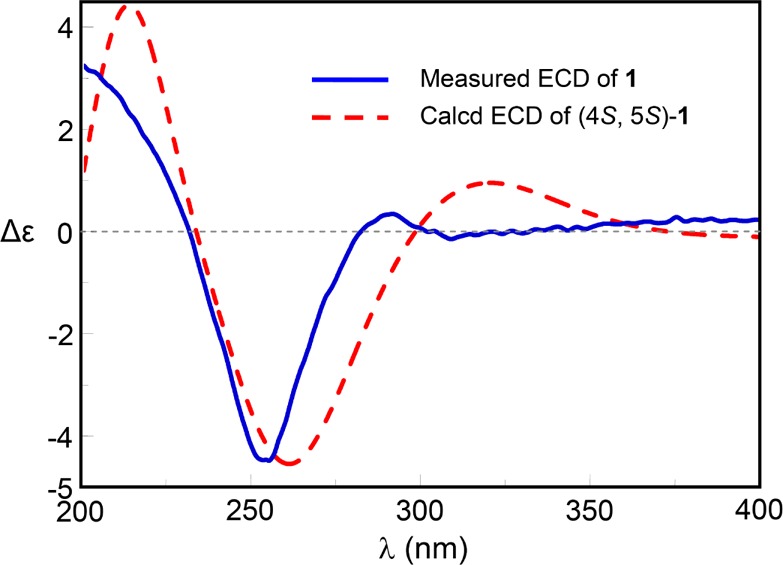
Measured and calculated ECD spectra for 1.
The molecular formula of spinoxazine B (2) was assigned as C13H16N2O3 based on the HRESIMS, which was only one oxygen less than that of 1. Careful comparison of its 1H and 13C NMR spectra (Table 1) with those of 1 showed that methylene signals at δC/H 24.7/2.21 and 1.71 in 2 replaced the corresponding oxygenated methine signals at δC/H 64.1/4.43 in 1. The COSY correlations of H-8/H-4/H-5/H-6 and the key HMBC correlations of H-8 to C-4/C-5 and H-4/H-5 to C-6/C-7 also supported this change in 2 (Figure 1). The ECD Cotton effect at λmax (Δε) 267 (−2.6) was nearly identical to 1 (Figure 4), indicating the same absolute configuration.
Bohemamine D (3) was isolated as a colorless oil. Its molecular formula was C13H18N2O3 based on the HRESIMS data. The UV spectrum showed similar absorptions to bohemamines at 252, 283, and 332 nm.6 Comparison of its NMR spectra with those of known bohemamine analogues indicated that the 1H and 13C NMR data of 3 (Table 2) were similar to NP25302 (9).6c The signal of the C-7 methyl group in NP25302 was no longer present, and δC at C-7 was shifted deshielded to 95.3, leading to the assignment of a C-7 hydroxy group. The molecular formula and the key HMBC correlations of H-8 to C-4/C-5 and H-6 to C-4/C-5/C-7/C-1 (Figure 1) also confirmed the structure of 3. In order to determine its relative configuration, a methylation reaction was carried out using iodomethane and sodium hydride to generate 3a (Figure S2). In the NOESY spectrum of compound 3a, the signal between H-8 and 7-OCH3 could be observed, which indicated the relative configuration of 3 as shown. The octant rule for cyclopentenone8,15 was used to determine the absolute configuration (Figure 6). The conformation of (4S,7S)-3 as shown in Figure 6 was placed in accordance with the octant rule model. The functional group at C-7 lying in the back lower left area was responsible for the negative Cotton effect (n−π*), which was consistent with the Cotton effect at 335 nm (Δε −19.0). Moreover, the ECD helicity rule of α,β-unsaturated ketones16 was used to further confirm the absolute configuration of compound 3. The P-helicity of the O=C–Cα=Cβ torsion angle correlates the positive Cotton effect (π–π*) at 297 nm (Figure 6).
Table 2. 1H (600 MHz) and 13C (100 MHz) NMR Data for Compounds 3–5 in DMSO-d6.
|
3 |
4 |
5 |
||||
|---|---|---|---|---|---|---|
| no. | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) |
| 1 | 198.9, C | 201.5, C | 203.2, C | |||
| 2 | 89.1, CH | 5.42, s | 93.5, CH | 5.41, s | 93.1, CH | 5.58, s |
| 3 | 168.4, C | 166.7, C | 168.0, C | |||
| 4 | 55.2, CH | 3.71, qd (6.7, 3.7) | 62.9, CH | 3.76, qd (6.9, 3.3) | 62.0, CH | 4.40, ddd (6.7, 2.2, 1.4) |
| 5 | 32.7, CH2 | 1.91, m; 1.74, m | 87.0, CH | 3.92, brs | 128.0, CH | 5.74, dd (6.0, 2.2) |
| 6 | 32.0, CH2 | 1.82, m; 1.71, m | 77.3, CH | 3.70, brs | 133.9, CH | 5.67, dd (6.0, 1.4) |
| 7 | 95.3, C | 78.4, C | 80.2, C | |||
| 8 | 22.0, CH3 | 1.30, d (6.6) | 23.2, CH3 | 1.31, s | 24.8, CH3 | 1.22, s |
| 9 | 16.4, CH3 | 1.20, d (6.9) | 17.9, CH3 | 1.19, d (6.6) | ||
| 1′ | 164.7, C | 163.9, C | 164.0, C | |||
| 2′ | 117.9, CH | 6.13, m | 117.7, CH | 5.99, m | 117.5, CH | 5.98, m |
| 3′ | 156.5, C | 156.0, C | 156.6, C | |||
| 4′ | 27.3, CH3 | 1.90, d (1.2) | 27.3, CH3 | 1.89, d (1.3) | 27.3, CH3 | 1.90, d (1.2) |
| 5′ | 20.1, CH3 | 2.13, d (1.2) | 20.0, CH3 | 2.14, d (1.3) | 20.0, CH3 | 2.15, d (1.2) |
| NH | 9.90, s | 10.31, s | 10.49, s | |||
Figure 6.

ECD spectra and the representation of the octant rule and helicity rule for the cyclopentenone moiety of 3 (a) and 9 (b).
Bohemamine E (4) was also thought to be an analogue of bohemamines due to the similar UV absorptions at 252, 280, and 335 nm.6 The molecular formula was determined as C14H20N2O4 by HRESIMS, with a H2O unit more than bohemamine (12). The 1D NMR spectrum was similar to 12 except for the shifts of C-5 and C-6. So, we deduced that the epoxide ring in 12 was hydrolyzed to yield compound 4. COSY and HMBC data (Figure 1) confirmed this diol structure. NOE correlations between H-5 and H-9, between H-4 and H-8, and between H-6 and H-8 were observed in the 2D NOESY, suggesting the relative configuration of 4 as shown (Figure 3).
The molecular formula of bohemamine F (5) was determined to be C14H18N2O2 from the protonated molecule peak at m/z 247.1441 [M + H]+ in the positive-ion HRESIMS spectrum, which was two hydrogen atoms less than that of NP25302 (9). Comparison of its 1H and 13C NMR spectra (Table 2) with those of 9 revealed that the signals of two methylene groups (C-5 and C-6) were replaced by two coupled olefinic methine signals (C-5: δC/H 128.0/5.74, dd, J = 6.0, 2.2; C-6: δC/H 133.9/5.67, dd, J = 6.0, 1.4). This structure can be confirmed by COSY correlations of H-9/H-4/H-5/H-6 and the key HMBC correlations from H-9 to C-4/C-5, H-6 to C-4/C-5/C-7/C-1, and H-8 to C-6/C-7. The relative configurations of C-4 and C-7 could be determined by the NOE correlation between H-8 and H-4.
The molecular formulas of bohemamines G–I (6–8) were determined as C15H22N2O2, C14H20N2O2, and C16H18N2O2 based on their HRESIMS spectra at m/z 263.1754 [M + H]+, 249.1596 [M + H]+, and 271.1443 [M + H]+, respectively. Examination of their 1H and 13C NMR spectra (Table 3) showed that they had the same pyrrolizidine skeleton as 9, with variation only in the amide moiety. For compound 6, the COSY correlations of H-2′/H-3′/H-4′ and H-5′/H-4′/H-6′ and the key HMBC correlations from H-2′ to C-1′, H-3′ to C-1′/C-4′, H-4′ to C-6′, H-5′ to C-6′, and H-6′ to C-3′ suggested the presence of a 4-methylpent-2-enamide side chain (Figure 1). The large coupling constant between H-2′ and H-3′ (J = 15.5 Hz) indicated the E- configuration. Compound 7 was determined to be an isomer of 9 (Table 3), where the amide moiety is a 2-methylbut-2-enamide group. This structure was supported by the COSY correlation of H-3′/H-4′ and the HMBC correlations from H-3′ to C-1′/C-5′, H-4′ to C-2′/C-3′, and H-5′ to C-1′ (Figure 1). The E-configuration for this side chain was determined by the NOE correlation between H-3′ and 3-NH (Figure 3). The NMR data were also consistent with those reported in the literature.7b In the 1D NMR spectrum of compound 8 (Table 3), the signals of a monosubstituted phenyl group were observed, which were further supported by 2D correlations (Figure 1). The NOE correlations between H-9 and H-5′β/H-6′β and between H-8′ and H-4′/H-5′α/H-6′α could be observed in the 2D NOESY spectra of compounds 6–8, which revealed that they had the same relative configuration as shown (Figure 3).
Table 3. 1H (600 MHz) and 13C (100 MHz) NMR Data for Compounds 6–8 in DMSO-d6.
|
6 |
7 |
8 |
||||
|---|---|---|---|---|---|---|
| no. | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) |
| 1 | 204.0, C | 203.8, C | 203.9, C | |||
| 2 | 93.5, CH | 5.54, s | 93.3, CH | 5.46, s | 93.9, CH | 5.60, s |
| 3 | 166.3, C | 166.9, C | 167.1, C | |||
| 4 | 54.2, CH | 3.97, qd (6.6, 6.6) | 54.3, CH | 4.03, qd (6.6, 6.6) | 54.4, CH | 4.10, qd (6.6, 6.6) |
| 5 | 34.5, CH2 | 2.39, m; 1.74, m | 34.4, CH2 | 2.37, m; 1.71, m | 34.5, CH2 | 2.41, m; 1.75, m |
| 6 | 27.6, CH2 | 1.67, dd (12.5, 6.2); | 27.7, CH2 | 1.67, dd (12.4, 6.3); | 27.8, CH2 | 1.71, dd (12.3, 6.2); |
| 1.52, dd (12.1, 6.2) | 1.53, dd (12.2, 6.5) | 1.56, dd (12.0, 6.9) | ||||
| 7 | 73.1, C | 73.3, C | 73.4, C | |||
| 8 | 24.7, CH3 | 1.15, s | 24.6, CH3 | 1.16, s | 24.6, CH3 | 1.20, s |
| 9 | 16.7, CH3 | 1.00, d (6.4) | 16.8, CH3 | 1.01, d (6.5) | 16.9, CH3 | 1.09, d (6.4) |
| 1′ | 163.7, C | 167.5, C | 165.8, C | |||
| 2′ | 120.3, CH | 6.19, dd (15.5, 1.5) | 131.5, C | 133.4, C | ||
| 3′ | 154.3, CH | 6.91, dd (15.5, 6.3) | 133.9, CH | 6.50, qd (6.8, 1.4) | 128.2, CH | 7.91, d (7.5) |
| 4′ | 30.3, CH | 2.50, m | 14.1, CH3 | 1.79, d (6.8) | 128.5, CH | 7.55, t (7.6) |
| 5′ | 21.1, CH3 | 1.04, d (6.8) | 12.3, CH3 | 1.80, s | 132.5, CH | 7.64, t (7.4) |
| 6′ | 21.1, CH3 | 1.04, d (6.8) | 10.28, s | 128.5, CH | 7.55, t (7.6) | |
| 7′ | 128.2, CH | 7.91, d (7.5) | ||||
| NH | 10.49, s | 10.92, s | ||||
The absolute configuration of compound 9 has been determined as (4S,7S) by total synthesis,5c which can be confirmed with the octant rule for cyclopentenone (Figure 6).8,15 The ECD helicity rule of α,β-unsaturated ketones can also be used to confirm this conclusion. M-Helicity of the O=C–Cα=Cβ torsion angle of compound 9 was consistent with the negative Cotton effect (π–π*) at 283 nm (Figure 6), which suggested the absolute configuration of 9 as (4S,7S). The ECD spectra of compounds 4–8 showed the same Cotton effects as those of 9 (Figure 7), indicating the same core configurations.
Figure 7.
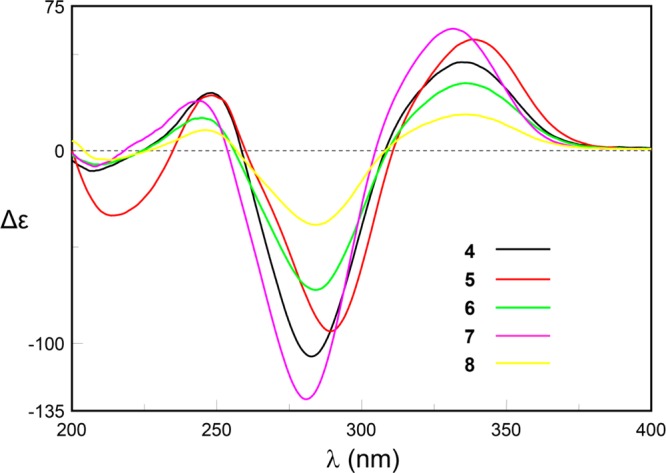
ECD spectra of 4–8.
A plausible biogenetic pathway for spinoxazines A and B (1 and 2) and bohemamine D (3) is postulated (Scheme 2). l-Glutamine undergoes decarboxylation and oxidation to generate the primary amine intermediate a, followed by condensation with two acetyl-CoA units and reduction to yield intermediate b.17 Dual nucleophilic addition of the amine to the amide and methyl ketone carbonyls, respectively, followed by elimination and reduction would provide the pyrrolizidine core d. Oxidation of the bridgehead carbon would result in the hemiaminal intermediate e, which undergoes N-acylation to yield compound 3.8,18 Dehydrogenation of the pyrrolizidine core would give the ring cleavage intermediate f,19 which could undergo a pericyclic reaction to form the 1,3-oxazin-6-one compound spinoxazine B (2). Further oxidation would generate spinoxazine A (1).
Scheme 2. Plausible Biogenetic Pathway of Compounds 1–3.

Compounds 1–8 did not show cytotoxicity to non-small-cell lung cancer cell lines HCC366, A549, HCC44, and HCC1171. In addition, they showed no antibacterial activities against Pseudomonas aeruginosa and Bacillus subtilis.
Experimental Section
General Experimental Procedures
Optical rotations were recorded with an AUTOPOL AP IV-6W polarimeter equipped with a halogen lamp (589 nm). UV spectra were recorded on a Shimadzu UV-1601 UV–vis spectrophotometer. ECD spectra were measured on a JASCO J-815 spectrometer. IR spectra were obtained on a PerkinElmer Spectrum 1000 FT-IR spectrometer. 1H and 2D NMR spectroscopic data were recorded at 600 MHz in DMSO-d6 or CDCl3 solution on a Varian System spectrometer, and chemical shifts were referenced to the corresponding residual solvent signal (δH/C 2.50/39.52 for DMSO-d6 and δH/C 7.26/77.16 for CDCl3). 13C NMR spectra were acquired at 100 MHz on a Varian System spectrometer. High-resolution ESI-TOF mass spectra were provided by The Scripps Research Institute (La Jolla, CA, USA). Low-resolution LC/ESIMS data were measured using an Agilent 1200 series LC/MS system with a reversed-phase C18 column (Phenomenex Luna, 150 mm × 4.6 mm, 5 μm) at a flow rate of 0.7 mL/min. Preparative HPLC was performed on an Agilent 1200 series instrument with a DAD detector, using a reversed-phase C18 column (Phenomenex Luna, 250 × 10.0 mm, 5 μm), a phenyl-hexyl column (Phenomenex Luna, 250 × 10.0 mm, 5 μm), or a CN column (Phenomenex Luna, 250 × 10.0 mm, 5 μm). Sephadex LH-20 (GE Healthcare) and ODS (50 mm, Merck) were used for column chromatography. Artificial seawater was used in microbial fermentations as described in a previous reference.20
Collection and Phylogenetic Analysis of Strain SNB-048
Streptomyces sp. strain SNB-048 was isolated from a sand sample collected from a Bahamian tidal flat. The sediment was desiccated and stamped onto agar plates using gauze 1 acidic media (10 g starch, 1 g NaNO3, 0.5 g K2HPO4, 0.5 g MgSO4, 0.5 g NaCl, 0.01 g FeSO4, 1 L seawater, 15 g agar, pH adjusted to 5.3 with phosphate buffer). Bacterial colonies were selected and streaked to purity using the same agar media. Analysis of the strain by 16S rRNA revealed 99.9% identity to Streptomyces spinoverrucosus. The sequence is deposited in GenBank under accession no. KR091963.
Cultivation and Extraction of SNB-048
Bacterium SNB-048 was cultured in 20 × 2.8 L Fernbach flasks each containing 1 L of a seawater-based medium (10 g starch, 4 g yeast extract, 2 g peptone, 1 g CaCO3, 40 mg Fe2(SO4)3·4H2O, 100 mg KBr) and shaken at 200 rpm at 27 °C. After 7 days of cultivation, sterilized XAD-7-HP resin (20 g/L) was added to adsorb the organic products, and the culture and resin were shaken at 200 rpm for 2 h. The resin was filtered through cheesecloth, washed with deionized H2O, and eluted with acetone. The acetone-soluble fraction was dried in vacuo to yield 1.2 g of extract. In order to get more material, another cultivation of 10 L was done under the same condition. From this cultivation, 460 mg of extract was obtained.
Purification
The extract (1.2 g) of the first cultivation was fractionated by flash column chromatography on ODS (50 μm, 30 g), eluting with a step gradient of MeOH and H2O (10:90–100:0), and 12 fractions (Fr1.1–Fr1.12) were collected. Fractions 7 and 8 (403.7 mg) were combined and separated into six fractions (Fr1.7.1–Fr1.7.6) on Sephadex LH-20, eluting with MeOH. Fr1.7.3 (151.2 mg) was further separated into 16 fractions (Fr1.7.3.1–Fr1.7.3.16) by flash column chromatography on ODS (50 μm, 30 g), eluting with a step gradient of MeOH and H2O (10:90–100:0). Fr1.7.3.6 (6.3 mg) was purified by HPLC on a phenyl-hexyl column (Phenomenex Luna, 250 × 10.0 mm, 5 μm, 2.5 mL/min) using a gradient solvent system from 20% to 100% CH3CN (0.1% formic acid) over 20 min to yield compound 4 (1.4 mg, tR = 9.5 min). Fr.7.3.8 (17.5 mg) was purified by HPLC on a phenyl-hexyl column (Phenomenex Luna, 250 × 10.0 mm, 5 μm, 2.5 mL/min) using a gradient solvent system from 20% to 85% CH3CN (0.1% formic acid) over 20 min to afford compound 3 (1.9 mg, tR = 11.1 min). Fr1.7.3.9 (8.1 mg) was also separated by HPLC on a phenyl-hexyl column (Phenomenex Luna, 250 × 10.0 mm, 5 μm, 2.5 mL/min) using a gradient solvent system from 20% to 85% CH3CN (0.1% formic acid) over 20 min to afford compound 7 (0.9 mg, tR = 12.5 min). Fr1.7.3.10 (31.7 mg) was separated by a reversed-phase HPLC (Phenomenex Luna, C18, 250 × 10.0 mm, 5 μm, 2.5 mL/min) using a gradient solvent system from 20% to 80% CH3CN (0.1% formic acid) over 20 min to yield compound 9 (3.8 mg, tR = 13.4 min) and Fr1.7.3.10.4 (12.6 mg, tR = 14.3 min). Fr1.7.3.10.4 (12.6 mg) was further purified by HPLC on a CN column (Phenomenex Luna, 250 × 10.0 mm, 5 μm, 2.5 mL/min) using a gradient solvent system from 20% to 100% CH3CN (0.1% formic acid) over 20 min to afford compounds 5 (3.3 mg, tR = 11.5 min) and 8 (2.3 mg, tR = 12.0 min). Fr1.7.3.11 (9.1 mg) was purified by HPLC on a phenyl-hexyl column (Phenomenex Luna, 250 × 10.0 mm, 5 μm, 2.5 mL/min) using a gradient solvent system from 20% to 100% CH3CN (0.1% formic acid) over 20 min to afford compound 6 (1.8 mg, tR = 13.6 min). Fr1.7.5 (21.7 mg) was purified by reversed-phase HPLC (Phenomenex Luna, C18, 250 × 10.0 mm, 5 μm, 2.5 mL/min) using a gradient solvent system from 20% to 90% CH3CN (0.1% formic acid) over 15 min to afford Fr1.7.5.1 (1.2 mg, tR = 12.2 min). Fr1.7.5.1 (1.2 mg) was further purified by HPLC on a phenyl-hexyl column (Phenomenex Luna, 250 × 10.0 mm, 5 μm, 2.5 mL/min) using a gradient solvent system from 20% to 100% CH3CN (0.1% formic acid) over 20 min to yield compound 1 (0.8 mg, tR = 13.4 min). The extract (460 mg) of the second cultivation was fractionated by flash column chromatography on ODS (50 μm, 30 g), eluting with a step gradient of MeOH and H2O (10:90–100:0), and eight fractions (Fr2.1–Fr2.8) were collected. Fr2.4 (21.1 mg) separated into nine fractions (Fr2.4.1–Fr2.4.9) on Sephadex LH-20, eluting with MeOH. Fr2.4.6 (3.5 mg) was purified by reversed-phase HPLC (Phenomenex Luna, C18, 250 × 10.0 mm, 5 μm, 2.5 mL/min) using a gradient solvent system from 30% to 100% CH3CN (0.1% formic acid) over 20 min to yield compound 2 (0.6 mg, tR = 15.7 min).
Spinoxazine A (1):
white powder, [α]23D +17 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 254 (4.04), 318 (3.79) nm; ECD (c = 0.87 mM, MeOH) λmax (Δε) 291 (+0.5), 254 (−4.7) nm; IR (NaCl disk) νmax 3398, 2982, 2925, 1740, 1594, 1541, 1394, 1272, 1258, 1208, 1187, 1102, 1071, 830 cm–1; 1H and 13C NMR, Table 1; HRESIMS m/z 265.1183 [M + H]+ (calcd for C13H17N2O4, 265.1183).
Spinoxazine B (2):
white powder, [α]23D +6 (c 0.03, MeOH); UV (MeOH) λmax (log ε) 255 (4.10), 320 (3.82) nm; ECD (c = 1.33 mM, MeOH) λmax (Δε) 333 (+0.5), 267 (−2.6) nm; IR (NaCl disk) νmax 2918, 2850, 1739, 1734, 1593, 1540, 1387, 1254, 1184, 830 cm–1; 1H and 13C NMR, Table 1; HRESIMS m/z 249.1235 [M + H]+ (calcd for C13H17N2O3, 249.1234).
Bohemamine D (3):
colorless oil, [α]23D −34 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 252 (4.05), 283 (3.79), 332 (3.69) nm; ECD (c = 0.40 mM, MeOH) λmax (Δε) 336 (−19.0), 297 (+24.5) nm; IR (NaCl disk) νmax 3430, 2100, 1645, 1506, 1394, 1206, 1135, 1058, 793, 733 cm–1; 1H and 13C NMR, Table 2; HRESIMS m/z 251.1391 [M + H]+ (calcd for C13H19N2O3, 251.1390).
Bohemamine E (4):
colorless oil, [α]23D −7 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 252 (4.01), 280 (3.78), 335 (3.70) nm; ECD (c = 0.25 mM, MeOH) λmax (Δε) 335 (+45.8), 282 (−106.8), 248 (+29.9), 207 (−10.5) nm; IR (NaCl disk) νmax 3422, 2092, 1638, 1201, 1130, 734 cm–1; 1H and 13C NMR, Table 2; HRESIMS m/z 281.1496 [M + H]+ (calcd for C14H21N2O4, 281.1496).
Bohemamine F (5):
colorless oil, [α]23D +422 (c 0.16, MeOH); UV (MeOH) λmax (log ε) 246 (4.02), 288 (3.77), 338 (3.66) nm; ECD (c = 0.41 mM, MeOH) λmax (Δε) 339 (+57.6), 290 (−93.7), 248 (+28.7), 207 (−26.2) nm; IR (NaCl disk) νmax: 3432, 2101, 1643, 1506, 1361, 1228, 1134, 1099, 756 cm–1; 1H and 13C NMR, Table 2; HRESIMS m/z 247.1441 [M + H]+ (calcd for C14H19N2O2, 247.1441).
Bohemamine G (6):
colorless oil, [α]23D +17 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 245 (4.09), 284 (3.81), 337 (3.71) nm; ECD (c = 0.38 mM, MeOH) λmax (Δε) 335 (+34.9), 284 (−72.3), 244 (+16.8), 209 (−7.1) nm; IR (NaCl disk) νmax 3428, 2100, 1504, 1132, 677 cm–1; 1H and 13C NMR, Table 3; HRESIMS m/z 263.1754 [M + H]+ (calcd for C15H23N2O2, 263.1754).
Bohemamine H (7):
colorless oil, [α]23D +37 (c 0.03, MeOH); UV (MeOH) λmax (log ε) 244 (4.09), 284 (3.81), 337 (3.71) nm; ECD (c = 0.40 mM, MeOH) λmax (Δε) 331 (+63.2), 281 (−129.0), 244 (+25.7), 207 (−8.2) nm; IR (NaCl disk) νmax 3429, 2099, 1638, 1506, 1260, 1174, 1121, 734 cm–1; 1H and 13C NMR, Table 3; HRESIMS m/z 249.1596 [M + H]+ (calcd for C14H21N2O2, 249.1598).
Bohemamine I (8):
colorless oil, [α]23D +39 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 248 (4.02), 282 (3.78), 336 (3.69) nm; ECD (c = 0.37 mM, MeOH) λmax (Δε) 335 (+18.7), 284 (−38.4), 246 (+10.7), 214 (−4.9) nm; IR (NaCl disk) νmax 3431, 2100, 1645, 1506, 1258, 1137, 794, 723, 704 cm–1; 1H and 13C NMR, Table 3; HRESIMS m/z 271.1443 [M + H]+ (calcd for C16H19N2O2, 271.1441).
NP25302 (9):
[α]23D +104 (c 0.10, MeOH), literature6c [α]24D +116 (c 0.11, MeOH); ECD (c = 0.40 mM, MeOH) λmax (Δε) 335 (+54.3), 283 (−115.9), 248 (+27.4), 210 (−12.2) nm.
Methylation of 3
To a solution of 3 (1.5 mg) in DMF (anhydrous, 0.5 mL) was added 2.0 mg of NaH. After allowing it to stir at 0 °C for 0.5 h, 20 μL of CH3I was added into the reaction mixture. It was stirred at room temperature for another 0.5 h. Then, a saturated solution of NH4Cl (2.0 mL) was added to quench the reaction. The product was extracted with EtOAc (3 × 3.0 mL) and purified by reversed-phase HPLC (Phenomenex Luna, C18, 250 × 10.0 mm, 2.5 mL/min, 5 μm) using a gradient solvent system from 20% to 100% CH3CN (0.1% formic acid) over 20 min to afford compound 3a (0.5 mg, tR = 14.4 min). Compound 3a: white powder; 1H NMR (600 MHz, CDCl3) δ 5.99 (m, 1H, H-2′), 5.05 (s, 1H, H-2), 3.62 (m, 1H, H-4), 3.29 (s, 3H, 3-NCH3), 3.27 (s, 3H, 7-OCH3), 2.17 (s, 3H, H-5′), 2.17 (m, 1H, H-5), 2.09 (m, 1H, H-6), 2.00 (m, 1H, H-5), 1.90 (s, 3H, H-4′), 1.88 (m, 1H, H-6), 1.28 (d, J = 6.8, 3H, H-8); 13C and 2D NMR, Figure S2; HRESIMS m/z 279.1704 [M + H]+ (calcd for C15H23N2O3, 279.1703).
Acknowledgments
We acknowledge the following grants for funding this project: Welch Foundation I-1689 and NIH R01 CA149833. J.B.M. is a Chilton/Bell Foundation Endowed Scholar.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.5b00604.
13C and 2D NMR data of 3a, calculated 13C NMR shifts for compound 1, computation methods of NMR and ECD, HRESIMS and NMR spectra for compounds 1–8 and 3a, a description of the bioassay protocols used (PDF)
The authors declare no competing financial interest.
Dedication
Dedicated to Professors John Blunt and Murray Munro, of University of Canterbury, for their pioneering work on bioactive marine natural products.
Supplementary Material
References
- a Tidjani S.; Okusa P. N.; Zellagui A.; Banuls L. M.; Stévigny C.; Duez P.; Rhouati S. Nat. Prod. Commun. 2013, 8, 439–440. [PubMed] [Google Scholar]; b Hessel S.; Gottschalk C.; Schumann D.; These A.; Preiss-Weigert A.; Lampen A. Mol. Nutr. Food Res. 2014, 58, 995–1004. 10.1002/mnfr.201300707. [DOI] [PubMed] [Google Scholar]
- Smith L. W.; Culvenor C. C. J. J. Nat. Prod. 1981, 44, 129–152. 10.1021/np50014a001. [DOI] [PubMed] [Google Scholar]
- a Chou M. W.; Wang Y. P.; Yan J.; Yang Y. C.; Beger R. D.; Williams L. D.; Doerge D. R.; Fu P. P. Toxicol. Lett. 2003, 145, 239–247. 10.1016/S0378-4274(03)00293-5. [DOI] [PubMed] [Google Scholar]; b Li N.; Xia Q.; Ruan J.; Fu P. P.; Lin G. Curr. Drug Metab. 2011, 12, 823–834. 10.2174/138920011797470119. [DOI] [PubMed] [Google Scholar]
- Grote R.; Zeeck A.; Stümpfel J.; Zähner H. Liebigs Ann. Chem. 1990, 1990, 525–530. 10.1002/jlac.1990199001100. [DOI] [Google Scholar]
- a Hu J. F.; Wunderlich D.; Thiericke R.; Dahse H. M.; Grabley S.; Feng X. Z.; Sattler I. J. Antibiot. 2003, 56, 747–754. 10.7164/antibiotics.56.747. [DOI] [PubMed] [Google Scholar]; b Snider B. B.; Duvall J. R.; Sattler I. Tetrahedron Lett. 2004, 45, 6725–6727. 10.1016/j.tetlet.2004.07.055. [DOI] [Google Scholar]; c Duvall J. R.; Wu F.; Snider B. B. J. Org. Chem. 2006, 71, 8579–8590. 10.1021/jo061650+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Doyle T. W.; Nettleton D. E.; Balitz D. M.; Moseley J. E.; Grulich R. E. J. Org. Chem. 1980, 45, 1324–1326. 10.1021/jo01295a037. [DOI] [Google Scholar]; b Nettleton D. E. Jr.; Balitz D. M.; Doyle T. W.; Bradner W. T.; Johnson D. L.; O’Herron F. A.; Schreiber R. H.; Coon A. B.; Moseley J. E.; Myllymaki R. W. J. Nat. Prod. 1980, 43, 242–258. 10.1021/np50008a003. [DOI] [PubMed] [Google Scholar]; c Zhang Q.; Schrader K. K.; Elsohly H. N.; Takamatsu S. J. Antibiot. 2003, 56, 673–681. 10.7164/antibiotics.56.673. [DOI] [PubMed] [Google Scholar]; d Bugni T. S.; Woolery M.; Kauffman C. A.; Jensen P. R.; Fenical W. J. Nat. Prod. 2006, 69, 1626–1628. 10.1021/np0602721. [DOI] [PubMed] [Google Scholar]
- a Aoyagi Y.; Manabe T.; Ohta A.; Kurihara T.; Pang G. L.; Yuhara T. Tetrahedron 1996, 52, 869–876. 10.1016/0040-4020(95)00928-0. [DOI] [Google Scholar]; b Snider B. B.; Duvall J. R. Org. Lett. 2005, 7, 4519–4522. 10.1021/ol0518784. [DOI] [PubMed] [Google Scholar]
- Fu P.; MacMillan J. B. Org. Lett. 2015, 17, 3046–3049. 10.1021/acs.orglett.5b01328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga K.; Aoyama S.; Xi W.; Arimoto H.; Yamaguchi K.; Yamada K.; Tsuji T.; Yamada A.; Uemura D. Heterocycles 2000, 52, 1033–1036. 10.3987/COM-99-S124. [DOI] [Google Scholar]
- Kita M.; Miwa R.; Widianti T.; Ozaki Y.; Aoyama S.; Yamada K.; Uemura D. Tetrahedron Lett. 2007, 48, 8628–8631. 10.1016/j.tetlet.2007.10.024. [DOI] [Google Scholar]
- a Pericàs M. A.; Serratosa F.; Valentí E.; Font-Altaba M.; Solans X. J. Chem. Soc., Perkin Trans. 2 1986, 961–967. 10.1039/p29860000961. [DOI] [Google Scholar]; b Alajarín M.; Vidal A.; Sánchez-Andrada P.; Tovar F.; Ochoa G. Org. Lett. 2000, 2, 965–968. 10.1021/ol0056168. [DOI] [PubMed] [Google Scholar]; c Alajarín M.; Sánchez-Andrada P.; Cossío F. P.; Arrieta A.; Lecea B. J. Org. Chem. 2001, 66, 8470–8477. 10.1021/jo015922e. [DOI] [PubMed] [Google Scholar]; d Bornemann H.; Wentrup C. J. Org. Chem. 2005, 70, 5862–5868. 10.1021/jo050429e. [DOI] [PubMed] [Google Scholar]
- a Zheng J.; Zhu H.; Hong K.; Wang Y.; Liu P.; Wang X.; Peng X.; Zhu W. Org. Lett. 2009, 11, 5262–5265. 10.1021/ol902197z. [DOI] [PubMed] [Google Scholar]; b Fu P.; Yang C.; Wang Y.; Liu P.; Ma Y.; Xu L.; Su M.; Hong K.; Zhu W. Org. Lett. 2012, 14, 2422–2425. 10.1021/ol3008638. [DOI] [PubMed] [Google Scholar]
- Konno T.; Meguro H.; Tuzimura K. Tetrahedron Lett. 1975, 16, 1305–1308. 10.1016/S0040-4039(00)72656-6. [DOI] [Google Scholar]
- Stephens P. J.; Pan J. J.; Krohn K. J. J. Org. Chem. 2007, 72, 7641–7649. 10.1021/jo071183b. [DOI] [PubMed] [Google Scholar]
- a Djerassi C.Optical Rotatory Dispersion: Applications to Organic Chemistry; McGraw-Hill: New York, 1960. [Google Scholar]; b Velluz L.; Grand M. L.; Grosjean M.. Optical Circular Dichroism: Principles, Measurements, and Applications; Academic Press: New York, 1965. [Google Scholar]; c Lin S.; Shi T.; Chen K.; Zhang Z.; Shan L.; Shen Y.; Zhang W. Chem. Commun. 2011, 47, 10413–10415. 10.1039/c1cc12079d. [DOI] [PubMed] [Google Scholar]
- a Snatzke G. Angew. Chem., Int. Ed. Engl. 1968, 7, 14–25. 10.1002/anie.196800141. [DOI] [Google Scholar]; b Cerda-García-Rojas C. M.; Coronel A.; del C.; de Lampasona M. E. P.; Catalán C. A. N.; Joseph-Nathan P. J. Nat. Prod. 2005, 68, 659–665. 10.1021/np050004n. [DOI] [PubMed] [Google Scholar]; c Hao Z.-Y.; Liang D.; Luo H.; Liu Y.-F.; Ni G.; Zhang Q.-J.; Li L.; Si Y.-K.; Sun H.; Chen R.-Y.; Yu D.-Q. J. Nat. Prod. 2012, 75, 1083–1089. 10.1021/np300095c. [DOI] [PubMed] [Google Scholar]
- For bohemamine analogues 4–15, the first key intermediate in their biogenetic pathway would be 4-amino-3-oxo-pentanamide.
- Ober D.; Kaltenegger E. Phytochemistry 2009, 70, 1687–1695. 10.1016/j.phytochem.2009.05.017. [DOI] [PubMed] [Google Scholar]
- a Staskun B. J. Org. Chem. 1988, 53, 5287–5291. 10.1021/jo00257a015. [DOI] [Google Scholar]; b Hu Y.; Dietrich D.; Xu W.; Patel A.; Thuss J. A.; Wang J.; Yin W. B.; Qiao K.; Houk K. N.; Vederas J. C.; Tang Y. Nat. Chem. Biol. 2014, 10, 552–554. 10.1038/nchembio.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M. D.; Selvin R. C.; Claus W.; Guillard R. R. L. J. Phycol. 1987, 23, 633–638. 10.1111/j.1529-8817.1987.tb04217.x. [DOI] [Google Scholar]
- While this manuscript was under review, the Kwon laboratory reported compounds 1 and 2. See:Kim M. C.; Lee J. H.; Shin B.; Subedi L.; Cha J. W.; Park J-S.; Oh D-C.; Kim S. Y.; Kwon H. C. Org. Lett. 2015, 17, 5024–5027. 10.1021/acs.orglett.5b02495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.