

Abstract
RNA-binding proteins (RBPs) control the regulation of gene expression in eukaryotic genomes at post-transcriptional level by binding to their cognate RNAs. Although several variants of CLIP (crosslinking and immunoprecipitation) protocols are currently available to study the global protein–RNA interaction landscape at single-nucleotide resolution in a cell, currently there are very few tools that can facilitate understanding and dissecting the functional associations of RBPs from the resulting binding maps. Here, we present Seten, a web-based and command line tool, which can identify and compare processes, phenotypes, and diseases associated with RBPs from condition-specific CLIP-seq profiles. Seten uses BED files resulting from most peak calling algorithms, which include scores reflecting the extent of binding of an RBP on the target transcript, to provide both traditional functional enrichment as well as gene set enrichment results for a number of gene set collections including BioCarta, KEGG, Reactome, Gene Ontology (GO), Human Phenotype Ontology (HPO), and MalaCards Disease Ontology for several organisms including fruit fly, human, mouse, rat, worm, and yeast. It also provides an option to dynamically compare the associated gene sets across data sets as bubble charts, to facilitate comparative analysis. Benchmarking of Seten using eCLIP data for IGF2BP1, SRSF7, and PTBP1 against their corresponding CRISPR RNA-seq in K562 cells as well as randomized negative controls, demonstrated that its gene set enrichment method outperforms functional enrichment, with scores significantly contributing to the discovery of true annotations. Comparative performance analysis using these CRISPR control data sets revealed significantly higher precision and comparable recall to that observed using ChIP-Enrich. Seten's web interface currently provides precomputed results for about 200 CLIP-seq data sets and both command line as well as web interfaces can be used to analyze CLIP-seq data sets. We highlight several examples to show the utility of Seten for rapid profiling of various CLIP-seq data sets. Seten is available on http://www.iupui.edu/∼sysbio/seten/.
Keywords: RNA-binding proteins, CLIP (crosslinking and immunoprecipitation), gene set enrichment, functional enrichment, genotype–phenotype, post-transcriptional networks
INTRODUCTION
Genes are transcribed into RNAs but these RNAs are not mature, especially in eukaryotic organisms where there are several layers of post-transcriptional regulation. Therefore, they must be processed before they are translated into protein products in the ribosomes. These post-transcriptional processes include 5′ capping, 3′ polyadenylation, splicing, and possibly RNA editing. Processes such as 5′ capping and 3′ polyadenylation ensure that ends of the RNA are protected during their maturation and their stability regulated (Shatkin and Manley 2000). The splicing process joins different parts of a protein coding sequence together as a typical eukaryotic gene includes exons separated by long noncoding sequences (i.e., introns) (McManus and Graveley 2011). It also provides the ability to produce different protein products (by joining different exons) as a result of alternative splicing of the same gene in eukaryotes. RNA editing is another mechanism of post-transcriptional regulation that results in the alteration of one or more nucleotides in the RNA molecule (Samuel 2003). These processes, as well as transport, degradation, and translation of the RNAs, are mediated by RNA-binding proteins (RBPs) (Gerstberger et al. 2014; Neelamraju et al. 2015). In cells, RNA is found to be assembled with RBPs and other proteins forming ribonucleoprotein complexes (RNPs) (Janga 2012). For example, the SR protein SF2/ASF acts from alternative splicing to translation of an RNA (Kim et al. 2009). Moreover, some heterogeneous nuclear ribonucleoproteins (hnRNPs) are known to participate in RNA splicing, 3′-end processing, transcriptional regulation, and immunoglobulin gene recombination (Chaudhury et al. 2010).
Understanding these dynamic post-transcriptional regulatory networks requires the study of interactions between RNAs and RBPs. For this purpose, crosslinking and immunoprecipitation (CLIP) and related experimental protocols have been devised. All CLIP protocols involve RNA–RBP ultraviolet (UV) crosslinking followed by immunoprecipitation against an RBP of interest (Ule et al. 2003). There are several CLIP protocols: CLIP-seq, PAR-CLIP, HITS-CLIP, and iCLIP. CLIP-seq protocol involves sequencing the cDNA library created from the RNA, which was previously purified by proteinase digestion after UV crosslinking and immunoprecipitation (Konig et al. 2011). For instance, PAR-CLIP (photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation) is a modified CLIP-seq technology that involves the use of photoreactive ribonucleoside analogs. These analogs can be ultraviolet crosslinked to interacting RBPs and are modified upon crosslinking. Hence, they can be used to separate RNAs bound by the RBP of interest from the background unbound RNAs (Spitzer et al. 2014). HITS-CLIP (high-throughput sequencing of RNA isolated by crosslinking and immunoprecipitation) is another CLIP protocol that overcomes the limitation in the low number of tags by yielding a higher number of tags for the same cost (Darnell 2010). iCLIP (individual-nucleotide resolution UV crosslinking and immunoprecipitation) is yet another CLIP protocol that provides genome scale, high-resolution, and specificity methods to enable analysis of cDNAs that are truncated at the RNA–RBP crosslink sites (Huppertz et al. 2014). Several computational methods have been developed for peak detection indicating the extent of binding from the data produced by these protocols. A common first step in all these frameworks before the peak detection is to map all the reads to the genome/transcriptome using algorithms such as Bowtie, RMAP, Novoalign (http://www.novocraft.com/products/novoalign/), and TopHat (Langmead et al. 2009; Smith et al. 2009; Trapnell et al. 2009; Kim et al. 2013). After mapping, cluster detection is performed, where a read belongs to a cluster if it overlaps at least one nucleotide with another read from the cluster. At this step, in order to filter noise, reads with a length greater than a determined threshold and clusters with a minimum number of unique reads can be selected for peak detection. The most common approach for peak detection is to analyze cluster distribution profiles by improving the signal-to-noise ratio, and hence removing background and false positives. The software that use this strategy include WavClusteR, PARalyzer, Piranha, PIPE-CLIP, and dCLIP (Corcoran et al. 2011; Uren et al. 2012; Chen et al. 2014; Reyes-Herrera and Ficarra 2014; Wang et al. 2014; Comoglio et al. 2015).
Although these tools are available for post-processing CLIP-seq data, there is no specific tool to either perform an enrichment analysis on such data sets or to compare them for functional or phenotypic differences. Perhaps the only tool that can perform gene set enrichment analysis for ChIP-seq data sets and could be configured for CLIP-seq data sets is ChIP-Enrich (Welch et al. 2014). Although ChIP-seq and CLIP-seq protocols are fundamentally different at several levels including approaches used for cross-linking, reagents used for sequencing library preparation, efficiency of crosslinking, as well as the peak calling algorithms employed, ChIP-Enrich provides an option to perform enrichment analysis of CLIP-seq processed outputs. The principle of an enrichment analysis is to associate gene sets (i.e., groups of relevant genes; e.g., processes, phenotypes, or diseases) with a given study by using the fact that the cofunctioning genes should have a higher potential to be detected by the high-throughput technologies (e.g., CLIP protocols). Such an approach can make the analysis of large gene lists move from an individual gene-oriented view to a relevant gene group-based analysis (Huang da et al. 2009). Huang da et al. (2009) categorize the available enrichment analysis methods into three groups. First, the singular enrichment analysis (SEA) group: Enrichment P-value in these methods is calculated on each gene set from the preselected interesting gene list utilizing Fisher's exact test, χ2, or binomial statistical methods. In the second group, gene set enrichment analysis (GSEA) methods, a complete set of genes (without preselection) and corresponding experimental values are given, and they utilize Kolmogorov–Smirnov-like, t-test, permutation, or z-score statistical methods. The last group is modular enrichment methods, which are similar to SEA but hierarchy among gene sets or genes are considered into the enrichment P-value calculation by utilizing κ statistics and Czekanowski–Dice Pearson's correlation (Huang da et al. 2009). While these methods are available for functional analysis or functional enrichment of genes from microarray and RNA-seq with some efforts specific to RIP-chip data (Erhard et al. 2013), no methods are available that can consider the binding affinity or scores of an RBPs binding potential on an RNA from CLIP-seq protocols to identify/perform an enrichment analysis using both functional and gene set enrichment approaches. Since it is increasingly appreciated, and an array of new technologies such as RBP Bind-n-Seq (Lambert et al. 2014, 2015) and DO-RIP-seq (Nicholson et al. 2016) are being developed to study the binding affinities of RBPs on target sites, it becomes important to leverage the signal strength of binding from CLIP-seq profiles for downstream functional analysis. Seten is able to do so by assuming that the binding score resulting from a peak calling method is a proxy for the extent of regulatory control of the RBP on the target transcript.
The primary foundation of Seten (http://www.iupui.edu/~sysbio/seten/) is to identify and compare processes, phenotypes, and diseases associated with RNA-binding proteins from condition-specific CLIP-seq profiles, given binding profile data sets provided as BED (browser extensible data) files. Seten comes with a web interface (WI) developed in JavaScript and a command line interface (CLI) developed in Python. Seten WI provides an easy to use front end without the need for installation and a better visualization and comparison of the enrichment results. Seten CLI is able to analyze multiple data sets in a single command and both the interfaces can be configured with multiple options.
RESULTS
Overview of Seten
In Seten, for each input BED file that has at least the five columns, namely, chromosome, chromosome start, chromosome end, feature name, and score associated with the binding of an RBP resulting from running a peak calling algorithm on a genome-aligned CLIP-seq data set, a gene set enrichment analysis (GSEA) is performed against the gene set collections selected by the user (Fig. 1, see Materials and Methods). Both the web interface (WI) and command line interface (CLI) versions of Seten currently support the gene sets from BioCarta, KEGG, Reactome, Gene Ontology (GO) biological process, GO molecular function, GO cellular compartment, Human Phenotype Ontology (HPO), and MalaCards Disease Ontology for organisms including fruit fly, human, mouse, rat, worm, and yeast, with the CLI allowing the user to include additional gene set collections and organisms (Supplemental Table S1). To facilitate easy access and navigation of existing CLIP-seq data sets, Seten WI includes results from precomputed functional analysis of 68 human RBPs obtained from CLIPdb as well as 138 human RBPs profiled in the ENCODE project (Supplemental Tables S2, S3). In addition to providing precomputed integrated functional analysis of dozens of CLIP-seq experiments, Seten WI also provides a user-friendly interface to compare the resulting annotations across experiments and RBPs as exportable bubble charts.
FIGURE 1.

Flowchart showing an overview of Seten's implementation for doing gene set and functional enrichment analysis from CLIP-seq data sets. Peak-detected data sets from RBP-specific CLIP-seq studies, CLIPdb, and ENCODE projects (Yang et al. 2015; Van Nostrand et al. 2016) are obtained as BED files to provide input to Seten. We organized several gene set collections for multiple genomes including fruit fly, human, mouse, rat, worm, and yeast. Currently included gene set collections comprise pathway annotations (BioCarta, KEGG, and Reactome), Gene Ontology annotations (biological process, molecular function, cellular compartment), Human Phenotype Ontology (HPO—human only), and MalaCards Disease Ontology (human only) (Kanehisa and Goto 2000; Nishimura 2001; Milacic et al. 2012; Kohler et al. 2014; Rappaport et al. 2014; Fabregat et al. 2016; Kanehisa et al. 2016). Supplemental Table S1 shows the number of gene sets across gene set collections currently available across organisms in Seten. Scores associated with each gene from a BED file are employed for gene set enrichment analysis by organizing the scores according to the chosen scoring method. Scores mapped onto the genes are used to compute an enrichment using a competitive permutation test, and corrected P-values from multiple testing are reported. In contrast, the functional enrichment method only uses the associated genes and not the scores from BED files for enrichment analysis using Fisher's exact test and computes a false discovery rate.
Seten's gene set enrichment outperforms functional enrichment, with peak scores contributing to the discovery of true annotations
In order to evaluate the performance of Seten, we employed the gene set and functional enrichment implementations in Seten and compared their performance against “negative control” BED files generated using BEDTools (Quinlan and Hall 2010) for eCLiP peaks of RBPs (IGF2BP1, PTBP1, SRSF7 in K562 cell line) (see Materials and Methods). For each eCLIP data set and their corresponding negative controls, we benchmarked the performance of the gene set and functional enrichment implementations against the annotations identified using the CRISPR RNA-seq gold standard for the corresponding RBPs in the K562 cell line (see Materials and Methods). We ran Seten using default parameters (i.e., corrected P-value [<0.01], gene set size [<350], number of gene set hits per RBP [>10]) for both the eCLIP and each negative BED separately. The F1 score, which is the harmonic mean of precision and recall, was computed for respective Seten runs against CRISPR gold standard annotations (Materials and Methods). For random data, we repeated the process for five random negative controls for each RBP and report the average F1 score for each RBP, as shown in Figure 2. Our results show that gene set enrichment exhibits relatively higher performance than functional enrichment for both the Reactome and GO Biological Process annotations (Fig. 2). F1-scores were also found to be significantly higher for eCLIP data compared to the negative controls resulting from randomized scores or genomic coordinates, for the gene set enrichment method (Fig. 2). In contrast, for functional enrichment, although results compared to randomized coordinates were higher, there was no significant difference in F1-scores compared to the randomized peak scores, suggesting that while functional enrichment is not impacted by the scores, gene set enrichment implementation has a significant improvement due to the use of scores (Fig. 2). Overall, although we have employed the maximum score for each gene as the scoring method, our analysis demonstrates that Seten's gene set enrichment implementation is likely to outperform functional enrichment for inference of annotations from eCLIP profiles, by exploiting the scores that can act as proxy for the extent of binding.
FIGURE 2.
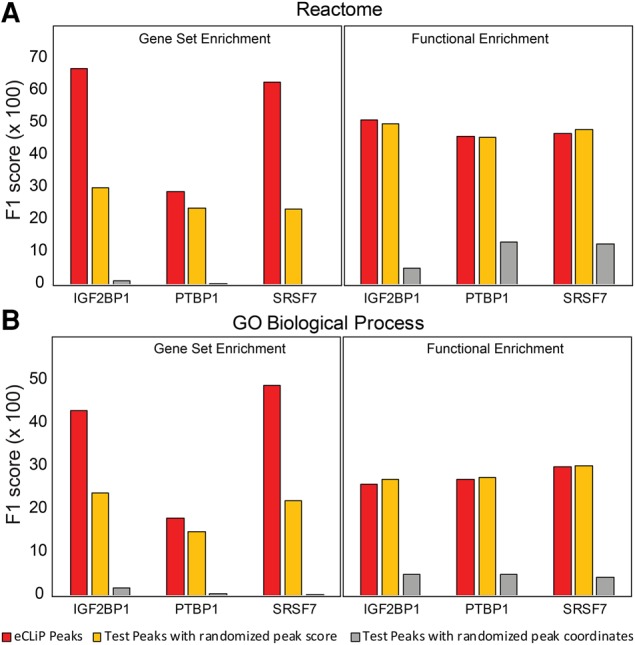
Comparison of Seten's gene set and functional enrichment methods against negative control. Histograms showing the performance comparison of Seten's gene set enrichment analysis (GSEA) and functional enrichment (FE) options along with their corresponding random BED files for each RBP, benchmarked against their CRISPR RNA-seq gold standard. F1 score, harmonic mean of precision and recall, represented on y-axis for each data set/option, was computed against CRISPR gold standard separately for (A) Reactome and (B) GO Biological Process by running Seten using eCLiP peaks (in red) and “negative control” peaks (test peaks with randomized peak score shown in orange, test peaks with randomized peak coordinates shown in gray). Negative control BED files for each RBP were generated using BEDTools, as described in Materials and Methods.
Seten can identify human phenotypic associations for RBPs profiled using diverse CLIP protocols
We ran Seten CLI on 206 human CLIP-seq data sets collected from CLIPdb and ENCODE to obtain gene set and functional enrichment results for multiple gene set collections (see Materials and Methods). We then filtered the gene set enrichment results for gene sets according to a corrected P-value (<0.01), gene set size (<350), number of gene set hits per RBP (>10), and limited the gene set collection to Human Phenotype Ontology (HPO) only. Since functional analysis on several of the RBPs using Gene Ontology (GO) and pathway level annotations has been performed in respective original CLIP studies, we preferred to focus our analysis in this section on the phenotypes identified by Seten using gene set enrichment, which was found to outperform the simple functional enrichment analysis. The results of these phenotype associations after the above-described filters have been applied are shown as a heatmap in Figure 3, generated by using the R package pheatmap (Kolde 2015). As can be seen in the heatmap, RBPs that belong to the same family were generally found to be clustered together, suggesting their phenotypic impact is likely similar. Among heterogeneous nuclear ribonucleoproteins (hnRNPs), hnRNP A1 and hnRNP U clustered together and were found to have the most number of HPO associations. hnRNP A1 nucleocytoplasmic shuttling activity has been reported to be required for normal myelopoiesis and BCR/ABL leukemogenesis (Iervolino et al. 2002). Our results from Seten for hnRNP A1 (K562 cell line/chronic myelogenous leukemia) indicate strong links to HPO gene sets such as erythroid hypoplasia (P-value = 0.001), acute leukemia (P-value = 0.001), and thrombocytosis (P-value = 0.001). hnRNP U has been shown to be linked with myeloid leukemia factor 1 (MLF1), which is an oncoprotein associated with hemopoietic lineage commitment and acute myeloid leukemia (Winteringham et al. 2006). In addition, hnRNP U has also been shown to regulate the β-transducin repeat-containing protein ubiquitin ligase (E3RS), which is linked to colorectal cancer (Davis et al. 2002; Ougolkov et al. 2004). Our results for hnRNP U include colon cancer (P-value = 0.0110) and acute leukemia (P-value = 0.0210). Additionally, we have obtained a recent iCLIP data set of FASTKD2 (FAS-activated serine/threonine kinase D2—in HEK293, human embryonic kidney cell line) where FASTKD2's role as an RBP is investigated (Popow et al. 2015) (see Materials and Methods). FASTKD2 is a recently discovered noncanonical RBP, which has been shown to be linked to mitochondrial encephalomyopathy (Ghezzi et al. 2008). We ran Seten CLI for this data set using default options, and the results from MalaCards Disease Ontology clearly indicated MELAS syndrome (P-value = 0.001) as one of the most significantly associated diseases. MELAS stands for mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes and is one of the family of mitochondrial cytopathies. MELAS is known to affect many of the body's systems, particularly the brain and nervous system (encephalo-) and muscles (myopathy), and is documented to be caused by mutations in the genes in mitochondrial DNA. Similarly, the FASTKD2 (K562) data set we obtained from ENCODE project also had MELAS syndrome as one of the significant hits (P-value = 0.0240) in its MalaCards Disease Ontology results.
FIGURE 3.

The inset of a heatmap (given in Supplemental Fig. S1) showing the clustering of RBPs based on their predicted human phenotypic associations from Seten. A total of 51 RBPs that had at least 160 phenotypic associations at a minimal gene set P-value of 0.01 were employed to generate this heatmap. Hierarchical clustering of the data on both axes revealed RBPs, which are likely to exhibit similar phenotypes as well as phenotypes shared by the RBPs included in this study. Only RBPs that had more than 10 gene sets associated and only gene sets that had less than 350 genes and exhibited a minimal P-value of 0.01 are included in this heatmap.
Cell type-specific differences in gene set associations can be identified by Seten
The above results also suggest that it is possible to not only identify the gene set associations of an RBP, but RBPs profiled in different cell lines and conditions can be compared for one or more gene set collections. Such a feature is available in Seten WI for both precomputed CLIP-seq data sets as well as for user uploaded BED formatted CLIP results. Seten can compare one or multiple gene set collections across conditions/cell lines of one or more RBPs to dynamically generate bubble charts for easy comparison of differences in the significance of associated gene sets. In both CLIPdb and ENCODE data sets, some RBPs have CLIP data from different cell lines, which allowed us to use Seten for comparing these cell type-specific data sets.
The two FASTKD2 data sets discussed in the previous section also exhibit cell line-specific differences as shown in Figure 4A. While the FASTKD2/K562 (human bone marrow cell line having chronic myelogenous leukemia) gene set enrichment results show pure red-cell aplasia (P-value = 0.001) and Diamond–Blackfan anemia (P-value = 0.001), the other FASTKD2/HEK293 does not exhibit these disease annotations.
FIGURE 4.

(A) The dynamically generated bubble chart from Seten WI, showing the comparison of significantly enriched MalaCards Disease Ontology terms for FASTKD2 in HEK293 and K562 cell lines. (B) The inset of a dynamically generated bubble chart from Seten WI (given in Supplemental Fig. S2), showing the comparison of significantly enriched MalaCards Disease Ontology terms for DDX6 in K562 and HepG2 cell lines. Only gene sets that had >5% of the total genes and exhibited a minimal P-value of 0.05 in one of the cell lines are included in this comparison. The radius of bubbles is computed as negative log10 (corresponding P-value).
DEAD-box helicase 6 (DDX6) is an RNA helicase found in P-bodies and stress granules and it functions in mRNA degradation and translation suppression (Wang et al. 2015). It has been shown to contribute to lymphoma genesis by deregulation of BCL6 (B-Cell CLL/Lymphoma 6) in nodal marginal zone lymphoma (Stary et al. 2013). It has also been shown to be required for efficient hepatitis C virus replication (Jangra et al. 2010). Figure 4B (a small subset of Supplemental Fig. S2) shows a bubble chart comparing the significance scores for MalaCards Disease Ontology term associations for DDX6/K562 (chronic myelogenous leukemia cell line) and DDX6/HepG2 (hepatocellular carcinoma cell line). As is evident from the chart, while monocytic leukemia (P-value = 0.0110) is specific to the K562 sample, fatty liver disease (P-value = 0.0140), liver cirrhosis (P-value = 0.001), and hepatoblastoma (P-value = 0.001) are specific to the HepG2 sample. However, we also see acute promyelocytic leukemia to be significant (P-value = 0.008) for DDX6/HepG2. This might be seen because DDX6 activation has been observed in acute leukemia (Poppe et al. 2004). Additionally, mantle cell lymphoma (P-value = 0.001 for DDX6/K562) and anaplastic large cell lymphoma (P-value = 0.005 for DDX6/K562 and 0.001 for DDX6/HepG2) appear as significant hits. Moreover, viral hepatitis is one of the significant hits for DDX6/HepG2 (P-value = 0.0150). These results suggest that Seten can be employed to study and navigate condition and cell line, as well as tissue-specific variations in the gene set associations for RBPs, starting from CLIP-seq data.
Seten's GO Biological Process and Reactome results agree with ChIP-Enrich gene set enrichment tool results
Since there are no existing tools to perform a gene set enrichment analysis on CLIP-seq data sets, in order to compare our results we used the ChIP-Enrich (C-E) gene set enrichment tool originally developed for ChIP-seq data sets by configuring its options to make it suitable for CLIP-seq data sets (Welch et al. 2014). We set the locus definition option to “Nearest gene” to assign all peaks to the nearest gene that is similar to our approach. We limited our comparison to GO Biological Process (GOBP) from Gene Ontology and Reactome from pathway databases. We filtered out the gene sets having more than 350 genes to be consistent with the default threshold in Seten. We ran both tools for several data sets from the ENCODE project. To compare the results, we filtered the enriched gene sets using the P-value threshold (corrected P-value <0.05 in the respective tools) and then ranked them separately. Finally, we took the overlapping gene sets between them and did a hypergeometric test to determine the significance of the overlap between the two approaches. We first compared Alanyl-tRNA Synthetase (AARS–K562) GOBP and Reactome results, which yielded a GOBP P-value of 6.15 × 10−14 and a Reactome P-value of 5.44 × 10−27 (hypergeometric test), indicating a significant agreement of the discovered processes and pathways between the two methods (Fig. 5A; Supplemental Table S2). We then compared the putative RNA-binding protein 15 (RBM15–K562) results for GOBP and Reactome gene set collections and obtained a GOBP P-value of 6.42 × 10−25 and a Reactome P-value of 2.70 × 10−49, suggesting a significant overlap (Fig. 5B; Supplemental Table S3). We finally compared Fragile X Mental Retardation 1 (FMR1–K562) GOBP and Reactome results, which yielded a GOBP P-value of 2.59 × 10−24 and a Reactome P-value of 6.90 × 10−35, indicating the reproducibility of the enriched processes/pathways between the methods (Fig. 5C; Supplemental Table S4).
FIGURE 5.

(A) The comparison of Seten and ChIP-Enrich using AARS–K562 data set for GO Biological Process and Reactome gene set enrichment analysis results. (B) The comparison of Seten and ChIP-Enrich using RBM15–K562 data set for GO Biological Process and Reactome gene set enrichment analysis results. (C) The comparison of Seten and ChIP-Enrich using FMR1–K562 data set for GO Biological Process and Reactome gene set enrichment analysis results.
Benchmarking of Seten and ChIP-Enrich against CRISPR RNA-seq reveals superior performance of Seten
Recent progress in utilizing CRISPR/Cas9 technologies for genome editing has enabled rapid sequencing-based profiling of genomic phenotypes (D'Agostino and D'Aniello 2017). Although the majority of the RBPs are known to be encoding for essential genes (Mittal et al. 2009), the ENCODE project has been successful in generating RNA-sequencing data of CRISPR/Cas9 based knockouts of several RBPs, including IGF2BP1, SRSF7, and PTBP1 in the human K562 cell line (The ENCODE Project 2017). Hence, to generate a gold standard set of functional annotations impacted by these RBPs and to benchmark both Seten and ChIP-Enrich tools against this common reference set for which both eCLIP and CRISPR data are available, we processed and organized the CRISPR RNA-seq data as described in Materials and Methods. By utilizing the functional annotations obtained from gene set enrichment analysis of the relative gene expression changes from CRISPR control versus knockout for each of these RBPs, as the gold standard, we compared the performance of both the tools against this reference by computing precision and recall (see Materials and Methods). As shown in Figure 6, for each of these three RBPs, Seten was found to exhibit significantly higher precision for both Reactome and GO Biological Process annotations compared to that observed for ChIP-Enrich (C-E). Seten exhibited an average precision of 72% and 58% for Reactome and GOBP gene sets. In contrast, C-E was found to show an average precision of 42% and 8%, respectively, indicating that Seten is more suitable for functional annotation of CLIP-seq data (Fig. 6). Comparison of the average recall values between the tools indicated that, while Seten exhibited higher recall than C-E for Reactome (51% versus 47%), an inverse trend was seen for GOBP annotations (32% versus 45%). A major contributor to the lower average recall of Seten is PTBP1, which was found to exhibit a relatively lower recall for both Reactome and GOBP annotations. In this context, it must be noted that not all RBP loss of binding events result in corresponding changes in RNA expression levels of their targets—a major assumption in the calculation of recall. This could be due to a number of reasons such as (i) redundancy in the functionality of RBPs, where a paralogous RBP might complement the function of the mutated RBP, (ii) RNA levels might not be impacted but protein levels might be impacted, and (iii) quality of the binding site might be low or the functional impact of the binding site might be minimal. Nevertheless, although the number of RBPs with both eCLIP and CRISPR data is currently limited, it is possible to conclude from this data that Seten achieves significantly higher precision and comparable recall as that of C-E.
FIGURE 6.

Benchmarking of predicted functional annotations from Seten and ChIP-Enrich against those identified from CRISPR-based RNA-seq data sets of RNA-binding proteins in K562 cell line. Precision and recall plots for IGF2BP1, SRSF7, and PTBP1 using Seten and ChIP-Enrich for the gene set collections (A) Reactome and (B) GO Biological Process. In both cases, gene set enrichment approach as implemented in the respective tools was utilized to generate functional annotations from eCLIP-based profiles, to compare their relative performance. Seten was found to exhibit a significantly higher precision and comparable recall to that observed for ChIP-Enrich.
It is important to note that currently there are very few high-resolution CRISPR data sets that stand orthogonal to CLIP-seq profiles. Also, since CRISPR screens are still in their infancy, it is unclear to what extent they strictly identify only the direct effects of regulatory molecules such as RBPs and not secondary off-target effects (D'Agostino and D'Aniello 2017). Hence, additional orthogonal approaches to probe and measure the genome-wide impact due to the loss/gain of function of an RBP are needed to comprehensively understand, model, and improve the functional annotations of RBPs using CLIP-seq profiles.
DISCUSSION
Functional enrichment is a common strategy to associate a particular list of genes with the preannotated gene sets. However, for data sets where a score can be given per gene in the list such as binding signals, which act as a proxy to indicate the extent of binding of RBPs to their targets, it is important to include those scores in the enrichment analysis for obtaining a better insight. While a crucial aspect of including such scores in functional analysis of post-transcriptional networks resulting from CLIP-seq protocols is the quality and resolution of the binding profiles of RBPs, it is important to note that these scores do depend on the depth of sequencing, efficiency of cross-linking method, as well as the ability of the peak calling algorithm to uncover the true positive sites. For instance, when the CLIP-seq protocols and/or the subsequent peak calling algorithms fail to produce a high-resolution binding profile, it is not possible to capture enriched gene sets based on the binding scores. We note that the published CLIP-seq data from individual research laboratories processed and made available via CLIPdb (Yang et al. 2015) are generally of lower sequencing depth and genomic coverage compared to those resulting from the ENCODE project. Hence, we anticipate that with enhanced CLIP protocols, such as those resulting from the ENCODE project (Van Nostrand et al. 2016), it is not only possible to accurately identify and quantify the extent of binding but to also uncover their differences between data sets to capture the processes, phenotypes, and diseases associated with RBPs to rationally design experiments and therapeutics for the specific subsystem under study. Indeed, analysis of the correlations between the corrected P-values obtained using gene set and functional enrichment methods, as implemented in Seten for various gene set collections and all available CLIP-seq data sets, resulted in a weak-to-moderate extent of correlation between the results, suggesting that different aspects might be captured by the respective methods (Supplemental Fig. S3). Nevertheless, benchmarking both gene set and functional enrichment methods as implemented in Seten against available CRISPR RNA-seq data sets revealed that scores significantly contribute to improved inference of true functional associations. Hence, as CLIP-seq protocols become more accurate in capturing the extent of binding and regulation of the post-transcriptional target gene, exploiting such signal information should significantly improve the downstream functional analysis pipelines.
Our framework presented here not only performs an enrichment analysis using the scores resulting from peak calling algorithms on CLIP-seq data sets, but it can also perform a comparison of the identified processes and phenotypes across a set of profiled RBPs both within and across conditions or tissue types being studied. Indeed, a comparison of the number of shared targets and binding regions for various RBPs that have been profiled using eCLIP protocol (Van Nostrand et al. 2016) in both the K562 and HepG2 cell lines, indicated that several RBPs, such as RBM22, DDX6, and TBRG4, exhibited significant differences in the targeted genes and their recognized binding regions, suggesting the possibility of extensive rewiring in post-transcriptional networks between cell types. Such variations in the post-transcriptional regulatory networks controlled by an RBP are likely to result in different functional outcomes, and tools like Seten fill the gap in our understanding of the downstream biological context resulting from such alterations.
Seten is implemented as a web interface (WI) using JavaScript and a command line interface (CLI) using Python (http://www.iupui.edu/~sysbio/seten/). Seten WI provides exportable visualizations of results as bar charts and bubble charts (in SVG format) and requires no installation or dependency except for an up-to-date browser. Seten CLI requires an installation and some dependencies, but thanks to the Python Package Index, a single command can take care of the installation. Using Seten CLI, multiple data sets can be analyzed using a single command.
MATERIALS AND METHODS
CLIP-seq data sets
To test Seten and construct a database of precomputed functional and gene set enrichment results, we used peak-detected data sets from CLIPdb and ENCODE projects (Yang et al. 2015; Van Nostrand et al. 2016). We downloaded human RBP data sets with peak calling scores from CLIPdb and merged multiple samples of an RBP for a cell line, which resulted in 68 unique RBP-cell line pairs (see Supplemental Table S5). Similarly, we also downloaded human RBP data sets along with their detected peaks from the ENCODE project in BigBed format and converted to BED format using UCSC BigBed tools (Kent et al. 2010). There are 138 unique RBP-cell line pairs after merging biological replicates of RBPs within a cell line in this data set (See Supplemental Table S6). Additionally, we obtained an iCLIP-based peak-detected data set for a noncanonical RBP FASTKD2, including three replicates that we merged and analyzed as a single data set (Popow et al. 2015). We merged the biological replicates or the data sets for the same RBP-cell line pairs by concatenating their corresponding BED files using Unix cat command in order to maximize the number of binding data available per RBP-cell line. In this study, the scores associated with a detected peak from a CLIP-seq experiment are also referred to as binding affinity scores of an RBP on the target RNA because they represent a proxy measure for the extent of binding on the transcript.
Gene set collections
Gene sets are groups of relevant genes that share the same pathway, function, or phenotype. We manually downloaded and organized gene set collections for fruit fly, human, mouse, rat, worm, and yeast. The gene set collections we obtained are pathway annotations (BioCarta, KEGG, and Reactome), Gene Ontology annotations (biological process, molecular function, cellular compartment), Human Phenotype Ontology (HPO—human only), and MalaCards Disease Ontology (human only) (Kanehisa and Goto 2000; Nishimura 2001; Milacic et al. 2012; Kohler et al. 2014; Rappaport et al. 2014; Fabregat et al. 2016; Kanehisa et al. 2016). The number of gene sets in gene set collections and the availability of organisms are given in Supplemental Table S1.
Obtaining distinct gene scores list
Binding sites from the input BED file are mapped onto their corresponding gene symbols using a mapping table downloaded from Ensembl for each available organism (Yates et al. 2016). After mapping is complete, in the case that multiple scores are available for a gene, we provide multiple methods to obtain a single score to represent that gene, which results in a distinct set of genes and their corresponding scores representing the extent of binding by an RBP. The available methods are maximum, minimum, mean, median, and sum. Therefore, for instance, if the selected method is sum, then the final score given to the corresponding gene will be the sum of all scores available for that gene. The default method we selected is maximum.
Gene set association analysis
We implemented a previously reported competitive method to apply gene set association analysis to transcription factor binding data sets to test whether an RBP preferentially targets to genes in a given gene set (Patra et al. 2015). This method finds the common genes between given RBP targets and genes in a given gene set and compares the scores of common genes to the scores of randomly permutated genes from RBP targets by a competitive test where the Mann–Whitney U test is used to test whether the median score of the common genes is significantly higher than that of randomly permutated genes (Mann and Whitney 1947). We provide options to set thresholds for maximum number of genes in a given gene set to allow more specific gene sets to be used (defaults to <350) and minimum number of common genes between RBP targets and genes in a given gene set (defaults to >5). Also, we provide an option to control the number of permutations to perform (defaults to 1000). At each permutation, the method checks if the P-value from the Mann–Whitney U test is significant using another option (defaults to < 0.05) and counts the significant tests. At the end, the final corrected P-value is computed as
Such corrected P-values resulting from gene set enrichment analysis are referred to as P-values in the manuscript for brevity.
Functional association analysis
We also implemented a functional association analysis using a two-sided Fisher's exact test (FET) for traditional functional enrichment (Fisher 1922). A correction method is used to correct the P-values obtained from functional enrichment analysis (FET). Currently, Seten's web interface has only one method, which is the false discovery rate method (FDR) or the Benjamini–Hochberg method (Benjamini and Hochberg 1995). Seten's command line interface includes several other methods for correcting the resulting P-values. Note that such correction methods are only available for functional enrichment analysis as the gene set enrichment method employs a different correction approach, as described above.
Processing CRISPR RNA-seq data sets of RBPs
Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 is a recently developed system for engineering genomes, which has transformed our ability to manipulate genes in cell lines and animal models (D'Agostino and D'Aniello 2017). In the ENCODE project, multiple RBPs have been screened using the CRISPR/Cas9 system followed by RNA-sequencing to better understand the downstream pathways impacted by the loss of function of an RBP. Hence, in order to generate a reference gold standard set of functional annotations that are affected by an individual RBP, and as a means of benchmarking the quality of the annotations predicted by Seten and ChIP-Enrich (C-E) from CLIP-seq data, we obtained RNA-sequencing data from nonspecific CRISPR control and those treated with gRNAs against three different RBPs, namely IGF2BP1, SRSF7, and PTBP1 in K562 cells (The ENCODE Project 2017). Since these RBPs had both eCLIP and CRISPR RNA-seq data sets available, they were ideal for performing a benchmarking analysis. This data set was composed of eight nonspecific CRISPR control RNA-seq data sets representing wild-type K562 cells and two replicate RNA-seq data sets each for the RBPs IGF2BP1, SRSF7, and PTBP1, wherein gRNAs were used to deplete the functional form of RBPs, enabling us to perform a quantitative differential expression analysis followed by gene set enrichment for various gene set collections using Seten, to develop a gold standard. In brief, we collected all the available RNA-seq data for CRISPR control and knockout data for multiple RBPs in the K562 cell line and processed the raw quality filtered (Phred score >30) sequence reads using HISAT (Kim et al. 2015) and StringTie (Pertea et al. 2015) pipeline with default parameters, to generate gene expression levels in transcripts per million (TPM) reads for all human annotated Ensembl genic features (Yates et al. 2016). The processed and gene expression quantified data were formatted into expression matrices and utilized for generating a reference set of functional annotations impacted by the respective RBPs, as described below.
Generation of gold standard set of functional annotations using CRISPR RNA-seq data sets of RBPs
Gene expression matrices comprising of CRISPR control and knockout for each RBP were used to compute a relative change in expression for each gene. Relative change in expression is defined as the ratio of the absolute change in the expression difference between the mean of replicates of control and knockout, respectively, divided by the mean expression level of the gene in the control RNA-seq data sets. By utilizing such a normalized relative change in expression of each gene across the entire genome for each combination of control and CRISPR knockout data sets of an RBP, we performed gene set enrichment analysis using Seten for both Reactome and GOBP gene set collections. This enabled the identification of gene sets enriched due to the loss of an RBP at a corrected P-value of 0.05 using the Seten's GSEA approach. Such gene sets have been defined in this study as the gold standard annotations for the RBP for the corresponding gene set collections. By utilizing these annotations, precision and recall values were computed for Seten and C-E to assess the performance of the tools. Precision was defined as the fraction of enriched gene sets from GSEA on the control versus CRISPR RNA-seq data for the respective RBPs that overlapped with the gene sets from Seten's or C-E's GSEA on CLIP-seq data at the same corrected P-values’ threshold of 0.05. Likewise, recall was defined as the fraction of gene sets identified by Seten's or C-E's GSEA on CLIP-seq data that overlapped with the enriched gene sets from GSEA on the control versus CRISPR RNA-seq data for the respective RBPs, at the same corrected P-value thresholds. Similarly, precision and recall were also computed for the negative control BED files described below for Seten's gene set or functional enrichment methods, which enabled the calculation of F1 scores to assess the relative performance of the methods and options.
Evaluation of Seten's performance against negative control
In order to evaluate the performance of the tool, we generated random BED files referred to as negative controls, corresponding to each RBP's eCLIP data set separately. We utilized BEDTools (Quinlan and Hall 2010) shuffle function with “chrom” (to ensure that each chromosome is equally represented in random BED files), “incl” (that keeps genomic features and assigns shuffled scores for peaks) and separately “excl” (that excludes the genomic features and assigns random genome-wide coordinates for each peak) parameters to generate two sets of arbitrary BED files. These two sets of negative control BED files were referred to as “test peaks with randomized peak score” and “test peaks with randomized peak coordinates.” We computed the F1 score, computed as the harmonic mean of precision and recall, to measure the performance of Seten against gold standard functional annotations described in the previous section, for three types of BED files, namely, original eCLIP peaks, test peaks with randomized peak score, and test peaks with randomized peak coordinates. This enabled us to assess the relative impact on the performance, for different options and to benchmark the annotations predicted by Seten for each of these types of BED files against the GSEA results obtained from the CRISPR RNA-seq gold standard described above. We repeated the analysis for three different RBPs that had both eCLIP and CRISPR RNA-seq data, namely IGF2BP1, PTBP1, and SRSF7 in K562 cell line. For test peak data, we repeated the analysis against five random BED files for each RBP and reported the average F1 scores.
Software availability
Seten WI (Web Interface)
Seten WI server is accessible on http://www.iupui.edu/~sysbio/seten/. Its source code, which can be used for initiating a local instance of Seten WI, is available via the GitHub repository: https:// github.com/gungorbudak/seten.
Seten CLI (Command Line Interface)
Seten CLI is a Python package and can be installed via the package manager or can be built from its source. Its GitHub repository has detailed information about installing and using Seten CLI: https:// github.com/gungorbudak/seten-cli.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank members of the Janga laboratory for providing feedback and helpful suggestions in the course of the development of the tool.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.059089.116.
REFERENCES
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B (Methodol) 57: 289–300. [Google Scholar]
- Chaudhury A, Chander P, Howe PH. 2010. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: focus on hnRNP E1's multifunctional regulatory roles. RNA 16: 1449–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Yun J, Kim MS, Mendell JT, Xie Y. 2014. PIPE-CLIP: a comprehensive online tool for CLIP-seq data analysis. Genome Biol 15: R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comoglio F, Sievers C, Paro R. 2015. Sensitive and highly resolved identification of RNA-protein interaction sites in PAR-CLIP data. BMC Bioinformatics 16: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, Ohler U. 2011. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol 12: R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Agostino Y, D'Aniello S. 2017. Molecular basis, applications and challenges of CRISPR/Cas9: a continuously evolving tool for genome editing. Brief Funct Genomics. 10.1093/bfgp/elw038. [DOI] [PubMed] [Google Scholar]
- Darnell RB. 2010. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip Rev RNA 1: 266–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Hatzubai A, Andersen JS, Ben-Shushan E, Fisher GZ, Yaron A, Bauskin A, Mercurio F, Mann M, Ben-Neriah Y. 2002. Pseudosubstrate regulation of the SCFβ-TrCP ubiquitin ligase by hnRNP-U. Genes Dev 16: 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project. 2017. RNA-seq profiling of CRISPR/Cas9 based knockouts of RNA-binding proteins in human cell line K562. https://www.encodeproject.org/search/?type=Experiment&assay_title=CRISPR+RNA-seq&replicates.library.biosample.life_stage=adult.
- Erhard F, Dölken L, Zimmer R. 2013. RIP-chip enrichment analysis. Bioinformatics 29: 77–83. [DOI] [PubMed] [Google Scholar]
- Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F, McKay S, et al. 2016. The Reactome pathway Knowledgebase. Nucleic Acids Res 44: D481–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. 1922. On the interpretation of X2 from contingency tables, and the calculation of P. J R Stat Soc 85: 87–94. [Google Scholar]
- Gerstberger S, Hafner M, Tuschl T. 2014. A census of human RNA-binding proteins. Nat Rev Genet 15: 829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D, Saada A, D'Adamo P, Fernandez-Vizarra E, Gasparini P, Tiranti V, Elpeleg O, Zeviani M. 2008. FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am J Hum Genet 83: 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppertz I, Attig J, D'Ambrogio A, Easton LE, Sibley CR, Sugimoto Y, Tajnik M, König J, Ule J. 2014. iCLIP: protein-RNA interactions at nucleotide resolution. Methods 65: 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iervolino A, Santilli G, Trotta R, Guerzoni C, Cesi V, Bergamaschi A, Gambacorti-Passerini C, Calabretta B, Perrotti D. 2002. hnRNP A1 nucleocytoplasmic shuttling activity is required for normal myelopoiesis and BCR/ABL leukemogenesis. Mol Cell Biol 22: 2255–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janga SC. 2012. From specific to global analysis of posttranscriptional regulation in eukaryotes: posttranscriptional regulatory networks. Brief Funct Genomics 11: 505–521. [DOI] [PubMed] [Google Scholar]
- Jangra RK, Yi M, Lemon SM. 2010. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J Virol 84: 6810–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. 2016. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44: D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, Karolchik D. 2010. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26: 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Hur J, Jeong S. 2009. Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB Rep 42: 125–130. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler S, Doelken SC, Mungall CJ, Bauer S, Firth HV, Bailleul-Forestier I, Black GC, Brown DL, Brudno M, Campbell J, et al. 2014. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res 42: D966–D974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R. 2015. Package ‘pheatmap’. https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf.
- Konig J, Zarnack K, Luscombe NM, Ule J. 2011. Protein-RNA interactions: new genomic technologies and perspectives. Nat Rev Genet 13: 77–83. [DOI] [PubMed] [Google Scholar]
- Lambert N, Robertson A, Jangi M, McGeary S, Sharp PA, Burge CB. 2014. RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol Cell 54: 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert NJ, Robertson AD, Burge CB. 2015. RNA Bind-n-Seq: measuring the binding affinity landscape of RNA-binding proteins. Methods Enzymol 558: 465–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann HB, Whitney DR. 1947. On a test of whether one of two random variables is stochastically larger than the other. Ann Math Stat 18: 50–60. [Google Scholar]
- McManus CJ, Graveley BR. 2011. RNA structure and the mechanisms of alternative splicing. Curr Opin Genet Dev 21: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milacic M, Haw R, Rothfels K, Wu G, Croft D, Hermjakob H, D'Eustachio P, Stein L. 2012. Annotating cancer variants and anti-cancer therapeutics in reactome. Cancers (Basel) 4: 1180–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal N, Roy N, Babu MM, Janga SC. 2009. Dissecting the expression dynamics of RNA-binding proteins in posttranscriptional regulatory networks. Proc Natl Acad Sci 106: 20300–20305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelamraju Y, Hashemikhabir S, Janga SC. 2015. The human RBPome: from genes and proteins to human disease. J Proteomics 127: 61–70. [DOI] [PubMed] [Google Scholar]
- Nicholson CO, Friedersdorf MB, Keene JD. 2016. Quantifying RNA binding sites transcriptome-wide using DO-RIP-seq. RNA 23: 32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura D. 2001. BioCarta. Biotech Softw Internet Rep 2: 117–120. [Google Scholar]
- Ougolkov A, Zhang B, Yamashita K, Bilim V, Mai M, Fuchs SY, Minamoto T. 2004. Associations among β-TrCP, an E3 ubiquitin ligase receptor, β-catenin, and NF-κB in colorectal cancer. J Natl Cancer Inst 96: 1161–1170. [DOI] [PubMed] [Google Scholar]
- Patra P, Izawa T, Pena Castillo L. 2015. REPA: applying pathway analysis to genome-wide transcription factor binding data. IEEE/ACM Trans Comput Biol Bioinform 1–1. [DOI] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow J, Alleaume AM, Curk T, Schwarzl T, Sauer S, Hentze MW. 2015. FASTKD2 is an RNA-binding protein required for mitochondrial RNA processing and translation. RNA 21: 1873–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppe B, Vandesompele J, Schoch C, Lindvall C, Mrozek K, Bloomfield CD, Beverloo HB, Michaux L, Dastugue N, Herens C, et al. 2004. Expression analyses identify MLL as a prominent target of 11q23 amplification and support an etiologic role for MLL gain of function in myeloid malignancies. Blood 103: 229–235. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappaport N, Twik M, Nativ N, Stelzer G, Bahir I, Stein TI, Safran M, Lancet D. 2014. MalaCards: a comprehensive automatically-mined database of human diseases. Curr Protoc Bioinformatics 47: 1.24.1–1.24.19. [DOI] [PubMed] [Google Scholar]
- Reyes-Herrera PH, Ficarra E. 2014. Computational methods for CLIP-seq data processing. Bioinform Biol Insights 8: 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE. 2003. RNA editing minireview series. J Biol Chem 278: 1389–1390. [DOI] [PubMed] [Google Scholar]
- Shatkin AJ, Manley JL. 2000. The ends of the affair: capping and polyadenylation. Nat Struct Biol 7: 838–842. [DOI] [PubMed] [Google Scholar]
- Smith AD, Chung WY, Hodges E, Kendall J, Hannon G, Hicks J, Xuan Z, Zhang MQ. 2009. Updates to the RMAP short-read mapping software. Bioinformatics 25: 2841–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer J, Hafner M, Landthaler M, Ascano M, Farazi T, Wardle G, Nusbaum J, Khorshid M, Burger L, Zavolan M, et al. 2014. PAR-CLIP (photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation): a step-by-step protocol to the transcriptome-wide identification of binding sites of RNA-binding proteins. Methods Enzymol 539: 113–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stary S, Vinatzer U, Mullauer L, Raderer M, Birner P, Streubel B. 2013. t(11;14)(q23;q32) involving IGH and DDX6 in nodal marginal zone lymphoma. Genes Chromosomes Cancer 52: 33–43. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. 2003. CLIP identifies Nova-regulated RNA networks in the brain. Science 302: 1212–1215. [DOI] [PubMed] [Google Scholar]
- Uren PJ, Bahrami-Samani E, Burns SC, Qiao M, Karginov FV, Hodges E, Hannon GJ, Sanford JR, Penalva LO, Smith AD. 2012. Site identification in high-throughput RNA-protein interaction data. Bioinformatics 28: 3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. 2016. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13: 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Xie Y, Xiao G. 2014. dCLIP: a computational approach for comparative CLIP-seq analyses. Genome Biol 15: R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Arribas-Layton M, Chen Y, Lykke-Andersen J, Sen GL. 2015. DDX6 orchestrates mammalian progenitor function through the mRNA degradation and translation pathways. Mol Cell 60: 118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch RP, Lee C, Imbriano PM, Patil S, Weymouth TE, Smith RA, Scott LJ, Sartor MA. 2014. ChIP-Enrich: gene set enrichment testing for ChIP-seq data. Nucleic Acids Res 42: e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winteringham LN, Endersby R, Kobelke S, McCulloch RK, Williams JH, Stillitano J, Cornwall SM, Ingley E, Klinken SP. 2006. Myeloid leukemia factor 1 associates with a novel heterogeneous nuclear ribonucleoprotein U-like molecule. J Biol Chem 281: 38791–38800. [DOI] [PubMed] [Google Scholar]
- Yang YC, Di C, Hu B, Zhou M, Liu Y, Song N, Li Y, Umetsu J, Lu ZJ. 2015. CLIPdb: a CLIP-seq database for protein-RNA interactions. BMC Genomics 16: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, et al. 2016. Ensembl 2016. Nucleic Acids Res 44: D710–D716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.