

Abstract
Spinal muscular atrophy (SMA) is caused by mutations and/or deletions of the survival motor neuron gene (SMN1). Besides its function in the biogenesis of spliceosomal snRNPs, SMN might possess a motor neuron specific role and could function in the transport of axonal mRNAs and in the modulation of local protein translation. Accordingly, SMN colocalizes with axonal mRNAs of differentiated NSC-34 motor neuron-like cells. We recently showed that SMN depletion gives rise to a decrease in the axonal transport of the mRNAs encoding Annexin A2 (Anxa2). In this work, we have characterized the structural features of the Anxa2 mRNA required for its axonal targeting by SMN. We found that a G-rich motif located near the 3′UTR is essential for axonal localization of the Anxa2 transcript. We also show that mutations in the motif sequence abolish targeting of Anxa2 reporter mRNAs in axon-like structures of differentiated NSC-34 cells. Finally, localization of both wild-type and mutated Anxa2 reporters is restricted to the cell body in SMN-depleted cells. Altogether, our studies show that this G-motif represents a novel and essential determinant for axonal localization of the Anxa2 mRNA mediated by the SMN complex.
Keywords: Anxa2, RNA transport, SMA, SMN, cis-acting motif
INTRODUCTION
Spinal muscular atrophy (SMA) is a prominent autosomal recessive neuromuscular disease characterized by dysfunction of motor neurons of the spinal cord anterior horn, which leads to paralysis and, in severe cases, to death. For more than 98% of patients, the disease is caused by deletions or mutations in the SMN1 gene coding for the survival motor neuron protein (Lefebvre et al. 1997; Wirth 2000). The SMN protein is part of a large macromolecular complex together with other proteins known as Gemins (2–8) and unrip (Meister et al. 2002; Gubitz et al. 2004; Cauchi 2010).
The SMN complex is essential for the biogenesis of spliceosomal snRNP particles, which are required for splicing of nuclear pre-mRNAs (Chari et al. 2009; Matera and Wang 2014). Several studies have reported that SMN deficiency alters the stoichiometry of snRNAs and causes widespread and differential pre-mRNA splicing defects (Gabanella et al. 2007; Zhang et al. 2008; Boulisfane et al. 2011; Lotti et al. 2012). These results support the view that SMA might arise from the inefficient splicing of pre-mRNAs coding for proteins required for motor neuron's function and/or organization (Burghes and Beattie 2009; Coady and Lorson 2011; Li et al. 2014). However, whether such splicing defects are directly caused by SMN loss or are secondary effects of cellular dysfunction is still unknown (Bäumer et al. 2009; Garcia et al. 2013).
To explain the specific effect of SMN deficiency on motor neurons, it has also been proposed that SMN might have a motor-neuron specific function and that loss of this activity results in SMA. This view is consistent with studies reporting that SMN complexes, lacking Sm proteins but containing some or all Gemins, are found in cytoplasmic granules exhibiting rapid and bidirectional movements along axons of primary neurons (Zhang et al. 2003, 2006; Burghes and Beattie 2009; Rossoll and Bassell 2009). It has also been shown that in axons, SMN is associated with distinct RNA-binding proteins as, for example, hnRNP R and Q (Rossoll et al. 2002; Dombert et al. 2014), HuD and FMRP (Piazzon et al. 2008; Fallini et al. 2011; Hubers et al. 2011), as well as TDP-43 and FUS (Wang et al. 2011; Tsuiji et al. 2013; Yamazaki et al. 2013). These studies suggest an involvement of SMN in the regulation of the assembly of ribonucleoproteins responsible for the transport, stability, and/or local translation of axonal mRNAs (Talbot and Davies 2008; Akten et al. 2011; Fallini et al. 2011; Hubers et al. 2011). Consistent with this hypothesis, we recently identified numerous axonal mRNAs associated with the SMN complex in differentiated NSC-34 motor neuron-like cells using a genome-wide analysis (Rage et al. 2013). We found also that axonal localization of some mRNAs is altered in SMN-depleted cells, indicating that SMN deficiency might indeed lead to the mislocalization of mRNPs required for axonal outgrowth and/or function.
A key process for neurons is their ability to spatially and temporally translate mRNA. The local translation of transcripts represents a very important mechanism contributing to synaptic plasticity and arborization (Till et al. 2011; Jung et al. 2012), axonal growth (Campbell and Holt 2001; Hörnberg and Holt 2013), as well as axonal viability and neuronal development (Taylor et al. 2013). In this regard, numerous studies showed that disruption of SMN function causes presynaptic abnormalities and abnormal synaptic transmission with poorly arborized axons at the neuromuscular junction (NMJ) terminals (Jablonka et al. 2007; Kariya et al. 2008; Kong et al. 2009), suggesting that SMN might be directly involved in the regulation of axonal growth. However, the precise molecular determinants and mechanisms involved in the transport of mRNAs by the SMN complex into the axonal compartment are still unknown.
The localization of mRNA to axons and dendrites requires the presence of cis-acting elements generally found in the 3′ untranslated region (3′UTR) of the transcript, although such elements have also been found in the 5′UTR or the coding sequence (Holt and Bullock 2009; Martin and Ephrussi 2009). These cis-acting elements are recognized by RNA-binding proteins, and the resulting messenger ribonucleoproteins (mRNP) are packaged into RNA granules, which are subsequently transported to their final destination (Hirokawa 2006; Kiebler and Bassell 2006).
In this work, we examined the transport of the mRNA encoding the mouse Annexin A2 (Anxa2) that we previously showed to be SMN-dependent in differentiated NSC-34 motor neuron-like cells (Rage et al. 2013). Annexin A2 is a ubiquitous Ca2+-binding protein that is essential for actin-dependent vesicle transport and which has been shown to be a critical regulator of actin remodeling in the proximity of dynamic cellular membranes (Hayes et al. 2006; Grieve et al. 2012; Gabel et al. 2015). Based on the observations that SMN deficiency leads to alterations in actin organization, the characterization of the functional relationships between Anxa2 mRNA transport and SMN is of considerable interest. Using a series of deletion/point mutations and fluorescent in situ hybridization (FISH) experiments, we characterized a short motif overlapping the end of the Annexin A2 coding region and the 3′UTR. Our studies show that this sequence is highly conserved in the human Anxa2 mRNA and functions as a SMN-dependent axonal localization element.
RESULTS
The 3′-region of the Anxa2 mRNA is sufficient to localize a reporter RNA to axons in NSC-34 cells
Using microfluidic chambers (MFCs), we previously reported that SMN is required for the localization of Anxa2 mRNAs in the axonal compartment of differentiated NSC-34 motor neuron-like murine cells (Rage et al. 2013). It has been reported that some mRNAs that are known to be localized into dendritic processes contain guanine-quadruplex structures, and accordingly it was shown that these G-quadruplex structures function as dendritic mRNA targeting signals in mouse cortical neurons (Subramanian et al. 2011). To analyze whether mouse Anxa2 mRNAs may contain motifs capable of forming G-quadruplex structures responsible for their axonal localization, we used the Quadfinder prediction software and the web-based server QGRS-mapper (Kikin et al. 2006; Scaria et al. 2006). As shown in Figure 1A, using default parameters, six candidate sequences with a likelihood of G-quadruplex formation were identified in mouse. The predicted G-quadruplex sequences have different G-scores, and an 18-nucleotide (nt) long sequence, located at the end of the coding region at position 1080-1097, has an overlap of 3 nt within the 3′UTR region (Supplemental Fig. 1A). Given that elements that confer subcellular localization are often located in the 3′UTR of mRNAs (Kiebler and Bassell 2006), we focused our analysis on this sequence (that we called G-motif) and tested whether it could be important for axonal targeting of the mouse Anxa2 mRNA.
FIGURE 1.

Identification of putative G-rich motifs in the mouse Anxa2 sequence. (A) The motifs predicted in the mouse Anxa2 sequences by the Quadfinder software are sorted by their position, length, and G-score. (B) Schematic view of the constructs used in this study. The indicated fragments of the mouse Anxa2 mRNA were cloned into the pcDNA3-LacZ-24xMS2 vector containing the LacZ gene and 24 MS2 binding sites. All constructs possess the same SV40 polyadenylation signal sequence. Expression of all fusion sequences was driven by the human cytomegalovirus (CMV) promoter. The names of the reporters are indicated on the left. Note that the diagram is not to scale.
To this end, we constructed different reporters carrying subdomains of the Anxa2 mRNA in the pcDNA3-lacZ-24xMS2 vector (Fusco et al. 2003). Various cDNA sub-fragments from the Anxa2 sequences were inserted into the plasmid at the 3′-end of the MS2 binding sites (Fig. 1B). As positive and negative controls for axonal localization, we used the FL-wt construct carrying the full-length Anxa2 region and the FL-rev reporter expressing the Anxa2 mRNA in the antisense orientation, respectively. These vectors, expressing chimeric reporter mRNAs, were transfected into murine motor neuron-like NSC-34 cells and FISH experiments were performed using probes directed against the LacZ coding region to examine localization of reporter mRNAs. As expected, punctuate staining was observed along axons for the FL-wt construct, indicating that the LacZ-MS2-Anxa2 reporter mRNA is efficiently transported in NSC-34 axons (Fig. 2A). Similar punctuate staining could also be observed in axonal processes upon transfection of the G-motif + 3′UTR or the G-motif construct restricted to the 18 nt (Fig. 2). In contrast, transfection of the vectors carrying the 3′UTR (3′UTR) or the region encompassing the 5′UTR and the coding sequence but lacking the G-motif (5′UTR + CS), resulted in a significant decrease of reporter mRNA abundance in axons of NSC-34 cells (Fig. 3A, panels B and C). When the FL-rev construct was transfected, only a few spots were also observed using the same probes (Fig. 3A, cf. panels A and D).
FIGURE 2.

The G-motif is sufficient to target reporter mRNA in axons of differentiated NSC-34 cells. (A) Representative pictures of cells expressing the indicated constructs. FISH was performed using LacZ probes after 3 d of differentiation. Scale bar, 20 µm. Insets on the right are enlarged images of boxed sections. (B) Quantitative analysis of localization of reporter RNAs in axons as illustrated in A. Scatter plots show the number of mRNA spots localized in the distal segment of axons with the FL-wt (n = 19), G-motif + 3′UTR (n = 36), and G-motif (n = 11) reporters (n corresponds to the number of cells that were analyzed). Mean and SEM are shown.
FIGURE 3.

The G-motif is required to target reporter mRNA in axons of differentiated NSC-34 cells. (A) Representative pictures of cells expressing the indicated constructs. FISH was performed using LacZ probes after 3 d of differentiation. Scale bar, 10 µm. Insets on the bottom of each panel are enlarged images of boxed sections. (B) Quantitative analysis of localization of reporter RNAs in axons as illustrated in A. Scatter plots show the number of mRNA spots localized in the distal segment of axons with the FL-wt (n = 19), 5′UTR + CS (n = 19), 3′UTR (n = 45), and FL-rev (n = 29) reporters (n corresponds to the number of cells that were analyzed). Mean and SEM are shown. Statistical significance was calculated using the Wilcoxon test. (***) P < 0.001.
Quantitative analysis, performed using the Imaris software as previously described (Rage et al. 2013), showed that an equivalent number of spots (1.66 ± 0.2 spots/µm for FL-wt, 1.66 ± 0.15 for G-motif + 3′UTR, and 1.8 ± 0.3 for G-motif) are observed in the axonal compartment of NSC-34 cells for constructs carrying the G-motif (Fig. 2B), while the absence of this region dramatically decreases the transport of reporter mRNAs into these processes (0.15 ± 0.02 spot/µm for 5′UTR + CS, 0.12 ± 0.01 for 3′UTR, and 0.09 ± 0.01 for FL-rev) (Fig. 3B).
The G-motif in Anxa2 mRNAs does not form a G-quadruplex structure
RNA G-quadruplexes have been reported in the past and the high occurrence of G-quadruplexes in 3′UTR regions of RNA has led to the hypothesis that they play an important role as localization and translational regulators (Huppert et al. 2008; Subramanian et al. 2011; Bugaut and Balasubramanian 2012). We thus tested whether the G-motif (1080-1097) of Anxa2 might also form a G-quadruplex, using a reverse transcription assay based on the monitoring of cation-dependent pauses during RT progression (Schaeffer et al. 2001). The formation of a G-quadruplex structure, which is stabilized in the presence of K+, was assessed by folding the RNA and performing fluorescent primer extension in the presence of various cations. The fluorescent cDNAs were separated by capillary electrophoresis and the electrophoregrams were analyzed with the QuShape software. In the presence of K+, no specific RT stop was observed for Anxa2 RNA in the predicted G4 forming sequences whereas strong stops are observed in the well-characterized N19 fragment from the Fmr1 RNA (Supplemental Fig. 2).
The folding of the region encompassing the G-motif into a G-quadruplex structure was also investigated by computational analysis using the Vienna RNA package, which allows the prediction of the formation of G-quadruplexes alongside other possible competing secondary structures (http://rna.tbi.univie.ac.at; Lorenz et al. 2013). The calculations executed by this package confirmed that the G-motif of Anxa2 does not form a stable G-quadruplex structure in contrast to a control sequence known to adopt a G-quartet conformation (Menon and Mihailescu 2007; Lorenz et al. 2013; data not shown). Similarly, no G-quadruplex folding could be detected for the additional candidate sequences found in the mouse Anxa2 mRNA (Fig. 1A).
Partial conservation of the G-motif in Anxa2 mRNAs from different organisms
Given the peculiar position of the G-motif at the end of the coding region of the Anxa2 mRNA in mouse, we tested whether this motif is also present in the Anxa2 transcripts of human and other organisms. To this end, we performed multiple sequence alignment using the MAFFT server, and as shown in Figure 4A and Supplemental Figure 1, the G-motif is strongly conserved in human, but only partial conservation can be observed evolutionarily. Interestingly, the G-motif region in human does not present G-quadruplex features when scanned by the QGRS-mapper (Supplemental Fig. 1). Folding analyses performed with the RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) and mfold (http://unafold.rna. albany.edu/?q=mfold/rna-folding-form) web servers, however, indicate that the G-motif of mouse and human can potentially form a stem–loop structure which is not conserved in the other species (Fig. 4B). Despite the fact that the free energy associated to this structure is not very favorable (ΔG = 0.70 kcal/mol as predicted by mfold), such a stem–loop could nevertheless be stabilized by protein binding.
FIGURE 4.
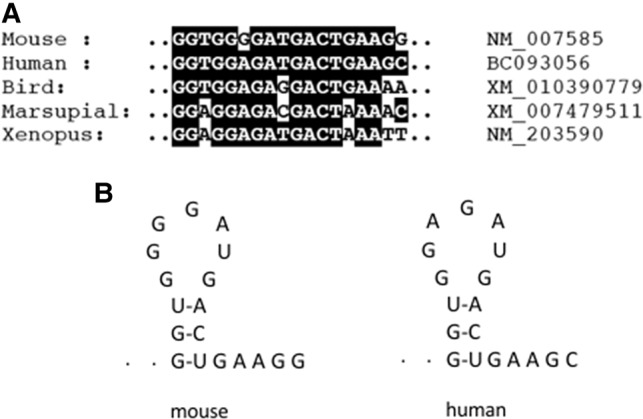
Evolutionary conservation of a G-rich region in Anxa2 mRNAs. (A) Multiple sequence alignment of the region encompassing the stop codon in Anxa2 mRNA of indicated species. Accession numbers are listed next to the name of the organisms. (B) Proposed secondary structures of mouse and human G-motifs as predicted by mfold.
Interestingly, axonal localization was also observed following transfection of a reporter gene, carrying the corresponding human G-motif + 3′UTR region into human SH-5YSY cells (Supplemental Fig. 3). In contrast, axonal transport of the reporter mRNA is abolished upon deletion of the region encompassing the human G-motif. As determined by quantification analyses, the deletion of the human G-motif gives rise to a fivefold decrease of axonal localization of the reporter mRNA (2.11 ± 0.28 versus 0.43 ± 0.09 spots/µm, Supplemental Fig. 3C).
The G-motif plays an essential role for the localization of Anxa2 mRNA in axons
Our initial experiments (Fig. 2) showed that reporter mRNAs carrying the mouse G-motif are able to be transported into axons, while its absence in the 3′UTR construct or in other noncontaining G-motif constructs such as 5′UTR + CS or FL-rev abolishes the localization (Fig. 3). This difference is not due to a variation in the expression of reporter genes, because similar steady-state mRNA levels are observed for both G-motif + 3′UTR and noncontaining G-motif constructs, as shown by RT-qPCR experiments (Supplemental Fig. 4A).
To firmly conclude that the G-motif is functionally relevant in addressing the mouse Anxa2 mRNA into axons, we performed an analysis of the corresponding region by first mutating all G residues (positions 1, 2, 4, 7, 10, 17, and 18), precluding thereby the formation of the potential stem–loop structure (Fig. 5C). The mutations in the G-motif sequence were introduced into the mouse FL-wt construct to give the FL-mut1 reporter. After transfection into NSC-34 cells, FISH experiments showed that mutations in the G-motif abolished the localization of the reporter mRNA into axons (0.22 ± 0.02 versus 1.66 ± 0.2 spots/µm, Fig. 5), demonstrating the importance of the G-motif in the transport of the mouse Anxa2 mRNA. Quantitation analyses show that the localization of the FL-mut1 reporter is decreased by 87% compared to the FL-wt construct (Fig. 5B).
FIGURE 5.

G-motif mutations impair axonal localization of Anxa2 mRNA reporters. (A) Representative pictures of cells expressing the indicated constructs. FISH was performed using LacZ probes after 3 d of differentiation. Scale bar, 10 µm. Insets on the bottom of each panel are enlarged images of boxed sections. (B) Quantitative analysis of localization of reporter RNAs in axons as illustrated in A. Scatter plots show the number of mRNA spots localized in the distal segment of axons with the FL-mut1 (n = 30; P < 0.0001), FL-mut2 (n = 13; P = 0.0002), FL-mut3 (n = 9; P = 0.0039), and FL-mut4 (n = 12; P = 0.001) reporters (n corresponds to the number of cells that were analyzed). Mean and SEM are shown. Statistical significance was calculated using the Wilcoxon test. (C) Mutations introduced in the G-motif of FL-mut1 to mut4 reporters are indicated in red. The position of the bases involved in the potential stem is indicated by (((and))) under the wild-type sequence.
Whether a stem–loop structure in the G-motif is required for the localization of the reporter mRNA in axons was then tested with three other constructs (Fig. 5C). In FL-mut2, two additional mutations were introduced in FL-mut1 in order to allow reformation of a 3-base pair (bp) stem as predicted for the wild-type sequence. In FL-mut3, the wild-type sequence of the stem was maintained while the sequence of the potential loop was mutated as in FL-mut1. As shown in Figure 5A, axonal localization of both reporter mRNAs was significantly decreased (0.16 ± 0.04 spot/µm and 0.32 ± 0.07 spot/µm for FL-mut2 and FL-mut3, respectively) as compared to the reporter containing the wild type G-motif (1.66 ± 0.2 spots/µm). These results support the notion that the G-motif does not fold into a stem–loop structure. This is further strengthened by the results obtained with the FL-mut4 reporter where only the first 3 nt of the G-motif are mutated, thus disrupting formation of the potential stem. As shown in Figure 5A, axonal localization of the corresponding mRNA is significantly decreased (0.55 ± 0.1 spot/µm), although not as much as what is observed for the FL-mut1, mut2, and mut3 reporters. Altogether, these observations confirmed that the sequence, rather than the structure, is the key determinant of the G-motif properties.
SMN depletion affects the localization of reporters carrying the G-motif of Anxa2
We previously showed that SMN is required for transport of endogenous Anxa2 mRNA into axons of differentiated NSC-34 cells (Rage et al. 2013). To test whether the LacZ-MS2 reporter mRNA containing only the G-motif and the 3′UTR of Anxa2 (G-motif + 3′UTR construct in Fig. 1) similarly requires SMN to be localized into the axonal processes, we first tested whether the reporter colocalizes with SMN. FISH experiments combined with immunofluorescence of the endogenous SMN protein demonstrate that colocalization with SMN occurs with mean value of 15.8% ± 1.5% for the FL-wt reporter and 16.4% ± 2.4% for the G-motif + 3′UTR construct (Fig. 6A,B).
FIGURE 6.
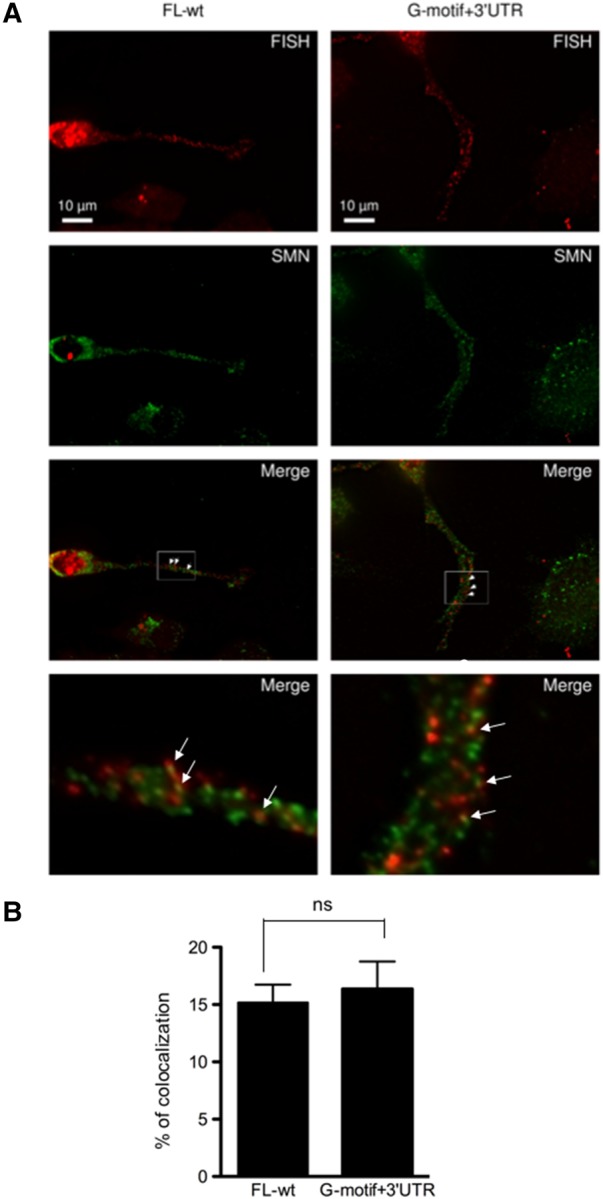
SMN colocalizes with the FL-wt and G-motif + 3′UTR region of Anxa2 mRNA. (A) Endogenous SMN protein (green spots) colocalizes with the full-length Anxa2 mRNA (FL-wt) and with the G-motif + 3′UTR containing reporter mRNAs (red spots). Differentiated NSC-34 cells were fixed and processed for FISH using MS2 antisense probes and for IF with anti-SMN antibodies. Arrowheads indicate colocalizing spots. Scale bar, 10 µm. Insets on the bottom of each panel are enlarged images of boxed sections. (B) Quantification of the percentage of mRNA spots colocalizing with SMN spots. Mean and SEM are shown. Statistical significance was calculated using the Student t-test (ns, not significant; n = 24, n corresponds to the number of cells that were analyzed).
To further analyze the functional relationships between SMN and the G-motif of Anxa2, we used the shSMN-NSC-34 cell line to deplete SMN after induction with doxycycline as described previously (Rage et al. 2013). As shown in Figure 7A, FISH experiments indicate that the number of spots containing the G-motif and the FL-wt mRNAs is decreased upon SMN depletion in axons of differentiated NSC-34 cells. Quantitation analyses indicate that the induction of shRNA resulted in a 70% and 90% decrease of the number of spots for the FL-wt (0.23 ± 0.02 versus 1.42 ± 0.13) and G-motif + 3′UTR transcripts (0.18 ± 0.02 versus 1.76 ± 0.2), respectively (Fig. 7B). This result is in agreement with what we previously observed with endogenous Anxa2 RNA (Rage et al. 2013). Again, the observed decrease is not a consequence of a difference in the expression levels of the FL-wt and G-motif + 3′UTR transcripts, since qRT-PCR of total RNA purified from noninduced and induced cells demonstrates that similar amounts of reporter mRNAs are expressed (Supplemental Fig. 4B). Moreover, the mean length of axons in differentiated cells is similar in both induced and noninduced cells, while the number of spots containing SMN is decreased upon shSMN expression, as expected (Supplemental Fig. 5). These results demonstrate that the G-motif represents an important cis-acting element required for the transport of the Anxa2 mRNA by the SMN complex.
FIGURE 7.
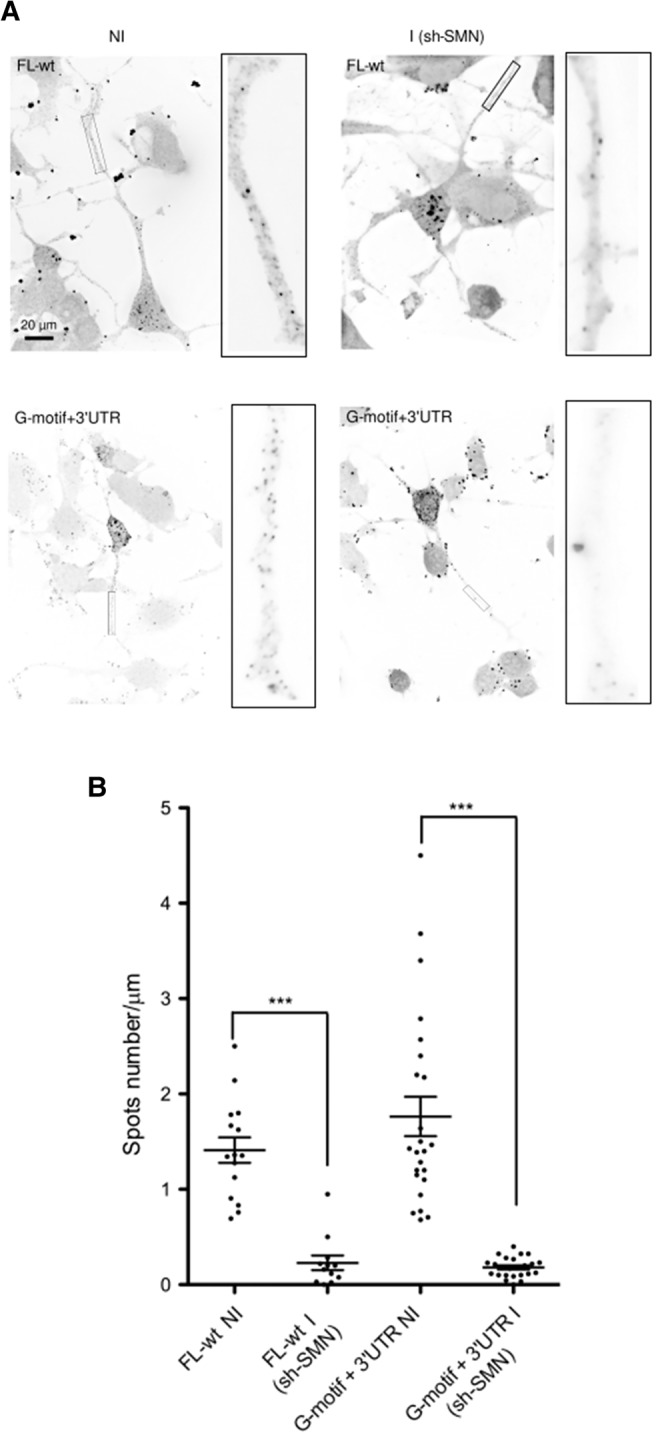
Localization of the FL-wt and G-motif + 3′UTR reporter mRNAs in axons of differentiated NSC-34 cells is SMN-dependent. (A) Spots corresponding to the indicated mRNAs in axons of noninduced (NI) or induced (I) shSMN-NSC-34 cells are shown. Representative images of FISH experiments show the RNA spots localized in axons before and following SMN depletion. Scale bar, 20 µm. Insets on the right of each panel are enlarged images of boxed sections. (B) Quantitative analysis of localization of reporter RNAs in axons as illustrated in A. Scatter plots show the number of mRNA spots localized in the distal segment of axons with the FL-wt construct before (NI, noninduced, n = 19) and after depletion of SMN (I, induced; sh-SMN, n = 19, P = 0;001), and with the G-motif + 3′UTR reporter (NI, n = 24; I, n = 24, P < 0.0001). Mean and SEM are shown. Statistical significance was calculated using the Wilcoxon test.
DISCUSSION
In this study, we have identified a cis-acting element that mediates the localization of the Anxa2 mRNA into the axonal compartment of NSC-34 motor neuron-like cells. This element is located in a region encompassing the open reading frame stop codon and the beginning of the 3′UTR and is conserved in the human Anxa2 mRNA, highlighting its functional importance.
Bioinformatic analyses of the mouse Anxa2 mRNA sequence revealed the presence of several potential G-quadruplex motifs known to act as signals for dendrite targeting (Subramanian et al. 2011). However, it has been reported that the majority of human RNA quadruplex-forming sequences adopt alternative secondary structures (Lorenz et al. 2013). The use of the algorithm described by Lorenz et al. (2013) showed that neither the G-motif encompassing the stop codon, nor the other potential G-quadruplex sequences found in the mouse Anxa2 mRNA, are able to form a stable G-quadruplex. This is also the case for motifs found in human Anxa2 mRNA.
Our studies also showed that the Anxa2 mRNA does not contain classical cis-acting elements known to be involved in RNA localization and whose lengths range from a few nucleotides to over 1 kb (Eliscovich et al. 2013). Examples of such regulatory sequences are the 11-nt-long A2RE motif in the MBP mRNA, the 62-nt stem–loop motif in the BC1 RNA, or the 70-nt-long CPE region of the αCaMKII mRNA (Jambhekar and Derisi 2007). No clear patterns of known axonal zipcode sequences have emerged, and the G-motif that we identified in Anxa2 thus represents an additional structure used for subcellular localization of mRNAs.
Our mutational studies indicate that the sequence of the G-motif, rather than the integrity of a potential stem–loop structure, is essential for function. Database searches of the 18-nt-long motif did not allow identification of additional mRNAs carrying this primary sequence, neither in mouse nor in human. However, we cannot rule out that the size of the motif required for axonal localization is shorter than 18 nt. If so, database searches with shorter sequences should lead to the identification of mRNA subsets sharing the same motif. Along this line, it is interesting to point out that the localization of the FL-mut4 reporter, where the first 3 nt of the G-motif are mutated, is significantly decreased as compared to that of the wild-type reporter (Fig. 5). This indicates that the cis-acting sequence required for axonal localization is likely located in the 5′ part of the G-motif.
Further experiments should help to identify the trans-acting factor(s) responsible for Anxa2 mRNP localization. Indeed, given that SMN itself lacks any known RNA-binding domains, it is likely that association with the Anxa2 mRNA occurs through RNA-binding proteins (RBPs) interacting with SMN. The identity of the RNA-binding protein(s) recognizing the G-motif of the Anxa2 mRNA is an important issue, and among RBPs known to associate with SMN, FMRP, hnRNP-R, HuD, and TDP-43 are found in axons (Hörnberg and Holt 2013). These proteins are, however, unlikely to be candidates for recognizing the G-motif of Anxa2 since it does not correspond to known binding sites of these RBPs. Nevertheless, further FISH experiments combined with immunofluorescence studies are needed to clarify this issue.
Consistent with the emerging view that SMN participates in axonal transport of mRNPs, SMN is found in granules in neuronal processes (Zhang et al. 2003; Fallini et al. 2012). Using various neural cells, several studies showed that, in addition to SMN, these granules contain some but not all Gemins, suggesting the presence of diverse SMN-containing multiprotein complexes in neuronal processes (Sharma et al. 2005; Zhang et al. 2006; Todd et al. 2010a,b). It would be of great interest to define more precisely which Gemins subcomplexes are associated with SMN granules containing Anxa2 mRNA. Such analysis is currently unrealizable in mouse due to the lack of a full panel of antibodies recognizing mouse Gemins. It is interesting to note that SMN depletion strongly affects the localization of FL-wt and G-motif + 3′UTR constructs, even more than expected in regard to the percentage of association between these transcripts and SMN. As the global expression of the transfected plasmids is not changed upon SMN depletion, a possible explanation already discussed in Rage et al. (2013) is that SMN is required for the assembly of Anxa2 mRNPs but does not necessarily interact with all of them during their axonal transport process.
The Annexin A2 protein belongs to a conserved family of proteins expressed in many organisms from protists to higher eukaryotes including plants (Hayes et al. 2007). Many members share biochemical activities and appear for the most part to be cellular Ca2+ effectors involved in several functions, like membrane trafficking, exocytosis, endocytosis, or membrane–cytoskeleton interactions (Monastyrskaya et al. 2007). Surprisingly, Anxa2 knockout mice are viable and healthy and this could indicate redundant function of annexins, although it has not been shown that overexpression of one family member compensates for loss of another (Ling et al. 2004).
It is interesting to note that Annexin A2 interacts with polymerized and monomeric actin and specifically inhibits elongation of actin filament at the barbed ends (Hayes et al. 2006). It has been shown that it plays an essential role in maintaining the plasticity of the actin cytoskeleton associated to membranes (Grieve et al. 2012). In this regard, numerous studies showed that SMN deficiency leads to alterations in microtubule and actin organization, suggesting that disorganization of the cytoskeleton might contribute to SMA pathogenesis (Rossoll et al. 2003; Bowerman et al. 2009; Fallini et al. 2011). Consistently, the PLS3 gene, coding for the plastin3 protein, has been identified as a protective gender-specific modifier of SMA (Oprea et al. 2008). Plastin 3 is a conserved calcium binding, actin-bundling/stabilizing protein that is expressed in various tissues and its overexpression rescues axon outgrowth defects in SMN-deficient zebrafish embryos and in cultured motor neurons from a SMA mouse model (Hao le et al. 2012; Ackermann et al. 2013). Invertebrate PLS3 orthologs also act as modifiers in C. elegans and Drosophila models of SMA, indicating that plastin-associated pathways are directly or indirectly connected to SMN function (Dimitriadi et al. 2010). These observations clearly warrant further studies to test whether modulating expression of Anxa2 might rescue axon outgrowth defects observed in SMN-deficient cells. Such studies should not only bring more insights into the functional relationships between transport of the Anxa2 mRNA and the SMN complex, but could also shed light on the role of the SMN complex in the homeostasis of the actin cytoskeleton. Importantly, they could potentially reveal how defects in actin dynamics might contribute to SMA pathogenesis.
MATERIALS AND METHODS
Cell culture and transfection
The murine shSMN NSC-34 cell line was maintained in DMEM supplemented with 10% fetal bovine serum (tetracycline-free, Biowest), penicillin (50 U/mL), streptomycin (50 µg/mL), and 300 ng/mL puromycin. Differentiation of the cells was obtained by growing them in the presence of low serum concentration (1%) for 5 to 7 d. The induction of shRNA expression has been performed using 3µg/mL doxycycline (Sigma-Aldrich), as previously described (Rage et al. 2013).
Human SH-SY5Y cells were maintained in DMEM supplemented with 10% heat inactivated fetal bovine serum, penicillin (50 U/mL), and streptomycin (50 µg/mL). Of note, 10 µM retinoic acid (RA) was added to this medium for differentiation during 4 d prior transfection.
Reporters were transfected using lipofectamin 2000 (Life Science) reagent with modification of the protocol as follows. Half of the culture medium was retrieved from cells before transfection, complemented with fresh medium and kept to 37°C. After 2 h transfection, this medium was added to the cells for 24 h. DNA and lipofectamin mix was performed in DMEM basal medium. The construction of the stable cell line carrying the shSMN expression construct has been described previously (Rage et al. 2013).
Plasmid construction
The different mouse Anxa2 cDNA fragments were obtained following RT-PCR amplification of total RNA from NSC-34 cells. The amplified sequences were cloned into the NheI–NotI restriction site of the pcDNA3-LacZ-24xMS2 expression vector.
Four constructs were generated, named FL-wt, G-motif + 3′UTR, 3′UTR, and 5′UTR + CS (Fig. 1). The human G-motif + 3′UTR and 3′UTR plasmids carrying the corresponding fragment were first cloned into the TOPO-TA cloning kit (Promega) after PCR amplification and then subcloned into NheI–NotI restriction sites of pCMV-LacZ-24xMS2 vector.
The G-motif reporter was obtained following insertion of a double-stranded oligonucleotide containing the G-motif alone and NheI–NotI restriction sites into the pCMV-LacZ-24xMS2 vector.
Construction of the FL-mut1-4 reporters was performed using the QuikChange Site-Directed Mutagenesis kit (Stratagene) and 60-mer oligonucleotides essentially according to the manufacturer's instructions.
G-quadruplex-forming structure detection
The presence of a G-quadruplex structure in the Anxa2 mRNA was assessed by reverse transcriptase-mediated primer extension following a protocol adapted from Schaeffer et al. (2001). Briefly, the Anxa2 cDNA sequence subcloned into pGEM-T easy vector was PCR amplified with the following primers (T7:5′-GACTGACTTAATACGACTCACTATAGGG-3′; M13Rev: 5′-CACACAGGAAACAGCTATGACC-3′). The purified PCR product was used as matrix in an in vitro transcription reaction (HiScribe T7 High Yield RNA Synthesis Kit). Purified RNA (100 ng) was mixed together with 5′-NED labeled M13Rev primer (1,25 µM) and heat denatured for 5 min at 65°C followed by 1 min at 90°C. Then the oligonucleotide was allowed to anneal to the template RNA for 10 min at 20°C in HB-folding buffer (50 mM HEPES pH 7.0, 100 mM NaCl or KCl or LiCl, 167 nM EDTA). Primer extension was initiated by addition of enzyme mix (Tris–HCl 50 mM [pH 8.5], MgCl2 6 mM, KCl 40 mM), 2.5 mM dNTP, and 4 units SuperScript III (Invitrogen). Reactions were performed at 37°C for 30 min. Sequencing reactions used a modified dNTP set containing 1 mM of each dNTP, except the one corresponding to the ddNTP (33 µM) that was adjusted to 0.25 mM. Primers were labeled with 6-fam or Vic fluorophores. At the end of the reaction, each elongation product was mixed with two sequencing reactions (6-fam: ddG and VIC: ddT), precipitated with 100% ethanol and 2 µg of glycogen, then washed with 70% ethanol and resuspended in 10 µL of highly deionized formamide (Life Technologies). Samples were loaded on an Applied Biosystems 3130XL capillary electrophoresis DNA sequencer. Electrophoregrams were generated and analyzed with the QuShape software (Deigan et al. 2009; Karabiber et al. 2013).
Quantification of mRNA
Total RNA from transfected cells was extracted using the RNeasy Mini Kit (Qiagen) according to manufacturer's instructions. RNAs were analyzed by RT-PCR as follows. One microgram of total RNA was reverse transcribed using 200 U M-MLV reverse transcriptase (Invitrogen) in the presence of 2.5 mM N6 random primers and 0.5 mM dNTP. cDNA (3µL) of 1/20 RT mix was used as a template for real time PCR using a Mx3000P thermocycler (Agilent) with a home-made SYBR Green QPCR master mix (Lutfalla and Uze 2006). The sequences of the LacZ primers were the following: Fw 5′CAGTCGTTTGCCGTCTGAAT3′ and Rev 5′CAACGAGACGTCACGGAAAA3′. PCR reactions were performed in 10 µL in the presence of 150 nM primers. Thermal cycling parameters were 10 min at 95°C, followed by 40 cycles at 95°C for 30 sec, 64°C for 30 sec, and 72°C for 30 sec. Data were analyzed and relative amounts of specifically amplified cDNA were calculated with MxPro software (Agilent). The mouse β-2 microglobulin was used as an internal reference (Fw 5′TATGCTATCCAGAAAACCCCTCAA3′ and Rev 5′GTATGTTCGGCTTCCCATTCTC3′).
FISH and IF experiments
Amine-modified oligonucleotides specific for the LacZ or MS2 sequences were labeled with amine-reactive compound (Cy3, Invitrogen, Molecular Probes). A mix of fourteen probes for lacZ was used for each in situ hybridization. Cells were fixed with 4% paraformaldehyde in 1× PBS and then permeabilized with 70% ethanol overnight at 4°C. NSC-34 cells were hybridized at 37°C (40% formamide for lacZ probes or 10% for MS2, 10% dextran sulfate, 10 µg of yeast tRNA, 1× SSC, 0.2% BSA, 0.2% VRC, and 10 ng of each probe) overnight and washed twice in 40% or 10% formamide, 1× SSC for 1 h and then in 1× SSC for 30 min. When noted, FISH was followed by immunofluorescence studies, which were performed as described previously (Rage et al. 2013).
Image acquisition and processing
Images were acquired on a DMRA microscope equipped for epifluorescence and with a 100× PlanApo objective. Digital images were recorded with a 12-bit C4795-NR CCD camera (Hamamatsu). The camera and microscope were controlled by MetaMorph software and images were treated as previously described (Rage et al. 2013).
For quantitative analyses of the RNA granules and SMN protein colocalization, we used the Spot function of Imaris (Bitplane), which allows to consider only the signal present in foci on three-dimensional images (Rage et al. 2013). The significance of the colocalization data was determined with the unpaired Student t-test (Prism software). Image acquisition and analysis were performed on workstations of the Montpellier RIO Imaging facility of the CNRS campus.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the CNRS and by grants of the ARSLA to F.R. and the AFM (no. 17760) to J.S. We thank Edouard Bertrand and members of his team for helpful discussions. We also thank the Montpellier RIO Imaging facility for help with the imaging experiments. K.R. was supported by a fellowship of the Lebanese Association.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.056788.116.
REFERENCES
- Ackermann B, Kröber S, Torres-Benito L, Borgmann A, Peters M, Hosseini Barkooie SM, Tejero R, Jakubik M, Schreml J, Milbradt J, et al. 2013. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet 22: 1328–1347. [DOI] [PubMed] [Google Scholar]
- Akten B, Kye MJ, Hao le T, Wertz MH, Singh S, Nie D, Huang J, Merianda TT, Twiss JL, Beattie CE, et al. 2011. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci 108: 10337–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumer D, Lee S, Nicholson G, Davies JL, Parkinson NJ, Murray LM, Gillingwater TH, Ansorge O, Davies KE, Talbot K. 2009. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet 5: e1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulisfane N, Choleza M, Rage F, Neel H, Soret J, Bordonné R. 2011. Impaired minor tri-snRNP assembly generates differential splicing defects of U12-type introns in lymphoblasts derived from a type I SMA patient. Hum Mol Genet 20: 641–648. [DOI] [PubMed] [Google Scholar]
- Bowerman M, Anderson CL, Beauvais A, Boyl PP, Witke W, Kothary R. 2009. SMN, profilin IIa and plastin 3: a link between the deregulation of actin dynamics and SMA pathogenesis. Mol Cell Neurosci 42: 66–74. [DOI] [PubMed] [Google Scholar]
- Bugaut A, Balasubramanian S. 2012. 5′-UTR RNA G-quadruplexes: translation regulation and targeting. Nucleic Acids Res 40: 4727–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghes AH, Beattie CE. 2009. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 10: 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DS, Holt CE. 2001. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron 32: 1013–1026. [DOI] [PubMed] [Google Scholar]
- Cauchi RJ. 2010. SMN and Gemins: ‘we are family’ … or are we?: insights into the partnership between Gemins and the spinal muscular atrophy disease protein SMN. Bioessays 32: 1077–1089. [DOI] [PubMed] [Google Scholar]
- Chari A, Paknia E, Fischer U. 2009. The role of RNP biogenesis in spinal muscular atrophy. Curr Opin Cell Biol 21: 387–393. [DOI] [PubMed] [Google Scholar]
- Coady TH, Lorson CL. 2011. SMN in spinal muscular atrophy and snRNP biogenesis. Wiley Interdiscip Rev RNA 2: 546–564. [DOI] [PubMed] [Google Scholar]
- Deigan KE, Li TW, Mathews DH, Weeks KM. 2009. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci 106: 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitriadi M, Sleigh JN, Walker A, Chang HC, Sen A, Kalloo G, Harris J, Barsby T, Walsh MB, Satterlee JS, et al. 2010. Conserved genes act as modifiers of invertebrate SMN loss of function defects. PLoS Genet 6: e1001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombert B, Sivadasan R, Simon CM, Jablonka S, Sendtner M. 2014. Presynaptic localization of Smn and hnRNP R in axon terminals of embryonic and postnatal mouse motoneurons. PLoS One 9: e110846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliscovich C, Buxbaum AR, Katz ZB, Singer RH. 2013. mRNA on the move: the road to its biological destiny. J Biol Chem 288: 20361–20368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Zhang H, Su Y, Silani V, Singer RH, Rossoll W, Bassell GJ. 2011. The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(A) mRNA in primary motor neuron axons. J Neurosci 31: 914–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, Rossoll W. 2012. Spinal muscular atrophy: the role of SMN in axonal mRNA regulation. Brain Res 1462: 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco D, Accornero N, Lavoie B, Shenoy SM, Blanchard JM, Singer RH, Bertrand E. 2003. Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr Biol 13: 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabanella F, Butchbach MER, Saieva L, Carissimi C, Burghes AHM, Pellizzoni L. 2007. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One 2: e921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel M, Delavoie F, Demais V, Royer C, Bailly Y, Vitale N, Bader MF, Chasserot-Golaz S. 2015. Annexin A2-dependent actin bundling promotes secretory granule docking to the plasma membrane and exocytosis. J Cell Biol 210: 785–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia EL, Lu Z, Meers MP, Praveen K, Matera AG. 2013. Developmental arrest of Drosophila survival motor neuron (Smn) mutants accounts for differences in expression of minor intron containing genes. RNA 19: 1510–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieve AG, Moss SE, Hayes MJ. 2012. Annexin A2 at the interface of actin and membrane dynamics: a focus on its roles in endocytosis and cell polarization. Int J Cell Biol 2012: 852430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubitz AK, Feng W, Dreyfuss G. 2004. The SMN complex. Exp Cell Res 296: 51–56. [DOI] [PubMed] [Google Scholar]
- Hao le T, Wolman M, Granato M, Beattie CE. 2012. Survival motor neuron affects plastin 3 protein levels leading to motor defects. J Neurosci 32: 5074–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MJ, Shao D, Bailly M, Moss SE. 2006. Regulation of actin dynamics by annexin 2. EMBO J 25: 1816–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MJ, Longbottom RE, Evans MA, Moss SE. 2007. Annexinopathies. Subcell Biochem 45: 1–28. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. 2006. mRNA transport in dendrites: RNA granules, motors, and tracks. J Neurosci 26: 7139–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Bullock SL. 2009. Subcellular mRNA localization in animal cells and why it matters. Science 326: 1212–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hörnberg H, Holt C. 2013. RNA-binding proteins and translational regulation in axons and growth cones. Front Neurosci 7: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubers L, Valderrama-Carvajal H, Laframboise J, Timbers J, Sanchez G, Côté J. 2011. HuD interacts with survival motor neuron protein and can rescue spinal muscular atrophy-like neuronal defects. Hum Mol Genet 20: 553–579. [DOI] [PubMed] [Google Scholar]
- Huppert JL, Bugaut A, Kumari S, Balasubramanian S. 2008. G-quadruplexes: the beginning and end of UTRs. Nucleic Acids Res 36: 6260–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonka S, Beck M, Lechner BD, Mayer C, Sendtner M. 2007. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J Cell Biol 179: 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambhekar A, Derisi JL. 2007. Cis-acting determinants of asymmetric, cytoplasmic RNA transport. RNA 13: 625–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Yoon BC, Holt CE. 2012. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci 13: 308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karabiber F, McGinning JL, Favorov OV, Weeks KM. 2013. QuShape: rapid, accurate, and best-practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. RNA 19: 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR. 2008. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet 17: 2552–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebler MA, Bassell GJ. 2006. Neuronal RNA granules: movers and makers. Neuron 51: 685–690. [DOI] [PubMed] [Google Scholar]
- Kikin O, D'Antonio L, Bagga PS. 2006. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res 34: W676–W682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Wang X, Choe DW, Polley M, Burnett BG, Bosch-Marcé M, Griffin JW, Rich MM, Sumner CJ. 2009. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci 29: 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. 1997. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16: 265–269. [DOI] [PubMed] [Google Scholar]
- Li DK, Tisdale S, Lotti F, Pellizzoni L. 2014. SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin Cell Dev Biol 32: 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Q, Jacovina AT, Deora A, Febbraio M, Simantov R, Silverstein RL, Hempstead B, Mark WH, Hajjar KA. 2004. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J Clin Invest 113: 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Qin J, Höner zu Siederdissen C, Tanzer A, Amman F, Hofacker IL, Stadler PF. 2013. 2D meets 4G: G-quadruplexes in RNA secondary structure prediction. IEEE/ACM Trans Comput Biol Bioinform 10: 832–844. [DOI] [PubMed] [Google Scholar]
- Lotti F, Imlach WL, Saieva L, Beck ES, Hao le T, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, et al. 2012. An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151: 440–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfalla G, Uze G. 2006. Performing quantitative reverse-transcribed polymerase chain reaction experiments. Methods Enzymol 410: 386–400. [DOI] [PubMed] [Google Scholar]
- Martin KC, Ephrussi A. 2009. mRNA localization: gene expression in the spatial dimension. Cell 136: 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera AG, Wang Z. 2014. A day in the life of the spliceosome. Nat Rev Mol Cell Biol 5: 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G, Eggert C, Fischer U. 2002. SMN-mediated assembly of RNPs: a complex story. Trends Cell Biol 12: 472–478. [DOI] [PubMed] [Google Scholar]
- Menon L, Mihailescu MR. 2007. Interactions of the G quartet forming semaphorin 3F RNA with the RGG box domain of the fragile X protein family. Nucleic Acids Res 35: 5379–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monastyrskaya K, Babiychuk EB, Hostettler A, Rescher U, Draeger A. 2007. Annexins as intracellular calcium sensors. Cell Calcium 41: 207–219. [DOI] [PubMed] [Google Scholar]
- Oprea GE, Kröber S, McWhorter ML, Rossoll W, Müller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B. 2008. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320: 524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazzon N, Rage F, Schlotter F, Moine H, Branlant C, Massenet S. 2008. In vitro and in cellulo evidences for association of the survival of motor neuron complex with the fragile X mental retardation protein. J Biol Chem 283: 5598–5610. [DOI] [PubMed] [Google Scholar]
- Rage F, Boulisfane N, Rihan K, Neel H, Gostan T, Bertrand E, Bordonné R, Soret J. 2013. Genome-wide identification of mRNAs associated with the protein SMN whose depletion decreases their axonal localization. RNA 19: 1755–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossoll W, Bassell GJ. 2009. Spinal muscular atrophy and a model for survival of motor neuron protein function in axonal ribonucleoprotein complexes. Results Probl Cell Differ 48: 289–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossoll W, Kröning AK, Ohndorf UM, Steegborn C, Jablonka S, Sendtner M. 2002. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum Mol Genet 11: 93–105. [DOI] [PubMed] [Google Scholar]
- Rossoll W, Jablonka S, Andreassi C, Kröning AK, Karle K, Monani UR, Sendtner M. 2003. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of β-actin mRNA in growth cones of motoneurons. J Cell Biol 163: 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaria V, Hariharan M, Arora A, Maiti S. 2006. Quadfinder: server for identification and analysis of quadruplex-forming motifs in nucleotide sequences. Nucleic Acids Res 34: W683–W685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer C, Bardoni B, Mandel JL, Ehresmann B, Ehresmann C, Moine H. 2001. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J 20: 4803–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lambrechts A, Hao le T, Le TT, Sewry CA, Ampe C, Burghes AH, Morris GE. 2005. A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp Cell Res 309: 185–197. [DOI] [PubMed] [Google Scholar]
- Subramanian M, Rage F, Tabet R, Flatter E, Mandel JL, Moine H. 2011. G-quadruplex RNA structure as a signal for neurite mRNA targeting. EMBO Rep 12: 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Davies KE. 2008. Is good housekeeping the key to motor neuron survival? Cell 133: 572–574. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Wu J, Tai HC, Schuman EM. 2013. Axonal translation of β-catenin regulates synaptic vesicle dynamics. J Neurosci 33: 5584–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till SM, Li HL, Miniaci MC, Kandel ER, Choi YB. 2011. A presynaptic role for FMRP during protein synthesis–dependent long-term plasticity in Aplysia. Learn Mem 18: 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AG, Shaw DJ, Morse R, Stebbings H, Young PJ. 2010a. SMN and the Gemin proteins form sub-complexes that localise to both stationary and dynamic neurite granules. Biochem Biophys Res Commun 394: 211–216. [DOI] [PubMed] [Google Scholar]
- Todd AG, Morse R, Shaw DJ, Stebbings H, Young PJ. 2010b. Analysis of SMN-neurite granules: core Cajal body components are absent from SMN-cytoplasmic complexes. Biochem Biophys Res Commun 397: 479–485. [DOI] [PubMed] [Google Scholar]
- Tsuiji H, Iguchi Y, Furuya A, Kataoka A, Hatsuta H, Atsuta N, Tanaka F, Hashizume Y, Akatsu H, Murayama S, et al. 2013. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med 5: 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Brent JR, Tomlinson A, Shneider NA, McCabe BD. 2011. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J Clin Invest 121: 4118–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B. 2000. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat 15: 228–237. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Chen S, Yu Y, Yan B, Haertlein TC, Carrasco MA, Tapia JC, Zhai B, Das R, Lalancette-Hebert M, et al. 2013. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep 2: 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HL, Pan F, Hong D, Shenoy SM, Singer RH, Bassell GJ. 2003. Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J Neurosci 23: 6627–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Xing L, Rossoll W, Wichterle H, Singer RH, Bassell GJ. 2006. Multiprotein complexes of the survival of motor neuron protein SMN with Gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci 26: 8622–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. 2008. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.