

Amankulor et al. created a syngeneic pair mouse model for mutant IDH1 (muIDH1) and wild-type IDH1 (wtIDH1) gliomas and demonstrated that IDH1 mutations caused down-regulation of leukocyte chemotaxis, resulting in repression of the tumor-associated immune system.
Keywords: IDH mutation, immuno-oncology, glioma
Abstract
Gliomas harboring mutations in isocitrate dehydrogenase 1/2 (IDH1/2) have the CpG island methylator phenotype (CIMP) and significantly longer patient survival time than wild-type IDH1/2 (wtIDH1/2) tumors. Although there are many factors underlying the differences in survival between these two tumor types, immune-related differences in cell content are potentially important contributors. In order to investigate the role of IDH mutations in immune response, we created a syngeneic pair mouse model for mutant IDH1 (muIDH1) and wtIDH1 gliomas and demonstrated that muIDH1 mice showed many molecular and clinical similarities to muIDH1 human gliomas, including a 100-fold higher concentration of 2-hydroxygluratate (2-HG), longer survival time, and higher CpG methylation compared with wtIDH1. Also, we showed that IDH1 mutations caused down-regulation of leukocyte chemotaxis, resulting in repression of the tumor-associated immune system. Given that significant infiltration of immune cells such as macrophages, microglia, monocytes, and neutrophils is linked to poor prognosis in many cancer types, these reduced immune infiltrates in muIDH1 glioma tumors may contribute in part to the differences in aggressiveness of the two glioma types.
Gliomas can be divided into two groups, CpG island methylator phenotype (CIMP) and non-CIMP, with the survival of patients with CIMP gliomas being significantly longer (Noushmehr et al. 2010). CIMP gliomas have a high frequency of mutations in isocitrate dehydrogenase 1/2 (IDH1/2), and the oncometabolite 2-hydroxygluratate (2-HG), which is produced by IDH1/2 mutations, is the driver of the hyper-DNA methylation phenotype (Dang et al. 2009; Turcan et al. 2012). In addition, there are canonical mutations in non-CIMP gliomas, including chromosome gains and losses (ch7+/ch10−) that are completely different from those in CIMP gliomas (Ozawa et al. 2014; Bai et al. 2016; Ceccarelli et al. 2016). How the CIMP phenotype drives glioma progression is not fully understood yet; however, a recent study has shown that hypermethylation promotes insulator dysfunction and leads to the increased expression of platelet-derived growth factor receptor A (PDGFRA) (Flavahan et al. 2016). The reasons for the differences in survival between CIMP and non-CIMP gliomas are likely multifactorial but could in part be directly due to the effects of IDH mutations.
Non-CIMP wild-type IDH1/2 (wtIDH1/2) gliomas are more aggressive compared with their CIMP counterparts, and CIMP status is a more robust prognosticator for better clinical outcomes than histological grading (Hartmann et al. 2010). Most experimental studies on gliomas have focused on the more aggressive non-CIMP and wtIDH1/2 glioblastoma multiformes (GBMs), partially due to the difficulty in establishing primary CIMP+ glioma cell cultures and a lack of a definitive mouse model of CIMP gliomas (Sasaki et al. 2012).
Multiple factors contribute to aggressiveness in solid tumors, one of which is immune cell content. Reduction or enhancement of specific immune cell types has led to increased or decreased survival in mouse models of several cancer types (Quail and Joyce 2013; Kitamura et al. 2015; Shalapour and Karin 2015). Although the brain is thought to be an immune-privileged site, gliomas have a substantial component of immune cells, where macrophages and microglia can account for as many as 20%–30% of the total cells in some highly aggressive tumors (Badie and Schartner 2000; Kong et al. 2010). In addition to differences in immune cell content, our group has previously described marked differences in effector immune cell function across mutant IDH1 (muIDH1) and wtIDH1 gliomas (Zhang et al. 2016).
In order to better understand this aspect of glioma aggressiveness, we compared the immune cell components of muIDH1 and wtIDH1 gliomas. We found that human CIMP gliomas have significantly lower numbers of several immune cell types relative to non-CIMP tumors. We investigated the specific effects of IDH1 mutations on the immune cell content of gliomas in vivo using the RCAS/tva system to create an isogenic glioma pair driven by PDGF-driven mouse glioma models whose initiating events differed only in the presence or absence of muIDH1. The muIDH1 mouse gliomas showed many similarities to human muIDH1/CIMP gliomas, including elevated 2-HG, DNA methylation, and gene expression patterns as well as phenotypic characteristics, including reduced immunologic cell content. Overall, these data suggest that the longer survival time of CIMP glioma patients may be due in part to the IDH mutation and its effect on reducing a part of the tumor-associated immune system that enhances aggressiveness.
Results
Human muIDH1 gliomas have less immune infiltration than wtIDH1 gliomas
Gene expression signatures of wtIDH1 gliomas are strongly associated with an inflammation and immunologic response (Ceccarelli et al. 2016) and high levels of several chemokines and interleukins that may stimulate infiltration of regulatory immune cells. In order to determine whether the immune profile of muIDH1/CIMP is different from that of wtIDH1/non-CIMP gliomas, we performed FACS analysis using wtIDH1 (n = 10) and muIDH1 (n = 6) human glioma tissue samples. Of the 10 wtIDH1 tumors analyzed, nine tumors were glioblastomas (grade 4), and one tumor was a grade 3 astrocytoma. Of the six muIDH1/CIMP tumors analyzed, four tumors were grade 3 astrocytomas, one tumor was a grade 2 astrocytoma, and one tumor was a grade 3 oligodendroglioma. The data in Figure 1A demonstrate that muIDH1 human gliomas have significantly fewer overall CD45+ immune cells, and further subset analysis of immune cells indicated that the depletion of immune infiltrates was global, including microglia, macrophages, dendritic cells, B cells, and T cells, compared with that of wtIDH1 human gliomas (Fig. 1B).
Figure 1.

Human muIDH1 gliomas had repressed tumor-associated immune systems. (A,B) FACS analysis using wtIDH1 (n = 10) and muIDH1 (n = 6) human glioma tissues to quantify tumor-associated immune cells. Error bars show the standard error of mean (SEM). Analysis was done using Student's t-test. (C) Summary of the number of up-regulated and down-regulated genes between two glioma groups: wtIDH1 human low-grade gliomas (LGGs, black, n = 91), human muIDH1 astrocytomas (AS, red; n = 248), and human muIDH1 oligodendrogliomas (OD, blue; n = 169), (D) Summary of the number of gene ontology (GO) terms associated with up-regulated and down-regulated genes between two glioma groups. (E) Immune-related GO terms and corresponding P-values that are associated with the down-regulated genes in muIDH1 human gliomas.
To validate and expand on these results, we turned to The Cancer Genome Atlas (TCGA) to look for gene expression evidence that muIDH1 human gliomas may contain fewer immune cells and determine to what extent the difference in expression is immune-related (Brennan et al. 2013; The Cancer Genome Atlas Research Network 2015). We used TCGA data sets from previously annotated samples from Ceccarelli et al. (2016). For this comparison, we ruled out the general correlation between malignancy and IDH1 mutation status by excluding GBMs from our analysis. Grade 2 and 3 gliomas were subdivided into two genetic groups: wtIDH1 and muIDH1. We identified 91 wtIDH1/non-CIMP and 417 muIDH1/CIMP human low-grade gliomas (LGGs). Gliomas with 1p/19q codeletion and IDH1 mutations are defined as oligodendrogliomas (n = 169), and other muIDH gliomas without 1p/19q codeletion are classified as astrocytomas (n = 248) (Supplemental Fig. S1; Supplemental Table S1). We then used the publically available data sets from these tumors to investigate whether the same grade tumors had differential immune activation based on IDH1 mutation status.
We then identified the number of genes that were differentially expressed between the wtIDH1 and muIDH1 groups. Using a threshold of twofold or greater and a significant adjusted P-value (P-value < 0.05), we found 1297 genes to be reduced in expression, while 328 genes were increased in muIDH1 human LGGs relative to wtIDH1/non-CIMP human LGGs (Fig. 1C). Gene ontology (GO) analysis of these genes included terms related to developmental biology in both down-regulated and up-regulated genes (Fig. 1D). The REVIGO (reduce and visualize GO) (Supek et al. 2011) plot using the down-regulated and up-regulated genes showed how these GO terms were clustered and correlated (Supplemental Fig. S2). REVIGO summarizes GO terms by removing redundant GO terms. The remaining terms can be visualized in semantic similarity-based scatter plots as in Supplemental Figure S2. With this analysis, the numbers of GO terms were clustered together, indicating that certain biological pathways were repeatedly correlated with each set of genes. The up-regulated genes were associated mainly with various developmental processes, such as multicellular organismal processes, developmental processes, regulation of multicellular organismal processes, and regulation of hormone levels, but rarely showed correlations with other types of GO terms (Supplemental Fig. S2A; Supplemental Table S2). In contrast, the down-regulated gene sets were associated with various aspects of developmental biology, metabolism, and extracellular matrix (Supplemental Fig. S2B; Supplemental Table S3). In addition, we also identified that the down-regulated genes in muIDH1 human gliomas are associated with immune system processes. When muIDH1 gliomas were separated into muIDH1 astrocytomas and muIDH1 oligodendrogliomas, we found that GO terms related to chemotaxis and immune cell migration were linked to the down-regulated genes of muIDH1 astrocytomas and muIDH1 oligodendrogliomas (Fig. 1E). Given that chemokines stimulate immune cell migration to wounded sites, repression of immune infiltrates could be due to the down-regulation of chemotaxis-associated genes in muIDH1 human gliomas.
There are significant genomic structural differences between muIDH1 and wtIDH1 gliomas, and the above gene expression differences between these two tumor types could be due to many factors other than IDH mutation status. Therefore, in order to determine what component of this difference is due to the IDH mutation specifically, we created an in vivo experimental system in immune-competent animals where we could compare two isogenic glioma populations that differed only in IDH mutation status.
Generation of a muIDH1 RCAS/tva mouse glioma model
To investigate to what extent the global immune repression in muIDH1 gliomas is due to IDH1 mutations, we created isogenic muIDH1 and wtIDH1 mouse glioma models whose initiating events were identical except for muIDH1 expression. Given that IDH mutations alone are insufficient to induce gliomas in mice and primarily affect the extracellular matrix and blood vessel maturation (Sasaki et al. 2012), we used the RCAS/tva system to ectopically express muIDH1 (R132H) in PDGF-driven gliomas. RCAS retroviral vectors were used to transfer genes to specific cell types in vivo that express the receptor tva (Dai et al. 2001; Hambardzumyan et al. 2009) to develop a pair of isogenic mouse tumor models that differ only in IDH1 mutation status. Both glioma types are driven by PDGF in combination with p53 loss, mimicking the frequent p53 mutation in IDH mutant astrocytomas (Bolouri et al. 2016). We prepared three RCAS vectors expressing PDGF, wtIDH1-shp53, or muIDH1 (R132H)-shp53 (Fig. 2A). Next, we used three different Ntva mouse strains in which the tva receptor is expressed from the nestin promoter, allowing gene transfer to CNS progenitors Ntva_Ink4a/Arf−/−, Ntva_Ink4a/Arf+/−, and Ntva_Ink4a/Arf+/+ (Supplemental Fig. S3A,B; Hambardzumyan et al. 2009). Ink4a/Arf deficiency is strongly linked with higher-grade glioma formation in both human and mouse gliomas and is much more prominent in wtIDH1 than muIDH1 gliomas (Uhrbom et al. 2002). Mice were injected with RCAS–PDGF-producing DF1 cells together with either DF1 cells producing RCAS–wtIDH1-shp53 (wtIDH1) or RCAS–muIDH1-shp53 (muIDH1). Tumors were generated from these injections with identical genomic backgrounds, with the initiating events differing only in IDH1 mutation status. We euthanized mice when they showed signs of CNS pathology and harvested tumors. In a separate control experiment, we injected wtIDH1-expressing or muIDH1-expressing DF1 cells without PDGF-expressing DF1 cells. None of these mice developed tumors (data not shown), consistent with previous reports showing that IDH mutation is not sufficient to induce gliomas.
Figure 2.

muIDH1-expressing mouse gliomas were generated using RCAS/tva technology. (A) Three RCAS vectors expressing PDGFa (black), human wtIDH1-shp53 with H1 promoter (wtIDH1-H1-shp53; blue), and human muIDH1-shp53 with H1 promoter (muIDH1-H1-shp53; red). IDH1 R132H was used for muIDH1. (B) Western blotting using mouse glioma tissues to verify muIDH1 expression. Glioma tissues were harvested from each glioma group (n = 3) and subjected to Western blotting using the IDH1 R132H antibody. All three muIDH1 mouse gliomas expressed muIDH1, while wtIDH1 mouse gliomas lacked muIDH1 expression. (C) Images of wtIDH1 and muIDH1 mouse gliomas using hematoxylin and eosin (H&E) staining and immunohistochemistry. Immunohistochemistry showed that muIDH1 expression was limited to muIDH1 mouse tumors. Images taken with 1.25× objectives show whole mouse brain sections with tumors infiltrating into normal brain tissue.
We characterized the resulting tumors so that we could compare them with their human counterparts. To verify integration of muIDH1 in the genomic backbone of murine gliomas, tumor tissues from each group were harvested and subjected to tumor genotyping from Ntva_Ink4a/Arf−/− mice (Supplemental Fig. S3C,D). The results shown in Supplemental Figure S3D confirmed that tumor DNA had integration of either wtIDH1 or muIDH1 genes of RCAS vectors. The presence of muIDH1 protein was verified by Western blotting using the muIDH1 R132H antibody (Fig. 2B). In addition, immunohistochemistry for muIDH1 R132H staining also showed that muIDH1 was widely expressed throughout and within the muIDH1 mouse glioma while being absent in wtIDH1-overexpressing tumors in Ink4a/Arf−/− mice (Fig. 2C). We stained both Ink4a/Arf−/− muIDH1 and wtIDH1 tumors for hematoxylin and eosin (H&E), Olig2, Ki67, and CD31 and did not detect a difference in the grade or expression of these markers between the two tumor types (Fig. 2C; Supplemental Fig. S4A–C).
muIDH1 mouse gliomas have higher 2-HG and DNA methylation than wtIDH gliomas
Survival rates of mice in both the wtIDH1 and muIDH1 tumor groups showed strong correlations with Ink4a/Arf gene expression (Fig. 3A,B). Mice with wtIDH1 tumors showed significantly shorter survival compared with mice with muIDH1 tumors in both Ink4a/Arf+/+ and Ink4a/Arf+/− backgrounds. In mice with an Ink4a/Arf+/− background, those with muIDH1 tumors had a 56-d median survival time compared with 42 d for wtIDH1 tumors (P-value <0.0001). The tumors that formed in the Ink4a/Arf−/− background did not result in any differences in median survival between the wtIDH1 and muIDH1 groups, presumably due to the aggressiveness of tumors in this cohort. All subsequent experiments were performed using Ink4a/Arf−/− mice. We used these mice in order to ensure that both IDH1 wild-type and mutant tumors had a similar growth rate so that the observed effects were not due to different tumors sizes.
Figure 3.

muIDH1 mouse gliomas resembled muIDH1 human gliomas. (A) Survival plots of muIDH1 versus wtIDH1 mouse gliomas with different genomic backgrounds (Ntva_Ink4a/Arf+/+, Ntva_Ink4a/Arf+/−, and Ntva_Ink4a/Arf−/−) (Hambardzumyan et al. 2009). (B) Table showing median survivals and P-values. (C) 2-HG concentrations in murine gliomas. Metabolites from murine glioma tissues were extracted followed by DATAN derivatization. 2-HG was measured with liquid chromatography-mass spectrometry. Seven and 11 metabolite extracts were used for the wtIDH1 and muIDH1 mouse glioma groups, respectively. Error bars show the standard deviation (SD). Analysis was done using Student's t-test. (D) CpG island methylation analysis using reduced representation bisulfite sequencing (RRBS). n = 4 muIDH1; n = 2 wtIDH1.
To determine whether muIDH1 was functional in vivo, we used mass spectrometry to measure 2-HG levels in mouse glioma tissues isolated from Ink4a/Arf−/− mice. Previous reports demonstrated that muIDH1 human gliomas and muIDH1-expressing mouse brain tissues contain 100-fold higher 2-HG than wtIDH1 human gliomas and wtIDH1-expressing mouse tissues (Dang et al. 2009; Sasaki et al. 2012). Our muIDH1 mouse gliomas also presented ∼100-fold higher 2-HG than wtIDH1 mouse gliomas (Fig. 3C). To determine whether 2-HG concentration is sufficient to affect DNA methylation as in human gliomas, we performed reduced representation bisulfite sequencing (RRBS) to quantify methylation of CpG islands in gliomas in Ink4a/Arf−/− mice (Meissner et al. 2005). The obtained results showed increased DNA methylation of CpG islands of muIDH1 glioma DNA (Fig. 3D). Anti-5-methylcytosine (5-mC) immunohistochemical staining also showed that muIDH1 mouse gliomas have higher DNA methylation than wtIDH1 mouse gliomas (Supplemental Fig. S5). Finally, the overall gene expression pattern of muIDH1 mouse gliomas was measurably different from that of wtIDH1 mouse gliomas.
Differential activation of biological processes in muIDH1 mouse gliomas
We investigated whether the biology of our muIDH1 mouse gliomas resembled the tumor biology of muIDH1 human gliomas. Initially, we compared RNA expression patterns and identified genes with expression higher or lower in the Ink4a/Arf−/− muIDH1 mouse tumors relative to their wtIDH1 mouse counterparts. The REVIGO plot using the down-regulated genes shows clustered GO terms similar to those of the muIDH1 human gliomas, including immune system processes, developmental processes, and extracellular matrix organization (Supplemental Fig. S6B). The REVIGO plot shows that the up-regulated genes had associations with L-serine synthesis and negative regulation of nerve system development (Supplemental Fig. S6A). The PANTHER overrepresentation test using the down-regulated genes categorized GO terms as “regulation of liquid surface tension,” “biological adhesion,” and “developmental process” in addition to “immune system process” (Fig. 4A; Mi et al. 2013). However, the PANTHER overrepresentation test did not show any categorized GO terms with the up-regulated genes in these muIDH1 mouse gliomas. Comparison of mouse and human data identified 18 overlapping GO terms from the REVIGO test (Fig. 4B). The immune system process was the second most unique GO term associated with the down-regulated genes in human muIDH1 gliomas (Fig. 4C). Gene set enrichment analysis (GSEA) also showed that wtIDH1 mouse gliomas had strong associations with positive regulation of immune responses (Fig. 4D). The complete list of overlapped GO terms associated with the down-regulated genes is provided in Supplemental Table S4. The global lower gene expression in our muIDH1 mouse gliomas caused by IDH1 mutation is similar to that of muIDH1/CIMP human LGGs. In conclusion, our muIDH1 and wtIDH1 mouse glioma models mimic their human counterparts in several important aspects, including differential association with immune system processes.
Figure 4.
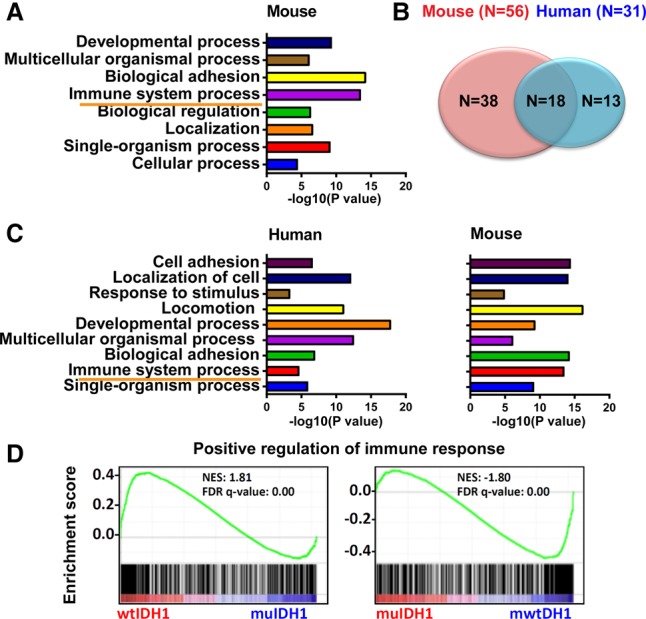
muIDH1 mouse gliomas resembled muIDH1 human gliomas. (A) PANTHER overrepresentation test using the down-regulated genes in muIDH1 mouse gliomas. (B,C) Overlaps of clustered GO terms between mouse and human muIDH1 gliomas. (D) GSEA using the human immune response gene set.
Fewer CD45+ cells are found in muIDH1 mouse gliomas
To validate the mouse gene expression data and investigate the phenotypic parallels between human and mouse muIDH1 tumors, we used flow cytometry to profile the immune cells present in normal mouse brain tissue and wtIDH1 and muIDH1 gliomas. We generated single-cell suspensions from brain tissue and evaluated immune cell content using the gating strategy depicted in Supplemental Figure S7. The first two gates were performed to exclude doublets. A fixable viability dye was used to exclude dead cells from the analysis. Consistent with human data, the major immune cells present in each Ink4a/Arf−/− muIDH1 and wtIDH1 mouse glioma were microglia and macrophages, defined as CD45loCD11b+ and CD45hiCD11b+, respectively. The muIDH1 mouse gliomas displayed significantly fewer CD45+ immune cells compared with wtIDH1 gliomas (Fig. 5A). Similar to human muIDH1 gliomas, we found reductions in microglia, macrophages, monocytes, and polymorphonuclear leukocytes (PMNs) in the muIDH1 tumors (Fig. 5B). Gene expression of muIDH1 mouse gliomas was also negatively associated with leukocyte and neutrophil migration (Fig. 5C).
Figure 5.
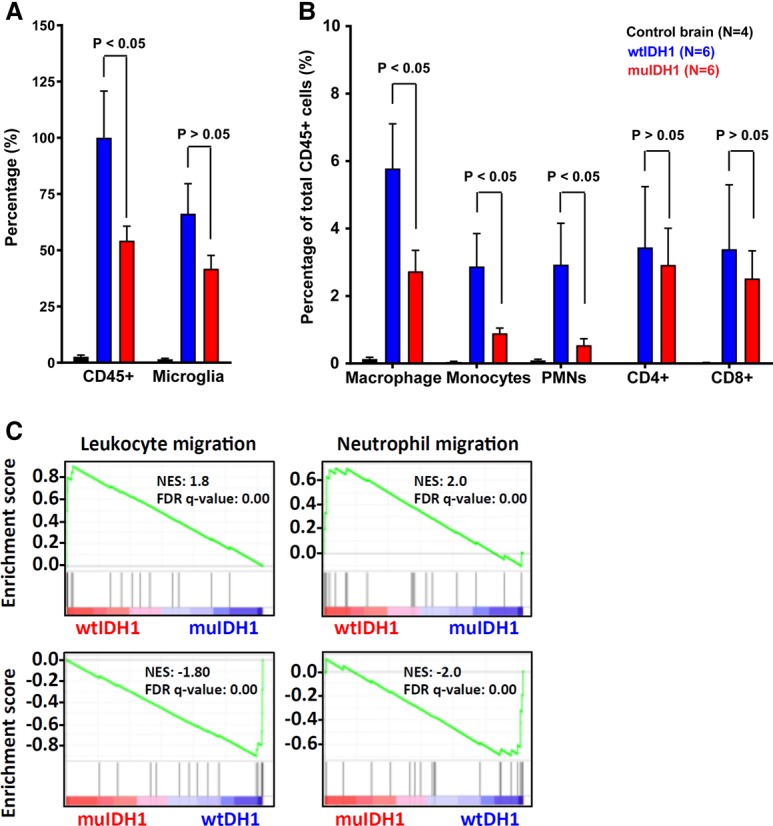
FACS analysis using muIDH1 and wtIDH1 mouse gliomas to quantify immune cells. (A) Normalized data showing total CD45+ cells and microglia relative to total CD45+ cells in wtIDH1 mouse gliomas. (B) Quantitation of each immune cell type. (C) GSEA plots showing that the gene expression of wtIDH1 mouse gliomas was positively associated with leukocyte migration, in contrast to muIDH1 mouse gliomas. Error bars show the SEM. Analysis was done using Student's t-test.
Neutrophil chemotaxis is impaired in muIDH1 mouse gliomas
The TCGA human glioma data set suggests that chemotaxis-regulating genes are down-regulated in muIDH1 human gliomas. Thus, we hypothesized that leukocyte migration may be functionally impaired in muIDH1 gliomas. Notably, GSEA of muIDH1 mouse gliomas revealed a negative correlation with chemotaxis, leukocyte chemotaxis, and neutrophil chemotaxis (Fig. 6A). We therefore investigated the extent of neutrophil migration in the presence of muIDH1 cells using a Boyden chamber. Neutrophils were plated on the top of a Boyden chamber, and then tissue homogenate from each Ink4a/Arf−/− tumor group was added to the bottom chamber. After 1 h of incubation to allow neutrophils to migrate, we normalized the migration index as 1 for muIDH1 mouse gliomas. As shown in Figure 6B, tissue homogenates of wtIDH1 mouse gliomas had approximately twice the migration index of muIDH1 mouse gliomas, suggesting that chemotaxis to muIDH1 gliomas was repressed. To investigate whether the reduced migration index was due to secretomes, we repeated the Boyden chamber experiment using conditioned medium from muIDH1 and wtIDH1 tumor-derived cells. The results showed that the conditioned medium from wtIDH1 tumor-derived cells had 1.7-fold higher chemotaxis than that from muIDH1 tumor-derived cells (Fig. 6C), which is parallel to the results shown in Figure 6B.
Figure 6.

Quantitative analysis of chemotaxis and chemokines. (A) GSEA plots showing that the gene expression of wtIDH1 mouse gliomas was positively associated with chemotaxis, leukocyte chemotaxis, and neutrophil chemotaxis, in contrast to muIDH1 mouse gliomas. (B) Migration index using wtIDH1 (n = 5) and muIDH1 (n = 9) tumor tissue lysates. (C) Migration index using conditioned medium from wtIDH1 (n = 5) and muIDH1 (n = 9) tumor-derived cells. (D) mRNA expression of CCL-2, CXCL-2, and C5 using quantitative PCR. (E) Protein expression of CCL-2, CXCL-2, and C5. Error bars show the SEM. Analysis was done using Student's t-test.
These data indicate that tissue homogenates from muIDH1 mouse gliomas may contain lower levels of neutrophil chemoattractants. Thus, we quantified mRNA and protein expression in tumor tissues using quantitative PCR (qPCR) and a Proteome Profiler mouse cytokine array kit. We discovered that a large number of cytokine protein expressions were down-regulated in muIDH1 mouse gliomas, including CCL-2, CCL-3, CXCL-1, CXCL-2, CXCL-4, CXCL-16, GM-CSF, IL-1ra, IL-2, IL-6, IL-16, etc. We also measured the mRNA expression of Ccl2, Cxcl2, and C5. These proteins are highly associated with migration of a large number of immune cells associated with innate immunity. CCL-2 is known for recruiting leukocytes, including monocytes, memory T cells, and dendritic cells, to sites of inflammation produced by either tissue injury or infection. CXCL-2 is chemotactic for PMNs, including neutrophils as well as monocytes and macrophages. C5a has chemotactic and anaphylatoxic properties, which are essential in innate immunity, but it is also linked with adaptive immunity. Gene expression of these three genes was down-regulated in muIDH1 tumors at the mRNA level (Fig. 6D) and protein level, as shown by a cytokine array (Fig. 6E).
We further investigated the expression of these chemokines in human glioma cells. We used three patient-derived wtIDH1 human glioma lines from three independent patients (U3039, U3046, and U3065) in addition to a patient-derived muIDH1 glioma line (TS603). As shown in Supplemental Figure S8, all three patient-derived wtIDH1 glioma lines showed significantly higher cytokine expression relative to the muIDH1 TS603.
Finally, we treated wtIDH1 or muIDH1 tumor-bearing Ntva_Ink4a/Arf+/− mice with an anti-Ly6g (1A8) or isotype control (2A3) antibody (500 µg per injection three times per week for 5 wk) to deplete the neutrophil populations. We observed no significant effect on muIDH1 tumors; however, we saw a significant survival benefit for wtIDH1 tumors (Supplemental Fig. S9).
In summary, our data suggest that decreased immune cell infiltration in IDH mutant gliomas may be partly responsible for the improved clinical outcomes observed in these patients.
Discussion
Compared with their wtIDH1/2/non-CIMP counterparts, muIDH1/2/CIMP tumors exhibit better clinical outcomes, including superior overall survival. However, the underlying factors responsible for these differential clinical outcomes are still unclear. One can speculate that differences in genomic structure between these two types of gliomas significantly contribute to these outcomes, but in vivo experimental systems that isolate the effects of IDH1/2 mutations will be needed in order to reach this conclusion definitively.
Historically, muIDH1/2/CIMP gliomas have been difficult to study for several reasons. First and foremost, muIDH1/2 glioma cell lines are rare. muIDH1/2 glioma cells are very difficult to culture in vitro, and forced overexpression of muIDH1/2 does not necessarily reflect the biology of these tumors (Piaskowski et al. 2011). Also, muIDH1/2 glioma cells have differential activation and deactivation of metabolism, and this phenotype may hinder proliferation of tumor cell growth in vitro (Tateishi et al. 2015; Cui et al. 2016). In addition, IDH1/2 mutations themselves do not drive the formation of gliomas. Our results showing a lack of glioma formation from IDH1 mutations alone or in combination with p53 or Ink4a/Arf loss are consistent with other efforts using viral gene transduction. Furthermore, transgenic expression of IDH1 mutations in the brain leads to defects in vascular and collagen formation but no tumors (Sasaki et al. 2012; Chen et al. 2014). Moreover, computational analysis using the TCGA human glioma data set provides limited information. Whereas the TCGA human glioma data set is useful for identifying genetic alterations between the two glioma subtypes, there is insufficient resolution to study particular components in the tumor microenvironment, such as stromal cells and immune infiltrates. Therefore, in order to study the specific biological impact of IDH1 mutation in gliomas, appropriate isogenic wtIDH1 and muIDH1 in vivo tumor models are required to rule out large genetic variations between wtIDH1/non-CIMP and muIDH1/CIMP gliomas.
PDGF is a significant component of gliomagenesis in humans and is the strongest driver of glioma formation in mice using RCAS/tva technology. In non-CIMP human gliomas, the earliest event in tumor generation is chromosomal nondisjunction, which results in gain of Ch7 (driven largely, but not entirely, by resultant PDGFA expression) and loss of Ch10 (driven partly by loss of phosphatase and tensin homolog [PTEN]) (Ozawa et al. 2014). CIMP tumors also show evidence of PDGF activation, including overexpression of both ligand and receptors. Therefore, unlike IDH1/2 mutations that show, at best, subtle oncogenic effects, forced PDGF expression provides the ability to create experimental gliomas that share many of the aspects of their human counterparts and are easily genetically modifiable. In our study, we used PDGF-driven gliomas as the base tumor formation and overexpressed IDH1 mutation to alter their phenotypes to CIMP-like gliomas. As shown in previous reports, muIDH1 expression alone is sufficient to establish a hypermethylated phenotype in astrocytes (Turcan et al. 2012). Therefore, concomitant overexpression of muIDH1 with PDGF in vivo should be able to induce muIDH1 gliomas, and we showed these tumors to have a hypermethylated phenotype. The major advantage of this muIDH1 mouse tumor model is the availability of the isotype control model, whose tumor-initiating events are identical to that of muIDH1 gliomas except for muIDH1 mutation expression.
These muIDH1 mouse gliomas showed many similarities to muIDH1 human gliomas, including similar histology, the production of similar levels of 2-HG, increased DNA methylation, and similar gene expression differences relative to wtIDH1 mouse tumors. The results from characterizing the impact of muIDH1 mutations on gene expression indicated that numerous genes associated with extracellular matrix formation, developmental biology, and metabolism were differentially expressed after muIDH1 expression compared with wtIDH1 expression. These biological differences mimic those observed in human muIDH1 gliomas. Comparison of expression profiles identified immune system-associated GO terms, such as leukocyte migration and chemotaxis, as down-regulated in muIDH1 gliomas. In addition, biological experiments using these muIDH1 mouse gliomas also showed an immunologic phenotype similar to that seen in muIDH1 human gliomas. These muIDH1 tumors show reduced numbers of many immune cell types, including microglia, macrophages, and PMNs, consistent with reduced production of chemokines in these tumors. These data are consistent with the immune-suppressed phenotype of CIMP gliomas being at least partially a result of IDH1 mutations and are also consistent with the longer survival time of patients with CIMP tumors compared with non-CIMP tumors, in part due to the IDH1 mutation itself and its effect on the immune component of the glioma.
This mouse model of IDH1 mutant gliomas will likely be useful for studying many aspects of CIMP glioma biology, since this isogenic IDH1 tumor pair reflects tumor biology altered by muIDH1 rather than any other genetic variations such as 1p19q codeletion and ch7+/ch10− loss. For instance, we observed that genes associated with extracellular matrix modification and formation (including those that encode collagens and integrins) were the most down-regulated in muIDH1 mouse tumors. As shown in this study, immune infiltrates and chemotaxis are regulated by IDH1 mutations. However, the complex genomic gains and losses seen in muIDH1 human astrocytomas and the characteristic loss of 1/19q observed in muIDH1 human oligodendrogliomas (The Cancer Genome Atlas Research Network 2015; Eckel-Passow et al. 2015; Suzuki et al. 2015; Ceccarelli et al. 2016) have not been recapitulated in any mouse model to date. This mouse model will also be useful to investigate the effect of specific inhibitors of muIDH1 on tumor progression. A recent study expressing wtIDH1 and muIDH1 in syngeneic glioma models has shown that the inhibition of muIDH1 using a specific inhibitor reversed the muIDH1 immune suppression (Kohanbash et al. 2017).
Immunotherapy is showing great potential in many cancer types, and there are currently several ongoing trials of immunotherapy strategies in gliomas. It is becoming clear that the immunologic state of all cancers, including gliomas, needs to be taken into account, and their complexities need to be understood in detail. It is possible that the increased myeloid cell content in non-CIMP gliomas contributes to lymphocyte suppression via myeloid-derived suppressor cell activity (Gabrilovich et al. 2012). Novel therapeutic strategies combining immune checkpoint blockade with myeloid cell depletion may prove effective for gliomas. Additionally, tumor-associated neutrophils are capable of promoting tumor growth through other mechanisms, including enhanced angiogenesis, matrix destruction, and increased tumor cell proliferation (Nozawa et al. 2006; Houghton et al. 2010; Coffelt et al. 2016). Here we show that systemic depletion of neutrophils slows down disease progression in wtIDH1 tumors but has no further effect on the progression of muIDH1 tumors, which already attract a lower number of neutrophils at basal levels. In addition, it will be important to study the effect of inhibitors of macrophage function (such as CSF1R antagonists) on wtIDH1 and muIDH1 tumors, and further studies will be required to clarify the specific roles of myeloid lineage cells in this context.
Materials and methods
Mouse brain tumors
Ntva_Ink4a/Arf+/+, Ntva_Ink4a-Arf+/−, and Ntva_Ink4a/Arf−/− mice were used to generate mouse gliomas. The genetic backgrounds of tva mice were FVB/N, C57BL6, BALB/C, and 129. To generate wtIDH1-expressing and muIDH1-expressing mouse gliomas, we used the RCAS/tva system as described previously (Holland and Varmus 1998; Holland et al. 1998; Shih and Holland 2006; Ozawa et al. 2014). Briefly, DF1 cells were maintained with 10% fetal bovine serum (FBS) in Dulbecco's modified Eagle medium (DMEM). Chicken fibroblasts were transfected with each RCAS viral plasmid using Fugene 6 transfection reagent (Roche) following the manufacturer's protocol. Next, PDGFa-expressing DF1 cells were mixed with either wtIDH1-shp53-expressing or muIDH1-shp53-expressing DF1 cells. These mixed DF1 cells were injected into Ntva_Ink4a/Arf+/+, Ntva_Ink4a–Arf+/−, and Ntva_ Ink4a/Arf−/− mice. Mice were monitored daily until they developed signs of illness, such as lethargy, poor grooming, weight loss, dehydration, macrocephaly, seizure, jumping, and/or paralysis. Kaplan-Meier analysis demonstrating symptom-free survival in murine gliomas was performed using the log-rank tests in Prism 6 software (GraphPad). Log rank P-values were measured with the Mantle-Cox test. A value of P < 0.05 was considered significant in this study.
Human LGG analysis
RNA sequencing raw counts and clinical information for 667 human LGGs were obtained using R/Bioconductor (http://genomebiology.com/content/5/10/R80) package AnnotationHub (version 2.3.16; https://bioconductor.org/packages/release/bioc/html/AnnotationHub.html). A total of 513 human LGGs for which the IDH1 mutation status was identified (using clinical variable column “IDH1 mutation found”) were used for further analysis. The CIMP/non-CIMP status of the 417 human LGGs was determined using Bolouri et al. (2016). Only those samples that were “muIDH1/CIMP” and “wtIDH1/non-CIMP” were considered for subsequent analyses.
Differentially expressed genes were determined between the muIDH1/CIMP (n = 417) and wtIDH1/non-CIMP (n = 91) human LGGs using DESeq2 (version 1.11.14; http://www.genomebiology.com/2014/15/12/550) with a cutoff of false discovery rate (FDR) <0.05 and fold change >2. GO-based enrichment tests were implemented using GOseq (version 1.23.0; http://www.genomebiology.com/2010/11/2/R14), which corrects for gene length bias. GO terms with corrected P-values <0.05 were considered significantly enriched (q-value <0.05). Up-regulated genes from various lists were enriched for Reactome and KEGG (Kyoto Encyclopedia of Genes and Genomes) Pathways using R/Bioconductor package ReactomePA (version 1.15.4) (Yu and He 2016) and clusterProfiler (version 2.5.4) (Yu et al. 2012), respectively.
2-HG measurements
2-HG was measured by the following procedure (Rakheja et al. 2011). Tumor tissues were washed with cold PBS, and cellular metabolism was quenched with ice-cold 80:20 MeOH:H2O. Samples were vortexed and either stored at −80°C or processed immediately. Cell suspensions were sonicated on ice and then centrifuged at 13,000 rpm for 10 min at 4°C. The supernatant was transferred to a new tube, and an internal standard (deuterated α-ketoglutarate; Sigma, D,L-2HG-D6) was added. Samples were lyophilized followed by derivatization in 50 µL of diacetyl-L-tartaric anhydride and 50 mg/mL methylene chloride:acetic acid (80:20) with incubation for 30 min at 75°C. After cooling at room temperature, the dry residue was dissolved in 0.5 mL of H2O. Samples were centrifuged again, and supernatants were analyzed by mass spectrometry. D-2HG and L-2HG standards (Sigma) ranging from 10 nM to 10 pM were used to create standard curves and quantify 2-HG in biological samples. The 2-HG values were normalized to protein concentration per sample.
RRBS
The RRBS method was modified from the previous protocol (Meissner et al. 2005). A 500-ng sample of DNA was digested overnight with MspI (New England Biolabs). The entire digestion reaction was then input directly into the KAPA hyperlibrary preparation protocol (KAPA Biosystems) and prepared through the adapter ligation step using SeqCap adapters (Roche-Nimblegen) and a 2.5× post-ligation Agencourt AMPure XP bead cleanup (Beckman Coulter). The adapter-ligated DNA was then size-selected using a size range of 160–340 base pairs (bp) on a 2% Pippin preparation gel (Sage Science) followed by bisulfite conversion using the Zymo EZ DNA Methylation Lightning kit with a 20-µL elution volume (Zymo Research). The converted DNA was then put back into the KAPA hyperlibrary preparation protocol, resuming at the library amplification step with some minor modifications. Specifically, amplification was performed with 19 PCR cycles using 2× KAPA Hifi HotStart Uracil+ ReadyMix (KAPA Biosystems). Post-amplification cleanup was performed with 0.8× Agencourt AMPure XP beads. Library size distributions were validated using the Agilent High-Sensitivity D1000 ScreenTape run on an Agilent 2200 TapeStation (Agilent Technologies, Inc.). Additional library quality control, blending of pooled indexed libraries, and cluster optimization were performed using Life Technologies-Invitrogen Qubit 2.0 Fluorometer (Life Technologies-Invitrogen). A pool comprised of eight libraries was sequenced over two lanes of an Illumina HiSeq 2500 run in Rapid Mode and using a single-read 67-bp (SR67) strategy. Image analysis and base calling were performed using Illumina's real-time analysis software version 1.18 followed by “demultiplexing” of indexed reads and generation of FastQ files using Illumina's bcl2fastq conversion software version 1.8.4 (http://support.illumina.com/downloads/bcl2fastq_conversion_software_184.html). Trim Galore version 0.3.7 was used for base call quality and adapter trimming of the reads followed by alignment to mm9 using Bimark version 0.13.11(Krueger and Andrews 2011). Methylation calls and differential methylation analyses were performed using the R package methylKit version 0.9.2 (Akalin et al. 2012).
Tissue processing
Tumor-bearing mouse brains were removed, fixed in 10% neutral-buffered formalin, and then embedded into paraffin blocks. The paraffin-embedded specimens were serially sectioned 5 µm in depth and slide-mounted. H&E staining was performed.
Immunohistochemistry
Automated stain processing (Discovery; Ventana Medical Systems, Inc.) was used for immunohistochemical detection with the manufacturer's standard protocol.
Tumor DNA analysis using PCR
RCAS vectors inserted with either wtIDH1 or muIDH1 used as controls. For an analysis of RCAS–DNA integration, three tumor groups were used: control, wtIDH1, and muIDH1 tumors. DNA was extracted from mouse tumor tissues using the DNeasy blood and tissue kit (Qiagen). PCR amplification was performed with IDH1-F (5′-ATGTCCAAAAAAATCAGTGGCGGTTCT-3′), IDH1-R (5′-TTAAAGTTTGGCCTGAGCTAGTTTGATC-3′), wtIDH1 or muIDH1 cDNA, and Taq DNA polymerase (Invitrogen, 10342020) on a MJ Research PTC-200 ThermoCycler. PCR conditions were 5 min at 94°C, denaturation for 45 sec at 94°C, annealing for 30 sec at 55°C, and extension for 90 sec at 72°C for 40 cycles followed by 10 min of final extension. PCR-amplified DNA was visualized using 1% agarose gel and then extracted using the QIAquick gel extraction kit (Qiagen, 28704) for capillary sequencing with the IDH1 sequencing primer (5′-CGGTCTTCAGAGAAGCCATT-3′). The PCR products and pENTR vector were incubated with PacI and NotI enzymes and purified using gel electrophoresis followed by ligation to insert wtIDH1 or muIDH1 cDNA into the pENTR vector. Gateway cloning was then performed to transfer the insert to RCAS vectors.
Human glioma tissue processing for FACS
Human glioma tissues were collected in accordance with the University of Pittsburgh Institutional Review Board. Freshly resected tumors from muIDH1 or wtIDH1 glioma patients were mechanically dissociated with Accutase (Sigma Aldrich) to form a single-cell suspension. The single-cell suspension was then loaded onto a Percoll gradient (Sigma Aldrich) and centrifuged at 2250 rpm for 20 min. Cells present in the Percoll interphase were collected and analyzed by flow cytometry. Staining was performed for the following markers: CD45-PECy7, CD3-APCCy7, CD4-PerCP-Cy5.5, CD8-FITC, CD25-BV421, FoxP3-PE, CD127-APC, CD20- AF700, CD11b-APCCy7, CD80-PECy7, CD86-BV421, CD163-APC, CD14-FITC, CD15-BV711, and CD11c-PerCP-Cy5.5. All antibodies were purchased from BD Biosciences. Cells were analyzed on a BD LSR II flow cytometer, and post-acquisition analysis was performed on FlowJo.
Mouse glioma tissue processing for FACS
Animals were sacrificed, and their brains were removed from the skull immediately after cardiac perfusion with 10 mL of PBS. Mouse brains were mechanically dissociated and subsequently gently forced through a 70-µm strainer followed by rinsing with 50 mL of Hank's balanced salt solution (HBSS) + 10% FBS. Cells were pelleted by centrifugation (500g for 10 min at 4°C) and immediately resuspended in RBC lysis buffer (5 mL per brain, 5 min on ice). RBC lysis was quenched by adding 3 vol of HBSS + 10% FBS, and cells were centrifuged at 500g for 10 min at 4°C. To remove myelin, cells were resuspended in 10 mL of 30% Percoll (GE Healthcare) and centrifuged at 700g for 10 min at room temperature without break. The myelin layer at the top was removed, and the cell pellet was resuspended in HBSS + 10%FBS, run through a 40-µm cell strainer, and centrifuged at 500g for 10 min at 4°C. The cell pellet was resuspended in cold PBS + 2% FBS to perform cell count and FACS staining.
Single-cell suspensions were incubated with mouse TruStain FcX prior to 30 min of immunostaining on ice using fluorochrome-conjugated antibodies (Supplemental Table S5). Dead cells were excluded with fixable viability dye (FVD) eFluor 780 (eBioscience) following the manufacturer's instructions. Stained cells with dye-conjugated antibodies listed in Supplemental Table S5 were washed, fixed with IC fixation buffer (eBioscience), and stored at 4°C until analysis. Samples were analyzed on a LSR II flow cytometer with FACSDiva software (BD). Compensation and analysis were performed with FlowJo software (TreeStar), and gates were defined by fluorescence minus one (FMO) controls.
RT–PCR (qPCR)
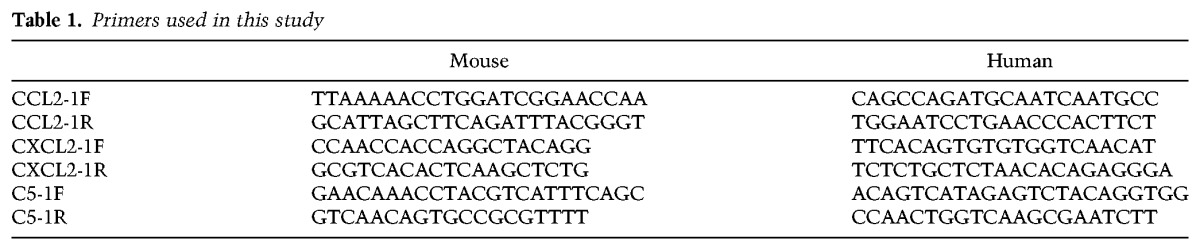
Total RNA was extracted from tumor tissues using RNeasy minikits (Qiagen) and was used to synthesize cDNA by using the SuperScript first strand synthesis system for RT–PCR (Invitrogen, 11904018) according to the manufacturer's protocol. SYBR Green real-time PCR was performed using primer sets, reagents, and protocols from Applied Biosystems in a 7900 HT Fast real-time PCR system (Applied Biosystems). Each sample was analyzed in quadruplicate. All primers used are shown in Table 1.
Table 1.
Primers used in this study
Antibodies for Western blotting and immunohistochemistry
The following primary antibodies were used for Western blots and immunohistochemical staining: Myc tag (Cell Signaling, 2272), PDGFA (Santa Cruz Biotechnology, sc-128), β-Actin (Sigma Aldrich, A1978), α-tubulin (Sigma Aldrich, T5168), wtIDH1 (Histobiotech, DIA-W09), muIDH1 (Histobiotech, DIA-H09), anti-Flag (Sigma Aldrich, F7425), anti-5-mC (Abcam, ab10805), and Olig2 (Millipore, AB9610).
Neutrophil isolation
Mice were injected with 4% sterile thioglycolate intraperitoneally. After 4 h, mice were injected with 4% sterile thioglycolate (Sigma-Aldrich, B2551) intraperitoneally. After 4 h, mice were euthanized, and the peritoneal space was flushed twice with 10 mL of sterile PBS to harvest neutrophils. Cells were washed in PBS and counted with a hematocytometer.
Preparation of protein homogenates
Brain tissue homogenates were prepared from frozen tissues from tumor-bearing and non-tumor-bearing mice. Brain tissue was minced, resuspended in RPMI-1640, and disintegrated using a tissue homogenizer. Homogenates were centrifuged at 5000 rpm for 5 min at 4°C, and the supernatant was collected.
Neutrophil depletion
For neutrophil depletion, tumor-bearing mice received intraperitoneal injections of 500 µg of 1A8 (Ly6g depletion) or 2A3 (control; both from Bio X Cell) antibody per mouse starting from day 21 after DF1 cell injection. Injections were given three times per week for 5 wk. Mice were monitored for signs of disease progression as described above.
MicroBoyden chamber chemotaxis assays
Neutrophil migration was evaluated using a 48-well microBoyden chamber (NeuroProbe) with a 5-µm pore size polycarbonate filter. Brain homogenate (2.5 µg and 30 µL) was added to the bottom wells, and neutrophils resuspended in 50 µL of RPMI-1640 (3 × 106 cells per milliliter) were added to the upper chamber and incubated for 1 h at 37°C. In place of the tumor homogenate, conditioned medium from tumor-derived cells was used overnight. Subsequently, cells were removed from the upper well, and the upper side of the filter was washed to remove nonmigrated cells. The migrated cells on the bottom side of the filter were fixed in ice-cold methanol and stained using Hema 3 (Thermo Fisher Scientific). The chemotactic index was defined as the number of migrated cells toward protein homogenates from tumor-bearing mice divided by the number of cells migrated toward protein homogenates prepared from normal brains.
Cytokine arrays
To screen cytokine expression, a Proteome Profiler mouse cytokine array kit, panel A (R&D, ARY006) was used following the manufacturer's protocol.
RNA expression analysis
Total RNA was extracted from tumor tissues using RNeasy minikits (Qiagen). Total RNA integrity was checked using an Agilent 2200 TapeStation (Agilent Technologies, Inc.) and quantified using a Trinean DropSense96 spectrophotometer (Caliper Life Sciences). High-quality RNA samples were converted to cDNA and biotin labeled for microarray analysis using Ambion's Illumina TotalPrep RNA amplification kit (Life Technologies). Labeled cRNAs were processed on a MouseWG-6v2 Expression BeadChip (Illumina, Inc.) and imaged using an Illumina iScan system.
Microarray data were assessed for quality followed by quantile normalization using the Bioconductor package lumi (Du et al. 2008). The data set was filtered initially by flagging probes that fell below a signal “noise floor,” which was established using the 75th percentile of the negative control probe signals within each array, retaining only those probes that were above threshold in all samples within a single group for a given comparison. We subsequently filtered the data set by using a variance filter using the “shorth” function of the Bioconductor package genefilter. Statistical analyses was performed using the Bioconductor package limma (Smyth 2005), and a FDR method was applied to correct for multiple testing (Reiner et al. 2003). Differential expression was defined as |log2 (ratio)|> = 0.585 (±1.5-fold) with the FDR set to 5%.
Statistical analyses
For the classification of TCGA samples into muIDH1/CIMP and wtIDH1/non-CIMP gliomas, we extracted sample data from Supplemental Table S1 from Ceccarelli et al. (2016). For the CIMP/non-CIMP classification, we used Bolouri et al. (2016).
Kaplan-Meier survival curves were prepared using GraphPad Prism 6 (GraphPad Software) and analyzed using log-rank (Mantel-Cox) tests.
GO analysis was conducted on differentially expressed genes between the wtIDH1 and muIDH1 groups using the R/Bioconductor package “goseq.” The REVIGO (Supek et al. 2011) Web interface was used to reduce and visualize these GO categories in a two-dimensional space as a scatter plot, derived by applying multidimensional scaling to a matrix of the GO terms’ semantic similarities. PANTHER (Mi et al. 2013) was used for analyzing gene lists of down-regulated genes in human and mouse muIDH1 tumors for overrepresented GO terms.
Supplementary Material
Acknowledgments
We thank C. Sather and A. Marty of the Fred Hutchinson Cancer Research Center Genomics Shared Resource for assistance with the RRBS assay, and R. Basom of the Fred Hutchinson Cancer Research Center Bioinformatics Shared Resource for assistance with next-generation sequencing data analysis. This work was supported by National Cancer Institute grants U54CA163167, R01CA195718, R01CA60882, and U54CA19346 (to E.C.H.).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.294991.116.
References
- Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE. 2012. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 13: R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badie B, Schartner JM. 2000. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery 46: 957–961. [DOI] [PubMed] [Google Scholar]
- Bai H, Harmanci AS, Erson-Omay EZ, Li J, Coskun S, Simon M, Krischek B, Ozduman K, Omay SB, Sorensen EA, et al. 2016. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet 48: 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolouri H, Zhao LP, Holland EC. 2016. Big data visualization identifies the multidimensional molecular landscape of human gliomas. Proc Natl Acad Sci 113: 5394–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. 2013. The somatic genomic landscape of glioblastoma. Cell 155: 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2015. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372: 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. 2016. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164: 550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Nishimura MC, Kharbanda S, Peale F, Deng Y, Daemen A, Forrest WF, Kwong M, Hedehus M, Hatzivassiliou G, et al. 2014. Hominoid-specific enzyme GLUD2 promotes growth of IDH1R132H glioma. Proc Natl Acad Sci 111: 14217–14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffelt SB, Wellenstein MD, de Visser KE. 2016. Neutrophils in cancer: neutral no more. Nat Rev Cancer 16: 431–446. [DOI] [PubMed] [Google Scholar]
- Cui D, Ren J, Shi J, Feng L, Wang K, Zeng T, Jin Y, Gao L. 2016. R132H mutation in IDH1 gene reduces proliferation, cell survival and invasion of human glioma by downregulating Wnt/β-catenin signaling. Int J Biochem Cell Biol 73: 72–81. [DOI] [PubMed] [Google Scholar]
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. 2001. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev 15: 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. 2009. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462: 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Kibbe WA, Lin SM. 2008. lumi: a pipeline for processing Illumina microarray. Bioinformatics 24: 1547–1548. [DOI] [PubMed] [Google Scholar]
- Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV, et al. 2015. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med 372: 2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE. 2016. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529: 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. 2012. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12: 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. 2009. Modeling adult gliomas using RCAS/t-va technology. Transl Oncol 2: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann C, Hentschel B, Wick W, Capper D, Felsberg J, Simon M, Westphal M, Schackert G, Meyermann R, Pietsch T, et al. 2010. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 120: 707–718. [DOI] [PubMed] [Google Scholar]
- Holland EC, Varmus HE. 1998. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc Natl Acad Sci 95: 1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EC, Hively WP, DePinho RA, Varmus HE. 1998. A constitutively active epidermal growth factor receptor cooperates with disruption of G(1) cell-cycle arrest pathways to induce glioma-like lesions in mice. Gene Dev 12: 3675–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, Stolz DB, Land SR, Marconcini LA, Kliment CR, et al. 2010. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med 16: 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Qian BZ, Pollard JW. 2015. Immune cell promotion of metastasis. Nat Rev Immunol 15: 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, Chheda ZS, Downey KM, Watchmaker PB, Beppler C, et al. 2017. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest 127: 1425–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong LY, Wu AS, Doucette T, Wei J, Priebe W, Fuller GN, Qiao W, Sawaya R, Rao G, Heimberger AB. 2010. Intratumoral mediated immunosuppression is prognostic in genetically engineered murine models of glioma and correlates to immunotherapeutic responses. Clin Cancer Res 16: 5722–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. 2005. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 33: 5868–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi HY, Muruganujan A, Casagrande JT, Thomas PD. 2013. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc 8: 1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. 2010. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17: 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa H, Chiu C, Hanahan D. 2006. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci 103: 12493–12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC. 2014. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 26: 288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piaskowski S, Bienkowski M, Stoczynska-Fidelus E, Stawski R, Sieruta M, Szybka M, Papierz W, Wolanczyk M, Jaskolski DJ, Liberski PP, et al. 2011. Glioma cells showing IDH1 mutation cannot be propagated in standard cell culture conditions. Brit J Cancer 104: 968–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. 2013. Microenvironmental regulation of tumor progression and metastasis. Nat Med 19: 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakheja D, Mitui M, Boriack RL, DeBerardinis RJ. 2011. Isocitrate dehydrogenase 1/2 mutational analyses and 2-hydroxyglutarate measurements in Wilms tumors. Pediatr Blood Cancer 56: 379–383. [DOI] [PubMed] [Google Scholar]
- Reiner A, Yekutieli D, Benjamini Y. 2003. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19: 368–375. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, Cairns RA, McCracken S, Wakeham A, Haight J, et al. 2012. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev 26: 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalapour S, Karin M. 2015. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest 125: 3347–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH, Holland EC. 2006. Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer Lett 232: 139–147. [DOI] [PubMed] [Google Scholar]
- Smyth GK. 2005. Limma: linear models for microarray data. In Bioinformatics and computational biology solutions using R and Bioconductor (ed. Gentleman VCR, et al. ), pp. 397–420. Springer, New York. [Google Scholar]
- Supek F, Bosnjak M, Skunca N, Smuc T. 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. Plos One 6: e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, Shimamura T, Niida A, Motomura K, Ohka F, et al. 2015. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet 47: 458–468. [DOI] [PubMed] [Google Scholar]
- Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebe F, Lelic N, Wiederschain D, Bedel O, Deng GJ, Zhang BL, et al. 2015. Extreme vulnerability of IDH1 mutant cancers to NAD plus depletion. Cancer Cell 28: 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al. 2012. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483: 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC. 2002. Ink4a–Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res 62: 5551–5558. [PubMed] [Google Scholar]
- Yu GC, He QY. 2016. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol Biosyst 12: 477–479. [DOI] [PubMed] [Google Scholar]
- Yu GC, Wang LG, Han YY, He QY. 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16: 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Rao A, Sette P, Deibert C, Pomerantz A, Kim WJ, Kohanbash G, Chang Y, Park Y, Engh J, et al. 2016. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol 18: 1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.