

In this study, Ho et al. research the mechanism by which TDG-dependent DNA demethylation occurs in a rapid and site-specific manner. Their findings demonstrate two distinct but complementary mechanisms by which XPC influences gene regulation by coordinating efficient TDG-mediated DNA demethylation along with active transcription during somatic cell reprogramming.
Keywords: DNA methylation, epigenetics, pluripotency, reprogramming
Abstract
Faithful resetting of the epigenetic memory of a somatic cell to a pluripotent state during cellular reprogramming requires DNA methylation to silence somatic gene expression and dynamic DNA demethylation to activate pluripotency gene transcription. The removal of methylated cytosines requires the base excision repair enzyme TDG, but the mechanism by which TDG-dependent DNA demethylation occurs in a rapid and site-specific manner remains unclear. Here we show that the XPC DNA repair complex is a potent accelerator of global and locus-specific DNA demethylation in somatic and pluripotent stem cells. XPC cooperates with TDG genome-wide to stimulate the turnover of essential intermediates by overcoming slow TDG–abasic product dissociation during active DNA demethylation. We further establish that DNA demethylation induced by XPC expression in somatic cells overcomes an early epigenetic barrier in cellular reprogramming and facilitates the generation of more robust induced pluripotent stem cells, characterized by enhanced pluripotency-associated gene expression and self-renewal capacity. Taken together with our previous studies establishing the XPC complex as a transcriptional coactivator, our findings underscore two distinct but complementary mechanisms by which XPC influences gene regulation by coordinating efficient TDG-mediated DNA demethylation along with active transcription during somatic cell reprogramming.
DNA methylation, specifically 5-methylcytosine (5mC), is highly correlated with gene silencing and is an essential epigenetic regulator of transcription that must be both faithfully maintained and dynamically regulated throughout development (Bird 2002; Smith et al. 2014). Similar to the wholesale epigenetic reprogramming that occurs during zygotic fertilization on paternal DNA and during primordial germ cell specification, somatic cell reprogramming requires global and locus-specific removal of somatic DNA methylation in order to reactivate the transcriptional network for pluripotency (Reik 2001). Indeed, efficient removal of 5mC at gene regulatory elements is essential for the proper resetting of the “epigenetic memory” of a somatic cell into a pluripotent state, and failure to do so compromises the fidelity of the resulting induced pluripotent stem cells (iPSCs) (De Carvalho et al. 2010; Kim et al. 2010; Polo et al. 2010; Ohi et al. 2011).
Despite several decades of research, the precise mechanism underlying DNA demethylation remains elusive. It is generally accepted that mammalian DNA demethylation is a multistep process initiated by the deamination and/or oxidation of 5mC. Spontaneous or enzymatic deamination of 5mC produces thymine (T), which can be further oxidized to 5-hydroxymethyluracil (5hmU) by the TET family of hydroxylases (Pfaffeneder et al. 2014). However, TETs can also catalyze the iterative oxidation of 5mC to generate 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and finally 5-carboxylcytosine (5caC) (Tahiliani et al. 2009; Ito et al. 2010, 2011; He et al. 2011). These structurally diverse intermediates can subsequently be repaired through the base excision repair (BER) pathway.
Although the pathways through which 5mCs can be actively removed appear to be quite diverse, current prevailing models favor the repair by thymine DNA glycosylase (TDG), which can catalyze the base excision of all known derivatives of 5mC; namely T, 5hmU, 5fC, and 5caC (Neddermann et al. 1996; He et al. 2011; Maiti and Drohat 2011; Hashimoto et al. 2012). TDG is the only member of the uracil N-glycosylase family whose knockout leads to embryonic lethality due to developmental defects and associated aberrant DNA methylation patterns (Cortázar et al. 2011; Cortellino et al. 2011). Consistent with TDG as the predominant enzyme responsible for oxidative DNA demethylation, TDG-deficient mouse embryonic stem cells (ESCs) show a marked accumulation of 5fC and 5caC at gene regulatory elements genome-wide, underscoring that DNA methylation status is a balancing act in which clearance of 5mCs must be actively maintained to counteract DNA methylase activities in PSCs (Shen et al. 2013). Furthermore, TETs are required for global erasure of DNA methylation during zygotic fertilization and somatic cell reprogramming, indicating that the oxidation of 5mC by TETs and the ensuing repair by TDG/BER is a key enzymatic mechanism of DNA demethylation during development (Ito et al. 2010; Gu et al. 2011; Hu et al. 2014).
However, biochemical studies of TDG kinetics have shown that, following cleavage of a mismatched base, TDG binds the abasic site with unusually high affinity. This results in a slow dissociation rate of TDG, leading to a binding half-life of 10 h in vitro that effectively consigns TDG to operate as a single-turnover enzyme (Waters and Swann 1998; Waters et al. 1999). This remarkably stable interaction suggests that there must exist mechanisms to increase substrate turnover by TDG in vivo in a manner that is compatible with the fast kinetics associated with active DNA demethylation. Indeed, a number of studies have implicated DNA repair factors such as APE1, NEIL1/2, and XPC in promoting TDG activity in vitro (Waters et al. 1999; Shimizu et al. 2003; Schomacher et al. 2016). However, these studies did not address the fundamental mechanism by which these factors stimulate base excision by TDG, a catalytic process that can be broken down into three core steps: substrate recognition, base cleavage, and product dissociation. Furthermore, it is unknown whether these factors can stimulate the excision of not just classical mismatch base pairs such as T:G that can result from DNA damage but substrates that are structurally distinct and more relevant to the DNA demethylation pathway; namely, 5fC:G and 5caC:G. More critically, the relevance of these putative cofactors in TDG-dependent DNA demethylation in vivo has not been tested (Waters et al. 1999; Shimizu et al. 2003).
Depletion of TDG in ESCs leads to ectopic 5fC and 5caC signals at active gene enhancers bound by pluripotency transcription factors, suggesting that, despite a lack of sequence specificity, TDG may be recruited to these regulatory sites to maintain a hypomethylated state by an unknown mechanism. Interestingly, the XPC DNA repair complex, a DNA damage sensor in nucleotide excision repair (Nemzow et al. 2015), was shown recently to function as a critical transcriptional coactivator for stem cell-specific transcription factors OCT4 and SOX2 (Fong et al. 2011). Similar to TDG, the XPC complex occupies both distal enhancers and promoters that are also bound by OCT4, SOX2, and potentially other transcription factors expressed in ESCs (Cattoglio et al. 2015). Taken together, these data raise the intriguing possibility that the XPC complex, in addition to acting as a classical transcriptional coactivator, can regulate transcription by cooperating with TDG to stimulate active DNA demethylation at regulatory regions across the genome in PSCs.
Here, we report an unexpected role of the XPC complex in influencing the epigenetic landscape of 5mC in human dermal fibroblasts (HDFs) and PSCs independent of XPC's DNA repair activity. Using complementary in vivo single-molecule imaging and in vitro biochemical techniques, we showed that XPC promotes TDG activity by reducing product inhibition of TDG, thereby increasing substrate turnover. ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) and MeDIP-seq (methylated DNA immunoprecipitation [MeDIP] combined with high-throughput sequencing) analyses revealed that XPC colocalizes extensively with TDG at gene promoters and regulatory elements. This TDG–XPC association promotes locus-specific DNA demethylation genome-wide in XPC gain-of-function somatic cells and PSCs. Furthermore, we demonstrated that ectopic expression of XPC during somatic cell reprogramming significantly improves the quality of the resulting iPSCs. In short, XPC operates as a reprogramming facilitator by overcoming an early epigenetic barrier through preferential DNA demethylation in HDFs. Our findings thus suggest that the coordinated action of XPC and TDG in active DNA demethylation may be crucial for not only improving the efficiency and quality of somatic cell reprogramming but also understanding the dynamic nature of epigenetic regulation of transcription.
Results
XPC expression affects global DNA methylation independent of its DNA repair activity
To determine whether the XPC complex regulates DNA demethylation, we first manipulated XPC expression in both human somatic cells and ESCs and measured the amount of global 5mC. XPC loss of function through shRNA-mediated knockdown in H9 human ESCs (Fig. 1A) and HDFs (Supplemental Fig. S1A,B) and the Cas9-mediated knockout of XPC in H9 ESCs (Supplemental Fig. S1C,D) led to an increase in global 5mC, as determined by a 5mC-specific ELISA. This global increase of 5mC is consistent with a previous report using siRNAs against XPC in HeLa cells (Le May et al. 2010). We also confirmed these results in mouse ESCs, suggesting that this is not a human-specific phenomenon but may be a conserved function of XPC in regulating DNA demethylation across multiple mammalian cell types (Supplemental Fig. S1E–H).
Figure 1.

Global DNA methylation is inversely correlated with XPC expression independent of DNA repair activity. Relative global DNA methylation was assayed by 5mC-specific ELISA using genomic DNA from XPC knockdown H9 human ESCs (A) and HDFs overexpressing wild-type (WT) or DNA repair-deficient (W690S) human XPC (B). Relative global DNA methylation in HDFs was also assayed by 5mC dot blot (C) and MeDIP enrichment (D). Methylene blue (MB) staining was used to control for total DNA transferred to the membrane. (E,F) TDG cleavage activity of a 5′-labeled 37mer double-stranded oligonucleotide DNA (0.2 µM) in the presence or absence of decreasing amounts of wild-type or W690S mutant XPC (0.2–0.4 µM). The 37mer dsDNA contains either a 5fC:G (E) or 5caC:G (F) base pair as substrate for TDG. Uncleaved intact 37mer (U) and its cleaved product (C) were separated on a denaturing PAGE gel. Representative gels are shown. The intensity of the cleaved product indicates the efficiency of base excision by TDG and is calculated in the graphs as the percentage of total labeled substrate [=C/(U + C)]. Error bars represent the standard deviation. n = 3. (***) P < 0.001; (**) P < 0.01; (*) P < 0.05, calculated by two-way ANOVA.
Remarkably, overexpression of the XPC complex (XPC–RAD23B–CETN2) or the XPC subunit alone led to a dramatic decrease in global 5mC when assayed by ELISA, dot blot, and MeDIP using an antibody specific for 5mC (Fig. 1B–D). Since the ectopic expression of the XPC subunit alone is sufficient to reduce global 5mC similar to that of the heterotrimeric complex and since overexpressed RAD23B and CETN2 subunits have no effect on their own (Fig. 1B; Supplemental Fig. S1I), XPC is likely the active subunit for promoting DNA demethylation. More importantly, we observed a similar reduction in global 5mC levels even when a DNA-binding-impaired and repair-defective mutant of XPC identified in a xeroderma pigmentosum patient (XPC W690S) was overexpressed in HDFs (Fig. 1B,C; Bunick et al. 2006; Maillard et al. 2007; Yasuda et al. 2007). Taken together, these results suggest that XPC is limiting in HDFs and that the DNA repair activity of XPC is dispensable and functionally separable from its role in DNA demethylation. We surmise that the slightly less pronounced effect of mutant XPC on DNA demethylation is likely due to the limiting levels at which we were able to overexpress the W690S mutant XPC proteins in HDFs (Supplemental Fig. S1J). This is consistent with previous reports showing that the missense mutation destabilizes XPC (Yasuda et al. 2007). It is worth noting that we did not observe a significant change in doubling time or growth rate of HDFs upon XPC overexpression (Supplemental Fig. S2), suggesting that stimulation of DNA demethylation by XPC is by an active process as opposed to passive, replication-dependent dilution of 5mC content.
To address the in vivo relevance of other putative cofactors implicated in DNA demethylation, such as APE1 and NEIL1/2, we performed analogous gain- and loss-of-function studies in HDFs and measured their global 5mC levels. We focused on APE1 and NEIL2 because we failed to detect NEIL1 expression in HDFs (data not shown). In contrast to what we observed with XPC, we found that acute depletion or overexpression of APE1 or NEIL2 in HDFs did not significantly alter global DNA methylation levels (Supplemental Fig. S3). While we cannot exclude the possibility that APE1 and NEIL proteins may still play some role in regulating DNA demethylation in vivo, it appears to be minor. Our results suggest that global 5mC level is exquisitely sensitive to changes in the expression level of XPC but not APE1 or NEIL2. Collectively, our results uncovered a novel function of the XPC complex as a potent facilitator of DNA demethylation in vivo.
A major pathway for active 5mC demethylation in mammalian cells is mediated by enzymatic oxidation of 5mC and the ensuing removal of these oxidized intermediates by TDG (Cortázar et al. 2007; Kohli and Zhang 2013). To test whether XPC can stimulate TDG-dependent removal of key demethylation intermediates of 5mC (namely, 5fC and 5caC), we performed TDG glycosylase assays in vitro using these substrates with and without purified recombinant XPC complex. We found that XPC is able to stimulate the glycosylase activity of recombinant human TDG on a 5′-labeled doubled-stranded oligonucleotide containing 5fC or 5caC (Fig. 1E,F; Supplemental Fig. S4A). We focused on the 5fC and 5caC substrates, given their importance in TET/TDG-mediated oxidative demethylation, but further showed that the XPC-mediated stimulation of TDG activity is similar across all known 5mC demethylation intermediates (T, 5hmU, 5fC, and 5caC) (Supplemental Fig. S4B,C). Furthermore, the addition of the DNA repair-deficient XPC mutant (XPC W690S) to the TDG glycosylase assays also stimulated base excision by TDG, consistent with our in vivo findings that the DNA repair activity of XPC is not required for promoting DNA demethylation (Fig. 1E,F; Supplemental Fig. S4B,C). These data suggest a model in which XPC promotes DNA demethylation at least in part by directly stimulating TDG glycosylase activity.
Genome-wide analyses reveal extensive colocalization of XPC with TDG in ESCs
TDG knockout in mouse ESCs leads to the accumulation of 5fC and 5caC at proximal and distal regulatory elements (Shen et al. 2013), which are enriched in motifs for hypoxia-inducible factor 1A (HIF1A), ESRRB, OCT4, and SOX2 (Lu et al. 2015). These results suggest that TDG may act with core ESC-specific transcription factors to regulate DNA demethylation at their target genes. Similarly, RAD23B, a subunit of the XPC complex, has been shown to cobind with OCT4 and SOX2 at regulatory elements in mouse ESCs (Fong et al. 2011; Cattoglio et al. 2015). Taken together, these data raise the possibility that XPC and TDG may colocalize in the mouse ESC genome. To evaluate the extent to which XPC and TDG interact in vivo, we compared a recently published biotin–TDG ChIP-seq data set (Neri et al. 2015) with our previously published ChIP-seq data set on endogenous RAD23B in mouse ESCs (Fong et al. 2011; Cattoglio et al. 2015).
Our analysis revealed a striking ∼93% overlap between TDG and RAD23B ChIP-seq peaks as identified by MACS2 (Fig. 2A). Further analysis of the ChIP-seq data sets revealed a strong enrichment of TDG and RAD23B at the transcription start sites (TSSs) of many genes (Fig. 2B). As we predicted, TDG and RAD23B co-occupy enhancer and promoter elements of several pluripotency genes, including Nanog, Oct4 (Pou5f1), and Tcf3, and of housekeeping genes such as Actb (Fig. 2C; Supplemental Fig. S5). Although there is overwhelming evidence suggesting that XPC and TDG act preferentially at promoters and regulatory regions, it is intriguing that two DNA repair proteins with no known sequence specificity would bind in an apparent sequence-specific manner. We speculate that the recruitment of TDG and XPC to gene promoters and enhancers is likely mediated by their interactions with sequence-specific transcription factors (Fong et al. 2011; Cattoglio et al. 2015).
Figure 2.
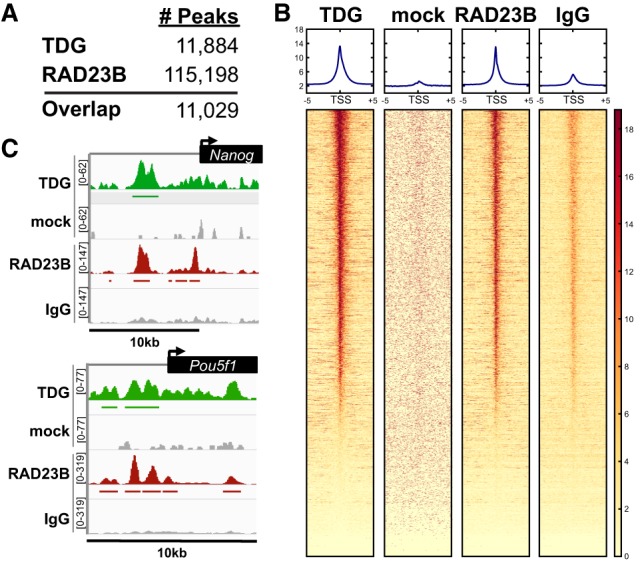
TDG is enriched at enhancers and promoters genome-wide and colocalizes with the XPC complex subunit RAD23B. (A) The number of ChIP-seq peaks identified using MACS2 for each data set. Approximately 93% of biotin–TDG (Neri et al. 2015) ChIP-seq peaks overlap with RAD23B in mouse ESCs. (B) Heat map of TDG- and RAD23B-bound regions over a ±5-kb window around the TSSs of genes. Genes in each heat map are sorted according to descending levels of TDG ChIP-seq signal. (C) Integrative Genomics Viewer (IGV)-computed ChIP-seq tracks are plotted as (number of reads) × [1,000,000/(total read count)] for pluripotency genes Nanog and Pou5f1. Mock and normal IgG were used as specificity controls for the TDG and RAD23B ChIP, respectively.
Proper erasure of DNA methylation in the somatic cell genome during iPSC induction is a major barrier to reactivation of genes essential for reacquisition of pluripotency. The highly dynamic and relatively rapid nature of DNA demethylation makes it particularly sensitive to perturbations in enzymes that catalyze this process. Indeed, both TDG and XPC are required for somatic cell reprogramming (Fong et al. 2011; Hu et al. 2014). The pronounced effect of XPC on reducing 5mC levels and its apparent cobinding with TDG across the genome—particularly at key genes that confer pluripotency—suggest that XPC could help reshape the epigenetic landscape in induced somatic cells conducive for productive reprogramming. We reasoned that early induced, partially reprogrammed iPSCs (pre-iPSCs) would likely be most sensitive to XPC expression because their epigenome is highly malleable and metastable (Stadtfeld et al. 2008). Therefore, to examine the potential impact of XPC expression on DNA demethylation during this critical early phase of reprogramming, we performed MeDIP-seq. MeDIP-seq allows us to specifically profile the 5mC status in XPC gain-of-function HDFs and pre-iPSCs on a genome-wide scale (Down et al. 2008; Bock et al. 2010; Harris et al. 2010). HDFs were transduced with control or XPC-expressing lentiviruses (Fig. 3A). Following selection and expansion of the infected HDFs, we induced reprogramming via episomal vectors expressing OCT4, SOX2, KLF4, L-MYC, LIN28A, GFP, and an shRNA against p53 (Okita et al. 2011). Sequencing libraries were made from either uninduced HDFs or FACS-sorted GFP-positive pre-iPSCs (Supplemental Fig. S6A).
Figure 3.

Ectopic expression of XPC results in loss of DNA methylation genome-wide in both HDFs and pre-iPSCs. (A) Design of MeDIP-seq experiments. Reprogramming factors are cotransfected with a GFP-expressing plasmid to allow for sorting of transfected cells. (B) Up-regulation of XPC expression results in lower enrichment over background and fewer peaks called in MeDIP-seq analyses for both uninduced HDFs (left) and pre-iPSCs (right; 7 d post-induction). The number of peaks identified for each data set using MACS2 is shown below the graphs. (C) Distribution of MeDIP-seq reads by their distance ±5 kb from the TSSs of RefSeq genes, input subtracted. (D) Motif discovery of pre-iPSC MeDIP peaks enriched in the control but not the XPC gain-of-function data set.
In agreement with our bulk methylation experiments (Fig. 1B–D), XPC overexpression results in a loss of DNA methylation in both HDFs and pre-iPSCs. In XPC gain-of-function cells, not only does DNA methylation occur in fewer regions compared with control wild-type cells (29,895 vs. 205,466 in HDFs and 85,456 vs. 230,179 in pre-iPSCs), but overall methylation levels are also lower than in wild-type cells, as shown by reduced MeDIP enrichment over background (Fig. 3B). Moreover, when we analyzed the reads in relation to their distance from the closest TSS, we observed reduced MeDIP enrichment in XPC gain-of-function cells compared with wild-type cells at nearly all regions ±5 kb from the TSS, with the exception of the proximal promoter (±250 base pairs [bp] from the TSS), suggesting that demethylation occurs preferentially at upstream and downstream enhancer elements rather than at the promoter itself (Fig. 3C; Supplemental Fig. S6B). It is also worth noting that the amount of methylated DNA fragments recovered from XPC-overexpressing HDFs, expressed as MeDIP fragment coverage, is remarkably similar to pre-iPSCs ±5 kb from the TSS, suggesting that the methylation levels in XPC gain-of-function HDFs have already been reduced to that of pre-iPSC levels (Fig. 3C). In other words, ectopic expression of XPC alone may create a DNA methylation landscape more favorable toward iPSC generation by lowering the epigenetic threshold for somatic cell reprogramming.
Recent methylome studies have reported dynamic methylation changes occurring preferentially at promoters and regulatory elements of silent and poised genes (Shen et al. 2013; Song et al. 2013; Lu et al. 2015). Although XPC overexpression is sufficient to cause an apparent widespread reduction of 5mC levels in the genome, it remains possible that XPC, by interacting with sequence-specific transcription factors such as OCT4 and SOX2 (Fong et al. 2011; Cattoglio et al. 2015), may also drive local DNA demethylation at specific gene promoters and enhancers to prime gene transcription.
To identify these potential local XPC-driven DNA demethylation events, we performed an unbiased analysis of motifs that are preferentially demethylated upon XPC overexpression in pre-iPSCs. We identified a number of sequence motifs for transcription factors involved in cell cycle regulation, mitogenesis, and metabolism; namely, HIF1, MYC, and CREB1 but, surprisingly, not OCT4 or SOX2 (Fig. 3D; Supplemental Fig. S6C; Supplemental Table S1). It is currently unclear why we failed to detect motif enrichment for OCT4 and SOX2. However, it has been shown that XPC can be recruited to active gene promoters by other transcription factors (Le May et al. 2010). It has also been suggested that the ability of OCT4 and SOX2 to bind their cognate high-affinity target sites normally found in ESCs may be attenuated in HDFs and pre-iPSCs by closed chromatin and the prevalent but sequence-nonspecific interaction of SOX2 with nucleosomal DNA (Soufi et al. 2012). Nonetheless, our identification of specific classes of transcription factor-binding motifs is consistent with our hypothesis that XPC may act on specific gene loci to promote DNA demethylation and their subsequent reactivation during reprogramming. Incidentally, TDG deficiency has been shown to preferentially compromise DNA demethylation at regulatory regions enriched for HIF1 motifs (Lu et al. 2015). Taken together, these observations support the idea that XPC cooperates with TDG to catalyze genome-wide DNA demethylation at specific gene enhancers.
Single-particle tracking (SPT) of TDG shows interaction between TDG and XPC in vivo
To investigate the potential mechanism by which XPC promotes TDG-dependent DNA demethylation in vivo, we used SPT to study the effects of XPC on TDG-binding dynamics to DNA in living cells. We generated the fusion proteins SNAP-tagged XPC (SNAP-XPC) and Halo-tagged TDG (TDG-Halo), two orthogonal protein tags that allow for the covalent addition of bright organic fluorophores to facilitate single-molecule experiments. Both fusion proteins were correctly targeted to the nucleus upon transient transfection into U2OS cells (Supplemental Fig. S7A), which were chosen because they are an ideal cell type for live-cell imaging (Darzacq et al. 2007). The expression levels of XPC, TDG, and the predominant DNA methyltransferase in somatic tissues, DNMT1, in U2OS cells are comparable with HDFs (Supplemental Fig. S7B). In addition, XPC's effect on global DNA demethylation appears to be cell type-independent, thus suggesting a conserved mechanism (Fig. 1; Supplemental Fig. S1; Le May et al. 2010). Given that TDG activity is constrained primarily by its product inhibition and not substrate recognition (Waters and Swann 1998), we hypothesized that XPC could increase TDG activity by promoting substrate turnover and thus decrease TDG DNA-bound residence times.
In order to assess XPC-dependent changes in TDG binding, we first established a stable U2OS cell line with doxycycline-inducible expression of TDG-Halo. Cells were then transfected with a M.SssI-methylated luciferase reporter plasmid containing an SV40 minimal promoter (pGL4.13[Luc2/SV40]) as a substrate for TDG and cotransfected with either SNAP-XPC or the SNAP tag with a nuclear localization signal (SNAP-NLS) as a control. SNAP fusion proteins were then labeled with JF646-SNAP ligand and TDG-Halo with photoactivatable JF549-HaloTag ligand 24 h after transfection, at a time when the transfected cells began to express luciferase, indicating transcriptional reactivation of the reporter plasmid (Supplemental Fig. S7C). In parallel, TDG-Halo-expressing cells were also transduced with lentiviruses containing shRNAs against XPC (shXPC) or a nontargeting (NT) control. Sparse illumination with 405-nm light stochastically activated labeled TDG-Halo molecules, which were subsequently imaged using long exposure times (500 msec). This imaging modality causes bound molecules to appear as a diffraction-limited spot, while fast-moving unbound populations are “blurred out.” It is worth emphasizing that the number of detectable TDG-Halo diffraction-limited spots, also termed trajectories, increased dramatically when cells were transfected with methylated plasmid DNA but not its unmodified counterpart (Supplemental Fig. S7D). This suggests that the overwhelming majority of these trajectories represents the high-affinity interactions between TDG and the newly generated abasic sites on plasmid DNA following base excision. Furthermore, the number of TDG-Halo trajectories detected did not appreciably change as a function of XPC concentration (data not shown), indicating that increasing or decreasing XPC protein levels had no effect on substrate recognition by TDG.
Tracking of individual TDG molecules over consecutive frames allowed us to plot the probability of a long TDG-binding event—thereby generating a “survival curve” for the dwell times of TDG—in either the XPC gain- or loss-of-function background (Fig. 4A; Supplemental Fig. S7E–J; Supplemental Movie S1). Fitting of a two-component exponential decay model to TDG dwell times allowed us to address changes in “specific” and “nonspecific” binding events in response to changes in XPC protein levels in the nucleus. Overexpression of XPC reduced the specific binding time of TDG by nearly twofold (∼31.9 to ∼17.4 sec), while shRNA-mediated knockdown of XPC dramatically increased TDG-specific binding time approximately threefold longer than in control cells (∼33.7 to ∼93.9 sec) (Fig. 4B; Supplemental Fig. S7K). Taken together, these live-cell imaging data are consistent with a model in which XPC increases TDG dynamics by overcoming product inhibition of TDG during oxidative DNA demethylation.
Figure 4.
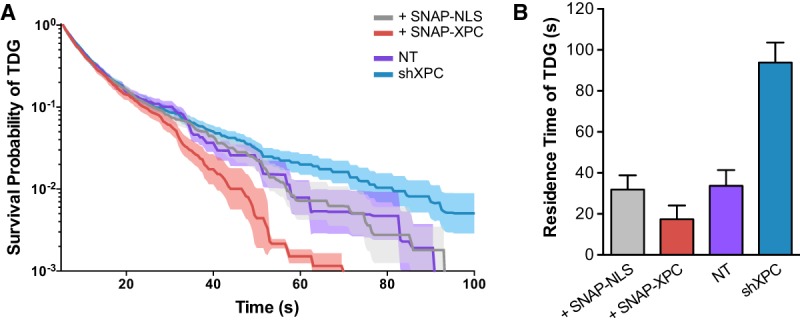
XPC modulates TDG-binding dynamics to DNA in vivo. (A) Survival probability of an individual TDG-Halo molecule being bound as a function of time. Individual molecules were tracked using single-molecule imaging over consecutive frames and binned by their total length in cells cotransfected with SNAP-NLS or SNAP-XPC or transduced with lentiviruses containing a control NT shRNA or shRNA against XPC (shXPC). Bold lines represent the mean probability; shaded regions denote the 95% confidence interval. (B) Quantification of TDG DNA-bound residence time using a two-exponential decay model after correction for photobleaching using Halo-H2B (see Supplemental Fig. S7K). Error bars depict the standard deviation of the model fit.
Functional characterization of the XPC–TDG interaction
Taking advantage of our reconstituted in vitro TDG glycosylase assay, we next sought to further delineate the XPC–TDG interaction by identifying their functional domains required for DNA demethylation. We purified a series of N-terminal and C-terminal truncations of XPC complexes as well as an internal truncation that removes a highly disordered mammalian-specific domain (Δ338–519) (Fig. 5A; Supplemental Fig. S8A; Bunick et al. 2006). When added to the in vitro glycosylase assay, the ΔN (residues 195–940) and Δ338–519 truncations of XPC were able to enhance TDG activity similarly to full-length XPC (Fig. 5B; Supplemental Fig. S8B). However, the C-terminal truncation of XPC (ΔC) was inactive (Fig. 5B), indicating an important function at the C terminus of XPC in regulating TDG activity. It is worth noting that the C-terminal truncation also disrupts XPC's ability to bind its subunit, CETN2 (but not RAD23B) (Popescu et al. 2003; Nishi et al. 2005). To eliminate the possibility that CETN2 contributes to XPC's activity in vitro, we showed that a heterodimer of XPC and RAD23B is sufficient to stimulate TDG activity (Fig. 5C; Supplemental Fig. S8C). Although the C-terminal domain of XPC appears to be important for optimal TDG activity in vitro, it is dispensable for OCT4/SOX2-dependent transcriptional activation (Fong et al. 2011; Cattoglio et al. 2015). Thus, the domains required for DNA repair, transcriptional coactivation, and regulation of TDG-dependent DNA demethylation all appear to be distinct.
Figure 5.

XPC-mediated stimulation of TDG activity requires the C terminus of XPC and N terminus of TDG. (A) Schematic representation of human XPC (wild-type [WT]) highlighting the DNA-binding domain (DBD) and various protein–protein interaction domains. XPC truncations used in this study—ΔN (195–940 amino acids), Δ338 (del338–519 amino acids), and ΔC (1–814 amino acids)—are also shown. (B) TDG cleavage activity of a 5′-labeled T:G mismatch-containing oligonucleotide dsDNA in the presence of wild-type XPC or various XPC truncation complexes (ΔN, Δ338, and ΔC). (C) The CETN2 subunit is dispensable for stimulation of TDG-mediated base excision. TDG cleavage activity of a 5′-labeled T:G mismatch DNA substrate in the presence of the XPC–RAD23B heterodimer or a heterodimer containing ΔC XPC and RAD23B. (D) Schematic representation of full-length human TDG (FL) and truncations used in the in vitro glycosylase assay. The domains shown previously to be essential for G:U and G:T mismatch repair are indicated (Gallinari and Jiricny 1996). (E) Glycosylase activity of TDG truncations in the presence or absence of wild-type XPC was assayed using 5fC:G (left) and 5caC:G (right) substrates. The concentrations used were 0.2 µM DNA, 0.4 µM XPC complexes, and 50 nM TDG. (F) Relative global DNA methylation assayed by 5mC-specific ELISA using genomic DNA purified from HDFs overexpressing wild-type or ΔC human XPC. Error bars represent the standard deviation. n = 3. (***) P < 0.001; (**) P < 0.01; (n.s.) nonsignificant, calculated by two-way ANOVA.
We next determined the protein domain of TDG required for the functional interaction between XPC and TDG. Serial truncations of TDG that remove portions of the N-terminal and C-terminal domains down to the catalytic core domain (residues 111–308) were purified to near homogeneity (Fig. 5D; Supplemental Fig. S8D,E). As expected, these truncated TDG proteins retain in vitro cleavage activity using physiologically relevant 5fC:G (Fig. 5E, left) or 5caC:G (Fig. 5E, right) DNA substrates. However, XPC failed to stimulate the glycosylase activity of TDG lacking the first 111 amino acids. Because stimulation was still observed using the 51- to 410-amino-acid TDG truncation mutant, our results suggest that residues 51–111 confer responsiveness to XPC stimulation (Fig. 5E; Supplemental Fig. S8F). Interestingly, this domain (51–111 amino acids) has also been implicated in the removal of some but not all mismatch substrates. For example, we showed that TDG lacking the 51- to 111-amino-acid region (111–410 amino acids) largely failed to catalyze the base excision of a 5hmU:G substrate yet retained significant activity toward 5fC and 5caC (Supplemental Fig. S8F). Indeed, this region encompasses a previously proposed “regulatory domain” that imparts TDG with the ability to discriminate various mismatch substrates and bind a host of regulatory factors (Gallinari and Jiricny 1996; Tini et al. 2002; Chen et al. 2003; Smet-Nocca et al. 2008). Therefore, the N-terminal region of TDG appears to be multifunctional in nature, and its activity may be modulated by interacting with different protein factors.
To corroborate our in vitro data demonstrating the requirement of the C-terminal domain of XPC in stimulating TDG activity, we next asked whether the ability of the ΔC XPC in promoting global DNA demethylation is also compromised. Indeed, overexpression of the ΔC XPC abolished the gain-of-function phenomenon that we observed with full-length XPC in decreasing global DNA methylation, indicating that the same domain in XPC critical for TDG stimulation in vitro is also required for DNA demethylation in vivo (Fig. 5F). Collectively, our results reveal the specific influence of XPC on TDG-mediated DNA demethylation in vivo.
XPC enhances human iPSC generation
Given the critical role of the XPC complex as a transcriptional coactivator for OCT4 and SOX2 in pluripotency gene activation, it is not surprising that our ability to generate both mouse and human iPSCs is diminished upon XPC depletion (Supplemental Fig. S9A,B; Fong et al. 2011). However, reprogramming of XPC loss-of-function cells unexpectedly halts prior to reactivation of endogenous NANOG, OCT4, and other pluripotency genes that normally takes place late in the reprogramming process (Stadtfeld et al. 2008). Therefore, XPC likely assumes an additional role in iPSC induction by facilitating reprogramming initiation. Based on the data presented in this study and a previous report showing that loss of TDG leads to a similar early arrest in reprogramming (Hu et al. 2014), we decided to investigate whether TDG-mediated DNA demethylation is an early roadblock in reprogramming that can be overcome by elevating XPC levels in HDFs and pre-iPSCs.
Baseline XPC expression is significantly lower in somatic cells compared with ESCs (Fong et al. 2011). Given that TDG substrate turnover and DNA demethylation are sensitive to XPC concentration, we hypothesize that low endogenous levels of XPC in somatic cells may act as a barrier or limiting factor in reprogramming. Thus, we set out to examine the effect of XPC overexpression on iPSC conversion. HDFs were transduced with control or XPC-expressing lentiviruses. Following selection and expansion of the HDFs, we induced reprogramming via episomal vectors, as described in Figure 6A (Okita et al. 2011). Flow cytometry analysis revealed that reprogrammed XPC gain-of-function cells contained a significantly higher proportion of iPSCs expressing TRA-1-60, a marker of late stage mature human iPSCs, 24 d post-induction (Fig. 6B). Importantly, the number of iPSC-like colonies obtained from control and XPC gain-of-function cells remains the same (Fig. 6C). These results suggest that XPC enhances TRA-1-60 expression not by increasing the number of iPSCs but by facilitating the derivation of more robust iPSCs that more closely resemble bona fide ESCs.
Figure 6.

XPC increases somatic reprogramming fidelity and iPSC self-renewal capacity. (A) Schematic diagram for human iPSC induction modified from methods in Okita et al. (2011). (B) Flow cytometry analysis of control and XPC complex (XPC–RAD23B–CETN2) gain-of-function cell populations 24 d post-induction for the late stage human iPSC marker TRA-1-60. (C) The average number of colonies obtained in the reprogramming experiment depicted in B. (D) iPSCs derived from control or XPC–RAD23B–CETN2-overexpressing HDFs were challenged with single-cell dissociation and allowed to recover for 3–4 d before staining with alkaline phosphatase (AP). The average number of AP+ colonies formed per 2.5 × 104 single cells plated. Error bars depict the standard deviation. n = 3. (*) P < 0.05, calculated by two-tailed Student's t-test.
Human PSCs survive poorly when passaged as single cells. They readily undergo apoptosis and differentiation upon single-cell dissociation, in part due to the metastable state of “primed” epiblast-like human PSCs (Ohgushi and Sasai 2011). To examine the self-renewal capacity of both control and XPC gain-of-function iPSCs, iPSCs were dissociated to single cells, subjected to colony-forming assays, and scored by the number of iPSC colonies formed that stained positive for alkaline phosphatase (AP), a marker of undifferentiated PSCs. iPSCs derived from XPC gain-of-function HDFs resulted in ∼2.7-fold more AP+ colonies following single-cell dissociation compared with the control (Fig. 6D). The increase in cell survival and renewal remained apparent when both control and XPC gain-of-function iPSCs were treated with the selective Rho-associated kinase inhibitor Y-27632, a potent inhibitor of dissociation-induced apoptosis (Watanabe et al. 2007; data not shown). These results suggest that the increase in cell survival in XPC gain-of-function iPSCs is unlikely to be due to enhanced suppression of apoptosis caused by single-cell dissociation. To understand at a molecular level the enhanced fidelity of these XPC gain-of-function iPSCs, RT-qPCR analyses on bulk iPSCs obtained 30 d post-induction were performed. We observed a mild but consistent increase in the expression of key pluripotency genes such as NANOG, OCT4, SOX2, and REX1 (ZFP42) in XPC gain-of-function iPSCs. The elevated expression level of these genes may help stabilize the pluripotent state in XPC gain-of-function iPSCs when challenged with conditions that favor the exit of pluripotency (Supplemental Fig. S9C). Furthermore, we found that XPC overexpression enhances the expression of both early and late iPSC markers SSEA-4 (data not shown) and TRA-1-60 (Supplemental Fig. S9D), respectively, as early as 7 d post-induction, suggesting that its positive effect on reprogramming manifests early. This is consistent with the early arrest phenotype observed when attempting to reprogram XPC-deficient HDFs (Supplemental Fig. S9A,B; Fong et al. 2011).
To discern whether it is the transcriptional coactivator or the DNA demethylation function of XPC that contributes to the enhanced expression of iPSC markers in XPC gain-of-function pre-iPSCs, we compared the ability of wild-type and ΔC XPC to promote cellular reprogramming. We found that the ΔC XPC truncation is inactive in stimulating TDG-dependent DNA demethylation (Fig. 5B,C,F) but is fully active as a transcriptional coactivator (Fong et al. 2011). Given that the positive effect of XPC on reprogramming occurs early (Supplemental Fig. S9D), we focused our analysis at the relatively early time point of 14 d post-induction. In contrast to wild-type XPC, ectopic expression of ΔC XPC completely failed to elevate the expression of SSEA-4 and TRA-1-60 (Supplemental Fig. S10), suggesting that stimulation of DNA demethylation by XPC appears to be critical in overcoming early barriers to reprogramming. Taken together, our results indicate that XPC overexpression leads to higher-fidelity reprogramming, likely by lowering the epigenetic threshold for the re-establishment of a more robust pluripotency transcriptional network in iPSCs.
Discussion
In stark contrast to previous biochemical studies, we showed that TDG residence times for binding to methylated DNA substrates in living cells is two orders of magnitude lower in vivo than in vitro. This fast off rate is more in line with previous observations that implicated DNA demethylation by a rapid and active mechanism (Mayer et al. 2000; Zhang et al. 2007; Bhutani et al. 2010; Ito et al. 2010; Cortázar et al. 2011; Cortellino et al. 2011; Gu et al. 2011; Shen et al. 2013; Hu et al. 2014). While a number of protein factors (e.g., APE1 and NEIL1/2) and post-translation modifications (Hardeland et al. 2002; Steinacher and Schär 2005; Smet-Nocca et al. 2011) have been proposed to facilitate TDG substrate turnover, their effects have been demonstrated largely in vitro. Here, using SPT in live cells, we showed that the rate-limiting step for TDG activity (that is, the dissociation of TDG from the abasic sites in the nucleus) is remarkably sensitive to changes in protein levels of XPC but not APE1 or NEIL2, suggesting that XPC is the predominant factor in vivo that enhances TDG-dependent DNA demethylation by alleviating product inhibition of TDG. This is supported by the cobinding of XPC and TDG genome-wide, particularly at gene regulatory regions that are often subjected to dynamic DNA demethylation by TDG. Our model is also confirmed by loss-of-function studies showing that depletion of XPC resulted in global and locus-specific changes in DNA methylation that are reminiscent of those observed in TDG-deficient mouse embryos (Cortázar et al. 2011; Cortellino et al. 2011).
We showed previously that the XPC complex functions as a critical transcriptional coactivator for OCT4, SOX2, and likely other transcription factors in PSCs. Therefore, it was perhaps not surprising that XPC is required for efficient iPSC generation (Fong et al. 2011). However, based on the findings in this study, our identification of XPC as a critical regulator of DNA demethylation better explains the early reprogramming arrest phenotype associated with XPC loss-of-function cells, which occurs at a stage prior to when the pluripotency transcriptional network is reactivated. Furthermore, we found that, contrary to wild-type XPC, the ectopic expression of a transcriptionally active but demethylation-defective ΔC XPC failed to enhance reprogramming fidelity. We therefore propose two distinct and potentially equally important molecular mechanisms for XPC to influence reprogramming: first as a regulator of DNA demethylation by modulating TDG activity, which is critical during early stages of reprogramming, and second by acting as a classical transcriptional coactivator to directly drive pluripotency gene expression during iPSC maturation. Indeed, there is strong evidence indicating that transcription factor binding, gene activation, and DNA demethylation are interdependent and highly intertwined events. For example, introduction of transcription factor-binding motifs into a methylated DNA element can lead to local demethylation. Likewise, removal of transcription factor-binding sites can lead to local methylation of regions otherwise unmethylated (Brandeis et al. 1994; Macleod et al. 1994; Kirillov et al. 1996; Siegfried et al. 1999; Hejnar et al. 2001). These observations also underscore the dynamic nature of DNA methylation, where, even at transcribed genes, active DNA demethylation at gene regulatory regions must occur in order to counteract the robust DNA methylation activities present in mammalian cells. It is therefore tempting to speculate that the recruitment of XPC to gene enhancers by sequence-specific transcription factors to stimulate transcription can also promote local DNA demethylation of critical regulatory elements such that they remain hypomethylated and permissive to transcription factor binding and gene activation. In support of this notion, our data and others’ showed that depletion of XPC or TDG in ESCs results in specific alterations of DNA methylation status at gene regulatory regions even though neither protein exhibits overt known sequence specificity. This epigenetic feedback loop may indeed be a prevalent two-pronged strategy to sustain gene transcription by recruiting transcriptional coactivators that can simultaneously antagonize the silencing effects of DNA methylases by promoting active DNA demethylation.
The somatic cell nucleus is largely refractory to direct reprogramming, in part due to its high levels of DNA methylation, a major epigenetic barrier to the reactivation of PSC-specific genes in somatic cells. Remarkably, overexpression of XPC alone in HDFs is sufficient to reduce DNA methylation to the low levels found in PSCs, suggesting that the epigenetic threshold for cellular reprogramming may be significantly lowered in these XPC gain-of-function HDFs. Unexpectedly, however, the reprogramming efficiency of these cells is identical to control HDFs. Genome-wide DNA demethylation induced by XPC expression did not endow more HDFs with the potential to give rise to iPSCs. Instead, we found that XPC gain-of-function iPSCs express higher levels of mature iPSC marker and key pluripotency genes and display enhanced clonogenic ability—characteristics that are indicative of enhanced fidelity in reprogramming. Our finding is instructive in that most factors previously shown to improve reprogramming, such as c-Myc overexpression or p53 depletion, often do so at the cost of reprogramming fidelity (Okita et al. 2007; Li et al. 2009; Marión et al. 2009; Araki et al. 2011). Our data revealed that sequence motifs such as those recognized by HIFs and MYC became preferentially demethylated in XPC gain-of-function pre-iPSCs. Pertinent to cellular reprogramming, these transcription factors are required to induce a requisite early metabolic switch from oxidative to glycolytic metabolism (Yoshida et al. 2009; Varum et al. 2011; Zhou et al. 2012; Mathieu et al. 2014). Furthermore, HIFs have also been implicated in the transcriptional regulation of key pluripotency genes, including NANOG and OCT4 (Gustafsson et al. 2005; Covello et al. 2006; Das et al. 2012; Petruzzelli et al. 2014). Therefore, in addition to its role as a stem cell coactivator, the XPC complex may promote the faithful rewiring of the somatic cell transcriptome toward pluripotency by coupling efficient DNA demethylation and robust ESC-specific transcription through the recruitment of TDG activity to sites bound by the XPC complex and transcriptional activators.
Materials and methods
DNA constructs and antibodies
cDNAs for human XPC and TDG were generated from total RNAs isolated from human NTERA-2 (NT2) cells. Mammalian expression plasmids were cloned using the pHAGE-EF1α-STEMCCA construct (Sommer et al. 2009), in which OCT4, KLF4, SOX2, and c-MYC were replaced with cDNAs for XPC, RAD23B, RAD23B–CETN2, or mCherry, which was used as a control (pHAGE-EF1α-XPC, pHAGE-EF1α-RAD23B, pHAGE-EF1α-RAD23B–CETN2, and pHAGE-EF1α-mCherry, respectively). For expressing full-length and truncated human TDG in Escherichia coli, N-terminal His6-tagged TDG was cloned into a pST44 polycistronic expression plasmid (Tan et al. 2005). Constructs for XPC expression in Sf9s were described previously (Fong et al. 2011). The TDG-HaloTag construct for single-molecule imaging experiments was subcloned into the pTRE3G plasmid (Clontech). shRNAs against XPC were cloned using the pLKO.1 system as described previously (Moffat et al. 2006); sequences are provided in Supplemental Table S2. The commercial antibodies used were as follows: anti-ACTB (A2228) from Sigma-Aldrich, anti-XPC (A301-122A) and anti-RAD23B (A302-306A) from Bethyl Laboratories, anti-CETN2 (15977-1-AP) from ProteinTech, anti-5mC (33D3) from Diagenode, and anti-SSEA4 (clone MC-813-70) and anti-TRA-1-60 (clone TRA-1-60-R) from Biolegends.
Cell culture
Normal adult HDFs were obtained from Lonza and cultured in DMEM high glucose with GlutaMAX (Life Technologies) supplemented with 10% fetal bovine serum (FBS; HyClone) and nonessential amino acids (Life Technologies). U2OS cells were cultured in DMEM low glucose with GlutaMAX (Life Technologies) supplemented with 10% FBS (HyClone). Human ESC line H9 (WiCell) was maintained in feeder-independent conditions using Synthemax SC-II substrate (Corning) and grown in mTeSR1 (Stem Cell Technologies). Medium was changed daily, and cell cultures were passaged using Dispase (Stemcell Technologies) according to the manufacturer's protocol. Mouse embryonic fibroblasts (MEFs) were prepared from embryonic day 13.5 (E13.5) CF-1 embryos (Charles River) and cultured in HDF medium. Inactivation of MEFs was accomplished using mitomycin C (Sigma-Aldrich).
Overexpression and shRNA-mediated knockdown of XPC by lentiviral transduction
For lentivirus production, mammalian expression plasmids were cotransfected with packaging vectors into 293T cells using Lipofectamine 2000 (Invitrogen). Supernatants were collected at 48 and 72 h after transduction, and viruses were pelleted by ultracentrifugation. Lentivirus titers were determined using the lentivirus-associated p24 ELISA kit (Cell Biolabs). HDFs were infected at a multiplicity of infection (MOI) of 5 in the presence of 4 µg/mL polybrene (Millipore) and replaced with fresh medium without polybrene after 24 h. All other cell types were infected in the presence of 8 µg/mL polybrene (Millipore).
Quantification of global methylation
Genomic DNA was purified using the DNeasy blood and tissue kit (Qiagen). Relative global methylation by 5mC-specific ELISA was determined using the MethylFlash methylated DNA 5mC quantification kit (Epigentek) according to the manufacturer's protocol. For 5mC dot blots, genomic DNA was denatured for 10 min at 95°C, quickly placed on ice, and neutralized with 0.1 vol of 6.6 M ammonium acetate. Denatured DNA was spotted onto a Hybond-N+ membrane (GE Healthcare). The DNA was subsequently UV cross-linked to the membrane and blocked in 10% milk, 1% BSA, and 0.1% Tween-20 in PBS. The member was successively incubated with anti-5mC antibody (clone 33D3, Diagenode) overnight at 4°C and then with HRP-conjugated anti-mouse IgG secondary antibody (Pierce) for 30 min at room temperature. Chemiluminescence was detected using the Western Lightning ECL+ detection system (Perkin Elmer). Methylene blue was used as a control for total DNA cross-linked to membrane.
ChIP-seq analysis
For TDG, we reanalyzed ChIP-seq data by Neri et al. (2015) obtained from an ESC line expressing a biotin-tagged TDG (GSE55660; bioTDG: SRR1184633; mock control: SRR1184632). For the XPC complex, we reanalyzed our previously published RAD23B ChIP-seq data set in mouse ESCs (GSE64040; RAD23B: SRR1702685; control IgG: SRR1702684). Raw reads were first quality checked with FastQC and aligned onto the mouse genome (mm10 assembly) using Bowtie (Langmead et al. 2009), allowing for two mismatches (-n 2) and no multiple alignments (-m 1). Enriched regions were visualized on the mm10 genome with the Integrative Genomics Viewer (IGV) (Robinson et al. 2011; Thorvaldsdottir et al. 2013) after creating tiled data files from alignment files (igvtools count --w 50 --e 200). Peaks were called with MACS (--pvalue = 1 × 10−3 --shiftsize = 60 --gsize = mm) against the mock control for TDG and the IgG data set for RAD23B.
To create heat maps, we used deepTools (version 2.4.1) (Ramírez et al. 2016). We first ran bamCoverage (--binSize 50 --normalizeTo1 × 2150570000 --extendReads 60 –ignoreDuplicates -of bigwig) and normalized read numbers to 1× sequencing depth, obtaining read coverage per 50-bp bins across the whole genome (bigWig format). We then used the bigWig files to compute read numbers ±5 kb around the TSSs of all annotated RefSeq genes (computeMatrix reference-point -R RefSeqGenes_mm10.bed --referencePoint = TSS --upstream 5000 --downstream 5000 --sortRegions = no). We sorted the output matrix by decreasing TDG enrichment, calculated as the total number of reads within ±1 kb around the TSS of each RefSeq gene. Finally, we created heat maps with the plotHeatmap tool (--averageTypeSummaryPlot = mean --colorMap='YlOrRd' --sortRegions = no).
MeDIP
Genomic DNA from HDFs or pre-iPSCs was sheared using the Covaris S2 focused ultrasonicator (10.0 duty power, 175 peak power, 200 cycles per burst, 430 sec in 130 µL of AE buffer). Size selection was performed using the Agencourt AMPure XP beads (Beckman Coulter) to obtain DNA fragments averaging 150–250 bp in length. End repair was performed using the NEBNext end repair module (New England Biolabs) followed by A-tailing with Klenow (New England Biolabs). TruSeq adapters were ligated to the sample DNA and subjected to another round of size selection to remove adapter dimers. DNA was denatured for 10 min at 95°C, quickly spun down, and placed on ice. Denatured sample DNA and anti-5mC antibody (clone 33D3, Diagenode) or mouse IgG (015-000-003, Jackson) were incubated together overnight at 4°C in MeDIP buffer (1 mM EDTA, 0.05% TritonX-100, in PBS). Input DNA was kept processed alongside the samples. The immunoprecipitate was added to precleared equilibrated M-280 anti-mouse IgG Dynabeads (Life Technologies) for 2 h at 4°C. Following the immunoprecipitation, DNA was recovered by proteinase K treatment and subsequently converted to dsDNA with KAPA HiFi HotStart polymerase (Kapa Biosystems) for four cycles. No sample DNA was recovered for IgG immunoprecipitations, indicating that very little background binding occurred. Sample DNA was subsequently subjected to another round of size selection to obtain fragment sizes averaging 200–400 bp in length, suitable for sequencing. Libraries were amplified using KAPA HiFi HotStart polymerase (Kapa Biosystems) for a final 10 cycles. Fragment size, purity, and concentration of the libraries were verified using the Agilent Technologies 2100 Bioanalyzer.
MeDIP-seq analysis
Input and MeDIP raw reads from wild-type and XPC-overexpressing HDFs and pre-iPSCs (GSE92445) were first quality checked with FastQC and aligned onto the human genome (hg19 assembly) using Bowtie (Langmead et al. 2009), allowing for two mismatches (-n 2) and no multiple alignments (-m 1). MeDIP enrichment over input was calculated by calling peaks with MACS2 (callpeak -g hs –nomodel; --extsize was set to 160 for HDFs and 250 for pre-iPSCs) (Feng et al. 2012).
The average MeDIP-seq enrichment around the TSSs of all RefSeq genes annotated in the human genome was computed with deepTools (version 2.4.1) (Ramírez et al. 2016). We first ran bamCompare (--binSize 50 --ratio subtract --normalizeTo1× 2451960000 --ignoreDuplicates -of bigwig; --extendReads was set to 160 for HDFs and 250 for pre-iPSCs) to normalize read numbers to 1× sequencing depth and subtract input reads, obtaining input-subtracted read coverage per 50-bp bins across the whole genome (bigWig format). We then used the bigWig files to compute read numbers in 250-bp bins ±5 kb around the TSSs of RefSeq genes (computeMatrix reference-point -R RefSeqGenes_hg19.bed --referencePoint = TSS --upstream 5000 --downstream 5000 ----sortRegion s= no --binSize 250 --averageTypeBins sum --skipZeros). We finally plotted the average read numbers by plotProfile (--averageType mean --plotType lines --refPointLabel TSS --perGroup).
For motif discovery analysis in pre-iPSCs, we first identified regions enriched in wild-type but not in XPC-overexpressing cells using Galaxy (operate on genomic intervals [on MACS2 called peaks]; join the intervals of two data sets side by side; 1-bp minimum overlap) (Afgan et al. 2016). We then used the MEME suite (Bailey et al. 2009) and determined which motifs were most centrally enriched ±250 bp around MACS2 called peaks with CentriMo (meme-chip –db JASPAR_CORE_2009_vertebrates_ MEME).
Expression and purification of recombinant XPC complexes and TDG
Expression and purification of recombinant XPC complexes from Sf9 cells were performed as described previously (Fong et al. 2011). For bacterial purification of recombinant TDG, pST44 expression plasmids were transformed into BL21-Codon Plus RIPL-competent cells (Agilent). Cultures were induced overnight at 18°C with 0.5 mM IPTG. Cell pellets were lysed in high-salt lysis buffer (HSLB; 25 mM HEPES at pH 7.9, 0.6 M NaCl, 0.6% TritonX-100, 0.05% NP-40, 10% glycerol, 10 mM 2-mercaptoethanol, protease inhibitors) and 0.5 mg/mL lysozyme. Sonicated lysates were cleared by ultracentrifugation, supplemented with 10 mM imidazole, and incubated with Ni-NTA resin for 16 h. Bound proteins were washed extensively with HSLB with 20 mM imidazole, equilibrated with 0.2 M NaCl HGN (25 mM HEPES at pH 7.9, 10% glycerol, 0.01% NP-40) with 20 mM imidazole, and eluted with 0.2 M NaCl HGN supplemented with 0.25 M imidazole. Eluted fractions were analyzed by SDS-PAGE followed by PageBlue staining. Peak TDG fractions were pooled and dialyzed to 0.1 M NaCl HGN. Dialyzed peak Ni-NTA fractions were applied to a Poros 20 HQ column (Applied Biosystems) and subjected to a linear gradient from 0.1 M to 0.6 M NaCl. Full-length TDG was eluted from the column at ∼0.15 M NaCl. Peak fractions from the Poros-HQ were pooled and dialyzed to 0.2 M NaCl HGN.
In vitro TDG glycosylase assay
5′-labeled Cy3 oligonucleotides were obtained containing a T, 5hmU, 5fC, or 5caC internal modification (Trilink Biotechnologies). Cy3-labeled oligonucleotides were annealed overnight and subsequently purified twice by sequential native PAGE. Reactions were performed in 10-µL volumes containing 20 mM HEPES (pH 7.9), 100 mM NaCl, 1 mM EDTA, 0.1% BSA, 0.01% NP-40, 0.5 pmol TDG, and 2 pmol DNA substrate for 45 min at 30°C. For reactions containing XPC, 2.0–4.0 pmol of purified XPC complexes was added simultaneously with TDG to the reactions. Samples were run on 15% denaturing PAGE containing 7 M urea at 200 V for 45–60 min. Cleavage products were visualized using the PharosFX Plus (Bio-Rad). Sequences of DNA oligonucleotides used are provided in Supplemental Table S2.
Inducible human TDG-HALO cell line generation
TDG-Halo-inducible cell lines were generated by cotransfection of pTRE3G-TDG-Halo and pEF1a-Tet3G (Clontech) into U2OS cells. Cells were selected for stable integration of the plasmids using 500 µg/mL G418, subsequently labeled with 500 µM JF549-HaloTag ligand (Grimm et al. 2015) in the presence of 10 µg/mL doxycycline, and subjected to FACS to identify TDG-Halo-expressing cells. Only cells that had >100-fold higher JF549 signal compared with uninduced cells were retained and expanded.
Single-particle imaging
The 4.6-kb plasmid pGL4.13[Luc2/SV40] (Promega) was methylated using 4.0 U of M.SssI (New England Biolabs) per microgram of DNA and incubated for 4 h at 37°C followed by ethanol precipitation. The plasmid contained ∼300 CpG motifs. TDG-Halo-inducible U2OS cells were transfected by nucleofection (Lonza) and plated on 25-mm glass coverslips. The medium was supplemented with 3 ng/mL doxycycline to induce expression of TDG-Halo at low levels (Supplemental Fig. S7B). Twenty-four hours following nucleofection, cells were labeled for imaging in 100–250 nM PA-JF549-HaloTag ligand and 100–250 nM JF646-SNAPTag ligand for 30 min in complete medium (Grimm et al. 2015, 2016). Labeled cells were then washed once with PBS and returned to phenol-free complete DMEM for a minimum of 30 min prior to imaging. During imaging, cells were maintained at 37°C and 5% CO2 using an incubation chamber and thermal control unit to control both chamber and objective temperature (Okolab).
Single-particle experiments were conducted on a custom-built Nikon TI microscope outfitted for simultaneous single-molecule data acquisition in two color channels as described previously (Teves et al. 2016; Hansen et al. 2017). Briefly, samples were imaged using a 100×/NA 1.49 oil immersion TIRF objective (Nikon apochromat CFI Apo TIRF 100× oil), a Perfect Focus System (Nikon), and a motorized mirror arm to facilitate HILO illumination (Tokunaga et al. 2008). Samples were illuminated using 405-nm (140 mW, OBIS coherent), 561-nm (1 W, genesis coherent), and 633-nm (1 W, genesis coherent) lasers, which were focused onto the back pupil plane of the objective via fiber and multinotch dichromatic mirror (405-nm/488-nm/561-nm/633-nm quad-band; Semrock, NF03-405/488/532/635E-25). Excitation intensity and pulse width were controlled through an acousto-optic transmission filter (AOTFnC-VIS-TN, AA Opto-Electronic) triggered using the camera's TTL exposure output signal. Fluorescence emission from the sample was separated into two parallel light paths with the use of a single-edge dichroic beam splitter (Semrock, Di02-R635-25x36) to direct the JF646 emission and the JF549 emission to two parallel EM-CCD cameras (Andor iXon Ultra 897). A 676/37-nm bandpass filter (Semrock, FF01-676/37-25) was placed in front of the first camera, and a 593/40-nm bandpass filter (Semrock, FF01-593/40-25) was used for the second camera. The cameras were synchronized using a National Instruments DAQ board (NI-DAQ PCI-6723). Two-color movies were acquired at a frame rate of 2 Hz for 750 frames, with excitation intensities set to maximize signal to noise while minimizing photobleaching in the range of 2–5 mW at the coverslip. Photo activation of PA-JF549 was achieved through 10-msec pulses of 405-nm light at the beginning of each frame.
Binding time analysis
Long exposure times of single molecules caused freely diffusing molecules to become “blurred out,” but bound molecules appeared as diffraction-limited spots (Chen et al. 2014). To analyze these bound spots, we used an adapted version of SLIMfast (Matlab), an implementation of the MTT particle-taking algorithm (Sergé et al. 2008; Normanno et al. 2015). Images were masked to limit analysis to only trajectories within the nucleus. Single molecules in each frame were detected using the following parameters: detection box, 9 px; emission max, 571 nm; localization error, 1 × 10−7; deflation loops, 0. Spots in successive frames were connected into trajectories using the following tracking settings: maximum expected D, 0.5; number of gaps allowed, one; maximum number of competitors, three.
After pooling all of the calculated trajectories from a given condition, we generated histograms of trajectory lengths and, from these data, generated curves of the survival probability (1 minus the cumulative distribution function) for each condition. In order to estimate the error of our measurements, we first determined which condition within a given experiment had the fewest trajectories (nmin) and performed bootstrap resampling without replacement using nmin/2 trajectories for each condition. We then repeated this resampling process 1000 times to find the most likely survival curve (mean) as well as determine the associated error, reported as the 95% confidence interval. Binding rates were extracted from the survival probability data by fitting to the following equation:
where −koff, emp, ns and −koff, emp, s are the empirically measured nonspecific and specific (respectively) binding times, and F is the fraction of trajectories from each. Because SPT experiments are dominated by short noisy trajectories, fitting was done only on trajectories lasting >11 frames, as measured empirically by determining the calculated −kemp, s as a function of first fitting frame (Supplemental Fig. S6C; for more details see Mazza et al. 2012; Hansen et al. 2017).
Finally, we corrected these fits for photobleaching. The length of each trajectory can be thought of as a function of both a photobleaching rate (kbleach) and an unbinding rate (koff, s): koff, emp, s =koff, s + kbleach. In order to measure the true off rate (koff, s), we performed SPT first on the histone H2B-Halo, assuming that H2B unbinding was negligible within the time scale of an imaging experiment, and thus trajectory lengths reported solely on kbleach. koff, s was then determined by subtracting kbleach from koff, emp, s. These H2B measurements were made separately prior to each day of imaging to account for any day-to-day fluctuations in the imaging setup. Binding times were reported as τ = 1/koff, s.
Human somatic cell reprogramming and flow cytometry
Human iPSC reprogramming was induced by the transfection of episomal vectors containing cDNAs for OCT4, SOX2, KLF4, L-MYC, LIN28A, and a shRNA against p53 (Okita et al. 2011) into HDFs using nucleofection (Lonza) according to the manufacturer's protocol. Following nucleofection, cells were plated in 60-mm2 dishes and cultured for 7 d in HDF medium. Cells were then dissociated and plated (2 × 104 to 4 × 104 cells per well of a six-well plate) on inactivated MEFs (2.5 × 105 cells per well of a six-well plate) in human iPSC medium containing knockout DMEM/F12 (Life Technologies), 20% knockout serum replacement (Life Technologies), 2 mM GlutaMAX (Life Technologies), nonessential amino acids (Life Technologies), 0.1 mM 2-mercaptoethanol (Sigma-Aldrich), and 4 ng/mL bFGF (Life Technologies). Reprogramming was assayed by AP staining (Millipore) or flow cytometry analysis on a BD LSRFortessa, performed according to the manufacturers’ protocols.
Supplementary Material
Acknowledgments
We thank X. Darzacq and A. Tangara for use of the microscope and technical assistance; A. Fischer and A. Killilea at the Cell Culture Facility (University of California at Berkeley), K. Heydari at the Li Ka Shing Flow Cytometry Facility (University of California at Berkeley), C. Inouye, S. Zheng, and C. Tsui for technical assistance; and S. Teves and all laboratory members for fruitful discussions and critical reading of the manuscript. Sequencing was performed at the Vincent J. Coates Genomics Sequencing Laboratory at University of California at Berkeley, supported by National Institutes of Health S10 Instrumentation grants S10RR029668 and S10RR027303. This work was supported by the Howard Hughes Medical Institute and California Institute for Regenerative Medicine Research grant RB4-06016 (to R.T.). J.J.H. and C.C. have been California Institute for Regenerative Medicine predoctoral/post-doctoral fellows (training program TG2-01164).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.295741.116.
References
- Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, et al. 2016. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res 44: W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki R, Hoki Y, Uda M, Nakamura M, Jincho Y, Tamura C, Sunayama M, Ando S, Sugiura M, Yoshida MA, et al. 2011. Crucial role of c-Myc in the generation of induced pluripotent stem cells. Stem Cells 29: 1362–1370. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME suite: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. 2010. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature 463: 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21. [DOI] [PubMed] [Google Scholar]
- Bock C, Tomazou EM, Brinkman AB, Müller F, Simmer F, Gu H, Jäger N, Gnirke A, Stunnenberg HG, Meissner A. 2010. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 28: 1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandeis M, Frank D, Keshet I, Siegfried Z, Mendelsohn M, Names A, Temper V, Razin A, Cedar H. 1994. Spl elements protect a CpG island from de novo methylation. Nature 371: 435–438. [DOI] [PubMed] [Google Scholar]
- Bunick CG, Miller MR, Fuller BE, Fanning E, Chazin WJ. 2006. Biochemical and structural domain analysis of xeroderma pigmentosum complementation group C protein. Biochemistry 45: 14965–14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoglio C, Zhang ET, Grubisic I, Chiba K, Fong YW, Tjian R. 2015. Functional and mechanistic studies of XPC DNA-repair complex as transcriptional coactivator in embryonic stem cells. Proc Natl Acad Sci 112: E2317–E2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Lucey MJ, Phoenix F, Lopez-Garcia J, Hart SM, Losson R, Buluwela L, Coombes RC, Chambon P, Schär P, et al. 2003. T:G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor α. J Biol Chem 278: 38586–38592. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang Z, Li L, Chen B-C, Revyakin A, Hajj B, Legant W, Dahan M, Lionnet T, Betzig E, et al. 2014. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156: 1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortázar D, Kunz C, Saito Y, Steinacher R, Schär P. 2007. The enigmatic thymine DNA glycosylase. DNA Repair 6: 489–504. [DOI] [PubMed] [Google Scholar]
- Cortázar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E, Wirz A, Schuermann D, Jacobs AL, Siegrist F, et al. 2011. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 470: 419–423. [DOI] [PubMed] [Google Scholar]
- Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, et al. 2011. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146: 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu C-J, Labosky PA, Simon MC, Keith B. 2006. HIF-2α regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev 20: 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH. 2007. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol 14: 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Bayat-Mokhtari R, Tsui M, Lotfi S, Tsuchida R, Felsher DW, Yeger H. 2012. HIF-2α suppresses p53 to enhance the stemness and regenerative potential of human embryonic stem cells. Stem Cells 30: 1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Carvalho DD, You JS, Jones PA. 2010. DNA methylation and cellular reprogramming. Trends Cell Biol 20: 609–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Gräf S, Johnson N, Herrero J, Tomazou EM, et al. 2008. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol 26: 779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, Liu XS. 2012. Identifying ChIP-seq enrichment using MACS. Nat Protoc 7: 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong YW, Inouye C, Yamaguchi T, Cattoglio C, Grubisic I, Tjian R. 2011. A DNA repair complex functions as an Oct4/Sox2 coactivator in embryonic stem cells. Cell 147: 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallinari P, Jiricny J. 1996. A new class of uracil–DNA glycosylases related to human thymine–DNA glycosylase. Nature 383: 735–738. [DOI] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, et al. 2015. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods 12: 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, English BP, Choi H, Muthusamy AK, Mehl BP, Dong P, Brown TA, Lippincott-Schwartz J, Liu Z, Lionnet T, et al. 2016. Bright photoactivatable fluorophores for single-molecule imaging. Nat Methods 13: 985–988. [DOI] [PubMed] [Google Scholar]
- Gu T-P, Guo F, Yang H, Wu H-P, Xu G-F, Liu W, Xie Z-G, Shi L, He X, Jin S, et al. 2011. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477: 606–610. [DOI] [PubMed] [Google Scholar]
- Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. 2005. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell 9: 617–628. [DOI] [PubMed] [Google Scholar]
- Hansen AS, Pustova I, Cattoglio C, Tjian R, Darzacq X. 2017. CTCF and cohesin regulate chromatin loop stability with distinct dynamics. Elife 6: 25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardeland U, Steinacher R, Jiricny J, Schär P. 2002. Modification of the human thymine–DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J 21: 1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, et al. 2010. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 28: 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, Zhang X, Cheng X. 2012. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res 40: 4841–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. 2011. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333: 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejnar J, Hajkova P, Plachy J, Elleder D, Stepanets V, Svoboda J. 2001. CpG island protects Rous sarcoma virus-derived vectors integrated into nonpermissive cells from DNA methylation and transcriptional suppression. Proc Natl Acad Sci 98: 565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Zhang L, Mao S-Q, Li Z, Chen J, Zhang R-R, Wu H-P, Gao J, Guo F, Liu W, et al. 2014. Tet and TDG mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell 14: 512–522. [DOI] [PubMed] [Google Scholar]
- Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. 2010. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466: 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science (80-) 333: 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LIR, et al. 2010. Epigenetic memory in induced pluripotent stem cells. Nature 467: 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirillov A, Kistler B, Mostoslavsky R, Cedar H, Wirth T, Bergman Y. 1996. A role for nuclear NF-κB in B-cell-specific demethylation of the Igκ locus. Nat Genet 13: 435–441. [DOI] [PubMed] [Google Scholar]
- Kohli RM, Zhang Y. 2013. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502: 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le May N, Mota-Fernandes D, Vélez-Cruz R, Iltis I, Biard D, Egly JM. 2010. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol Cell 38: 54–66. [DOI] [PubMed] [Google Scholar]
- Li H, Collado M, Villasante A, Strati K, Ortega S, Cañamero M, Blasco MA, Serrano M. 2009. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460: 1136–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Han D, Zhao BS, Song C-X, Zhang L-S, Doré LC, He C. 2015. Base-resolution maps of 5-formylcytosine and 5-carboxylcytosine reveal genome-wide DNA demethylation dynamics. Cell Res 25: 386–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod D, Charlton J, Mullins J, Bird AP. 1994. Spl sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev 8: 2282–2292. [DOI] [PubMed] [Google Scholar]
- Maillard O, Solyom S, Naegeli H. 2007. An aromatic sensor with aversion to damaged strands confers versatility to DNA repair. PLoS Biol 5: e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A, Drohat AC. 2011. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem 286: 35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez-Capetillo O, Serrano M, Blasco MA. 2009. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460: 1149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu J, Zhou W, Xing Y, Sperber H, Ferreccio A, Agoston Z, Kuppusamy KT, Moon RT, Ruohola-Baker H. 2014. Hypoxia-inducible factors have distinct and stage-specific roles during reprogramming of human cells to pluripotency. Cell Stem Cell 14: 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. 2000. Demethylation of the zygotic paternal genome. Nature 403: 501–502. [DOI] [PubMed] [Google Scholar]
- Mazza D, Abernathy A, Golob N, Morisaki T, McNally JG. 2012. A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res 40: e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. 2006. A Lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124: 1283–1298. [DOI] [PubMed] [Google Scholar]
- Neddermann P, Gallinari P, Lettieri T, Schmid D, Truong O, Hsuan JJ, Wiebauer K, Jiricny J. 1996. Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J Biol Chem 271: 12767–12774. [DOI] [PubMed] [Google Scholar]
- Nemzow L, Lubin A, Zhang L, Gong F. 2015. XPC: going where no DNA damage sensor has gone before. DNA Repair (Amst) 36: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri F, Incarnato D, Krepelova A, Rapelli S, Anselmi F, Parlato C, Medana C, Dal Bello F, Oliviero S. 2015. Single-base resolution analysis of 5-formyl and 5-carboxyl cytosine reveals promoter DNA methylation dynamics. Cell Rep 10: 674–683. [DOI] [PubMed] [Google Scholar]
- Nishi R, Okuda Y, Watanabe E, Mori T, Iwai S, Masutani C, Sugasawa K, Hanaoka F. 2005. Centrin 2 stimulates nucleotide excision repair by interacting with xeroderma pigmentosum group C protein. Mol Cell Biol 25: 5664–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normanno D, Boudarène L, Dugast-Darzacq C, Chen J, Richter C, Proux F, Bénichou O, Voituriez R, Darzacq X, Dahan M. 2015. Probing the target search of DNA-binding proteins in mammalian cells using TetR as model searcher. Nat Commun 6: 7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgushi M, Sasai Y. 2011. Lonely death dance of human pluripotent stem cells: ROCKing between metastable cell states. Trends Cell Biol 21: 274–282. [DOI] [PubMed] [Google Scholar]
- Ohi Y, Qin H, Hong C, Blouin L, Polo JM, Guo T, Qi Z, Downey SL, Manos PD, Rossi DJ, et al. 2011. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat Cell Biol 13: 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. 2007. Generation of germline-competent induced pluripotent stem cells. Nature 448: 313–317. [DOI] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K, et al. 2011. A more efficient method to generate integration-free human iPS cells. Nat Methods 8: 409–412. [DOI] [PubMed] [Google Scholar]
- Petruzzelli R, Christensen DR, Parry KL, Sanchez-Elsner T, Houghton FD. 2014. HIF-2α regulates NANOG expression in human embryonic stem cells following hypoxia and reoxygenation through the interaction with an Oct–Sox cis regulatory element. PLoS One 9: e108309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffeneder T, Spada F, Wagner M, Brandmayr C, Laube SK, Eisen D, Truss M, Steinbacher J, Hackner B, Kotljarova O, et al. 2014. Tet oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat Chem Biol 10: 574–581. [DOI] [PubMed] [Google Scholar]
- Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li Y, Shioda T, et al. 2010. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol 28: 848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu A, Miron S, Blouquit Y, Duchambon P, Christova P, Craescu CT. 2003. Xeroderma pigmentosum group C protein possesses a high affinity binding site to human centrin 2 and calmodulin. J Biol Chem 278: 40252–40261. [DOI] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. 2016. deepTools2: a next generation Web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W. 2001. Epigenetic reprogramming in mammalian development. Science 293: 1089–1093. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomacher L, Han D, Musheev MU, Arab K, Kienhöfer S, von Seggern A, Niehrs C. 2016. Neil DNA glycosylases promote substrate turnover by Tdg during DNA demethylation. Nat Struct Mol Biol 23: 116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergé A, Bertaux N, Rigneault H, Marguet D. 2008. Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat Methods 5: 687–694. [DOI] [PubMed] [Google Scholar]
- Shen L, Wu H, Diep D, Yamaguchi S, D'Alessio AC, Fung H-L, Zhang K, Zhang Y. 2013. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 153: 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Iwai S, Hanaoka F, Sugasawa K. 2003. Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO J 22: 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried Z, Eden S, Mendelsohn M, Feng X, Tsuberi BZ, Cedar H. 1999. DNA methylation represses transcription in vivo. Nat Genet 22: 203–206. [DOI] [PubMed] [Google Scholar]
- Smet-Nocca C, Wieruszeski J-M, Chaar V, Leroy A, Benecke A. 2008. The thymine–DNA glycosylase regulatory domain: residual structure and DNA binding. Biochemistry 47: 6519–6530. [DOI] [PubMed] [Google Scholar]
- Smet-Nocca C, Wieruszeski J-M, Léger H, Eilebrecht S, Benecke A. 2011. SUMO-1 regulates the conformational dynamics of thymine–DNA glycosylase regulatory domain and competes with its DNA binding activity. BMC Biochem 12: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Chan MM, Humm KC, Karnik R, Mekhoubad S, Regev A, Eggan K, Meissner A. 2014. DNA methylation dynamics of the human preimplantation embryo. Nature 511: 611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer CA, Stadtfeld M, Murphy GJ, Hochedlinger K, Kotton DN, Mostoslavsky G. 2009. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells 27: 543–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C-X, Szulwach KE, Dai Q, Fu Y, Mao S-Q, Lin L, Street C, Li Y, Poidevin M, Wu H, et al. 2013. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 153: 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Donahue G, Zaret KS. 2012. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151: 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Breault DT, Hochedlinger K. 2008. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell 2: 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinacher R, Schär P. 2005. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr Biol 15: 616–623. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. 2009. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (80-) 324: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Kern RC, Selleck W. 2005. The pST44 polycistronic expression system for producing protein complexes in Escherichia coli. Protein Expr Purif 40: 385–395. [DOI] [PubMed] [Google Scholar]
- Teves SS, An L, Hansen AS, Xie L, Darzacq X, Tjian R. 2016. A dynamic mode of mitotic bookmarking by transcription factors. Elife 5: 66464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tini M, Benecke A, Um S-J, Torchia J, Evans RM, Chambon P. 2002. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell 9: 265–277. [DOI] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, Sakata-Sogawa K. 2008. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods 5: 159–161. [DOI] [PubMed] [Google Scholar]
- Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA, Ramalho-Santos J, Van Houten B, Schatten G. 2011. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One 6: e20914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Nishikawa S, Muguruma K, et al. 2007. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 25: 681–686. [DOI] [PubMed] [Google Scholar]
- Waters TR, Swann PF. 1998. Kinetics of the action of thymine DNA glycosylase. J Biol Chem 273: 20007–20014. [DOI] [PubMed] [Google Scholar]
- Waters TR, Gallinari P, Jiricny J, Swann PF. 1999. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J Biol Chem 274: 67–74. [DOI] [PubMed] [Google Scholar]
- Yasuda G, Nishi R, Watanabe E, Mori T, Iwai S, Orioli D, Stefanini M, Hanaoka F, Sugasawa K. 2007. In vivo destabilization and functional defects of the xeroderma pigmentosum C protein caused by a pathogenic missense mutation. Mol Cell Biol 27: 6606–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S. 2009. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 5: 237–241. [DOI] [PubMed] [Google Scholar]
- Zhang F, Pomerantz JH, Sen G, Palermo AT, Blau HM. 2007. Active tissue-specific DNA demethylation conferred by somatic cell nuclei in stable heterokaryons. Proc Natl Acad Sci 104: 4395–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, et al. 2012. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J 31: 2103–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.