

Abstract
Introduction of a silyl group to one of the internal olefin positions in di-olefinic substrates is able to control effectively the resulting olefin geometry in ring-closing metathesis. The application of this method to a range of macrocyclic E-alkenyl siloxanes is described. Protodesilylation of alkenyl siloxane products yields novel Z-macrocycles.
Ring-closing metathesis (RCM) reactions1 have enabled advances in materials science,2 chemical biology,3 natural product,4 medicinal,5 and diversity-oriented synthetic chemistry.6 However, in efforts to synthesize macrocyclic compounds via RCM reactions, control of the stereochemistry of the resulting olefin is often problematic (Scheme 1, A). Since a variety of factors can determine the stereochemical outcome,1d, 7 general strategies that give rise to either Z or E products remain a significant challenge. Additionally, in the absence of a strong steric or electronic bias, the 1,2-disubstituted olefin in RCM products is difficult to modify regiospecifically, limiting the potential of further functionalization and diversification (Scheme 1, A). Post-metathesis functionalization has in general been limited to ‘symmetrical’ transformations such as hydrogenation, dihydroxylation,6d, 8 epoxidation8b, 9 and aziridination.10
Scheme 1.

Expansion of RCM through silyl group incorporation.
Two major advances that address the stereoselectivity of RCM reactions have been achieved. One approach involves ring-closing alkyne metathesis and selective reduction of the alkyne intermediate to yield Z or E olefins.11 However, this approach has only been applied to ring sizes of 12 and larger.12 Another approach is based on the development of Z-selective catalysts;13 however, it has not yet been reported for macrocyclic RCM reactions. Both strategies generate 1,2-disubstituted olefins, which are primarily limited to symmetrical post-metathesis transformations. To address these limitations, we investigated the use of vinylsiloxanes as substrates to access tri-substituted macrocyclic silyl alkenes.
We imagined that a silyl group at an internal position of one of the olefins could serve two purposes (Scheme 1, B). First, the sterics of the silyl group was expected to favor formation of the E-silyl alkene product. The Z-disubstituted olefin would then be obtained following protodesilylation. Second, the exocyclic silyl substituent was expected to enable functionalization of the RCM product, thus yielding specific tri-substituted olefins.14 In this report, we explore the ability of vinylsiloxanes to undergo productive RCMs, their influence on the stereochemistry of the resulting products, and their protodesilylation following ring closure.
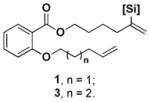
Although the influence of various silyl groups on RCMs has not been studied extensively, there are many examples of their use in cross metathesis (CM) reactions. Pietraszuk et al. demonstrated that the influence of the silyl groups on the yield of CM reactions is associated with electronic effects of the substituents on silicon.15 Silyl groups with electron withdrawing substituents such as EtO-, AcO-, Cl- gave better yields than with Me- and/or Ph- groups. Accordingly, although vinyltrialkylsilanes yielded a variety of five and six-membered rings they failed to produce medium and large rings.16 These observations are consistent with the few examples of RCM reactions leading to exocyclic vinylsilanes and vinylsiloxanes reported in the literature.17 With this knowledge, we next focused on vinylsiloxanes. We prepared several salicylate- derived substrates with one of the olefins bearing a triethoxysilyl group at the internal position. The vinylsiloxane substrates were obtained in excellent yields from the corresponding alkyne precursors through hydrosilylation.17a, 17c Using the second-generation Grubbs catalyst, a 14-membered salicylate macrocycle (2a) was formed, albeit with low yield (Scheme 2). When the same conditions were applied to substrate 3a, the yield of the 15-membered ring 4a dropped to 3% (Scheme 2). We used this demanding substrate to optimize the reaction conditions.
Scheme 2.

Initial successful case of macrocyclic RCM of a vinylsiloxane and the model substrate used for reaction optimization.
Among commercially available Ru-based metathesis catalysts, catalyst A was best able to increase the yield under the original reaction conditions (see supporting information for details). In contrast to the second-generation Grubbs catalyst, A bears one methyl group on the ortho-position of each phenyl ring, making it less sterically hindered and more reactive.18 After varying solvent, temperature, and concentration we found that optimal results (4a, 63%) were obtained using benzene or toluene as a solvent, temperatures of 35 °C and 20 mol% of catalyst (Scheme 2).
Under the optimal reaction conditions, unreacted starting material was still observed, indicating that the catalyst was deactivated before the reaction went to completion. To explore this observation further, catalyst stability studies were performed. It was determined that catalyst A decomposed at a similar rate on treatment of any olefin substrate, with or without the silyl group (see supporting information for details). In addition to the commonly observed decomposition pathways,19 Csp2-H bond insertion of the N-aryl ring on the NHC ligand can also lead to fast deactivation of catalyst A.20 Our results suggest that vinylsiloxane substrates do not intrinsically cause catalyst deterioration.
We next examined the influence of different silyl groups on the yield of the RCM reaction (Table 1). The results are in alignment with those of CM reactions. Generally, ethoxy substituents promote the formation of product and lead to higher yields compared to alkyl and/or aryl substituents (compare a–d with e, f). More ethoxy substituents are preferred over less (compare b with c). Pietraszuk and Fischer noted that electron-withdrawing substituents shut down an undesired pathway that leads to catalyst deactivation.21 We suggest that these unproductive processes are operative in macrocyclization reactions as well and require the appropriate siloxane group to suppress them. In contrast to the CM precedents, the sterics of the silyl group also influenced the reaction yield. This effect is seen with the more demanding substrates 3a–f, while substrates 1a–f show the same trend but to a lesser degree. When one of the ethoxy substituents was changed to a methyl group (3a versus 3b), the yield was improved while a change to a phenyl group (3b versus 3d) resulted in the yield being halved. Our results indicate that the diethoxymethylsilyl group delivers the best reaction outcomes by maintaining a balance between both steric and electronic effects.
Table 1.
Influence of the silicon substituents on the yield of RCM.
| Substrate | Entry | Silyl group | 1[a] | 3[a] |
|---|---|---|---|---|
|
a | Si(OEt)3 | 92 | 60 |
| b | Si(OEt)2Me | 95 | 76 | |
| c | Si(OEt)Me2 | 81 | 62[b] | |
| d | Si(OEt)2Ph | 69[b] | 35[b] | |
| e | SiMe2Ph | 54 (71[b]) | 32 | |
| f | SiEt3 | 10[b] | < 2 |
Isolated yield (%) unless otherwise indicated;
yield calculated based on 1H NMR analysis of reaction mixtures.
Using the diethoxymethylsilyl group, we synthesized macrocyclic alkenyl siloxane compounds with a range of ring sizes in moderate to excellent yields (Scheme 3). Diastereomers gave differing yields of purified products. For some substrates, silyl groups were incorporated into each of the alkene termini of the diene substrates. The trans-cyclohexanediols with silyl groups on different termini behaved similarly (12 and 14) while the silyl regioisomers of cis-cyclohexanediols behaved differently (13 and 15). To test the generality of this method in more complex molecules, two substrates inspired by previous work22 were prepared, incorporating vinylsiloxanes on either alkene termini. The 16-membered rings were both formed in moderate yields (18 and 21).
Scheme 3.

Isolated yields of the RCM of various substrates and protodesilylation of the alkenyl siloxane products (in parentheses).
As envisaged, the tri-substituted olefins in the macrocyclic products have the E-configuration (except for 17 and 21 that are mixtures of both stereoisomers with high E selectivity). This suggests that one of the two productive metallocyclobutane intermediates is preferably formed leading to the E-silyl alkene; however, further mechanistic studies are required. All corresponding Z-disubstituted olefins were obtained by protodesilylation with good to excellent yields while maintaining the geometry of the olefins.
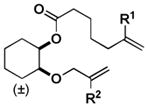
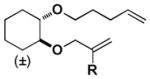
We next sought to understand the role of the silyl group in controlling the stereochemical outcome of the reaction. For comparison, all of the corresponding simple (non-silyl containing) RCM precursors were synthesized in order to determine the intrinsic stereoselectivity of the substrates (Table 2). Upon treatment with the optimized reaction conditions as well as typical RCM conditions using second generation Grubbs catalyst, Z selective (entries 1, 2, 6, 7, 9, 10, and 11), E selective (entries 4, 5, 8, 12, and 14), and non-selective outcomes (entry 13) were observed. The introduction of the silyl group was found to reinforce the intrinsic stereoselectivity and, more dramatically, for some substrates completely override it. This confirmed our initial hypothesis that a silyl group serves as an effective controlling group in macrocyclic RCM reactions.
Table 2.
Influence of the silyl group on the specificity and stereoselectivity of RCM reactions.
| Substrate | R | Cond. I | Cond. II | Substrate | R | Cond. I | Cond. II | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
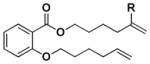
| 1 |
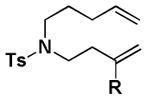
|
[Si][a] | Z[b] | - | 8 |
|
|
|
Z | - | |||||
| H | Z | Z | H | [Si] | Z | - | |||||||||
| H | H | c. mix. (19:81) | c. mix. (19:81) | ||||||||||||
| 2 |
|
[Si] | Z | - | 9 |
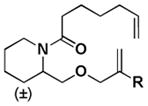
|
[Si] | Z | - | ||||||
| H | c. mix.[c] (Z)[d] | c. mix. (Z) | H | c. mix. (Z) | c. mix. (Z) | ||||||||||
| 3 |
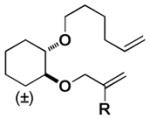
|
[Si] | Z | - | 10 |
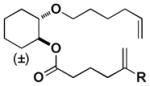
|
[Si] | Z | - | ||||||
| H | c. mix. | c. mix. | H | c. mix. (76:24) | c. mix. (76:24) | ||||||||||
| 4 |
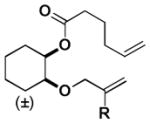
|
[Si] | Z | - | 11 |
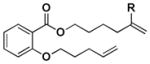
|
[Si] | Z | - | ||||||
| H | c. mix. (24:76) | c. mix. (24:76) | H | 81:19[e] | 80:20[f] | ||||||||||
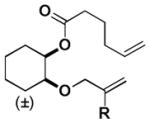
| 5 |
|
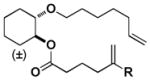
[Si] | Z | - | 12 |
|
[Si] | 90:10 | - | ||||||
| H | E | E | H | 28:72 | 24:76 | ||||||||||
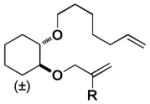
| 6 |
|
[Si] | Z | - | 13 |
|
[Si] | Z | - | ||||||
| H | c. mix. (Z) | c. mix. (Z) | H | 57:43[e] | 54:46[f] | ||||||||||
| 7 |
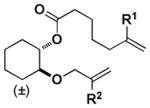
|
|
|
Z | - | 14 |
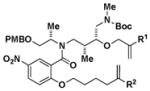
|
|
|
Z | - | ||||
| H | [Si] | Z | - | H | [Si] | 86:14 | - | ||||||||
| H | H | c. mix. (Z) | c. mix. (Z) | H | H | 36:64 | - | ||||||||
Cond. I: cat. A (20 mol%), toluene, 35 °C; Cond. II: Grubbs II (10 mol%), 1,4-benzoquinone (20 mol%), toluene, 35 °C;
For silylated substrates, Z:E assignment is based on the protodesilylated products;
Single stereoisomer reported for Z:E ratios >98:2, otherwise Z:E ratio given (determined by 1H NMR analysis of the crude);
c. mix.: complex mixture of products (cyclized and uncyclized oligomers);
stereochemistry of cyclized monomer reported in parentheses whenever its proportion within the complex mixture was sufficient for determination;
reactions performed at room temperature;
reactions performed in reflux DCM.
It is also noteworthy that several of the simple substrates gave rise to complex mixtures of products (Table 2, entries 2, 4, 6, 7, 8, 9, and 10), sometimes without detectable levels of cyclized monomer (entry 3). The LCMS analysis of the crude reaction indicated the formation of cyclized dimers and other polymeric by-products. This is a general problem for macrocyclic RCM reactions of simple olefins.23 While the reaction conditions for the simple substrates were not optimized, these results point to the ability of the silyl group to suppress the formation of undesired products.
Based on our data, we propose a model for the reaction pathways depicted in Scheme 4. Several unproductive pathways are involved in RCM reactions of simple olefins including re-opening of the monocyclized product (A−), cross-metathesis to generate an acyclic dimer or oligomer (B, C), and possible cyclization at either of these stages (C, D). In contrast, when the silyl group is incorporated in one of the olefins, pathway A leading to the desired product is no longer reversible. When we re-subjected the purified trisubstituted silyl alkene product to the reaction conditions, no reactivity was observed. Pathway B exists to generate the acyclic dimers, but only through the CM between the simple olefins – the 2-silyl alkene remains a spectator to CM under our reaction conditions.24 However, when re-subjected to the reaction conditions, the purified acyclic cross-dimers of the silylated substrates yielded macrocyclic products with conversion comparable to that starting from monomer.25 Additionally, since the 2-silyl alkene remains a spectator to CM, pathway C is shut down. Pathway D is also blocked because the formation of a tetrasubstituted alkene with two silyl groups is highly disfavored.
Scheme 4.

Possible reaction pathways of macrocyclic RCM with and without a 2-silyl group.
Overall, the silyl group is able to lower the reactivity of the attached olefin, thereby suppressing non-productive pathways while yielding the desired product. In accord with this analysis, for the substrates that gave low yields, only unreacted starting material, the acyclic cross-dimers, and a styrene derivative were observed along with the product. To explore the “trapping” role of the silyl group, the monocyclized Z-alkene compound 7b (not observed from the RCM of the simple diene, Table 2, entry 3) obtained from protodesilylation of compound 7 was subjected to reaction condition II. Not surprisingly it was almost completely consumed to generate dimers and oligomers (see supporting information). Owing to the fact that the silyl group can be removed, this method offers a means to cyclize recalcitrant substrates using RCM.
In summary, 8- to 16-membered macrocyclic rings containing E-trisubstituted silyl olefins and the corresponding Z-disubstituted olefins can be accessed using RCM of vinylsiloxane substrates. This study illustrates the use of a silyl group in controlling the stereoselectivity of RCM reactions in macrocyclic systems. Additionally, productive RCMs of vinylsiloxanes allow access to challenging simple olefin products that may be otherwise disfavored compared to acyclic or cyclic dimers and oligomers. As a second goal of this project, studies are underway to explore the use of alkenyl siloxanes as chemical handles for further functionalization and diversification of the RCM products.
Supplementary Material
Acknowledgments
We gratefully acknowledge Drs. Tuoping Luo, Drew Adams, Mingji Dai, Sivaraman Dandapani, Giovanni Muncipinto, Bhaumik Pandya, Eamon Comer, and Qiu Wang for helpful discussions, and Christopher Johnson and Joachim Azzi for their assistance with SFC/MS analyses. This work was funded by the NIGMS-sponsored Center of Excellence in Chemical Methodology and Library Development (Broad Institute CMLD; P50 GM069721). S.L.S. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
Supporting Information. Complete Ref. 14b and 22, experimental procedures, and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.(a) Arisawa M, Nishida A, Nakagawa M. J Organomet Chem. 2006;691:5109–5121. [Google Scholar]; (b) Chattopadhyay SK, Karmakar S, Biswas T, Majumdar KC, Rahaman H, Roy B. Tetrahedron. 2007;63:3919–3952. [Google Scholar]; (c) Deiters A, Martin SF. Chem Rev. 2004;104:2199–2238. doi: 10.1021/cr0200872. [DOI] [PubMed] [Google Scholar]; (d) Gradillas A, Perez-Castells J. Angew Chem Int Ed. 2006;45:6086–6101. doi: 10.1002/anie.200600641. [DOI] [PubMed] [Google Scholar]; (e) Martin SF. Pure Appl Chem. 2005;77:1207–1212. [Google Scholar]; (f) Rouge dSA, Kaiser CR, Ferezou J-P. Quim Nova. 2008;31:655–668. [Google Scholar]
- 2.Grubbs RH, Marsella MJ, Maynard HD. WO 98/30557 A1. Chem Abstr 1998. 1998;129:136661.
- 3.(a) Peng LF, Stanton BZ, Maloof N, Wang X, Schreiber SL. Bioorg Med Chem Lett. 2009;19:6319–6325. doi: 10.1016/j.bmcl.2009.09.089. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL, Taveras KM, Hyman JM, Lee SW, Koehler AN, Chen JK, Fox JL, Mandinova A, Schreiber SL. Nat Chem Biol. 2009;5:154–156. doi: 10.1038/nchembio.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369. [DOI] [PubMed] [Google Scholar]
- 5.(a) Garcia-Fandino R, Aldegunde MJ, Codesido EM, Castedo L, Granja JR. J Org Chem. 2005;70:8281–8290. doi: 10.1021/jo050568w. [DOI] [PubMed] [Google Scholar]; (b) Troin Y, Sinibaldi ME. Targets Heterocycl Syst. 2009;13:120–146. [Google Scholar]
- 6.(a) Jimenez-Hopkins M, Hanson PR. Org Lett. 2008;10:2223–2226. doi: 10.1021/ol800649n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kotha S, Mandal K, Tiwari A, Mobin SM. Chem Eur J. 2006;12:8024–8038. doi: 10.1002/chem.200600540. [DOI] [PubMed] [Google Scholar]; (c) Kotha S, Singh K. Eur J Org Chem. 2007:5909–5916. [Google Scholar]; (d) Krishna PR, Reddy PS. J Comb Chem. 2008;10:426–435. doi: 10.1021/cc700171p. [DOI] [PubMed] [Google Scholar]; (e) Lu K, Huang M, Xiang Z, Liu Y, Chen J, Yang Z. Org Lett. 2006;8:1193–1196. doi: 10.1021/ol060221v. [DOI] [PubMed] [Google Scholar]; (f) Mukherjee P, Roy SJS, Sarkar TK. Org Lett. 2010;12:2472–2475. doi: 10.1021/ol100557f. [DOI] [PubMed] [Google Scholar]; (g) Oikawa M, Naito S, Sasaki M. Tetrahedron Lett. 2006;47:4763–4767. [Google Scholar]; (h) Spiegel DA, Schroeder FC, Duvall JR, Schreiber SL. J Am Chem Soc. 2006;128:14766–14767. doi: 10.1021/ja065724a. [DOI] [PubMed] [Google Scholar]; (i) Zhou A, Rayabarapu D, Hanson PR. Org Lett. 2009;11:531–534. doi: 10.1021/ol802467f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Hung AW, Ramek A, Wang Y, Kaya T, Wilson JA, Clemons PA, Young DW. Proc Natl Acad Sci U S A. 2011;108:6799–6804. doi: 10.1073/pnas.1015271108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Prunet J. Angew Chem Int Ed. 2003;42:2826–2830. doi: 10.1002/anie.200301628. [DOI] [PubMed] [Google Scholar]; (b) Lee CW, Grubbs RH. Org Lett. 2000;2:2145–2147. doi: 10.1021/ol006059s. [DOI] [PubMed] [Google Scholar]
- 8.(a) Enders D, Lenzen A, Backes M, Janeck C, Catlin K, Lannou MI, Runsink J, Raabe G. J Org Chem. 2005;70:10538–10551. doi: 10.1021/jo0518093. [DOI] [PubMed] [Google Scholar]; (b) Miles JAL, Mitchell L, Percy JM, Singh K, Uneyama E. J Org Chem. 2007;72:1575–1587. doi: 10.1021/jo0620258. [DOI] [PubMed] [Google Scholar]
- 9.(a) Fellows IM, Kaelin DE, Martin SF. J Am Chem Soc. 2000;122:10781–10787. [Google Scholar]; (b) Meng D, Bertinato P, Balog A, Su DS, Kamenecka T, Sorensen EJ, Danishefsky SJ. J Am Chem Soc. 1997;119:10073–10092. [Google Scholar]
- 10.(a) Hanessian S, Guesne S, Chenard E. Org Lett. 2010;12:1816–1819. doi: 10.1021/ol100427v. [DOI] [PubMed] [Google Scholar]; (b) Yadav JS, Satheesh G, Murthy CVSR. Org Lett. 2010;12:2544–2547. doi: 10.1021/ol100755v. [DOI] [PubMed] [Google Scholar]
- 11.(a) Fürstner A, Radkowski K. Chem Commun. 2002:2182–2183. doi: 10.1039/b207169j. [DOI] [PubMed] [Google Scholar]; (b) Fürstner A, Stelzer F, Rumbo A, Krause H. Chem Eur J. 2002;8:1856–1871. doi: 10.1002/1521-3765(20020415)8:8<1856::AID-CHEM1856>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]; (c) Lacombe F, Radkowski K, Seidel G, Fürstner A. Tetrahedron. 2004;60:7315–7324. [Google Scholar]; (d) Fürstner A, Guth O, Rumbo A, Seidel G. J Am Chem Soc. 1999;121:11108–11113. [Google Scholar]
- 12.(a) Bunz UHF, Kloppenburg L. Angew Chem Int Ed. 1999;38:478–481. doi: 10.1002/(SICI)1521-3773(19990215)38:4<478::AID-ANIE478>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]; (b) Fürstner A, Davies PW. Chem Commun. 2005:2307–2320. doi: 10.1039/b419143a. [DOI] [PubMed] [Google Scholar]
- 13.(a) Vougioukalakis GC, Grubbs RH. J Am Chem Soc. 2008;130:2234–2245. doi: 10.1021/ja075849v. [DOI] [PubMed] [Google Scholar]; (b) Flook MM, Jiang AJ, Schrock RR, Muller P, Hoveyda AH. J Am Chem Soc. 2009;131:7962–7963. doi: 10.1021/ja902738u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Meek SJ, O’Brien RV, Llaveria J, Schrock RR, Hoveyda AH. Nature. 2011;471:461–466. doi: 10.1038/nature09957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trisubstituted vinyl bromides generated from RCM reactions of vinyl bromides can also serve this purpose. However, the application was quite limited. See: Polito L, Cravini M, Poletti L, Lay L. Synth Commun. 2006;36:2203–2209.Paone DV, et al. J Med Chem. 2007;50:5564–5567. doi: 10.1021/jm070668p.
- 15.Pietraszuk C, Fischer H, Rogalski S, Marciniec B. J Organomet Chem. 2005;690:5912–5921. [Google Scholar]
- 16.Trialkylsilanes using trimethylsilyl, tert-butyldimethylsilyl, or phenyldimethylsilyl group were prepared for ring closure. The reactions led to moderate to good yield for 5- and 6-membered rings. Larger rings (8-membered and above) completely failed to close.
- 17.Trost BM, Ball ZT. J Am Chem Soc. 2001;123:12726–12727. doi: 10.1021/ja0121033.Schuman M, Gouverneur V. Tetrahedron Lett. 2002;43:3513–3516.Trost BM, Ball ZT. J Am Chem Soc. 2005;127:17644–17655. doi: 10.1021/ja0528580.Alternatively, for some examples of RCM reactions leading to endocyclic silanes or siloxanes see: Denmark SE, Muhuhi JM. J Am Chem Soc. 2010;132:11768–11778. doi: 10.1021/ja1047363.Lee YJ, Schrock RR, Hoveyda AH. J Am Chem Soc. 2009;131:10652–10661. doi: 10.1021/ja904098h.
- 18.Stewart IC, Ung T, Pletnev AA, Berlin JM, Grubbs RH, Schrodi Y. Org Lett. 2007;9:1589–1592. doi: 10.1021/ol0705144. [DOI] [PubMed] [Google Scholar]
- 19.Vougioukalakis GC, Grubbs RH. Chem Rev. 2010;110:1746–1787. doi: 10.1021/cr9002424. [DOI] [PubMed] [Google Scholar]
- 20.No decompostition studies have been reported on this particular catalyst. However, Csp2-H bond insertion of the N-aryl ring on the NHC ligand has been reported before. See: Hong SH, Chlenov A, Day MW, Grubbs RH. Angew Chem Int Ed. 2007;46:5148–5151. doi: 10.1002/anie.200701234.
- 21.Catalyst deactivation was shown to occur via β-Si migration. See: Pietraszuk C, Fischer H. Chem Commun. 2000:2463–2464.Pietraszuk C, Fischer H. Organometallics. 2001;20:4641–4646.
- 22.Marcaurelle LA, et al. J Am Chem Soc. 2010;132:16962–16976. doi: 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Fürstner A, Langemann K. J Org Chem. 1996;61:3942–3943. doi: 10.1021/jo960733v. [DOI] [PubMed] [Google Scholar]; (b) Lee CW, Grubbs RH. J Org Chem. 2001;66:7155–7158. doi: 10.1021/jo0158480. [DOI] [PubMed] [Google Scholar]; (c) Monfette S, Fogg DE. Chem Rev. 2009;109:3783–3816. doi: 10.1021/cr800541y. [DOI] [PubMed] [Google Scholar]
- 24.No CM product between the vinylsiloxane and the simple olefin was observed under the optimal reaction conditions. For the defination of “spectator”, see: Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882.
- 25.The acyclic cross-dimers (E and Z) of substrate 3a were resubmitted to condition I. The reaction yielded 4a in 60% yield (by 1H NMR).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.