

Abstract

Histone proteins are subject to dynamic post-translational modifications (PTMs) that cooperatively modulate the chromatin structure and function. Nearly all functional PTMs are found on the N-terminal histone domains (tails) of ∼50 residues protruding from the nucleosome core. Using high-definition differential ion mobility spectrometry (FAIMS) with electron transfer dissociation, we demonstrate rapid baseline gas-phase separation and identification of tails involving monomethylation, trimethylation, acetylation, or phosphorylation in biologically relevant positions. These are by far the largest variant peptides resolved by any method, some with PTM contributing just 0.25% to the mass. This opens the door to similar separations for intact proteins and in top-down proteomics.
Histone proteins (H1, H2A, H2B, H3, and H4) are extensively enzymatically modified by post-translational modifications (PTM) such as N-methylation (me), N-acetylation (ac), or O-phosphorylation (p) that regulate chromatin structure and function by recruiting proteins involved in replication, transcription, DNA repair, and chromatin compaction. Interpreting the PTM-mediated protein language necessitates analyses of intact histones or their large domains preserving the co-occurring multisite PTM information.1 In particular, the N-terminal domains (tails) protruding from the nucleosome core are strongly enriched in PTMs and can be cleaved off by endoproteinase Glu-C enzyme.2,3
The staggering challenge here is disentangling numerous isomeric proteoforms that feature the same set of PTMs in different positions (PTM localization variants). While liquid chromatography (LC) techniques such as WCX-HILIC can fractionate histones by me or ac content, many variants still coelute.4−6 Tandem mass spectrometry (MS/MS) by any mechanism could characterize the individual variants, but fails for mixtures of three or more variants, since those with internal PTM positions yield no unique fragments.7,8
A growing alternative or complement to LC is ion mobility spectrometry (IMS) based on gas-phase transport properties. Linear IMS approaches (e.g., drift tube or traveling-wave) separate ions by mobility (K) at a moderate electric field (E).9 While these can resolve many isomers including some localization variants,10 their power in conjunction with MS is constrained by the intrinsically tight correlation between the mass-to-charge ratio (m/z) and collision cross section within a chemical class. That correlation is much looser for differential or field asymmetric waveform IMS (FAIMS) that sorts ions by the difference of mobilities at high and low field strength.11−13
In FAIMS, a gas flow moves ions through a gap between two electrodes. A periodic asymmetric field established there (with a short segment of high positive E and long segment of low negative E) pushes ions toward one of the electrodes, depending on the difference between the mobilities in the two segments. A fixed compensation voltage (CV) superposed on the waveform can equilibrate a given species and let it through the gap into MS analyzer.14,15 The resolution is maximized by strong homogeneous field in planar gaps, precise waveforms and He/N2 or H2/N2 buffers.16−18 Another path to higher FAIMS resolution is adding gas modifiers (usually organic volatiles) that cluster with ions in the low-field segment and desorb upon collisional heating in the high-field segment. However, this approach was mostly demonstrated for small singly charged molecules rather than peptides or proteins.19,20 One problem with extending it to multiply charged ions such as peptides generated by electrospray ionization (ESI) is the proton transfer to vapor modifiers that have higher proton affinity than N2, He, or H2.21 Some proposed means to mitigate that22 would be explored in the future. Here we employ the He/N2 buffers established in proteomic analyses using FAIMS.
The variants separated by any method need to be identified. Assigning the FAIMS spectral peaks using standards can be realistic in targeted applications, but global analyses and discovery of novel proteoforms call for peptide identification by MS/MS. Large multiply charged modified peptides are optimally sequenced by electron transfer dissociation (ETD) that severs the backbone, but not weaker PTM links, producing informative c- and z-fragments,23 which enable detailed analyses of complete histone H3 tails.24,25
Previous IMS and FAIMS studies focused on the localization variants of peptides in the bottom-up range, with the largest separated species being methylated or acetylated H3 and H4 mini-tails with ∼20–25 residues.26,27 The ETD characterization of FAIMS-resolved variants was limited to even smaller peptides (∼1.5 kDa).7,8 Such segments cannot reveal the full PTM information encoded in histones. The position of a single PTM would intuitively affect the 3-D geometries of larger peptides less, diminishing their resolution by IMS methods. Here we demonstrate widely applicable FAIMS separation of complete isomeric 5.4 kDa histone tails that contain nearly all histone PTMs–a major capability for proteomics and epigenetic research.
Experimental Section
Materials and Reagents
Reagents were from Sigma, except 2-Cl-(Trt)-Cl resin (GenScript), 9-fluorenylmethyl carbazate (TCI chemicals), Fmoc-Lys(Me, Boc)-OH (AnaSpec), Fmoc-Lys(Me)3-OH (GL Biochem), Fmoc-Lys(Ac)-OH, Fmoc-Ser (PO(OBzl)OH)-OH, Fmoc-Thr(PO(OBzl)OH)-OH, and Fmoc-Tyr(PO(OBzl)OH)-OH (Merck) and VA-04 (J&K). The HPLC column was XSelect CSH C18 Prep Column 10 × 250 mm, 130 Å, 5 μm bead diameter (Waters).
Synthesis of Histone Tail Standards
These standards were obtained by producing two peptides of 25 residues each via Fmoc SPPS and joining them via native chemical ligation (NCL).28 The Fmoc hydrazide beads were prepared in-house with 0.49 mmol/g loading. The part I (ARTKQTARKSTGGKAPRKQLATKA) was synthesized on Fmoc-hydrazine 2CTC resin, the part II (CRKSAPATGGVKKPHRYRPGTVALR) was produced on Wang resin with Glu residue attached. The amounts of 10 μmole at a time were made on Intavis ResPep SPPS machine using the program specified in the SI. The peptides were detached from resin by the cleavage solution (80% TFA, 2.5% H2O, 2.5% DTT, 15% triisopropylsilane) and precipitated by adding 10× volumes of ice-cold diethyl ether. The resulting part I contained a hydrazine group at its C-terminus, while the part II started with Cys. Both parts were purified on the Waters XSelect CSH C18 column using Akta Purifier 900 (HPLC conditions in the SI).
In the NCL step, the parts I and II were resuspended at 10 mg in 100 μL of 6 M GndCl (pH 2.0), with 3.4 mg MPAA added for part II. The hydrazine in part I was converted to an azide by oxidation for 20 min at −15 °C on an acetone–ice bath by adding 10 μL of 0.5 M NaNO2. The two solutions were then combined and pH adjusted to 7.0 using 6 M NaOH. After 4–6 h, the reaction was stopped by adding 20 uL of 0.5 M TCEP (pH 7.0) for 20 min. The product was purified on a PD-mini trap column (GE-healthcare) and dried under vacuum. Desulfurization was performed using the Va-044 azo initiator.29
FAIMS Operation
Histone tails standards were analyzed employing our FAIMS device with 1.9 mm gap width, coupled to the Thermo LTQ XL ion trap with electrospray ionization (ESI) source and electron transfer dissociation (ETD) stage via a slit aperture/ion funnel interface.8,16 The waveform amplitude (dispersion voltage) was 4 kV, the CV scan rate was 0.33 V/min, and the flow of carrier gas (65% He/35% N2) was 2 L/min. Samples were delivered for analysis using 20 μM i.d. glass emitters prepared by chemical etching.30 Raw data was processed using MSconvert (Proteowizard package)31 and a custom R script.
FAIMS/ETD Analyses
The ETD data were recorded by successively setting the CVs to apexes of major peaks in FAIMS and fragmenting transmitted ions. The initial FAIMS/ETD work has involved 2+ and 3+ precursors, which produced the 1+ fragments only, and used single reaction time (tETD) of 120 ms.7,8,32 Present larger peptides with much higher z yield multiply charged fragments with z up to 5. With the limited resolving power of ion trap, rich ETD MS/MS spectra of large peptides include many mass interferences. Longer tETD permit more consecutive ETD steps, yielding different fragments with overall lower z. To disentangle those interferences and raise the informative content of spectra, all species were fragmented at tETD of 15, 30, and 150 ms. For utmost specificity, we performed MS/MS in the zoom and ultrazoom modes. As these are less sensitive than the normal mode, each ETD spectrum was acquired for 10 min. The final ETD MS/MS spectra are combinations of regions extracted from the spectra with different tETD.
Results and Discussion
Variant Separations
To evaluate FAIMS separation we produced 18 proteoforms of human H3.1 tail (residues 2–51, monoisotopic mass of 5350 Da) with PTMs in biologically relevant positions (Table 1) by combination of Fmoc solid phase peptide synthesis and native chemical ligation using novel hydrazide chemistry.28
Table 1. PTM Localizations in H3 Tails (ART3K4QT6ARK9S10TGGK14APRK18QLATK23AARK27S28APATGGVK36KPHRY41RPGTVALRE).
| PTM | positions |
|---|---|
| Me | K4, K9, K23 |
| me3 | K4, K9, K23, K27, K36 |
| Ac | K9, K14, K18, K27, K36 |
| P | T3, T6, S10, S28, Y41 |
The modified variants (5 μM solutions in water with 0.1% formic acid) were initially infused individually, and then their FAIMS spectra (plots of signal vs CV) were overlaid. To correct for uncontrolled fluctuations of operating parameters (primarily the ambient pressure and temperature), the CV axes of individual FAIMS spectra were linearly scaled using an internal calibrant - the unmodified tail (1 μM). For transferability across devices with unequal gap widths, the CV scale is expressed as the compensation field (EC).
Figure 1 illustrates the experimental concept. Trimethylated tails form charge states z = 8–12 (Figure 1A), each exhibiting a unique FAIMS spectrum (Figure 1B,C). Overall, the 10+ peaks were most intense and permitted maximum discrimination for monomethylated, acetylated, and phosphorylated variants (Figure 2A–C, S1). Only five variants (K4me, K9me, K23me3, K9ac, K27ac) of the 18 total exhibited secondary features at >20% of the base peak heights at this charge. We baseline-resolved all variants for me, two groups for me3 (K4/K36, K9/K23/K27), three groups for ac (K9, K14/K27, K18/K36), and four for p (S10, S28/Y41, T3, T6). Since binary variant mixtures can be fully characterized using ETD,7,8 the only case not completely solved by FAIMS/ETD here at z = 10 is the ternary K9me3/K23me3/K27me3 mixture. We confirmed the robustness and reproducibility of the experimental procedure by ensuring that the spectra for binary and ternary variant mixtures matched the superimposed spectra for individual species (Figures 2D–F, S2, and S3).
Figure 1.

ESI MS spectrum for a trimethylated histone tail (A) and FAIMS spectra for z = 10 (B) and 11 (C).
Figure 2.
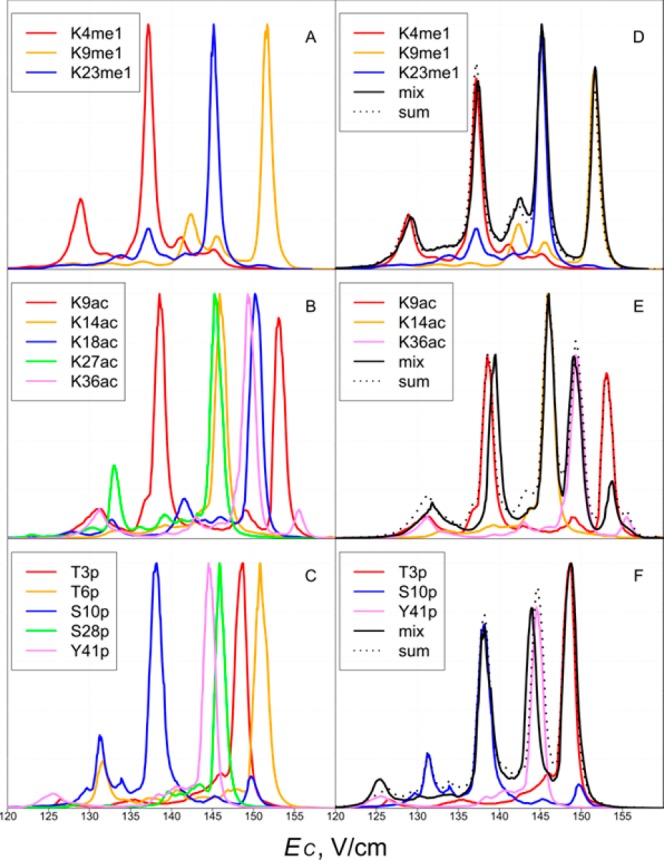
FAIMS spectra for monomethylated, acetylated, and phosphorylated histone tails at z = 10 (A–C), same for the spectra for selected tertiary mixtures (D–F). The spectra for individual variants are normalized in (A)–(C), vertically scaled to match the features for the mixture in (D)–(F). The sums of spectra for all variants are also shown in (D)–(F).
Orthogonality of Separation across Charge States
The separations in different charge states are mutually independent (orthogonal),26,27,33 hence, the variants overlapping at one z are often resolved at another. For example, the overlaps at z = 10 are fully resolved at other charge states: K4me3/K36me3 at z = 8, 11, and 12; K9me3/K23me3 and K9me3/K27me3 at z = 11; K14ac/K27ac at z = 11, 12; K18ac/K36ac and S28p/Y41p at z = 9, 12 (Figure S1). The only partly unresolved binary isomeric pair was K23me3/K27me3, with the best (half-maximum) resolution at z = 11 (Figure 1C). As the result, the me3 variants were best separated at z = 11 (Figure 1C).
To assess said orthogonality systematically, we tabulated the EC values for tallest peaks of all variants with trimethylation, acetylation, and phosphorylation PTMs in every charge state (Table S1) and computed their pairwise correlations (Table S2). For example, the plot of EC for five me3 variants at z = 10 versus those at z = 11 reveals a linear correlation of R2 = 0.01 (Figure S4a), and the average R2 for all 30 charge state pairs (10 pairs for every PTM) is 0.25, suggesting no significant correlation. This conclusion is confirmed by rigorous statistical tests (see SI) and clearly seen in Figure S4b.
Separation of Acetylated and Trimethylated Peptides
Attachment of me3 (42.047 Da) and ac (42.011 Da) to lysine residues is mutually exclusive and has different functional outcomes. For example, (i) H3K9me3 or H3K27me3 are repressing marks, whereas H3K9ac or H3K27ac activate transcription,34,35 (ii) H3K36me3 occurs predominantly in the coding regions of genes and participates in transcriptional elongation, whereas H3K36ac is localized in the promoters and facilitates transcription initiation.36 Baseline MS resolution of the tails with me3 and ac requires resolving power of ∼3 × 105 (and ∼8 × 105 for whole H3) in theory, and is commonly precluded by spectral congestion in practice. However, me3 and ac may well favor distinct 3-D geometries, enabling FAIMS separation. Of our three tail pairs with me3 or ac on same residue, two (PTM on K27 or K36) were resolved baseline and one (PTM on K9) nearly so (Figures 3 and S5).
Figure 3.
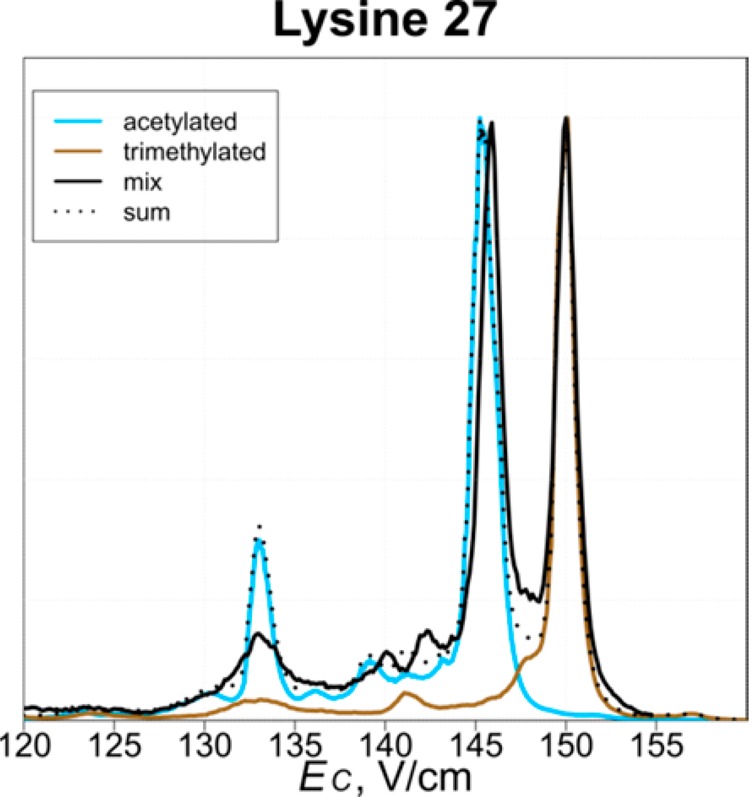
Normalized FAIMS spectra for the tails trimethylated or acetylated at K27 and their mixture.
Confirmation by ETD
The ETD data unequivocally prove the variant resolution seen at the precursor level. For instance, all fragments observed at high S/N ratio corroborate the separation of T3p/S10p/Y41p mixture (Figure 4).
Figure 4.

FAIMS/ETD analysis of tertiary mixture of phosphorylated tails: FAIMS spectrum (left) and selected ETD spectral windows (from the data at various tETD) comprising the informative fragments with schematic sequences on top (right).
Parallel results for other representative binary and tertiary mixtures (K4me1/K9me1/K23me1; K4me3/K27me3; K9ac/K14ac/K36ac; K9ac/K18ac/K27ac; T6p/S10p/S28p) show the general utility of procedure (Figure S6). For utmost specificity, we performed MS/MS in the zoom mode. While the ultrazoom mode delivers yet greater MS resolution, a limited scan range of 100 m/z units and low signal demand long acquisitions; we obtained one spectrum to prove feasibility (Figure S7).
Conclusions
We have demonstrated near-universal complete separation of PTM localization variants (involving methylation, acetylation, and phosphorylation at distinct residues) for peptides above 5 kDa, where PTMs amount to 0.25–1.5% of the peptide mass. The maximum EC difference between principal peaks is ∼10–15% with all PTMs (e.g., for K4me/K9me at 10+, K4me/36me3 at 9+, K9/K14ac at 11+, or T6p/S10p at 10+), same as or only moderately below the values for methylated or acetylated mini-tails (2–3 kDa).26,27 Hence, the separation space narrows quite gradually (if at all) for longer peptides, encouraging us to extend this FAIMS approach to yet larger peptides and intact proteins. In fact, the 5.4 kDa histone tails explored here approach the size of small proteins (e.g., insulin at 5.8 kDa). In principle, the orthogonality of separations between charge states expands the overall FAIMS peak capacity for polypeptides in proportion to the number of charge states (n) output by the ion source. With regular ESI, n scales roughly as the (peptide mass)1/2 or faster,37 increasing from 2–3 for typical tryptic peptides (∼1–1.5 kDa) to at least 5 for present tails to ∼10–15 for smaller proteins (∼10–20 kDa).38 That number can be augmented by generating higher z via supercharging and lower z via proton stripping.39,40 This trend largely underlies the success of this work and will facilitate the separation of localization variants for proteins. The main problem here is the ubiquitous conformational multiplicity (at charge states besides 10+ here), which would be more important for real samples with unequal variant abundances. We will seek to reduce that multiplicity by optimizing the ESI and FAIMS conditions.
Biological histone samples exhibit much higher complexity and dynamic range than the mixtures of standards in this study. The true variant distribution is convoluted in the analysis with nonuniformities of ionization efficiency, transmission through the FAIMS gap, and ETD yield in total and for specific informative fragments.8 This may facilitate or impede the detection of any variant in the presence of others. These discrimination effects are generally unequal across charge states, making the apparently redundant separations in multiple states helpful in practice.
While the filtering mechanism of FAIMS enables adding it to all MS instrument types, ion trapping platforms that effect extended accumulation and multistep fragmentation by ETD and other methods are eminently suitable. Analyses of real histone samples demand much higher MS peak capacity and mass accuracy provided by Fourier-Transform (FT) MS instruments in the Orbitrap or FTICR implementations. However, turbomolecular pumps are less effective for light gases (He or H2) with fast Brownian motion than all air components (N2, O2, Ar, CO2). That is manageable with FAIMS stages employing those gases coupled to ion traps operating at ∼10–5 Torr, but not to Orbitrap MS that needs ultrahigh vacuum (10–10 Torr).41 We are currently re-engineering the FAIMS stages and Orbitrap vacuum system to enable addition of high-resolution FAIMS using helium-rich buffers.
Comprehensive analyses also require an LC step in front to partition histone tails into fractions with same number of acetyl and methyl PTMs.4 The challenge for online LC/FAIMS integration is time scale mismatch: the typical chromatographic peak width is ∼10 s, while a full high-resolution FAIMS scan takes ∼10 min. One established solution is replacing a gradual FAIMS scan by stepping through discrete CVs.42 This approach best fits targeted applications, for example, distinguishing several peptide variants with known CVs such as present histone tails. In external convolution, the same LC gradient is repeated with different fixed CVs in a mode resembling selected reaction monitoring in MS.43,44 In internal convolution, the CV is rapidly stepped through several values (with dwell times of a few s) in a mode resembling multiple reaction monitoring while LC peak elutes.43,45,46 An alternative is off-line LC fractionation followed by direct infusion. We foresee the integration of high-definition FAIMS with 2-D LC and FTMS to allow unprecedentedly comprehensive characterization of proteomes and proteoforms.
Acknowledgments
We thank Drs. Y. C. Huang and Y. M. Li for guiding our synthetic work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b00379.
Description of procedures for histone tail synthesis, additional FAIMS spectra for further variants and charge states, and statistical methods for evaluation of the orthogonality between FAIMS and MS dimensions (PDF).
Author Contributions
§ The two senior authors of this work.
This research was funded by a grant from the VILLUM Foundation to the VILLUM Center for Bioanalytical Sciences at University of Southern Denmark (O.N.J.), a Lundbeck Foundation Postdoctoral Fellowship (P.S.), and NIH COBRE (P30 GM110761) and NSF CAREER (CHE-1552640) grants (A.S.). A.S. also holds a faculty appointment at the National Research Nuclear University MEPhI (Moscow Engineering Physics Institute), Russia.
The authors declare the following competing financial interest(s): Alexandre Shvartsburg has interest in Heartland MS that makes FAIMS and ion funnel systems utilized in this work.
Supplementary Material
References
- Jensen O. N. Nat. Rev. Mol. Cell Biol. 2006, 7 (6), 391. 10.1038/nrm1939. [DOI] [PubMed] [Google Scholar]
- Molden R. C.; Garcia B. A. Curr. Protoc. Protein Sci. 2014, 77 (7), 1. 10.1002/0471140864.ps2307s77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S. D.; Ueberheide B. M.; Liu Y.; Tackett A. J.; Diaz R. L.; Shabanowitz J.; Chait B. T.; Hunt D. F.; Allis C. D. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (7), 2086. 10.1073/pnas.0610993104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N. L.; DiMaggio P. A.; Plazas-Mayorca M. D.; Baliban R. C.; Floudas C. A.; Garcia B. A. Mol. Cell. Proteomics 2009, 8 (10), 2266. 10.1074/mcp.M900238-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S.; Schwämmle V.; Ruminowicz C.; Hansen T. A.; Wu X.; Helin K.; Jensen O. N. Proteomics 2014, 14 (19), 2200. 10.1002/pmic.201400084. [DOI] [PubMed] [Google Scholar]
- Tvardovskiy A.; Wrzesinski K.; Sidoli S.; Fey S. J.; Rogowska-Wrzesinska A.; Jensen O. N. Mol. Cell. Proteomics 2015, 14 (12), 3142. 10.1074/mcp.M115.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Y.; Creese A. J.; Horner J. A.; Cooper H. J. Rapid Commun. Mass Spectrom. 2009, 23 (13), 1963. 10.1002/rcm.4101. [DOI] [PubMed] [Google Scholar]
- Baird M. A.; Shvartsburg A. A. J. Am. Soc. Mass Spectrom. 2016, 27 (12), 2064. 10.1007/s13361-016-1498-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanucara F.; Holman S. W.; Gray C. J.; Eyers C. E. Nat. Chem. 2014, 6 (4), 281. 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- Ibrahim Y. M.; Shvartsburg A. A.; Smith R. D.; Belov M. E. Anal. Chem. 2011, 83 (14), 5617. 10.1021/ac200719n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guevremont R.; Barnett D. A.; Purves R. W.; Vandermey J. Anal. Chem. 2000, 72 (19), 4577. 10.1021/ac0000271. [DOI] [PubMed] [Google Scholar]
- Tang K.; Li F.; Shvartsburg A. A.; Strittmatter E. F.; Smith R. D. Anal. Chem. 2005, 77 (19), 6381. 10.1021/ac050871x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba J.; Bonneil E.; Pomiès C.; Eng K.; Thibault P. J. Proteome Res. 2009, 8 (7), 3355. 10.1021/pr801106a. [DOI] [PubMed] [Google Scholar]
- Guevremont R. J. Chromatogr. A 2004, 1058 (1–2), 3. 10.1016/S0021-9673(04)01478-5. [DOI] [PubMed] [Google Scholar]
- Shvartsburg A. A.Differential Ion Mobility Spectrometry; CRC Press: Boca Raton, FL, 2008. [Google Scholar]
- Shvartsburg A. A.; Li F.; Tang K.; Smith R. D. Anal. Chem. 2006, 78 (11), 3706. 10.1021/ac052020v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvartsburg A. A.; Danielson W. F.; Smith R. D. Anal. Chem. 2010, 82 (6), 2456. 10.1021/ac902852a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvartsburg A. A.; Seim T. A.; Danielson W. F.; Norheim R.; Moore R. J.; Anderson G. A.; Smith R. D. J. Am. Soc. Mass Spectrom. 2013, 24 (1), 109. 10.1007/s13361-012-0517-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider B. B.; Covey T. R.; Coy S. L.; Krylov E. V.; Nazarov E. G. Anal. Chem. 2010, 82 (5), 1867. 10.1021/ac902571u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves R. W.; Ozog A. R.; Ambrose S. J.; Prasad S.; Belford M.; Dunyach J.-J. J. Am. Soc. Mass Spectrom. 2014, 25 (7), 1274. 10.1007/s13361-014-0878-z. [DOI] [PubMed] [Google Scholar]
- Blagojevic V.; Chramow A.; Schneider B. B.; Covey T. R.; Bohme D. K. Anal. Chem. 2011, 83 (9), 3470. 10.1021/ac200100s. [DOI] [PubMed] [Google Scholar]
- Blagojevic V.; Koyanagi G. K.; Bohme D. K. J. Am. Soc. Mass Spectrom. 2014, 25 (3), 490. 10.1007/s13361-013-0800-0. [DOI] [PubMed] [Google Scholar]
- Mikesh L. M.; Ueberheide B.; Chi A.; Coon J. J.; Syka J. E. P.; Shabanowitz J.; Hunt D. F. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764 (12), 1811. 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H. R.; Sidoli S.; Haldbo S.; Sprenger R. R.; Schwämmle V.; Pasini D.; Helin K.; Jensen O. N. Anal. Chem. 2013, 85 (17), 8232. 10.1021/ac401299w. [DOI] [PubMed] [Google Scholar]
- Jung H. R.; Pasini D.; Helin K.; Jensen O. N. Mol. Cell. Proteomics 2010, 9 (5), 838. 10.1074/mcp.M900489-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvartsburg A. A.; Zheng Y.; Smith R. D.; Kelleher N. L. Anal. Chem. 2012, 84 (10), 4271. 10.1021/ac300612y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvartsburg A. A.; Zheng Y.; Smith R. D.; Kelleher N. L. Anal. Chem. 2012, 84 (15), 6317. 10.1021/ac301541r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.-C.; Chen C.-C.; Li S.-J.; Gao S.; Shi J.; Li Y.-M. Tetrahedron 2014, 70 (18), 2951. 10.1016/j.tet.2014.03.022. [DOI] [Google Scholar]
- Zheng J.-S.; Tang S.; Qi Y.-K.; Wang Z.-P.; Liu L. Nat. Protoc. 2013, 8 (12), 2483. 10.1038/nprot.2013.152. [DOI] [PubMed] [Google Scholar]
- Kelly R. T.; Page J. S.; Luo Q.; Moore R. J.; Orton D. J.; Tang K.; Smith R. D. Anal. Chem. 2006, 78 (22), 7796. 10.1021/ac061133r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessner D.; Chambers M.; Burke R.; Agus D.; Mallick P. Bioinformatics 2008, 24 (21), 2534. 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridon G.; Bonneil E.; Muratore-Schroeder T.; Caron-Lizotte O.; Thibault P. J. Proteome Res. 2012, 11 (2), 927. 10.1021/pr200722s. [DOI] [PubMed] [Google Scholar]
- Shvartsburg A. A.; Singer D.; Smith R. D.; Hoffmann R. Anal. Chem. 2011, 83 (13), 5078. 10.1021/ac200985s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J.; Rice J. C.; Strahl B. D.; Allis C. D.; Grewal S. I. Science 2001, 292 (5514), 110. 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- Koch C. M.; Andrews R. M.; Flicek P.; Dillon S. C.; Karaöz U.; Clelland G. K.; Wilcox S.; Beare D. M.; Fowler J. C.; Couttet P.; James K. D.; Lefebvre G. C.; Bruce A. W.; Dovey O. M.; Ellis P. D.; Dhami P.; Langford C. F.; Weng Z.; Birney E.; Carter N. P.; Vetrie D.; Dunham I. Genome Res. 2007, 17 (6), 691. 10.1101/gr.5704207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S. A.; Rao B.; Garcia B. A.; Hake S. B.; Diaz R. L.; Shabanowitz J.; Hunt D. F.; Allis C. D.; Lieb J. D.; Strahl B. D. J. Biol. Chem. 2007, 282 (10), 7632. 10.1074/jbc.M607909200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez de la Mora J. Anal. Chim. Acta 2000, 406 (1), 93. 10.1016/S0003-2670(99)00601-7. [DOI] [Google Scholar]
- Hoaglund-Hyzer C. S.; Counterman A. E.; Clemmer D. E. Chem. Rev. 1999, 99 (10), 3037. 10.1021/cr980139g. [DOI] [PubMed] [Google Scholar]
- Iavarone A. T.; Jurchen J. C.; Williams E. R. Anal. Chem. 2001, 73 (7), 1455. 10.1021/ac001251t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelimov K. B.; Jarrold M. F. J. Am. Chem. Soc. 1997, 119 (13), 2987. 10.1021/ja962914k. [DOI] [Google Scholar]
- Makarov A.; Denisov E. J. Am. Soc. Mass Spectrom. 2009, 20 (8), 1486. 10.1016/j.jasms.2009.03.024. [DOI] [PubMed] [Google Scholar]
- Cooper H. J. J. Am. Soc. Mass Spectrom. 2016, 27, 566. 10.1007/s13361-015-1326-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creese A. J.; Shimwell N. J.; Larkins K. P. B.; Heath J. K.; Cooper H. J. J. Am. Soc. Mass Spectrom. 2013, 24 (3), 431. 10.1007/s13361-012-0544-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swearingen K. E.; Hoopmann M. R.; Johnson R. S.; Saleem R. A.; Aitchison J. D.; Moritz R. L. Mol. Cell. Proteomics 2012, 11 (4), M111.014985. 10.1074/mcp.M111.014985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venne K.; Bonneil E.; Eng K.; Thibault P. Anal. Chem. 2005, 77 (7), 2176. 10.1021/ac048410j. [DOI] [PubMed] [Google Scholar]
- Canterbury J. D.; Yi X.; Hoopmann M. R.; MacCoss M. J. Anal. Chem. 2008, 80 (18), 6888. 10.1021/ac8004988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.