

Abstract
The cellular prion protein, PrPC, is attached by a glycosylphosphatidylinositol anchor to the outer leaflet of the plasma membrane. Its misfolded isoform PrPSc is the causative agent of prion diseases. Conversion of PrPC into PrPSc is thought to take place at the cell surface or in endolysosomal organelles. Understanding the intracellular trafficking of PrPC may, therefore, help elucidate the conversion process. Here we describe a time-resolved fluorescence energy transfer (FRET) assay reporting membrane expression and real-time internalization rates of PrPC. The assay is suitable for high-throughput genetic and pharmaceutical screens for modulators of PrPC trafficking. Simultaneous administration of FRET donor and acceptor anti-PrPC antibodies to living cells yielded a measure of PrPC surface density, whereas sequential addition of each antibody visualized the internalization rate of PrPC (Z′ factor >0.5). RNA interference assays showed that suppression of AP2M1 (AP-2 adaptor protein), RAB5A, VPS35 (vacuolar protein sorting 35 homolog), and M6PR (mannose 6-phosphate receptor) blocked PrPC internalization, whereas down-regulation of GIT2 and VPS28 increased PrPC internalization. PrPC cell-surface expression was reduced by down-regulation of RAB5A, VPS28, and VPS35 and enhanced by silencing EHD1. These data identify a network of proteins implicated in PrPC trafficking and demonstrate the power of this assay for identifying modulators of PrPC trafficking.
Keywords: cell surface, endocytosis, fluorescence resonance energy transfer (FRET), high-throughput screening (HTS), prion disease
Introduction
The infectious agent causing prion diseases (1) is a misfolded and aggregated isoform (PrPSc) of the host-encoded cellular prion protein (PrPC).5 The primary translation product of PrPC is cotranslationally translocated to the rough endoplasmic reticulum (2), where it undergoes the addition of N-linked oligosaccharide chains, the formation of an intramolecular disulfide bond, and the attachment of a glycosylphosphatidylinositol (GPI) anchor (3). Distal sorting delivers PrPC to the plasma membrane where it resides in lipid rafts (4). Mature PrPC is then recycled between the plasma membrane and the endocytic compartment (5).
Several processes have been reported to participate in the endocytosis of PrPC (6–8). PrPC located in lipid rafts may be internalized by caveolin/dynamin-dependent endocytosis. PrPC is transported via caveosomes to early endosomes or to lysosomes for degradation. Alternatively, internalization of PrPC may be mediated by a clathrin-dependent process (9–11) whereby PrPC migrates to clathrin-coated pits and is then transported to early endosomes. Dynamin-independent endocytosis pathways may also be involved. This process may be crucial for the conversion of normal PrPC into PrPSc, as the conformational change seems to take place at the cell surface and/or in the endocytic pathway. Real-time measurements of PrPC internalization are, therefore, of high interest and might eventually help identify therapeutic targets against prion diseases.
Fluorescence resonance energy transfer (FRET)-based methods are widely used for the detection of proteins (12) and to validate the proximity of proteins in cells and tissues (13). FRET is a quantum-mechanical process that transmits energy from an excited donor molecule to an acceptor molecule, resulting in red-shifted fluorescence emission by the latter (14). Time-resolved FRET methods based on the use of europium and allophycocyanin (APC) as donor and acceptor, respectively, allows for homogeneous-phase assays in the absence of background fluorescence (15). The long fluorescence lifetime of europium leads to a high signal-to-noise ratio and is thus suited to high-throughput applications.
In the present study we applied the FRET technology to the development of a robust and sensitive quantification assay for PrPC internalization. The assay was designed to detect either cell-surface expression of human PrPC or the internalization rate of PrPC in living cells. For the detection of PrPC on the cell surface, anti-PrP antibodies were added simultaneously, whereas the addition of the two FRET components at different time points enabled the measurement of the endocytosis rate of surface-resident PrPC by signal subtraction. Furthermore, we show that this assay can be used to identify drugs and genes that modulate PrPC surface expression and PrPC endocytosis.
Results
A homogeneous-phase TR-FRET immunoassay for PrP
We first developed a homogeneous TR-FRET immunoassay for the detection of recombinant mouse PrP (residues 23–231; recPrP) and PrPC in cell lysates. The principle of the assay is shown in Fig. 1. We tested several candidate antibodies from an in-house PrP-targeted antibody collection designated as POM antibodies (16) and the previously characterized 3F4 antibody (17) for their efficiency in this type of assay. Some of these antibodies target linear epitopes in the N-terminal, unstructured “flexible tail” of PrP, whereas others recognize discontinuous, conformational epitopes within the C-terminal globular domain of PrP (18–20).
Figure 1.
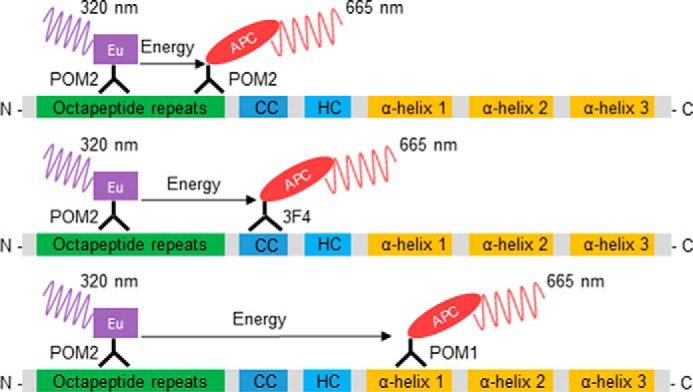
Schematic representation of the antibody-binding sites for PrP detection in the homogeneous TR-FRET assay. The assay is based on the in-solution binding of two fluorophore-labeled POM antibodies to identical or unique epitopes on PrP. POM antibodies were either labeled with europium chelate (Eu) donor fluorophore or APC-labeled acceptor. The antibody pairs POM2-Eu/POM2-APC (upper), POM2-Eu/3F4-APC (middle), and POM2-Eu/POM1-APC (lower) were used in the assays. POM1 binds to α-helix 1 in the C-terminal globular domain, POM2 binds to the octapeptide repeats (residues 51–91, mouse sequence) in the unfolded N-terminal part of PrP, and 3F4 binds to the charged cluster (CC). The close proximity of the donor and acceptor antibodies upon binding to PrP leads to a fluorescent TR-FRET signal at 665 nm after excitation at 320 nm, which is proportional to the PrP concentration. HC, hydrophobic core region.
Antibodies were coupled to the europium chelate (Eu) donor or the APC acceptor fluorophores. Various permutations of antibodies in both donor and acceptor functions were tested for their capability to detect recombinant and normal PrPC in solution. We predicted, and confirmed experimentally, that repetitive binding of the POM2 antibody to the multiple octapeptide repeats of PrP would allow for simultaneously utilization as both the donor and acceptor antibody (Fig. 1).
Among the different antibody FRET pairs, the antibody combination POM1-Eu/POM2-APC exhibited a satisfactory dynamic range over 3 logs (125 pg/ml–100 ng/ml) for the detection of recPrP diluted in crude Hpl Prnp−/− cell lysate, an immortalized hippocampal cell derived from Prnp−/− mice (21) (Fig. 2A), with signal linearity between 0.125 and 8 ng/ml (Fig. 2B). A lower detection limit (LDL) of ≤0.125 ng/ml was determined for the detection of recPrP.
Figure 2.

TR-FRET immunoassays for the detection of recombinant PrP and PrPC in crude cell lysates. A, dilution range of recPrP using the antibody tandem pair POM1-Eu/POM2-APC. Recombinant PrP (128–0.125 ng/ml) was serially diluted in 1 mg/ml Hpl Prnp−/− cell lysate in polystyrene 384-well microtiter plates and detected by TR-FRET. The sensitivity (LDL) of the assay is ≤0.125 ng/ml. B, dynamic range of recPrP detection. The assay shows high linearity (R2 = 0.93) at concentrations of 0.5–8 ng/ml for the detection of recPrP. C, dilution range of PrPC in murine N2aPK1 cell lysates using the antibody pair POM1-Eu/POM2-APC. Cells were lysed in RIPA buffer and serially diluted (1024–1 μg/ml) in 1 mg/ml Hpl Prnp−/− cell lysate detected by TR-FRET. The sensitivity (LDL) of the assay is ≤20 μg/ml crude cell extract. D, dynamic range of PrPC detection. The assay shows a high linearity (R2 = 0.92) in a concentration range between 500 and 30 μg/ml for the detection of PrPC. Each dilution was performed in 10 replicate measurements (duplicates). Error bars in the linear regression blots represent the standard deviation (±S.D.) of six replicate measurements. Student's t test: *, p < 0.05; **, p < 0.01; ****, p < 0.0001.
We next assessed the specificity of the assay for PrPC. We prepared serial dilutions of murine N2a PK1 cell lysates in Hpl Prnp−/− cell lysates followed by FRET measurement using POM1-Eu/POM2-APC for the detection of PrPC. PrPC was detected in lysates across a wide dynamic range (16–500 μg/ml) (Fig. 2C) with excellent linearity (Fig. 2D). An LDL of ≤ 20 μg/ml of crude cell extract was determined for the detection of PrPC in cells.
The performance of the TR-FRET-based immunoassay was further characterized by various validation parameters and their defined acceptance criteria: the coefficient of variance (%CV), the signal-to-noise ratio, and the Z′ factor (22), a widely used indicator for high-throughput suitability. A Z′ factor >0.5 is generally considered sufficient for cell-based high-throughput campaigns. These data are listed in Table 1. Hence, the above data demonstrate a robust and accurate performance of the assay with high specificity, which makes it suitable for the detection of both recPrP and PrPC in high-throughput assays.
Table 1.
Performance of the homogeneous FRET assay for recPrP (upper table) and PrPC from murine N2aPK1 cell lysates (lower table). The assay was evaluated according to three parameters: coefficient of variation (%CV), signal-to-noise ratio (S/N) and the Z′ factor. Acceptable values were: %CV values < 10% S/N > 8 and Z′ > 0.5.

Design and development of cell surface PrPC FRET immunoassays
PrPC is present at the surface of neurons and on various non-neuronal tissues and leukocytes (23). To adapt our assay to human cells, we first assessed PrPC expression in several cell lines using a PrPC-specific ELISA (supplemental Fig. S1). The human lung epithelial cell line A549 and HeLa cells were identified as human cell lines with the highest total PrPC expression. To identify the most efficacious antibody pair enabling detection of PrPC by FRET, several POM antibodies were again screened in various combinations of FRET donors and acceptors for A549 cells (supplemental Fig. S2). The FRET pair POM2-Eu/POM2-APC, which binds to the octapeptide repeats (Fig. 1), yielded the highest FRET signals within the POM antibody library. As this antibody pair also yielded a good signal for HeLa cells (supplemental Fig. S2B), we selected it for further use in the assay.
We then determined the optimal cell number to be used in the assay by plating different numbers of HeLa and A549 cells, respectively, into 384-well plates. On the following day cells were washed with ice-cold Tris-Krebs buffer and labeled with POM2-Eu/POM2-APC for 30 min at 4 °C. A cell number-dependent FRET signal was detected in a large dynamic range for both human cell lines without (homogeneous time-resolved FRET (HTRF) mode), which is more suited for automated high-throughput screening applications and after washing out unbound antibodies (TR-FRET mode) (Fig. 3A), whereas no signal was obtained for PrP-deficient cells (Hpl Prnp−/−) used as negative control. These results confirmed the specificity of the assay.
Figure 3.

FRET immunoassay for the detection of PrPC on the cell surface of intact human cells. A, schematic representation of the cell-surface PrPC FRET detection assay. The FRET signal was assessed either in the HTRF mode or after a washing step (TR-FRET mode) to remove unbound chromophore-labeled antibodies. B and F, cell number dilution range in HTRF mode. A549 and HeLa cells were serially diluted (sextuplicates) in DMEM medium starting from 32,000 to 30 cells/well in polystyrene 384-well microtiter plates. Hpl Prnp−/− cells were used as negative controls. C and G, linear range of PrPC cell-surface detection of A549 and HeLa cells in the HTRF mode, respectively. D and H, cell number dilution range of A549 and HeLa cells in TR-FRET mode. E and I, linear range of PrPC cell-surface detection of A549 and HeLa cells in the TR-FRET mode, respectively. Error bars represent the standard deviation (±S.D.) of 6 replicate measurements. Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
For A549 cells, a linear dynamic range was found between 500 and 4000 cells/well (R2 = 0.69) (Fig. 3, B and C) in HTRF mode. The linear correlation was improved (R2 = 0.94) in TR-FRET mode (Fig. 3, D and E). Similar results were obtained when titrating numbers of HeLa cells using the same antibody pair (Fig. 3, F and H). Again, essentially linear dynamic ranges were found from 500 to 8000 cells/well in the HTRF mode (R2 = 0.90) and in the TR-FRET mode (R2 = 0.98) (Fig. 3, G and I). For both cell lines and in both measurement modes, 500 cells were sufficient to generate a statistically reliable FRET signal. Conventional assay performance acceptance criteria (Z′ factors of >0.5, coefficient of variance values of <10%, signal-to-noise ratio values of >8) were achieved for HeLa cells in the range of 8,000 to 32,000 cells in TR-FRET and HTRF mode and for A549 in the range of 2,000 to 32,000 cells in TR-FRET and 32K cells in HTRF mode (Table 2). These data validated the assay as a robust and accurate assay for the detection of cell surface PrPC on intact cells under these conditions.
Table 2.
Assay performance of the cell-surface FRET assay in the HTRF mode (without washing steps) and TR-FRET mode (after washing) in A549 (upper tables) and HeLa cells (lower tables) was assessed by three parameters: coefficient of variation (%CV), signal-to-noise ratio (S/N) and the Z′ factor. Acceptable values were: %CV values < 10%, S/N > 8 and Z′ > 0.5. K: cell number in thousands.

Manipulation of human PrPC cell-surface expression on living cells using HTRF and TR-FRET
We then assessed the applicability of the assay to the detection of cell-surface PrPC. 500 intact, viable cells were preincubated with varying concentrations of unlabeled POM2 antibody. After removing unbound POM2 by washing, cells were incubated with labeled POM2-Eu/POM2-APC. Results in the HTRF (Fig. 4A) and TR-FRET mode (Fig. 4B) showed an increase in the FRET signal intensity with decreasing concentrations of unlabeled POM2 antibody, as expected from binding site saturation by competing antibodies.
Figure 4.

Suitability test of the FRET immunoassay for the manipulation of PrPC cell-surface expression. A, saturation binding curves of PrPC. A constant number of HeLa cells was incubated with unlabeled POM2 antibody at different concentrations followed by FRET signal detection with POM2-Eu/POM2-APC in HTRF mode. B, saturation binding curves of PrPC after removing unbound POM-2 antibodies (TR-FRET mode). C and D, release of cell-surface PrPC by PI-PLC treatment. A constant number of A549 cells was labeled with POM2-Eu/POM2-APC followed by PI-PLC digestion at various concentrations. PrPC cell-surface FRET signal was measured after PI-PLC treatment at the cell surface (C) as well as in the cell culture medium (D). E, inhibition of PrPC cell-surface expression by BFA treatment. A constant number of HeLa cells was exposed to different BFA concentrations. PrPC cell-surface expression was measured by POM2-Eu/POM2-APC in the HTRF. F, same as E, but in the TR-FRET mode. G, siRNA mediated PRNP knockdown. A549 cells were treated with different concentrations of PRNP siRNA and control siRNAs for 3 days. PrPC cell-surface expression was detected with the POM2-Eu/3F4-APC FRET antibody pair. Scrambled and GAPDH siRNAs as well as Hpl Prnp−/− cells were used as negative and positive controls, respectively. Error bars represent the standard deviation (±S.D.) of six replicate measurements. Student's t test: ***, p < 0.001; ****, p < 0.0001.
To control the level of PrPC cell-surface expression on intact cells, the GPI anchor of PrPC was enzymatically cleaved with phosphatidylinositol-specific phospholipase C (PI-PLC). A549 cells were initially labeled with POM2-Eu/POM2-APC and exposed to different concentrations of PI-PLC. FRET signals of cell-surface retained and released PrPCs were measured after transferring the supernatant into a new plate (Fig. 4, C and D). With increasing PI-PLC concentrations, we found decreasing levels of surface PrPC and elevated levels of PrPC in the supernatant. To monitor cytotoxicity of PI-PLC exposure on A549 cells, we established an alamarBlue® cell viability assay (supplemental Fig. S3). No effect of PI-PLC treatment on A549 cell viability was detectable (supplemental Fig. S3).
We next tested the performance of the assay in detecting the pharmacological manipulation of PrPC surface expression in living HeLa and A549 cells. Cells were treated with 0–10 μg/ml brefeldin A (BFA) diluted in 0.75% DMSO (24). BFA inhibits the transport of proteins from the endoplasmic reticulum to the Golgi apparatus and thus reduces their display at the cell surface. We found that a BFA concentration of 1 μg/ml in 0.75% DMSO decreased the PrPC expression level at the cell surface of ∼66% in the HTRF (Fig. 4E) and ∼75% in the TR-FRET mode (Fig. 4F) without affecting the cell viability.
Next, we investigated the applicability of our assay to the genetic manipulation of the cell-surface PrPC expression of intact A549 cells. We first used a small interfering RNA (siRNA) targeting the human PRNP gene. The PRNP siRNA was also fully functional as shown by qPCR (supplemental Fig. S5) and had no toxic effects on the cells as monitored by the alamarBlue assay (supplemental Fig. S4). We found that the siRNA efficiently down-regulated the expression of PrPC on the surface of living A549 cells in a concentration-dependent manner (p < 0.0001), whereas scrambled siRNAs used as negative controls had no significant effect on PrPC expression (Fig. 4G). Complete suppression was reached at a concentration of 50–100 nm siRNA, as shown by a comparison of the Net-FRET signal to those of Hpl Prnp−/−-deficient cells. These data demonstrate that the assay can be applied to high-throughput pharmacological and genetic PrPC cell-surface manipulation screens.
PrPC endocytosis measured by FRET
In a next step we modified the assay format toward a PrPC endocytosis assay to assess its suitability for the identification of modifiers controlling the cell-surface expression and endocytosis of PrPC. The principle of the modified assay is shown in Fig. 5A. In a first set of experiments, various antibody combinations and cell culture conditions were tested. Satisfactory results were obtained with A549 cells cultured in DMEM medium (without phenol red, antibiotics and FBS) during endocytosis using the antibody combination POM2-Eu/POM2-APC. After labeling A549 cells with POM2-Eu, free antibodies were removed by washing and endocytosis was induced at 37 °C after 5 min of preincubation. At specified time points (t = 0–70 min) POM2-APC was added to different wells (15 time points in 6 wells). After the last addition of POM2-APC at 70 min, FRET signals were measured for all wells (Fig. 5B). A gradual decrease of Eu-POM2-labeled cell-surface PrPC was observed over time; the decrease was almost linear between 0 and 30 min (R2 = 0.79; Fig. 5C). Similar data were also obtained for HeLa cells (supplemental Fig. S6A). We found that PrPC undergoes endocytosis also when antibodies are added in a reversed order (supplemental Fig. S6B). The endocytosis rate of PrPC was calculated for A549 cells according to the endocytosis index formula (see “Experimental procedures”). After 30 min of incubation, 80% of cell-surface-resident PrPC was endocytosed (Fig. 5D), which is in agreement with previously reported PrPC internalization rates (5).
Figure 5.

Principle of the endocytosis FRET immunoassay and detection of PrPC uptake by human cells. A, schematic representation of the endocytosis FRET assay. PrPC on the cell surface is first labeled with the donor FRET Eu-labeled antibody (t = 0; 4 °C). After removing excess antibody, cells are exposed to 37 °C to initiate the uptake of Eu-labeled cell-surface PrPC (t = 1). During the course of internalization, Eu-labeled cell-surface PrPC is endocytosed from the cell surface into the cell, and residual Eu-labeled cell-surface PrPC is measured by adding the acceptor FRET antibody at various time intervals (t = 1–4). The endocytosis rate of PrPC is calculated according to the endocytosis index formula (see “Experimental procedures”). B, time course of PrPC endocytosis. A549 cells were first labeled with POM2-Eu at 4 °C followed by the exposure to 37 °C to initiate the internalization of PrPC. At different time points (t = 0–70 min) POM2-APC was added, and the Net-FRET of the remaining cell-surface POM2-Eu-labeled PrPC was measured. C, linear range of PrPC internalization. Error bars represent the standard deviation (±S.D.) of six replicate measurements. D, determination of the internalization rate. The PrPC endocytosis index was calculated according to the formula in “Experimental procedures” and reached a half-life time of ∼25 min. E, time course of cell-surface PrPC A549 cells were labeled with POM2-Eu/POM2-APC at 4 °C followed by exposure to 37 °C. The Net-FRET of cell-surface PrPC was measured at different time points.
Pharmacological blocking of PrPC endocytosis pathways
We then used the endocytosis assay to gauge the effects of compounds inhibiting clathrin-mediated endocytosis (chlorpromazine/CPZ) and clathrin-independent uptake (methyl-β-cyclodextrin/MβCD) on internalization of PrPC in A549 cells (25). CPZ causes clathrin lattices to assemble on endosomal membranes and prevents the assembly of coated pits at the plasma membrane (26), whereas MβCD selectively extracts cholesterol from the plasma membrane (27). Cells were pretreated for 30 min with CPZ or MβCD in a dose-dependent manner and subsequently labeled with POM2-Eu (Fig. 6). After 30 min of incubation at 37 °C, POM2-APC was added, and inhibition of PrPC uptake was assayed by TR-FRET. Fig. 6A represents the internalization of PrPC in A549 cells in the presence of CPZ. CPZ decreased the entry of PrPC in A549 cells in a dose-dependent manner. A concentration of 0.28 μm CPZ was sufficient to block clathrin-mediated endocytosis of PrPC without affecting the cell viability of A549 cells as determined in an alamarBlue assay (Fig. 6B). On the other hand, a concentration of 5 mm MβCD inhibited the uptake of PrPC without any toxic effect on cell viability as monitored by an alamarBlue assay (Fig. 6, C and D). The combination of CPZ and any inhibitor blocking clathrin-mediated internalization did not cause any further decrease in the uptake of PrPC. These data support recent findings on MβCD in initiating PrPC endocytosis (28) and the importance of cholesterol for the endocytosis of PrPC (29) as well as the inhibitory effect of CPZ on the internalization of PrPC (30).
Figure 6.

Effect of pharmaceutical compounds on PrPC endocytosis. A, a constant number of A549 cells was prelabeled with POM2-Eu and treated with various concentrations of chlorpromazine. After 30 min of incubation, PrPC endocytosis was assessed by the addition of POM2-APC. B, cell viability was measured by the alamarBlue assay. C, same as A, but for MβCD. D, same as B, but for MβCD. Error bars represent the standard deviation (±S.D.) of six replicate measurements. Student's t test. *, p < 0.05; **, p < 0.01.
Identification of genes that differently impact cell-surface expression and endocytosis of PrPC
Next, the potential of the endocytosis assay toward genetic manipulations was tested by using seven hand-picked siRNAs targeting major proteins of the membrane-trafficking machinery and their downstream organelles (Fig. 7 and Table 3). We expected that some of these siRNAs would modify the endocytosis rate and cell-surface expression of PrPC, resulting in a reduced FRET signal. The efficacy of PRNP knock-down was again controlled using a siRNA targeting the PRNP gene, whereas untreated cells acted as a negative control. The functionality of the siRNAs was verified by quantitative PCR (supplemental Fig. S5), and potential toxic effects of the siRNAs to the cells were controlled by performing cell viability assays (supplemental Fig. S4).
Figure 7.

Manipulation of PrPC endocytosis rate and cell-surface expression by siRNA treatments. A549 cells were treated for 3 days with 50 nm siRNA. PrPC internalization (t0–t30) and cell-surface expression (t0) was measured by FRET using the antibody pair POM2-Eu/POM2-APC, and the endocytosis index (Table 4) was calculated according to the Equation 4 under “Experimental procedures”. Untreated and PRNP siRNA-treated cells were used as controls. Error bars represent the standard deviation (±S.D.) of six replicate measurements. Student's t test.
Table 3.
Overview of the tested siRNAs on PrPC endocytosis rate and cell-surface level
For the study of PrPC endocytosis and cell-surface expression, seven siRNAs acting in protein recycling pathways and their downstream organelles were selected.
| Abbreviation | Description |
|---|---|
| AP2M1 | Encodes the adaptor protein 2 (AP-2), which is involved in clathrin-dependent endocytosis |
| EHD1 | Encodes the EH domain-containing protein 1, plays a role in endocytosis |
| GIT2 | Encodes the ARF GTPase-activating protein GIT2 |
| M6PR | Encodes the mannose 6-phosphate receptor, interacts with AP1 clathrin adaptor complex |
| RAB5A | G-protein, required for the fusion of plasma membrane an early endosomes |
| VPS28 | Encodes the vacuolar protein sorting-associated protein 28 homolog, involved in endosomal sorting of cell-surface receptors |
| VPS35 | Encodes the vacuolar protein sorting-associated protein 35 homolog, involved in retrograde transport of proteins from endosomes to the trans-Golgi network |
We found that knockdown of the small GTPase protein RAB5a, which controls clathrin-dependent endocytosis (31–33) and early endosome dynamics (34), significantly reduced the cell-surface expression (t0) and strongly blocked the endocytosis (t0-t30) of PrPC (Fig. 7 and Table 4). Also. AP2M1 (AP-2 adaptor protein), a gene involved in clathrin-dependent endocytosis pathways (35), blocked the uptake of PrPC. Other genes, such as M6PR (mannose-6-phosphate receptor) (36) and VPS35 (vacuolar protein sorting 35 homolog) (37), which are involved in retrograde transport of proteins from endosomes to the trans-Golgi network, also suppressed the internalization of PrPC, and VPS35 also reduced PrPC cell-surface expression. Moreover, the VPS35 protein was recently shown to reduce the expression of α-synuclein in a prion-like seeding model in transgenic mice, thus protecting them from neurodegeneration (38). We also tested EHD1, a gene that encodes the EH domain-containing protein 1 that is involved in endocytosis (39). Suppression of EHD1 slightly increased the cell-surface expression (t0) level of PrPC (Fig. 7 and Table 4) without affecting endocytosis. For VPS28, a gene that encodes the vacuolar protein sorting-associated protein 28 homologue involved in endosomal sorting of cell-surface receptors (40), an increased endocytosis rate and reduced PrPC cell-surface expression was found. Silencing of GIT2, a gene that encodes the ARF GTPase-activating protein GIT2 (41), increased the uptake of PrPC without affecting PrP cell-surface level (Fig. 7). Hence, we were able to identify a number of genes involved in clathrin-dependent internalization of PrPC and its trafficking to early endosomes. This demonstrates the potential of the assay for comprehensive high-throughput screens of compounds and genes that target the internalization and endocytosis pathway of PrPC.
Table 4.
Effect of siRNA treatment on PrPC endocytosis rate and cell-surface expression
PrPC cell-surface expression, influence of siRNA treatment on PrPC cell-surface expression. Cells were incubated for 3 days with siRNAs. PrPC cell-surface level was detected with the POM2-Eu/POM2-APC antibody pair. PrPC cell-surface expression of siRNA-treated cells is expressed as -fold untreated cells. PrPC endocytosis, influence of siRNA treatment on the endocytosis rate of PrPC. Cells were incubated for 3 days with siRNAs and labeled with POM2-Eu at 4 °C. To induce endocytosis, cells were exposed to 37 °C. At time point t0 = 0 min and t1 = 30 min POM2-APC was added. The endocytosis rate was calculated as described under “Experimental procedures.”
| siRNA | PrPC cell-surface expression |
PrPC endocytosis |
||
|---|---|---|---|---|
| Mean ± S.D. (t0) | Normalized to untreated (t0) | Mean ± S.D. (t0—t30) | Endocytosis rate | |
| Untreated | 448.43 ± 103.37 | 1 ± 0.23 | 213.62 ± 110.26 | 0.48 ± 0.36 |
| PrP | 229.24 ± 108.70 | 0.51 ± 0.24 | 52.65 ± 118.97 | 0.23 ± 0.63 |
| AP2M1 | 443.52 ± 19.41 | 0.99 ± 0.04 | 40.64 ± 30.35 | 0.09 ± 0.07 |
| EHD1 | 493.18 ± 28.72 | 1.10 ± 0.06 | 254.44 ± 80.13 | 0.52 ± 0.19 |
| GIT2 | 459.97 ± 50.50 | 1.03 ± 0.11 | 228.70 ± 90.94 | 0.50 ± 0.25 |
| M6PR | 435.74 ± 82.25 | 0.97 ± 0.18 | 79.65 ± 102.08 | 0.18 ± 0.27 |
| RAB5A | 202.00 ± 89.66 | 0.45 ± 0.20 | −51.60 ± 115.41 | −0.26 ± 0.46 |
| VPS28 | 349.60 ± 125.23 | 0.78 ± 0.28 | 199.25 ± 136.61 | 0.57 ± 0.59 |
| VPS35 | 344.96 ± 122.54 | 0.77 ± 0.27 | 65.19 ± 137.30 | 0.19 ± 0.47 |
Discussion
The exposure of PrPC at the surface of target cells is a precondition to prion replication and to prion neurotoxicity (42, 43). Several studies have shown that masking PrPC with antibodies (44) or suppressing its expression with siRNA (45) can reduce prion replication and prolong the life of prion-infected mice. Therefore, an in-depth understanding of the factors controlling PrPC expression at the cell surface may pave the way to treating human prion diseases.
High-throughput screening platforms to identify modulators of the transcription, translation, and protein cycling turnovers that regulate cellular PrP may point to druggable targets, which could be used to selectively reduce human PrPC in the treatment of CJD patients (46). Being a single-pot method, FRET is suitable to high-throughput assay formats. However, FRET does not allow for the extensive signal amplification afforded by enzyme-linked immunoassays. We, therefore, wondered whether FRET-based PrPC detection in high-density microtiter plates would provide the analytical power necessary for this type of assay. Indeed, the assays described here provide high scalability, high specificity, a wide dynamic range, and sensitive detection that compare well with enzyme-linked immunosorbent assays (47). Moreover, the assays exhibit broad dynamic ranges and were found to be highly target-specific by spiking, competition, and blocking experiments and required only nanomolar quantities of antibodies. The large detection window and the Z′ factor finally allowed us to identify compounds and genes that differently impact the cell-surface expression and endocytosis rate of normal PrPC, thus demonstrating the practicality of the FRET assays for high-throughput screening applications in a 384-well format (46, 48).
We then studied the applicability of the FRET-based immunoassays for the simultaneously identification of compounds and genes that differently affect the level and internalization rate of cell-surface PrPC. We first studied the internalization of endogenously expressed human PrPC on cells. A cell-surface half-life time of ∼25 min of PrPC was determined, which is in agreement with previous results (5). No PrPC was detectable in the culture medium during the endocytosis experiment, confirming that PrPC is mainly endocytosed rather than released from the cell surface. We also observed no loss of FRET signal intensity over the whole time period (75 min under cell culture conditions) in the cell-surface PrPC FRET assay, demonstrating that the PrP-mAbs-Eu complex is stable in our experimental settings. We further concluded from the good agreement of our internalization rate with previously published results (5) that the stability of the PrP-mAbs-Eu complex is also not significantly affected in endosomal compartments and that the binding of POM2, even if it might modulate interactions of PrPC with other membrane proteins, had no significant effect on PrP endocytosis. In control experiments, MβCD and CPZ inhibited endocytosis of PrPC without affecting cell viability at the chosen concentrations as already reported earlier (31, 49).
We tested the suitability of the FRET-based endocytosis assay for the identification of cell-surface and endocytosis modulators of PrPC. The siRNAs repressing RAB5A and AP2M1 (encoding the μ subunit of the AP-2 complex) showed the strongest inhibition of PrPC internalization. These data suggest clathrin-mediated endocytosis of PrPC, as hypothesized by earlier studies showing that AP-2 co-localizes with PrPC (10).
RAB5a suppression brought about a strong reduction in PrPC cell-surface expression and completely blocked the endocytosis of PrPC after 30 min. Similar results were reported by Magalhaes et al. (46), who showed that green fluorescent protein (GFP)-tagged PrPC, but not GFP-GPI, is endocytosed via Rab5-containing early endosomes (50). Knockdown of VPS35 and M6PR also suppressed endocytosis of PrPC, albeit less prominently. This suggests additional physical or epistatic interactions of these genes with PrPC. Finally, knockdown of VPS28 resulted in an increased uptake of PrPC by reducing PrPC cell-surface expression.
In summary, we provide proof-of-concept for FRET-based assays that are suited as high-throughput screening platforms to identify small molecules or compounds that genetically or pharmacologically block or induce PrPC endocytosis. The assays can be used to measure the internalization rate, to characterize the different endocytosis pathways, and to perform large-scale RNAi screenings to identify genes regulating PrPC expression, biosynthesis, and endocytosis. The knowledge gained from these experiments is likely to lead to the discovery of novel compounds for the treatment of prion diseases and perhaps even for other protein misfolding and aggregation diseases, which share pathways with those active in prion diseases.
Experimental procedures
Cell culture
N2a PK1 (a subclone of neuroblastoma N2a cells) and Hpl Prnp−/− cells (21) were cultured in 75-cm2 tissue culture flasks in Opti-MEM medium supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% penicillin/streptomycin. Human HeLa and A549 cells were cultured in DMEM medium supplemented with 10% (v/v) fetal bovine serum and 1% penicillin/streptomycin. In FRET experiments, all cell lines were cultured in medium without FBS, antibiotics, and phenol red. To determine cell numbers and cell viability, resuspended cells were stained with trypan blue and counted in a Neubauer chamber.
FRET antibody labeling
Monoclonal POM antibodies were labeled in-house for FRET applications. The donor fluorophore was europium chelate (Eu-W1024 ITC chelate, AD0096, PerkinElmer Life Sciences). Coupling to proteins occurs at alkaline pH via reaction of lysine residues and free N termini with the aromatic isothiocyanate group of the europium chelate. To remove possible contaminants interfering with labeling, antibodies were dialyzed overnight in 1 ml of 100 mm sodium carbonate buffer (Na2CO3, pH 9–9.3). Dialysis cassettes (Slide-A-Lyzer®, Thermo scientific) with a 7-kDa cut-off were used. Dialysis was done under stirring in a volume of 2 liters of 100 mm Na2CO3, pH 9–9.3, at 4 °C overnight. Dialysis buffer was changed after 4–6 h.
Antibodies were concentrated by Centricon concentrators (Millipore). Protein concentration was adjusted by the Bradford or BCA assay to a concentration of ∼5 mg/ml. A molar excess of 24× of europium chelate over IgG was added into the protein solution on ice and incubated in 100 mm Na2CO3 overnight at 4 °C. Separation of the labeled protein from non-reacted chelate was performed by size exclusion chromatography (Superdex 200 column, GE Healthcare). Elution from the column was done with 50 mm Tris-HCl, pH 7.8, + 0.9% sodium chloride. Sample fractions of 500 μl were collected. Fractions were pooled and concentrated with Micron centrifugal filters (Amicon). Labeling ratio and concentration of labeled proteins were assessed by a europium standard solution (PerkinElmer Life Sciences) and NanoDrop measurement, respectively. APC (AnaTagTM Labeling Kit 72111, Anaspec) was used as acceptor fluorophore for conjugation to POM antibodies. Maleimide groups of APC reacted with sulfhydryl groups on the target antibody to form a covalent bond. POM antibodies were concentrated to 2–10 mg/ml in a volume of 100 μl with Centricon concentrators (Millipore). Antibodies were reduced with 20 μl of dithiothreitol per ml of IgG solution for 30 min without agitation at room temperature. Depending on reaction volume, reduced antibodies were desalted either by spin or gravity columns. Protein concentration was assessed by NanoDrop measurement. For the conjugation reaction, 1.5 mg of activated APC per mg of reduced IgG was added to the reduced antibodies solution and incubated for 1 h at room temperature with agitation. Free thiol groups were blocked by adding DMSO and N-ethylmaleimide for 30 min at room temperature. To remove free APC molecules from the antibody solution, reaction mixture was purified via a protein G-Sepharose column (Sigma). The column was washed with water and 10 volumes of 20 mm phosphate buffer, pH 7, to equilibrate. Samples were added and washed with 5 volumes of 20 mm phosphate buffer, pH 7. Flow-through was collected. The column was eluted with 0.1 m glycine pH 2.3 and diluted with 1 m Tris HCl, pH 8. APC-labeled POM antibodies were characterized by NanoDrop (A280 and A650).
Time-resolved fluorescence measurements
The Wallac EnVision multilabel reader (PerkinElmer Life Sciences) was used for all FRET experiments using protocols installed by the manufacturer. A nitrogen laser device (337 nm) excites Eu, which activates APC in a distance-dependent manner. Fluorescence emissions were monitored both at 615 nm for Eu and at 665 nm for APC. An integration time of 100 or 400 μs was used after a time delay ranging from 50 to 100 μs to remove short-lived background signals.
Data analysis
The distance-dependent energy transfer between the chromophore-conjugated antibodies determines the FRET signal intensity in the presence of PrP and is henceforth reported as “net FRET” signal. Net FRET signals are the number of APC counts depending on FRET events and calculated by using the equation,
| (Eq. 1) |
where “counts” are the raw FRET-dependent signals in APC and Eu channel minus the background fluorescence. The proportionality factor P defines spectral overlap compensations and is measured by Equation 2.
| (Eq. 2) |
Eublank and APCblank correspond to all reagents except Eu or APC in the Eu or APC channel, respectively. In addition, cells deficient of PrP were used as negative controls.
To show the reliability of FRET-based immunoassays for high-throughput applications, the statistical parameter Z′ factor (22) was calculated by the formula,
| (Eq. 3) |
where S.D. is the standard deviation, and μ is the mean of the positive and negative controls. An assay is considered to be excellent for high-throughput applications if the Z′ factor is equal or superior to 0.5.
PrPC cell-surface FRET assay
Cells were plated 1 day before into 96-well or 384-well microtiter plates (PerkinElmer Life Sciences) in cell culture medium without phenol red at the desired density. Cells were washed once, and FRET antibody labeling was performed at 4 °C containing 1 nm Eu-labeled and 5 nm APC-labeled antibodies in Tris/Krebs buffer (20 mm Tris, pH 7.4, 118 mm NaCl, 5.6 mm glucose, 1.2 mm KH2PO4, 1.2 mm MgSO4, 4.7 mm KCl, 1.8 mm CaCl2, 1% BSA). FRET signals were measured with the Wallac EnVision reader (homogeneous TR-FRET assay) with a time delay of ∼100 μs between the excitation and fluorescence measurements. Thereafter, plates were washed with Tris/Krebs buffer, and the FRET signal was measured again (TR-FRET assay).
Pharmacological treatment of cells was performed with brefeldin A diluted in DMSO for 18 h of incubation at 37 °C. DMSO concentration was kept constant at 0.75%. Cells were washed once with ice-cold Tris/Krebs buffer and labeled with FRET antibodies as mentioned above. Enzymatic treatment of cells was done with PI-PLC. PI-PLC was dialyzed in PBS overnight, and protein concentration was assessed by the Bradford assay (Bio-Rad). Digestion was carried out either at 4 °C or at 37 °C for 1 h with shaking (450 rpm). The supernatant was transferred into a new plate and medium, and cells were labeled with FRET antibodies. The neuronal Hpl Prnp−/− cell line was used as the negative control for assay development and optimization.
PrPC endocytosis FRET assay
Cells were plated and cultured for 1 day in 96- or 384-well microtiter plates (PerkinElmer Life Sciences) at the desired density. Cells were washed once and labeled with 1 nm Eu-labeled POM antibodies in Tris/Krebs buffer for 30 min at 4 °C. After washing out unbound antibodies, cells were preincubated for 5 min at 37 °C. At different time intervals, 5 nm APC-labeled antibodies were added in cell culture medium without phenol red. FRET signals were measured with a Wallac EnVision reader in the homogeneous FRET assay mode or after a washing step in the TR-FRET mode. The internalization rate of PrPC was calculated with Equation 4,
| (Eq. 4) |
To suppress PrPC endocytosis, A549 cells were treated with ascending doses of CPZ or MβCD and subsequently labeled with POM2-Eu. After 30 min of incubation at 37 °C, POM2-APC was added, and inhibition of PrPC uptake was assayed by TR-FRET.
siRNA treatment
To transfect siRNA into mammalian cells, LipofectamineTM 2000 (InvitrogenTM) was utilized. One day before transfection, 2500 cells in 80 μl of DMEM cell culture medium without antibiotics were seeded into a 96-well plate per well. siRNA-LipofectamineTM 2000 complexes were prepared as follows. 5 μl of siRNA (1 μm) was diluted in 5 μl of pre-warmed DMEM cell culture medium without serum and antibiotics and mixed gently. 0.1 μl of LipofectamineTM 2000 was diluted in 9.9 μl of prewarmed DMEM cell culture medium without serum and antibiotics. After an incubation of 5 min at room temperature, the diluted LipofectamineTM 2000 was combined with the diluted siRNA. To allow formation of siRNA-LipofectamineTM 2000 complexes, the combined solutions were mixed and incubated for 20 min at room temperature. 20 μl of siRNA-LipofectamineTM 2000 complexes were added to each well of the 96-well plate, and cells were cultured at 37 °C in a CO2 incubator for 72 h.
Quantitative PCR
Cell cultures were prepared and incubated as stated above. Cells were washed once with PBS, and total RNA was extracted using the RNeasy® mini kit (Qiagen) according to the manufacturer's protocol. RNA concentration and purity was determined by NanoDrop measurements. cDNA was synthesized from 0.5 μg of total RNA with QuantiTect reverse transcription kit (Qiagen) using random hexamers according to the manufacturer's protocol. Before cDNA synthesis, genomic DNA was eliminated. Successful cDNA synthesis and contamination of total RNA with genomic DNA was tested by PCR with a primer specific for GAPDH. Quantitative real-time PCR was performed using the SYBR Green master mix (Applied Biosystems) on an ABI Prism VIIA7 sequence detector (PerkinElmer Life Sciences). -Fold regulation was calculated relative to untreated cells after normalization to the GAPDH signal. The following primer pairs were used: VPS28 sense (NM_016208), 5′-TTG TTC CCA GGG GCT CCT AT-3′; antisense, 5′-ATC TTC CCG ACC GCG AG-3′); RAB5A sense (NM_004162), 5′-TTA GAA AAG CAG CCC CAA TG-3′; antisense 5′-GTA CTT CTG GGA GAG TCC GC-3′); AP2M1 sense (NM_004068), 5′-AAT CAT GGC GGC AGA TCA GT-3′; antisense 5′-ATC TTG GGA TCC GGA GAG TG-3′); GIT2 sense (NM_014776), 5′-AGA CAA ATC CAG GCT GCT GT-3′; antisense, 5′-GCG CCT CGT GGA AAT ACA GT-3′); EHD1 sense (NM_006795), 5′-AAG AGC AGG ATG ATG CGG T-3′; antisense, 5′-CCT GTC TGG AGA GAA GCA GC-3′); M6PR sense (NM_002355), 5′-ATT CTC TCA CTG CCA CAG CC-3′; antisense 5′-TGG CTA CTC CAG TTT CCC AC-3′); VPS35 sense (NM_018206), 5′-CAC TGA TAG TCT GGT GGG CA-3′; antisense, 5′-CAG CTT ACC AGC TGG CTT TT-3′; PRNP sense (NM_000311), 5′-AGT GTT CCA TCC TCC AGG C-3′; antisense 5′-GAG CTT CTC CTC TCC TCA CG-3′); GAPH sense (NM_002046), 5′-CAT GAG AAG TAT GAC AAC AGC CT; antisense 5′-AGT CCT TCC ACG ATA CCA AA GT-3′).
Cell viability assay
The alamarBlue® assay (Invitrogen) was utilized to assess cell viability according to the manufacturer's protocol. All experiments were performed in cell culture medium without antibiotics and phenol red. Cells were incubated with the alamarBlue dye at 37 °C, and the fluorescence intensity was measured with a plate reader after 1–6 h.
Author contributions
B. A. B. designed, performed, and analyzed the experiments and wrote the initial draft of the paper. R. M. provided technical assistance. P. L. and L. P. designed the experiments. S. H. and A. A. conceived, coordinated, and designed the final study, analyzed the data, and wrote the paper. All authors analyzed and reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Irina Abakumova and Clemence Tournaire for technical help.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S6.
- PrPC
- cellular prion protein
- recPrP
- recombinant mouse PrP
- GPI
- glycosylphosphatidylinositol
- APC
- allophycocyanin
- Eu
- europium
- LDL
- lower detection limit
- TR-FRET
- time-resolved FRET
- HTRF
- homogeneous TR-FRET
- PI-PLC
- phosphatidylinositol-specific phospholipase C
- BFA
- brefeldin A
- CPZ
- chlorpromazine
- MβCD
- methyl-β-cyclodextrin
- M6PR
- mannose 6-phosphate receptor
- VPS35
- vacuolar protein sorting 35 homolog
- AP-2
- adaptor protein 2.
References
- 1. Aguzzi A., and Calella A. M. (2009) Prions: protein aggregation and infectious diseases. Physiol. Rev. 89, 1105–1152 [DOI] [PubMed] [Google Scholar]
- 2. Prusiner S. B. (2004) Prion Biology and Diseases, p. 1050, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- 3. Prusiner S. B., Scott M. R., DeArmond S. J., and Cohen F. E. (1998) Prion protein biology. Cell 93, 337–348 [DOI] [PubMed] [Google Scholar]
- 4. Taylor D. R., and Hooper N. M. (2007) The low-density lipoprotein receptor-related protein 1 (LRP1) mediates endocytosis of the cellular prion protein. Biochem. J. 402, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shyng S. L., Huber M. T., and Harris D. A. (1993) A prion protein cycles between cell surface and an endocytic compartment in cultured neuroblastoma cells. J. Biol. Chem. 268, 15922–15928 [PubMed] [Google Scholar]
- 6. Harris D. A. (2003) Trafficking, turnover, and membrane topology of PrP. Br. Med. Bull. 66, 71–85 [DOI] [PubMed] [Google Scholar]
- 7. Prado M. A., Alves-Silva J., Magalhães A. C., Prado V. F., Linden R., Martins V. R., and Brentani R. R. (2004) PrPC on the road: trafficking of the cellular prion protein. J. Neurochem. 88, 769–781 [DOI] [PubMed] [Google Scholar]
- 8. Campana V., Sarnataro D., and Zurzolo C. (2005) The highways and byways of prion protein trafficking. Trends Cell Biol. 15, 102–111 [DOI] [PubMed] [Google Scholar]
- 9. Shyng S. L., Lehmann S., Moulder K. L., and Harris D. A. (1995) Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein, PrPC, in cultured cells. J. Biol. Chem. 270, 30221–30229 [DOI] [PubMed] [Google Scholar]
- 10. Sunyach C., Jen A., Deng J., Fitzgerald K. T., Frobert Y., Grassi J., McCaffrey M. W., and Morris R. (2003) The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 22, 3591–3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taylor D. R., Watt N. T., Perera W. S., and Hooper N. M. (2005) Assigning functions to distinct regions of the N terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J. Cell Sci. 118, 5141–5153 [DOI] [PubMed] [Google Scholar]
- 12. Morrison L. E. (1988) Time-resolved detection of energy transfer: theory and application to immunoassays. Anal. Biochem. 174, 101–120 [DOI] [PubMed] [Google Scholar]
- 13. Jares-Erijman E. A., and Jovin T. M. (2003) FRET imaging. Nat. Biotechnol. 21, 1387–1395 [DOI] [PubMed] [Google Scholar]
- 14. Selvin P. R. (2000) The renaissance of fluorescence resonance energy transfer. Nat Struct Biol. 7, 730–734 [DOI] [PubMed] [Google Scholar]
- 15. Bazin H., Trinquet E., and Mathis G. (2002) Time resolved amplification of cryptate emission: a versatile technology to trace biomolecular interactions. J. Biotechnol. 82, 233–250 [DOI] [PubMed] [Google Scholar]
- 16. Polymenidou M., Moos R., Scott M., Sigurdson C., Shi Y. Z., Yajima B., Hafner-Bratkovic I., Jerala R., Hornemann S., Wuthrich K., Bellon A., Vey M., Garen G., James M. N., Kav N., and Aguzzi A. (2008) The POM monoclonals: a comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS ONE 3, e3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bolton D. C., Seligman S. J., Bablanian G., Windsor D., Scala L. J., Kim K. S, Chen C. M., Kascsak R. J., and Bendheim P. E. (1991) Molecular location of a species-specific epitope on the hamster scrapie agent protein. J. Virol. 65, 3667–3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hornemann S., Korth C., Oesch B., Riek R., Wider G., Wüthrich K., and Glockshuber R. (1997) Recombinant full-length murine prion protein, mPrP(23–231): purification and spectroscopic characterization. FEBS Lett. 413, 277–281 [DOI] [PubMed] [Google Scholar]
- 19. Riek R., Hornemann S., Wider G., Billeter M., Glockshuber R., and Wüthrich K. (1996) NMR structure of the mouse prion proteindomain PrP(121–231). Nature 382, 180–182 [DOI] [PubMed] [Google Scholar]
- 20. Wüthrich K., and Riek R. (2001) Three-dimensional structures of prion proteins. Adv. Protein Chem. 57, 55–82 [DOI] [PubMed] [Google Scholar]
- 21. Sakudo A., Onodera T., and Ikuta K. (2007) Prion protein gene-deficient cell lines: powerful tools for prion biology. Microbiol. Immunol. 51, 1–13 [DOI] [PubMed] [Google Scholar]
- 22. Zhang J. H., Chung T. D., and Oldenburg K. R. (1999) A Simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 [DOI] [PubMed] [Google Scholar]
- 23. Aguzzi A., and Polymenidou M. (2004) Mammalian prion biology: one century of evolving concepts. Cell 116, 313–327 [DOI] [PubMed] [Google Scholar]
- 24. Nebenführ A., Ritzenthaler C., and Robinson D. G. (2002) Brefeldin A: deciphering an enigmatic inhibitor of secretion. Plant Physiol. 130, 1102–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vercauteren D., Vandenbroucke R. E., Jones A. T., Rejman J., Demeester J., De Smedt S. C., Sanders N. N., and Braeckmans K. (2010) The use of inhibitors to study endocytic pathways of gene carriers: optimization and pitfalls. Mol. Ther. 18, 561–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang L. H., Rothberg K. G., and Anderson R. G. (1993) Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 123, 1107–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klein U., Gimpl G., and Fahrenholz F. (1995) Alteration of the myometrial plasma membrane cholesterol content with β-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry 34, 13784–13793 [DOI] [PubMed] [Google Scholar]
- 28. Luo K., Li S., Xie M., Wu D., Wang W., Chen R., Huang L., Huang T., Pang D., and Xiao G. (2010) Real-time visualization of prion transport in single live cells using quantum dots. Biochem. Biophys. Res. Commun. 394, 493–497 [DOI] [PubMed] [Google Scholar]
- 29. Rodal S. K., Skretting G., Garred O., Vilhardt F., van Deurs B., and Sandvig K. (1999) Extraction of cholesterol with methyl-β-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol. Biol. Cell 10, 961–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sarnataro D., Caputo A., Casanova P., Puri C., Paladino S., Tivodar S. S., Campana V., Tacchetti C., and Zurzolo C. (2009) Lipid rafts and clathrin cooperate in the internalization of PrP in epithelial FRT cells. PLoS ONE 4, e5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lanzetti L., Rybin V., Malabarba M. G., Christoforidis S., Scita G., Zerial M., and Di Fiore P. P. (2000) The Eps8 protein coordinates EGF receptor signalling through Rac and trafficking through Rab5. Nature 408, 374–377 [DOI] [PubMed] [Google Scholar]
- 32. Martinu L., Santiago-Walker A., Qi H., and Chou M. M. (2002) Endocytosis of epidermal growth factor receptor regulated by Grb2-mediated recruitment of the Rab5 GTPase-activating protein RN-tre. J. Biol. Chem. 277, 50996–51002 [DOI] [PubMed] [Google Scholar]
- 33. McLauchlan H., Newell J., Morrice N., Osborne A., West M., and Smythe E. (1998) A novel role for Rab5-GDI in ligand sequestration into clathrin-coated pits. Curr. Biol. 8, 34–45 [DOI] [PubMed] [Google Scholar]
- 34. Stenmark H., Valencia A., Martinez O., Ullrich O., Goud B., and Zerial M. (1994) Distinct structural elements of rab5 define its functional specificity. EMBO J. 13, 575–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boucrot E., Saffarian S., Zhang R., and Kirchhausen T. (2010) Roles of AP-2 in clathrin-mediated endocytosis. PLoS ONE 5, e10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Le Borgne R., and Hoflack B. (1997) Mannose 6-phosphate receptors regulate the formation of clathrin-coated vesicles in the TGN. J. Cell Biol. 137, 335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bonifacino J. S., and Hurley J. H. (2008) Retromer. Curr. Opin. Cell Biol. 20, 427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dhungel N., Eleuteri S., Li L. B., Kramer N. J., Chartron J. W., Spencer B., Kosberg K, Fields J. A., Stafa K., Adame A., Lashuel H., Frydman J., Shen K., Masliah E., and Gitler A. D. (2015) Parkinson's disease genes VPS35 and EIF4G1 interact genetically and converge on α-synuclein. Neuron 85, 76–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rainey M. A., George M., Ying G., Akakura R., Burgess D. J., Siefker E., Bargar T., Doglio L., Crawford S. E., Todd G. L., Govindarajan V., Hess R. A., Band V., Naramura M., and Band H. (2010) The endocytic recycling regulator EHD1 is essential for spermatogenesis and male fertility in mice. BMC Dev. Biol. 10, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bishop N., and Woodman P. (2001) TSg101/mammalian VPS23 and mammalian VPS28 interact directly and are recruited to VPS4-induced endosomes. J. Biol. Chem. 276, 11735–11742 [DOI] [PubMed] [Google Scholar]
- 41. Premont R. T., Perry S. J., Schmalzigaug R., Roseman J. T., Xing Y., and Claing A. (2004) The GIT/PIX complex: an oligomeric assembly of GIT family ARF GTPase-activating proteins and PIX family Rac1/Cdc42 guanine nucleotide exchange factors. Cell. Signal. 16, 1001–1011 [DOI] [PubMed] [Google Scholar]
- 42. Brandner S., Isenmann S., Raeber A., Fischer M., Sailer A., Kobayashi Y., Marino S., Weissmann C., and Aguzzi A. (1996) Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 379, 339–343 [DOI] [PubMed] [Google Scholar]
- 43. Sonati T., Reimann R. R., Falsig J., Baral P. K., O'Connor T., Hornemann S., Yaganoglu S., Li B., Herrmann U. S., Wieland B., Swayampakula M., Rahman M. H., Das D., Kav N., Riek R., Liberski P. P., James M. N., and Aguzzi A. (2013) The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 501, 102–106 [DOI] [PubMed] [Google Scholar]
- 44. Heppner F. L., Musahl C., Arrighi I., Klein M. A., Rülicke T., Oesch B., Zinkernagel R. M., Kalinke U., and Aguzzi A. (2001) Prevention of scrapie pathogenesis by transgenic expression of anti-prion antibodies. Science 294, 178–182 [DOI] [PubMed] [Google Scholar]
- 45. Pfeifer A., Eigenbrod S., Al-Khadra S., Hofmann A., Mitteregger G., Moser M., Bertsch U., and Kretzschmar H. (2006) Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice. J. Clin. Invest. 116, 3204–3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karapetyan Y. E., Sferrazza G. F., Zhou M., Ottenberg G., Spicer T., Chase P., Fallahi M., Hodder P., Weissmann C., and Lasmézas C. I. (2013) Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc. Natl. Acad. Sci. U.S.A. 110, 7044–7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Triantaphyllidou I. E., Sklaviadis T., and Vynios D. H. (2006) Detection, quantification, and glycotyping of prion protein in specifically activated enzyme-linked immunosorbent assay plates. Anal. Biochem. 359, 176–182 [DOI] [PubMed] [Google Scholar]
- 48. Maurel D., Kniazeff J., Mathis G., Trinquet E., Pin J. P., and Ansanay H. (2004) Cell surface detection of membrane protein interaction with homogeneous time-resolved fluorescence resonance energy transfer technology. Anal. Biochem. 329, 253–262 [DOI] [PubMed] [Google Scholar]
- 49. Marella M., Lehmann S., Grassi J., and Chabry J. (2002) Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J. Biol. Chem. 277, 25457–25464 [DOI] [PubMed] [Google Scholar]
- 50. Magalhães A. C., Silva J. A., Lee K. S., Martins V. R., Prado V. F., Ferguson S. S., Gomez M. V., Brentani R. R, and Prado M. A. (2002) Endocytic intermediates involved with the intracellular trafficking of a fluorescent cellular prion protein. J. Biol. Chem. 277, 33311–33318 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.