

Abstract
Autophagy is a cellular surveillance pathway that balances metabolic and energy resources and transports specific cargos, including damaged mitochondria, other broken organelles, or pathogens for degradation to the lysosome. Central components of autophagosomal biogenesis are six members of the LC3 and GABARAP family of ubiquitin‐like proteins (mATG8s). We used phage display to isolate peptides that possess bona fide LIR (LC3‐interacting region) properties and are selective for individual mATG8 isoforms. Sensitivity of the developed sensors was optimized by multiplication, charge distribution, and fusion with a membrane recruitment (FYVE) or an oligomerization (PB1) domain. We demonstrate the use of the engineered peptides as intracellular sensors that recognize specifically GABARAP, GABL1, GABL2, and LC3C, as well as a bispecific sensor for LC3A and LC3B. By using an LC3C‐specific sensor, we were able to monitor recruitment of endogenous LC3C to Salmonella during xenophagy, as well as to mitochondria during mitophagy. The sensors are general tools to monitor the fate of mATG8s and will be valuable in decoding the biological functions of the individual LC3/GABARAPs.
Keywords: ATG8, immunofluorescence, LC3, phage display, selective autophagy
Subject Categories: Autophagy & Cell Death
Introduction
Autophagy is a highly conserved cellular pathway important for adaptation to stressful conditions, including starvation, pathogen invasion, protein aggregation, and oxidative stress (for a review, see Kroemer et al, 2010; Ohsumi, 2014). Under basal conditions, autophagy is dedicated to the continuous renewal of the intracellular pool of proteins, carbohydrates, lipids, and organelles, and thus also plays a critical role in cellular homeostasis (Klionsky & Codogno, 2013). While autophagy originally had been described as a non‐selective bulk process induced in response to starvation, it is now recognized that the degradation of damaged organelles, removal of protein aggregates, and elimination of intracellular pathogens are highly selective and tightly regulated processes that require cargo recognition and processing by the autophagic machinery (Stolz et al, 2014; Ktistakis & Tooze, 2016). To fulfill these diverse functions, the autophagic machinery includes adapters as well as cargo receptors, which are specific for individual selective autophagy pathways (Rogov et al, 2014; Stolz et al, 2014). Common for these two functional classes is their interaction with the central autophagy component ATG8 via their ATG8‐interacting motif (AIM), also called LC3‐interacting region (LIR) (Rogov et al, 2014). Members of the ATG8 family [six in humans: LC3A, LC3B, LC3C, GABARAP, GABARAPL1, and GABARAPL2; later referred to as mammalian ATG8s (mATG8s), or as LC3s and GABARAPs when referring to individual subfamilies] are ubiquitin‐like (UBL) proteins, which can be covalently modified with phosphatidylethanolamine resulting in their association with intracellular membranous structures (Kabeya et al, 2000). Although mATG8s are known to be crucial for the formation of functional isolation membranes (autophagic membrane that isolates cargo from cytosol) and subsequently autophagosomes by providing a binding platform for autophagy adaptors and receptors, little is known about the overlapping and specific functions of the individual family members (Rubinsztein et al, 2012). LC3/GABARAPs also have distinct, non‐overlapping intracellular localization and varying expression levels in different cell types and tissues (Le Grand et al, 2013; Hiyama et al, 2015; Koukourakis et al, 2015).
To date, GABARAPs were shown to be involved in intra‐Golgi transport and Golgi re‐assembly (Sagiv et al, 2000; Muller et al, 2002; Leil et al, 2004). Different functions for the LC3 and GABARAP subfamilies were subsequently described during the early and late stages of autophagosome formation, respectively (Weidberg et al, 2010). However, this functional difference between the two mATG8 subfamilies is not strict, as some early steps of the initiation of autophagy seem to be also dependent on GABARAPs (Alemu et al, 2012).
Even though some earlier studies reported specificity of mATG8 isoforms in selective autophagy pathways (Novak et al, 2010; von Muhlinen et al, 2012), only in recent years an increasing number of studies have concentrated on their individual characteristics and functions. Consistent with acting at later stages of autophagy, GABARAPs seem to be involved in facilitating membrane fusion (McEwan et al, 2015; Wang et al, 2015; Landajuela et al, 2016). Presumably, LC3s are more important for recruitment of cytosolic receptors, while GABARAPs in general facilitate transport and recruitment of membrane‐bound factors (Sagiv et al, 2000; Muller et al, 2002; Leil et al, 2004; Genau et al, 2015; Joachim et al, 2015).
One reason for the limitations to study individual mATG8s on endogenous level is the lack of appropriate tools, such as isoform‐specific antibodies or sensors. Gene replacement and tagging of endogenous proteins is becoming easier with recent developments in the CRISPR technology (Kaulich et al, 2015), but this technique needs to be applied to each studied cell line individually. Additionally, off‐targets on the genomic level and effects of even small tags on the protein function can never be completely excluded. We therefore aimed to create a set of fluorescent sensors that target individual mATG8s and would be applicable in all kinds of cell lines. By utilizing binding domains of known interactors, we and others have previously generated a set of chain‐specific ubiquitin sensors (Fig 1A) (Sims et al, 2012; van Wijk et al, 2012). In order to create specific sensors for individual mATG8 homologues, we used an unbiased peptide phage display approach to identify peptides that specifically bind mammalian ATG8 isoforms with high affinity. Peptide phage display is a powerful approach to identify specificity profiles of protein domains that recognize linear peptide motifs, and has been used to map the binding specificity of PDZ, SH3, and WW domains (Tonikian et al, 2008, 2009; Fowler et al, 2010). Additionally, it has been shown that peptide phage display has a high level of sensitivity that is able to detect the influence of single point mutations on the peptide‐binding profile of an engineered PDZ domain (Ernst et al, 2009, 2010). Here, we report the development of engineered peptides, which selectively bind individual mATG8s in vitro and in vivo. Starting from a first generation of ATG8 sensors (AS), we established different strategies to generate a second and third generation of improved sensors. As a proof of concept, we show that LC3C sensors are specifically recruited to mitochondria and Salmonella during mitophagy and xenophagy pathways, respectively.
Figure 1. Development of peptide‐based ATG8 sensors binding individual ATG8 proteins.
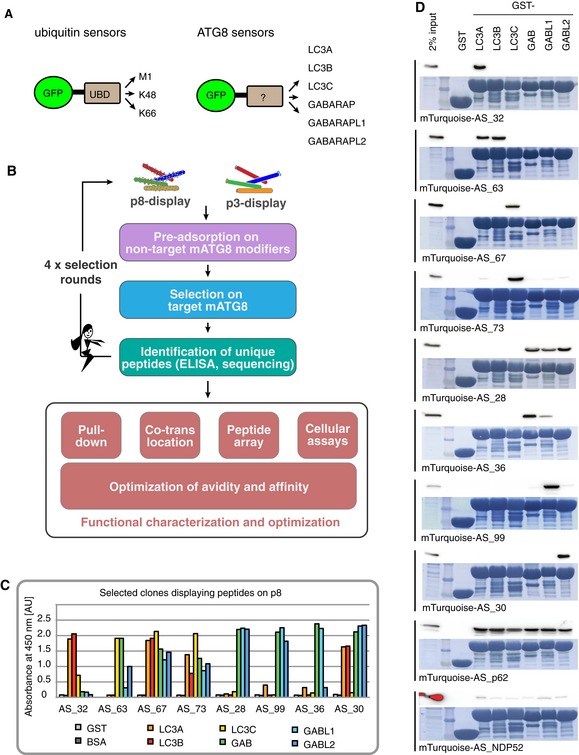
- mATG8‐specific peptides are developed analogous to chain‐specific ubiquitin sensors (van Wijk et al, 2012).
- Peptides are displayed on major coat protein p8 or minor coat protein p3, pre‐adsorbed on non‐target mATG8 (purple box), and selected on target mATG8 (blue box). Specific peptides are identified by ELISA and subsequently sequenced (green box). Efficacy and specificity of the selected sensors are determined by pull‐downs, intracellular co‐translocation assay, peptide‐binding arrays, and cellular assays. Sensors are further improved by rational optimization of avidity and binding affinity.
- Specific peptides for mATG8 are identified by phage ELISA on immobilized GST‐mATG8s and negative control proteins BSA and GST. Binding signal is shown as absorbance at 450 nm in arbitrary units [AU].
- Pull‐down of mTurquoise‐peptide fusions from cell lysates on GST‐mATG8s beads followed by immunoblotting with α‐GFP. As loading control, the protein staining with amido black is shown.
Results
Peptides selected by phage display discriminate between individual LC3/GABARAPs in vitro
The mammalian ATG8 family contains six members divided into two subfamilies: LC3s (LC3A, LC3B, LC3C) and GABARAPs [GABARAP; GABARAPL1 (GABL1); GABARAPL2/GATE 16 (GABL2)]. Using peptide phage display, we aimed to create a toolbox of peptides, which bind specifically to individual mATG8s. Initially, a library of random 16‐mer peptides fused to the M13 major coat protein p8 was constructed. The resulting library consisted of a pool of approximately 1011 unique phage particles, each displaying multiple copies of an individual peptide on its surface (Fig 1B). Display of numerous peptides on the major coat protein p8 results in a strong avidity effect that allows the selection of highly specific peptides optimized for binding to individual mATG8 proteins (Sidhu et al, 2000). To avoid cross‐reactivity of the resulting peptides, the phage pools have been extensively pre‐adsorbed on the five remaining, non‐cognate mATG8 family members, followed by positive selection on the targeted mATG8 isoform (Fig 1B). After selections, individual phage clones displaying peptide copies with a unique sequence were analyzed by ELISA for their binding specificity toward individual mATG8 family members and sequenced (representative ELISA results shown in Fig 1C).
To further analyze in vitro binding selectivity of the engineered peptides, all identified peptides from p8 display were cloned as mTurquoise fusion proteins in a mammalian expression vector and subjected to pull‐down analysis (Fig 1D and Appendix Fig S1). For control and comparison, we added in our analysis peptides comprising LIR motifs from two known autophagy receptors, p62 and NDP52 (Table EV1). NDP52 is known to specifically bind to LC3C as a full‐length protein (von Muhlinen et al, 2012). However, the peptide containing the LIR of NDP52 bound weakly to all GST‐tagged mATG8s to a similar extent (Fig 1D). Similarly, the p62 LIR peptide was shown to interact with all mATG8s, albeit with apparently much stronger affinity (Fig 1D) (Birgisdottir et al, 2013). This observation indicates that additional binding surfaces adjacent to the LIR could be required to mediate specific binding of autophagy receptors and adaptors to individual mATG8s. Such a mechanism to mediate specificity to GABARAP has been already described for ALFY (Lystad et al, 2014). In contrast to the control p62 peptide that binds to all six isoforms in the pull‐down experiment, we identified 34 peptides with varying degrees of specificity to individual mATG8s (Fig 1D and Appendix Fig S1). Notably, the fluorophore‐fused peptides showed higher levels of selectivity in pull‐down experiments compared to the ELISA with peptides displayed on the phage surface (compare Fig 1C and D). Based on their in vitro binding specificities, we have selected 13 peptides targeting individual mATG8 for further cellular and functional studies (Table EV1).
Interactions between engineered peptides and their target mATG8 are preserved in cells, and affinities can be further increased
In a next step, we tested the specificity of selected peptides in a cellular environment. For this purpose, we applied an annexin A4‐driven membrane co‐translocation assay (Piljic & Schultz, 2008). The engineered peptides were fused to the C‐terminus of an annexin A4‐mCherry (A4‐mCh) module and co‐expressed with individual EGFP‐mATG8s devoid of the C‐terminal Gly residue required for lipidation. The A4‐mCh–peptide fusion is recruited to membranes upon ionomycin‐induced calcium influx and causes co‐translocation of the EGFP‐mATG8 in case there is interaction between the two molecules (Fig 2A). As a positive control, we used the interaction between A4‐mCh‐AS_p62 peptide and EGFP‐LC3B (Fig EV1A). Three out of all tested peptides gave rise to a comparable or better co‐recruitment of their target mATG8 when compared to the p62‐derived peptide–LC3B interaction (Figs 2B and EV1A).
Figure 2. Cellular validation of ATG8 sensors.
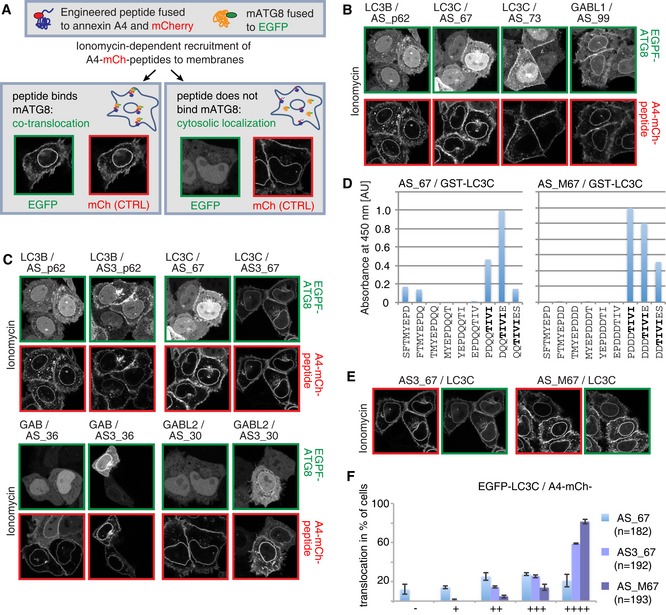
-
ASchematic illustration of annexin A4‐based co‐translocation assay of potential mATG8 binding peptides and their target mATG8 to membranes upon calcium influx induced by ionomycin treatment.
-
B–F(B, C, E) Annexin A4‐based co‐translocation assays of peptide binders and EGFP‐mATG8s. In comparison with single peptides (B), triplication of peptides (C) or introduction of negative charges adjacent to the binding motif (E) increases the co‐translocation extent of the peptide sensors with their target mATG8. Control assays with non‐target mATG8 are shown in Fig EV2. (D) Biotinylated octameric peptides representing a single amino acid walk‐through along the complete 16‐mer peptide AS_67 and AS_M67 are immobilized on streptavidin. Binding of GST‐LC3C fusion protein to the immobilized peptides is detected by a GST‐specific antibody (HRP‐conjugated) and shown as absorbance at 450 nm. Identified core motif shown in bold letters. AU = arbitrary unit. (F) Translocation efficiency of the unmodified peptide AS_67, triplicate AS3_67, and charge‐optimized peptide AS_M67 is classified as percentage of cells that show no translocation “−” to very good “++++” translocation with EGFP‐LC3C; n = total number of analyzed cells. Data are presented as mean of three biological replicates ± SD.
Figure EV1. Cellular validation of ATG8 sensors.
-
A–CAnnexin A4‐based co‐translocation assays of peptide binders and EGFP‐mATG8. Triplication of peptides (B) or introduction of negative charges in close proximity to the binding motif (C) increases the potential of peptide binders to co‐translocate their target mATG8.
-
DBiotinylated octameric peptides representing a single amino acid walk‐through along the complete 16‐mer peptide AS_67 and AS_M67 are immobilized on streptavidin. Binding of GST‐ATG8 fusion protein to the immobilized peptides is detected by a GST‐specific antibody (HRP‐conjugated) and shown as absorbance at 450 nm. AU = arbitrary unit.
To improve the binding strength of the engineered peptides, we generated constructs that encode triple repeats (AS3) of the engineered peptide sequences fused to the C‐terminus of the A4‐mCh module. This approach let us identify two additional in vivo mATG8 binders and also improved the co‐recruitment levels of the aforementioned positive peptides (Figs 2C and F, and EV1B). Importantly, the triplication of the peptide did not alter its target mATG8 preference (Fig EV2).
Figure EV2. Cellular validation of ATG8 sensors.
- Annexin A4‐based co‐translocation assays of peptide binders and EGFP‐mATG8. mATG8 peptide binders specifically co‐translocate their target‐mATG8, while non‐cognate mATG8s remain cytosolic upon ionomycin treatment.
- Quantification of the translocation potential of peptide binders for their target mATG8. Combined data from three biological replicates are presented. Number of counted cells per experiment > 50. Classification ranges from “−” (no translocation) to “++++” (very good); n = total number of counted cells. Error bars represent ± SD.
It has been shown previously that negatively charged residues in close proximity to the core LIR motif improve binding toward LC3s (Wild et al, 2011). In order to test whether introduction of negatively charged amino acids upstream of the binding motif would improve the performance of the sensor, we focused on the LC3C‐specific peptide AS_67 as a model system (Table EV1). First, we performed a peptide array analysis via a single amino acid walk‐through with octameric peptides along the complete 16‐mer. In this assay, we identified a CLIR‐like motif “TIVI” to be essential for interaction with LC3C (Fig 2D) (von Muhlinen et al, 2012). Subsequently, we mutated the original peptide sequence SFTMYEPDQQTIVIES (AS_67) to SFTMYEPDDDTIVIES (AS_M67), to provide additional negative charges N‐terminal to the core CLIR‐like motif. The AS_M67‐based A4‐mCh construct (containing only a single copy of the modified peptide) resulted in improved co‐translocation of LC3C comparable to AS3_67 (Figs 2E and F, and EV1C). Also in the presence of additional negative charges, the CLIR‐like motif “TIVI” remained the necessary determinant of AS_M67 binding to LC3C (Fig 2D).
As the p8‐derived peptides directed against LC3A and LC3B (AS_32, AS_55, AS_57, AS_59, AS_61, AS_63) did not show translocation in the annexin A4‐based assay (Table EV1), we performed additional phage display selections with increased selection stringency and an altered pre‐adsorption strategy. In the first step, we constructed a fully randomized 12‐mer peptide library displayed on M13 minor coat protein p3 (instead of p8), resulting in the display of only 1–3 peptide copies on the phage surface (Fig 1B). This library with a diversity of 1010 was used to select against LC3A and LC3B with pre‐adsorption only on GABARAPs and LC3C. With this approach, we favor the selection of peptides that are bispecific for LC3A and LC3B, the two closest family members that share ~80% identity. In this way, we identified a new peptide, named AS_AB2, which showed preferential binding to LC3A and LC3B in pull‐down assays (Table EV1). This peptide was not analyzed in the annexin assay, but directly tested for recruitment to LC3A/LC3B‐positive autophagosomes (Table EV1 and see below).
In summary, both strategies—the triplication of the peptide and charge optimization adjacent to the LIR motif—are suited for enhancing binding between peptides and mATG8s, by either inducing avidity or increasing affinity, respectively. Our results show that the engineered peptides can discriminate between mATG8s in the cellular environment. Additionally, their interaction strength is sufficient to follow ATG8 trafficking during autophagy, making them suitable for the generation of ATG8 sensors.
Engineered peptides are recruited to mATG8‐positive autophagosomes in vivo
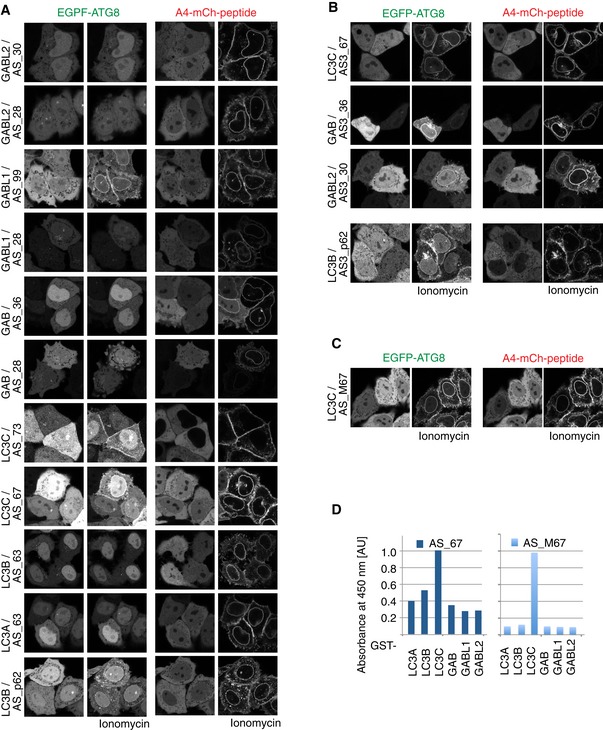
As a starting point for our in vivo sensor development, we compared the localization of selected peptides in cells overexpressing individual EGFP‐tagged mATG8s or endogenous mATG8s under basal and autophagy‐stimulated conditions (3 h KU‐0063784 + bafilomycin A1 treatment, later referred to as KU + Baf). As control, we used the triplicate repeat of the p62‐derived peptide fused to mCherry (mCh‐AS3_p62; Fig 3 and Table EV1) as a sensor for LC3B. While overexpressed EGFP‐LC3B as well as the p62‐derived sensor remained mainly cytosolic in non‐stimulated cells, they were co‐recruited to autophagosomes upon autophagy stimulation (Fig 3A). Importantly, mCh‐AS3_p62 was also recruited to autophagosomes when LC3B was present only on endogenous levels (Fig 3B), suggesting that the sensor approach is widely applicable and not limited to overexpressed target proteins.
Figure 3. ATG8 sensors are recruited to autophagosomes upon autophagy induction.
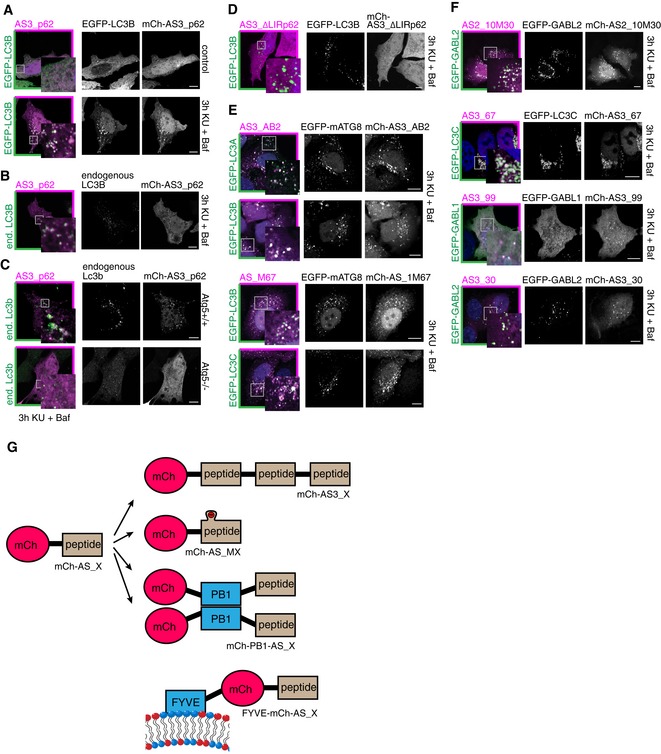
-
A, BRecruitment of the mCherry‐fused p62‐derived sensor AS3_p62 to autophagosomes positive for overexpressed EGFP‐LC3B (A) or endogenous LC3B (B) is visualized by immunofluorescence upon autophagy induction by 3 h KU + Baf treatment.
-
CRecruitment of AS3_p62 in Atg5+/+ or Atg5−/− MEFs to endogenous Lc3b.
-
DLack of recruitment of the p62‐derived sensor in the absence of the hydrophobic core residues of the LIR motif (∆LIR) to EGFP‐LC3B‐positive autophagosomes.
-
E, FATG8 sensors are recruited to autophagosomes labeled with respective target mATG8 proteins.
-
GExisting sensors can be further improved by triple repeats of the engineered peptide (mCh‐AS3_X), by flanking charges (mCh_AS_MX), by introduction of an oligomerization domain (mCh‐PB1‐AS_X), or by inducing membrane association (FYVE‐mCh‐AS_X).
To ensure that recruitment of the sensor to autophagosomes was indeed dependent on its interaction with mATG8s and to rule out that the sensor was recruited to autophagosomes in some other non‐specific way, we performed a similar analysis in WT and Atg5 KO mouse embryonic fibroblasts (MEFs). ATG5 is a key player in the lipidation process of LC3B, and cells lacking this protein fail to form ATG8‐positive autophagosomes (Kuma et al, 2004). While endogenous Lc3b was recruited to autophagosomes in WT MEFs upon autophagy stimulation, it remained cytosolic in Atg5 KO cells (Fig 3C). Similarly, mCh‐AS3_p62 remained cytosolic in Atg5 KO cells after autophagy induction, indicating Atg8 dependency for its recruitment to autophagosomes (Fig 3C).
Further tests on mCh‐AS3_p62 revealed that the sensor preferentially recognizes LC3A‐, LC3B‐, and GABARAPL2‐positive autophagosomes in cells (Fig EV3A and B). This preference for a subset of mATG8s in a cellular environment was not predictable from our in vitro pull‐down analysis, where AS_p62 bound all six mATG8s to a similar extent (Fig 1D). It is important to note that endogenous LC3B is continuously present in the cell lines used and contributes to the recruitment of the sensor to autophagosomes positive for overexpressed ATG8s. It is therefore possible that the specificity of this sensor for LC3A and LC3B is higher than the evaluation presented in Fig EV3B suggests.
Figure EV3. ATG8 sensors are specific for autophagosomes decorated with cognate mATG8.
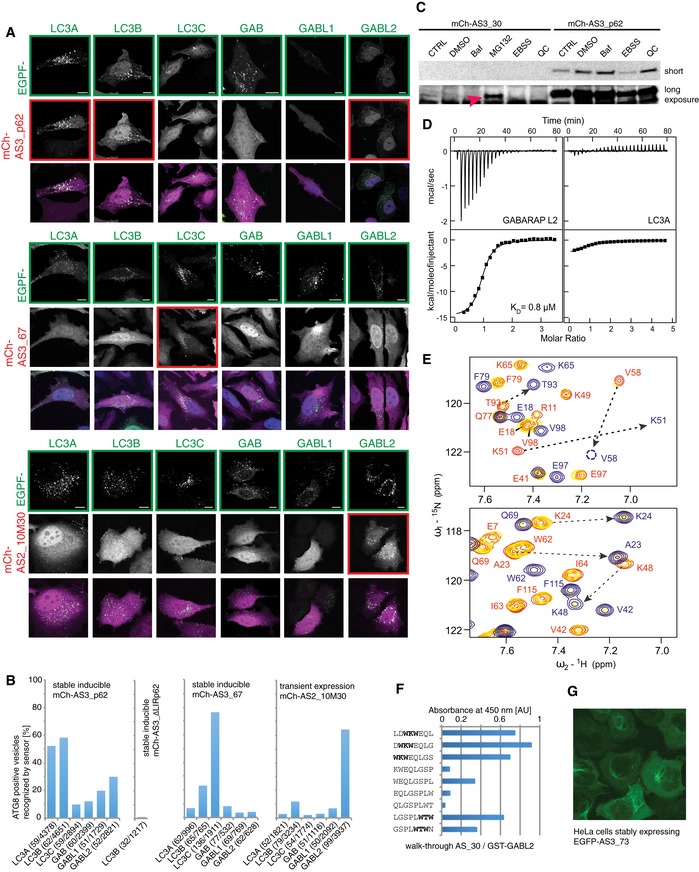
- mCh‐AS3_p62, mCh‐AS3_67, and mCh‐AS2_10M30 are assayed for specificity toward all six mATG8s. Experiments were performed in HeLa cells stably expressing indicated sensor after doxycycline induction. Scale bar: 10 μm.
- Quantification of experiments presented in (B) and Fig 3D. Number of counted cells/autophagosomes is provided for each experiment.
- Sensor mCh‐AS3_30 is instable. Stable inducible HeLa cell lines expressing indicated sensor were treated with indicated compounds and analyzed by immunoblot with antibodies against mCherry. MG132 treatment stabilizes the sensor, as it is apparent in the overexposed blot (red arrow).
- ITC experiments showed significant preference of the Ub19‐AS_30 fusion construct to GABARAPL2 (K D 0.8 μM, ΔH −15.0 kcal/mol, and ΔS −22.3 cal/mol/K) over LC3A (K D ˜10 μM, ΔH −3.4 kcal/mol, and ΔS +12.4 cal/mol/K). Top panels display the raw heat per injection, and the lower panels display the integrated heat per titration over molar ratio (protein: Ub19‐AS_30) with correction to heat of dilution. K D values determined in assumption of “one site interaction” are given for each experiment. These parameters were mirrored in NMR experiments presented in (E).
- Titration of Ub19‐AS_30 fusion construct to 15N‐labeled LC3B (upper plot) indicates weak interaction with intermediate exchange mode, while that for GABARAPL2 (lower plot) shows strong interaction in slow exchange mode. Representative sections of 1H‐15N HSQC spectra for 15N‐labeled proteins (LC3B and GABARAPL2) upon titration with Ub19‐AS_30 fusion construct are shown in overlay. Both plots show “fingerprint regions” of the proteins spectra (around HN resonance of K51 in LC3A and LC3B, which is K57 in LC3C and K48 in all GABARAP proteins). Resonances of LC3B and GABARAPL2 proteins in their free states and in the presence of 0.25 and 2.0 molar ratios of Ub19‐AS_30 fusion construct are red and yellow and blue, respectively. Resonance assignments are given where necessary in red for free state and blue for Ub19‐AS_30‐saturated state. Changes in positions of resonances due to interaction with AS_30 are highlighted with dashed arrows.
- Peptide array analysis of single amino acid walk‐through with octameric peptides along the complete 16‐mer of peptide binder AS_30 against GST‐GABL2. AU = arbitrary unit.
- Immunofluorescence of EGFP‐AS3_73 expressed in HeLa cells revealed recruitment of the sensor to subcellular structures under basal conditions.
Source data are available online for this figure.
The interaction between the autophagy receptor p62 and LC3/GABARAPs depends on its LIR motif (WTHL), particularly on the two hydrophobic residues at positions X1 and X4. Substitution of the four LIR residues by alanine in our p62‐derived sensor resulted in the construct mCh‐AS3_∆LIRp62 (Table EV1). This construct was no longer recruited to autophagosomes labeled with overexpressed LC3B (Figs 3D and EV3B), demonstrating the LIR dependence of the p62‐derived sensor mCh‐AS3_p62.
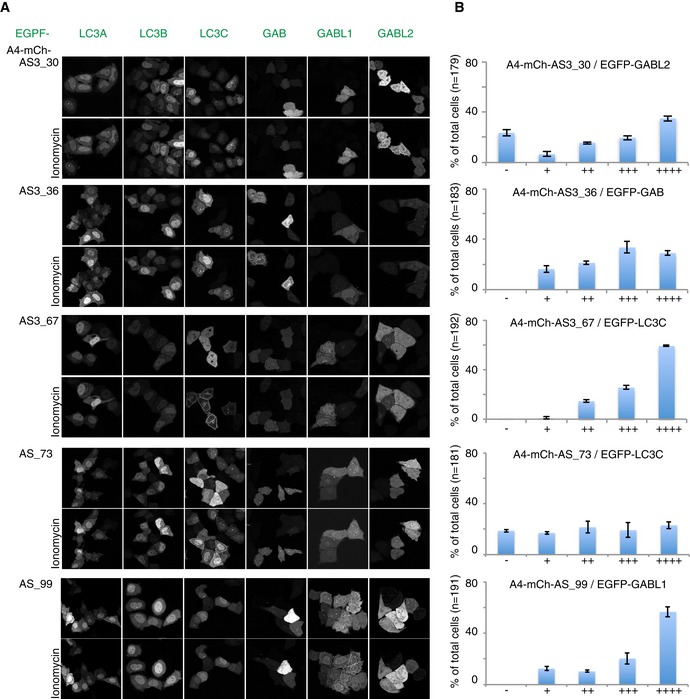
In the next step, we tested whether the engineered isoform‐specific sensors are also recruited to autophagosomes that are positive for their respective target mATG8 after autophagy induction. We transiently co‐transfected individual mCherry sensors (AS3_AB2, AS3_67, AS_M67, AS3_99, and AS3_30) with respective target EGFP‐mATG8s in HeLa cells and tested whether the sensors were recruited to mATG8‐positive autophagosomes. Individual sensors showed recruitment to autophagosomes positive for their overexpressed target mATG8 (Fig 3E and F). The most prominent example was AS3_67, a specific sensor for LC3C, that at low intracellular abundance was recruited to EGFP‐LC3C‐positive autophagosomes, while it did not recognize any other overexpressed EGFP‐ATG8 (Figs 3D, and EV3A and B).
Unfortunately, not all sensors showed such strong binding to their cognate modifiers in cells. Possible reasons are either their instability in cellular environment, insufficient affinity toward their target mATG8, or mislocalization of sensor to distinct structures under resting condition (Figs 3 and EV3C and G). For the GABARAPL1 sensor AS3_99, the used assay (KU + Baf treatment) does not seem to be the optimal condition to validate performance of the sensor: Overexpressed EGFP‐GABARAPL1 remained mainly cytosolic and failed to accumulate primarily on autophagosomes after autophagy induction by KU (Fig 3F). In another instance, the sensor AS3_30 was recruited to GABARAPL2‐positive structures in only few highly expressing cells (Fig 3F), but its consistent recruitment failed in a stable inducible cell line with low expression levels of the sensor. Further analysis revealed that the peptide AS_30 binds GABARAPL2 with 0.8 μM affinity in vitro, but AS3_30 undergoes rapid degradation by the proteasome, potentially driven by its unstructured C‐terminal peptide part (Fig EV3C–E).
Based on these results, we reasoned that a shortened peptide would be less sensitive to proteasomal degradation. The identification of the minimal core motif in AS_30 in a peptide‐binding assay (Fig EV3F) showed that a 10‐mer is sufficient to mediate binding to GABARAPL2. To further decrease the probability of creating a “loose end” that is recognized by the proteasome (Beskow et al, 2009; Verhoef et al, 2009), we used only a duplicate, not a triple repeat, of the peptide. Additionally, the affinity of the shortened peptide to GABARAPL2 was further increased by introduction of negatively charged amino acids close to the predicted LIR‐like binding motif. The resulting sensor was named AS2_10M30 (Table EV1). This optimized sensor was well expressed in cells and specifically recruited to its target GABARAPL2 (Figs 3F and EV3A and B). In summary, our experiments demonstrate that the development of mATG8‐isoform‐specific sensor is feasible and that the sensors can be used to trace individual mATG8s in cells.
Engineered sensors do not affect autophagy flux
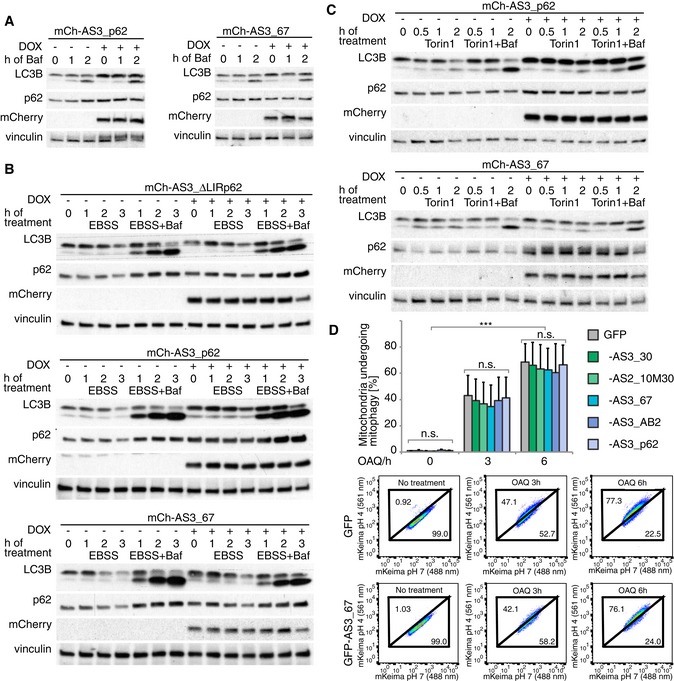
Since mATG8s have central functions in the initiation and formation of autophagosomes, a peptide‐based sensor could act as a competitor preventing binding of endogenous proteins to mATG8s. This could negatively influence the autophagy flux, for example, by blocking binding sites important for mATG8 lipidation. To address this question, we performed autophagy flux assays under basal and autophagy‐stimulated conditions in stable, doxycycline (DOX)‐inducible cell lines expressing the p62‐derived sensor mCherry‐AS3_p62 or the LC3C‐specific sensor mCherry‐AS3_67. To eliminate possible effects resulting from mCherry overexpression or DOX treatment, we generated a control cell line expressing the LIR‐defective version of the p62‐derived sensor, AS3_∆LIRp62, which does not bind mATG8s. DOX treatment in the cell line expressing the control construct mCherry‐AS3_∆LIRp62 had no effect on LC3B lipidation (LC3B II) under basal and autophagy‐induced conditions (Figs 4A–C, EV4 and EV5). However, block of lysosomal degradation with bafilomycin A1 led to faster accumulation of p62 and LC3B I indicating accelerated basal and starvation‐induced autophagy flux upon DOX‐induced protein expression (Fig EV5). Taking these observations into account, the p62‐derived sensor mCherry‐AS3_p62 and sensor mCh‐AS3_67 did not inhibit autophagy flux (Figs 4A–C, EV4 and EV5). While the LC3C‐specific sensor mCh‐AS3_67 only marginally affected p62 turnover, sensor mCh‐AS3_p62 clearly delayed p62 degradation indicating competition between the sensor and p62 for endogenous ATG8s.
Figure 4. Autophagy flux is unaffected by sensors.
-
A–CAnalysis of autophagy flux in stable inducible HeLa cell lines expressing mCherry fusion proteins of indicated ATG8 sensor upon doxycycline (DOX) induction: Autophagy flux is monitored in basal conditions (A), upon starvation (EBSS) (B), or after autophagy induction by Torin 1 (C) indirectly via lipidation of LC3B and turnover of the autophagy receptor p62.
-
DFACS‐based mitophagy flux analysis performed in HeLa cell line stably expressing HA‐Parkin, mt‐mKeima, and a GFP fusion of the indicated sensor. Cells were treated with OAQ (oligomycin, antimycin, Q‐VD) for the indicated times and analyzed by FACS for lysosomal‐positive mt‐mKeima (561 nm). The average of three independent experiments is presented. Representative FACS data from experiments (GFP control and AS3_67) are shown on the lower panels. Error bars indicate standard deviation. Significance calculation: two‐way ANOVA of one‐way ANOVA. n.s. = not significant. *** indicates P‐value of 0.0001.
Source data are available online for this figure.
Figure EV4. Autophagy flux in the absence and presence of ATG8 sensors.
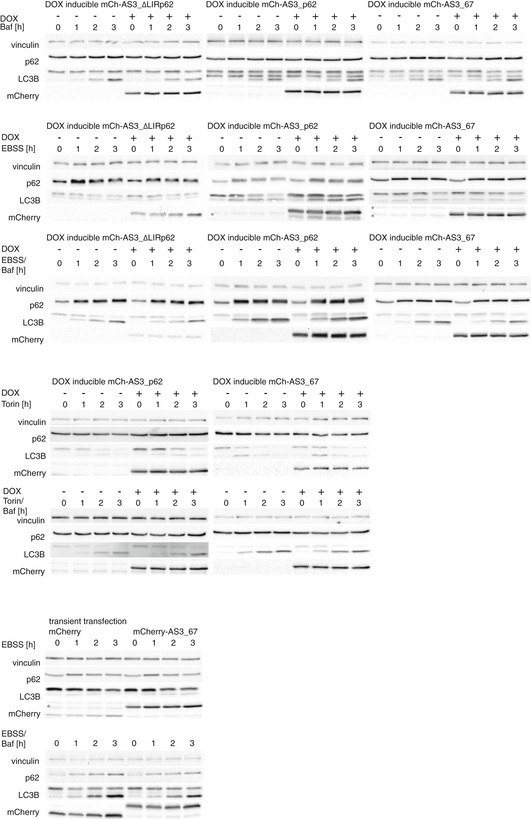
Analysis of autophagy flux in stable inducible HeLa cells expressing mCherry fusions of indicated ATG8 sensor upon doxycycline (DOX) induction or in HEK293T cells expressing indicated ATG8 sensor upon transient transfection. Representative images for quantifications of Fig EV5 are shown. Asterisk indicates unspecific binding of the antibody.
Figure EV5. Quantification of autophagy flux in the absence and presence of ATG8 sensors.
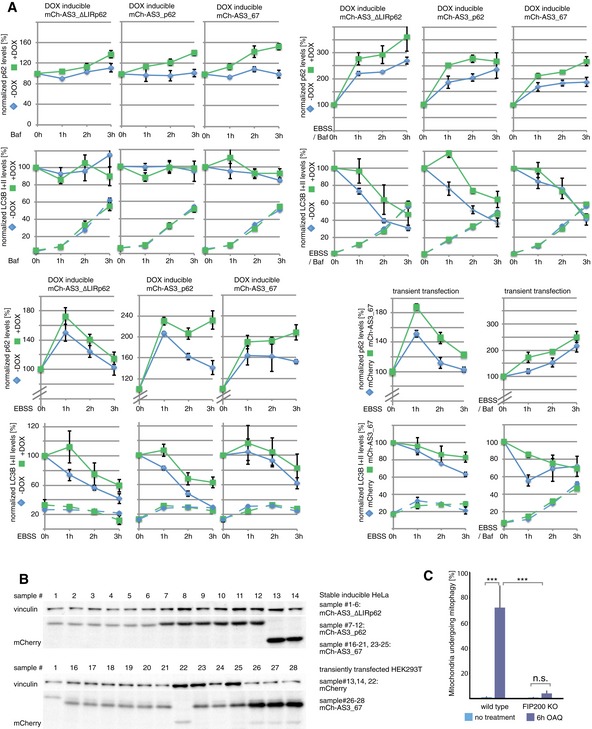
- Quantification of autophagy flux data shown in Fig EV4. Normalized levels of LC3B I and II are represented by continuous and dashed lines, respectively. Data present the mean of three independent experiments ± SEM.
- Comparison of the expression levels of sensors. 3 h samples of experiments presented in Fig EV4 were loaded and detected with anti‐tRFP antibody. Numbers above gel lanes assign individual samples to cells expressing indicated constructs.
- FACS‐based mitophagy flux analysis performed in wild‐type and FIP200 KO cells stably expressing HA‐Parkin and mt‐mKeima. Cells were treated with OAQ (oligomycin, antimycin, Q‐VD) for the indicated times and analyzed by FACS for lysosomal‐positive mt‐mKeima (561 nm). The average of three independent experiments is presented. Error bars indicate standard deviation. Significance calculation: one‐way ANOVA. n.s. = not significant. *** indicates P‐value of 0.0001.
Additionally, several sensors (AS3_30, AS2_10M30, AS3_67, AS2_10M67, AS3_73, AS3_AB2, AS3_p62) have been tested for their potential inhibitory effect on selective autophagic removal of mitochondria (mitophagy) (Figs 4D and EV4C; Table EV1). We used an assay that is based on the pH‐dependent changes in mKeima fluorescence during mitophagy induced by oligomycin/antimycin A (Bingol et al, 2014; Lazarou et al, 2015). Fused to a mitochondrial localization signal, the changes in mKeima‐fluorescence emission allow tracking of the translocation of damaged mitochondria from the cytosol to the lysosome by FACS: In an environment at pH 7, mKeima has an excitation maximum at 438 nm, whereas at pH 4 the maximum shifts to 550 nm. None of the stably expressed sensors had a significant effect on mitophagy flux over a period of 6 h, suggesting that they do not block the endogenous mitophagy machinery (Fig 4D). Taken together, the engineered mATG8‐specific sensors detect mATG8‐positive autophagosomes, but they do not seem to affect the endogenous machinery that controls autophagy flux.
Characterization of LC3C involvement in xenophagy pathways via improved LC3C‐targeting sensors
LC3C was suggested to be specifically involved in the autophagic clearance of Salmonella (xenophagy) (von Muhlinen et al, 2012). We were interested in monitoring the involvement of LC3C along this pathway by using sensor mCh‐AS3_67 that showed strong specificity toward LC3C both in vitro and in vivo (Table EV1; Figs 1D and EV3A and B).
A stable HeLa cell line expressing mCh‐AS3_67 upon DOX treatment was infected with the Salmonella strain SFH2. The genome of Salmonella SFH2 carries GFP as a reporter gene under the control of the uhpT promoter (Wild et al, 2011). Upon escape of bacteria from Salmonella‐containing vacuoles (SCVs) into the cytosol, expression of the reporter gene is activated due to the presence of glucose‐6‐phosphate in the host's cytosol. This enables the specific identification of cytosolic bacteria, which are in principle accessible to the autophagic machinery. Analysis by fluorescence microscopy of cells at an early infection stage (up to 90 min after infection) showed that we could capture the event of Salmonella escaping SCVs. Sensor mCh‐AS3_67 was very efficiently recruited to bacteria during these early events (Fig 5A): In five HeLa cells that contained Salmonella in the process of turning GFP‐positive (low GFP signal; Fig 5B, white arrow), all pathogen cells were labeled with mCh‐AS3_67. This signal overlapped with the signal of endogenous LC3C (Fig 5B, lower panels). Two cells contained more than only one Salmonella cell (i.e., five and eight bacteria, respectively). In these HeLa cells, sensor‐negative bacteria had already developed a bright green color, indicating prolonged residence in the cytosol (Fig 5B, compare white and green arrow). In advanced stages of bacterial infection (4 or 6 h after infection), co‐localization of mCh‐AS3_67 with Salmonella could not be detected despite the observation that a subpopulation of cytosolic bacteria was strongly positive for endogenous LC3C (Figs 5C and EV6D). In contrast to LC3C, endogenous LC3B accumulated in spots in close proximity to the bacteria (Fig 5C).
Figure 5. Performance of ATG8 sensors can be increased by introduction of the oligomerization domain PB1.
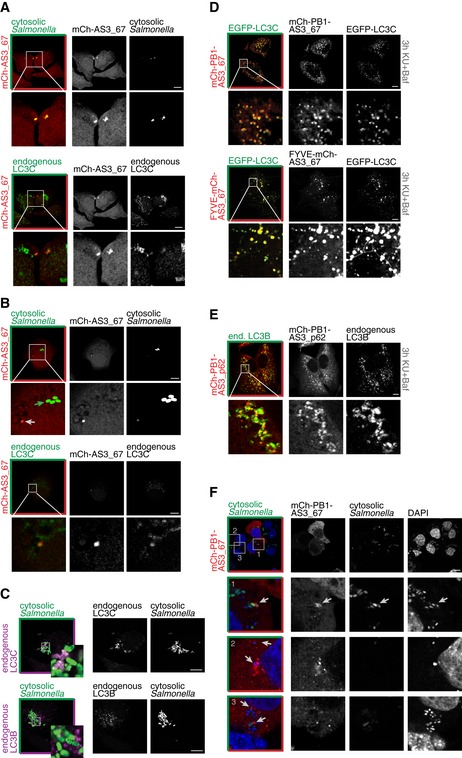
- LC3C‐specific sensor mCh‐AS3_67 is recruited to Salmonella (SFH2) escaping SCVs. Salmonella SFH2 strain is carrying a GFP‐reporter gene activated by availability of glucose‐6‐phosphate in the host's cytosol.
- Salmonella with prolonged cytosolic residence and strong activation of the GFP‐reporter gene (green arrow) are not recognized by the LC3C‐specific sensor mCh‐AS3_67.
- A subpopulation of hyperproliferative Salmonella in the cytosol is coated with endogenous LC3C. In contrast, endogenous LC3B is enriched in spots in close proximity to the cytosolic bacteria.
- Sensitivity of LC3C‐specific sensor mCh‐AS3_67 is increased upon fusion of membrane‐tethering FYVE domain or PB1 oligomerization domain.
- Introduction of the PB1 domain into mCh‐AS3_p62 increases its performance in recognizing endogenous LC3B.
- High‐performance sensor mCh‐PB1‐AS3_67 was transiently expressed in Salmonella‐infected HeLa cells to monitor xenophagy events.
Figure EV6. Specificity of ATG8 sensors.
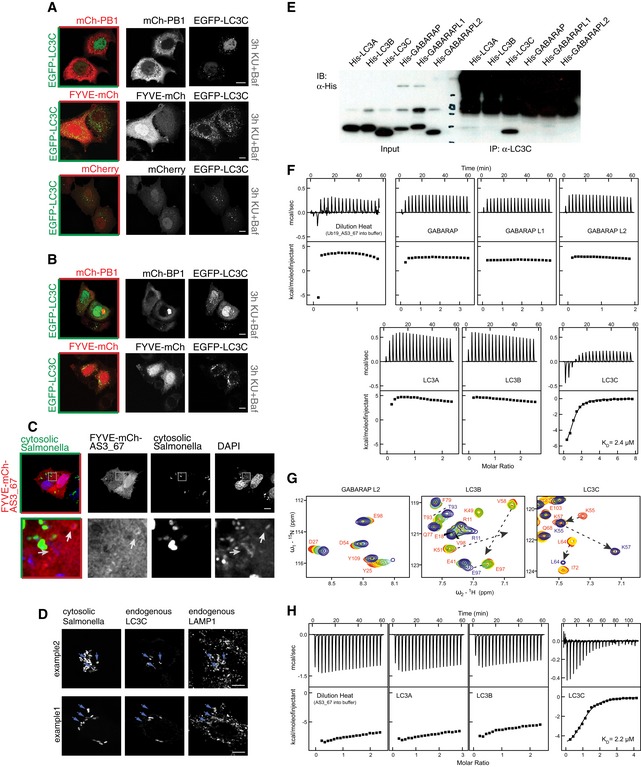
- A, B
-
CHigh‐performance sensor FYVE‐mCh‐AS3_67 was transiently expressed in Salmonella‐infected HeLa cells to monitor xenophagy events. Even though the sensor was recruited to cytosolic Salmonella to certain extent, its performance fell short of sensor mCh‐PB1‐AS3_67. Scale bar: 10 μm.
-
DA subpopulation of cytosolic Salmonella is coated with endogenous LC3C and engulfed by lysosomes in HeLa cells (marker: endogenous LAMP1). Arrows indicate examples of Salmonella located within lysosomes. Scale bar: 10 μm.
-
ELC3C‐specific antibody (Stadel et al, 2015) was incubated with purified, recombinant HIS6 fusion proteins of mATG8s and subsequently subjected to immunoprecipitation (IP) experiments with protein A–Sepharose. The specificity of the antibody for LC3C was confirmed. Unspecific signal in the high molecular weight area of the IP samples is caused by the LC3C antibody.
-
FITC experiments showed significant preference of Ub19‐AS_67 fusion construct to LC3C. Titrations of all other human ATG8 proteins with Ub19‐AS_67 fusion construct revealed weak interactions and do not differ significantly from Ub19‐AS_67 dilution heat. In contrast, titration of LC3C with Ub19‐AS_67 fusion construct shows a relatively strong interaction (K D 2.4 μM, ΔH −7.2 kcal/mol, and ΔS +1.4 cal/mol/K) comparable to that for p62 and NBR1 LIR interactions to LC3B (Rozenknop et al, 2011). Top panels display the raw heat per injection, and the lower panels display the integrated heat per titration over molar ratio (protein: Ub19‐AS_67).
-
GNMR titration of Ub19‐AS_67 fusion construct to 15N‐labeled GABARAPL2, LC3B, and LC3C proteins shows its specificity for LC3C. NMR titration of Ub19‐AS_67 fusion construct to 15N‐labeled GABARAPL2 (left plot) and LC3B (middle plot) indicates weak interactions with an intermediate exchange mode, while that for LC3C shows a stronger interaction in slow exchange mode. Representative sections of 1H‐15N HSQC spectra for 15N‐labeled proteins upon titration with Ub19‐AS_67 fusion construct are shown in overlay. All plots show “fingerprint regions” of the proteins spectra (around HN resonance of K51 in LC3B, K48 in GABARAPL2, and K57 in LC3C). Molar ratios of protein: Ub19‐AS_67 are rainbow‐color‐coded (1:0, 1:0.25, 1:0.5, 1:1, 1:2; red to blue) for each titration step. Changes in positions of resonances due to interaction with AS_67 are highlighted with dashed arrows.
-
HITC experiments showed significant preference of AS_67 peptide to LC3C. Similar to Ub19‐AS_67 fusion construct, de‐tagged AS_67 peptide did not show significant interaction with LC3A and LC3B proteins. Corresponding titration profiles are very similar to dilution heat of the peptides. In contrast, AS_67 peptide interacts with LC3C protein, and ITC analysis results are similar to Ub19‐AS_67 parameters of interaction (K D 2.2 μM, ΔH −5.4 kcal/mol, and ΔS +7.8 cal/mol/K). Top panels display the raw heat per injection, and the lower panels display the integrated heat per titration over molar ratio (protein: AS_67).
To validate the specificity of the antibody used for staining of endogenous LC3C, we performed pull‐down experiments with purified, recombinant mATG8s and showed that the antibody specifically immunoprecipitated LC3C (Fig EV6E). We further characterized binding characteristics of AS_67 to mATG8s by isothermal titration calorimetry (ITC) demonstrating that binding of the engineered sensor is specific for LC3C (Fig EV6F–H). We hypothesize that upon prolonged Salmonella infection, LC3C is shielded by endogenous autophagic protein complexes that outcompete the sensor for binding to LC3C. This is supported by nuclear magnetic resonance spectroscopy (NMR) data, which show the fingerprint region of LC3C upon AS_67 binding. As expected, the residues of the LIR binding region of LC3C are also involved in the interaction with AS_67 (Fig EV6G). Consequently, endogenous proteins that have a higher affinity can outcompete the transient interaction of the sensor mCh‐AS3_67 with LC3C.
To further improve the mCh‐AS3_67 sensor, we considered the following two options: (i) A membrane‐tethering domain fused to AS3_67 could serve as an additional anchor to promote binding of sensors to autophagosomal membranes, in addition to lipidated LC3C; and (ii) fusion to an oligomerization domain could also contribute to local enrichment of the AS3_67 sensor (Fig 3G). To this end, we fused mCh‐AS3_67 to the mutated FYVE domain of EEA1 as a membrane‐tethering domain (Hayakawa et al, 2004) or used the PB1 domain of p62 as an oligomerization domain (Wurzer et al, 2015). Constructs carrying the additional domain either upstream or downstream of mCherry were generated and tested for their efficiency in detecting LC3C‐positive structures under autophagy‐induced conditions (KU + Baf treatment, as in Fig 3D). The presence of the PB1 domain between mCherry and AS3_67 or N‐terminal addition of the FYVE domain significantly increased the sensitivity of the LC3C‐specific sensor AS3_67 (Fig 5D). The control constructs expressing only oligomerization or membrane‐tethering domains, mCh‐PB1 and FYVE‐mCh, respectively, showed largely cytosolic localization, similar as when expressing mCherry alone (Fig EV6A). Only a minor fraction of cells contained aggregated mCh‐PB1 or clusters, which were distinct from LC3C spots (Fig EV6B). Furthermore, combining the PB1 domain with the AS3_p62 sensor improved recognition of autophagosomes labeled with endogenous LC3B (Fig 5E).
Next, FYVE‐mCh‐AS3_67 and mCh‐PB1‐AS3_67 were tested for their ability to monitor LC3C‐positive cytosolic Salmonella (Fig 5F). In this experiment, we detected cytosolic bacteria via its GFP reporter, while the total pool of Salmonella was stained with DAPI. The sensor mCh‐PB1‐AS3_67 was recruited to Salmonella even at later stages of infection. In addition to autophagosomes forming directly around bacteria, the sensor stained vesicles in proximity to bacteria that potentially serve as membrane source or initiation points for new autophagosomes (Fig 5F). Also the FYVE‐mCh‐AS3_67 sensor was recruited to cytosolic Salmonella, but its recruitment was less prominent than with the PB1 construct (Fig EV6C). These findings indicate the efficacy of the improved LC3C sensors mCh‐PB1‐AS3_67 and FYVE‐mCh‐AS3_67 to monitor LC3C‐dependent xenophagy in cells.
Characterization of LC3C involvement in mitophagy pathways
Recent evidence indicates involvement of LC3C in the clearance of dysfunctional mitochondria (Lazarou et al, 2015). To further validate this finding, we applied our optimized LC3C sensor (mCh‐PB1‐AS3_67) to monitor mitophagy in vivo. EGFP‐tagged LC3C or LC3A was co‐expressed with mCh‐PB1‐AS3_67 in U2OS cells. Mitophagy was induced by CCCP treatment and the mitochondria were stained with MitoTracker Deep Red. EGFP‐LC3C coated a subpopulation of depolarized mitochondria, and the same subpopulation of mitochondria was decorated with LC3C‐specific sensor mCh‐PB1‐AS3_67 (Fig 6A). mCh‐PB1‐AS3_67 remained bound to a subpopulation of mitochondria in cells co‐expressing EGFP‐LC3A, indicating that the recruitment of the sensor was driven by endogenous LC3C (Fig 6B). EGFP‐LC3A showed a different localization pattern compared to EGFP‐LC3C and formed small puncta‐like structures in close proximity to the sensor‐coated mitochondria fraction (Fig 6B). Our findings demonstrate that endogenous LC3C is recruited to mitochondria during mitophagy and that this can be monitored by the sensor mCh‐PB1‐AS3_67. Indeed, endogenous LC3C seems to accumulate on mitochondria upon depolarization by CCCP and additional block of lysosomal degradation by bafilomycin A1 (Baf) treatment (Fig 6C). The signal of endogenous LC3C overlaps to a large extent with sensor mCh‐PB1‐AS3_67 and seems to accumulate within lysosomes (Fig 6D), indicating a function of LC3C in delivering dysfunctional mitochondria to the lysosome.
Figure 6. LC3C‐specific ATG8 sensor monitoring mitophagy.
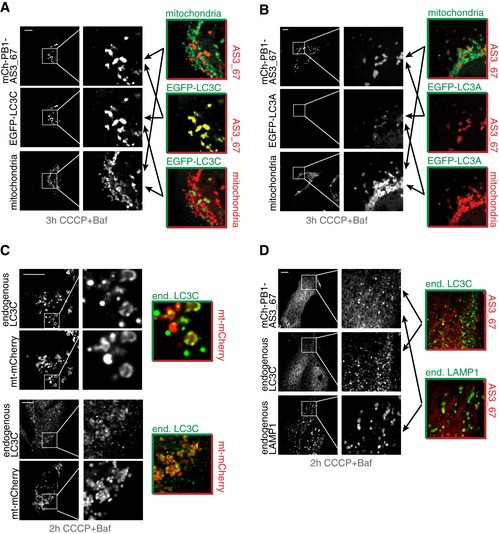
-
A, BLC3C‐specific sensor mCh‐PB1‐AS3_67 is recruited to a subset of mitochondria undergoing mitophagy. Mitochondria were labeled with MitoTracker Deep Red (Thermo Fisher). U2OS cells were transiently co‐transfected with indicated sensor and mATG8 proteins. Mitophagy was induced by CCCP treatment over 3 h. Lysosomal degradation of mitochondria undergoing mitophagy was blocked by the addition of bafilomycin A1 (Baf).
-
CUpon depolarization, endogenous LC3C is localized to mitochondria labeled with mt‐mCherry in U2OS cells. An example of rare events with nearly complete overlap of signals for endogenous LC3C and mitochondria (upper panel) and a representative image (lower panel) is given.
-
DOverlap of LC3C‐specific sensor signal with endogenous LC3C and endogenous LAMP1 in U2OS cells induced for mitophagy.
Discussion
In the present study, we developed peptide‐based mATG8 sensors that are able to monitor the localization of individual ATG8 proteins in mammalian cells and could be used to elucidate their distinct functions.
Phage display was used to isolate peptides that possess bona fide LIR (LC3‐interacting region) properties. Although many of the selected peptides have shown strong specificity for individual mATG8 proteins in vitro, their implementation as in vivo sensors to monitor endogenous mATG8 was not trivial, as the initial tests showed that the sensors lacked sufficient binding affinity. Consequently, we further optimized individual mATG8 sensors by peptide multiplication, charge optimization, and introduction of additional anchor domains to assist the binding to mATG8‐positive structures. In a next step, the sensors were subjected to extensive validation, starting from in vitro characterization, via analysis of their performance in a cellular environment, to an application in functional assays.
We adapted the phage display for the development of specific mATG8 binding peptides based on previous protocols to derive linear binding motifs with high affinity and high specificity (Skelton et al, 2003). Due to the avidity effect of displaying a peptide library on major coat protein p8, it is possible to detect subtle differences in biophysical constraints in peptide recognition modules (Sidhu et al, 2000; Ernst et al, 2009). Although members of the mATG8 family are structurally conserved, GABARAP, GABARAPL1, GABARAPL2, and LC3C are sufficiently different to allow the selection of specific binders. Surprisingly, the binding sites present on LC3A and LC3B seem to be too similar to give rise to a selective peptide; therefore, we focused our efforts on creating a peptide that is able to differentiate between LC3A/B and the remaining mATG8s. To this end, we used monovalent display on p3 in combination with extensive pre‐adsorption on LC3C and GABARAPs, which resulted in a high‐affinity peptide specifically binding to both LC3A and LC3B.
The selectivity of peptides for individual mATG8s was initially analyzed by ELISA of peptides displayed on phage particles (Fig 1C). It was followed by in vitro pull‐down assay of first‐generation sensors, consisting of a fluorophore fused to the respective peptides (Fig 1D and Appendix Fig S1). To our surprise, the specificity observed in phage ELISA and pull‐down experiment did not overlap, underlining the need for biochemical tests of monovalently expressed peptides. The initial analysis of peptides showed that their specificity for mATG8s in pull‐down assays was preserved in cellulo, yet most sensors of the first generation were poorly performing in cells (Figs 2 and EV1). To overcome this shortcoming, we applied different strategies to improve the sensitivity of our sensors, while attempting to preserve the specificity and prevent unwanted inhibitory effects (Fig 3G). In most cases, the performance of the sensors could be considerably improved by multiplication of the peptide sequence, thus increasing the avidity of the mCherry‐tagged sensor. Unexpectedly, multiplication of the GABARAPL2‐specific peptide AS_30 led to intracellular instability of the sensor mCh‐AS3_30 (Fig EV3C). We could alleviate this defect by the identification of the minimal binding motif and subsequent truncation of the peptide (Fig EV3).
Another approach to optimize binding was inspired by studies on the autophagy receptor optineurin where phosphorylation in close proximity to the LIR motif improves affinity to mATG8s (Wild et al, 2011; Rogov et al, 2013). Introduction of negatively charged amino acids close to the CLIR‐like motif of sensor AS_67 improved the interaction with LC3C of the resulting sensor AS_M67 (Fig 2F; Table EV1). However, introduction of negative charges also abrogated LC3C specificity and resulted in additional binding to LC3B (Fig 3E). Analogous to ubiquitin chains and their respective receptors, phosphorylation close to the LIR motif in many cases seems to not only increase the affinity, but also broaden the selectivity of binding (Wild et al, 2011; Farre et al, 2013; Heo et al, 2015; Richter et al, 2016).
All sensors that showed specificity and improved co‐translocation in the annexin assay (Figs 2, EV1 and EV2) were tested for recruitment to autophagosomes coated with overexpressed mATG8 proteins. In total, we have derived six sensory peptides, which are expressed as fusion proteins with a fluorophore. These were either peptide repeats or peptides that contain additional charged residues adjacent to the engineered LIR motif (Fig 3).
We tested the sensors mCh‐AS3_67 and mCh‐AS2_10M30 in greater detail and could demonstrate that they are indeed highly specific for LC3C and GABARAPL2, respectively (Fig EV3A and B). Surprisingly, the p62‐derived sensor mCh‐AS3_p62 exhibited a preferential binding to LC3A and LC3B, despite the initial pull‐down results suggesting that it is a pan‐specific sensor (Figs 1D and EV3A and B).
In this context, one also needs to point at an important limitation of using overexpressed mATG8s for selectivity testing in wild‐type cells: there is always a possibility that endogenously expressed target ATG8 participates in the sensor recruitment, thus masking its true binding specificity. Trustworthy analysis of the sensor efficacy with endogenously expressed ATG8s depends on the availability of reliable antibodies or KO cell lines for individual ATG8s. After extensive validation of commercially available antibodies we could only confirm the specificity of antibodies to LC3B and LC3C. Consequently, we could show that LC3B sensors mCh‐AS3_AB2 and mCh‐AS3_p62 are both recruited to autophagosomes coated with endogenous LC3B (Figs 3B and 5E, and data not shown) whereas the LC3C sensor mCh‐PB1‐AS3_67 was recruited to endogenous LC3C (Fig 6D).
In addition, the LC3C antibody was invaluable to assess the usage of the generated sensors in vivo. We used LC3C sensor AS3_67 to study the poorly understood role of LC3C in selective autophagy pathways. The function of LC3C in autophagy has been mainly assigned to xenophagy and, recently, to COPII‐dependent ER export as well as to mitophagy (von Muhlinen et al, 2012; Lazarou et al, 2015; Stadel et al, 2015). Here, we show that sensor AS3_67 is recruited to Salmonella escaping from SCVs into the cytosol (Fig 5). This is consistent with the current view that LC3C is responsible for the efficient recruitment of NDP52 to damaged SCVs, thereby facilitating the sequestration of the bacteria into autophagosomes (von Muhlinen et al, 2012; Verlhac et al, 2015). However, the mCh‐AS3_67 sensor was unsuccessful in tracking endogenous LC3C at later stages of Salmonella infection. Thus, in order to extend the application range of the LC3C sensor for xenophagy study, we further improved sensor mCh‐AS3_67 by introducing either the oligomerization domain PB1 of p62 or the membrane‐binding domain FYVE as assisting domains (Fig 3G). In particular, the oligomerization domain of p62 increased the sensitivity of the sensor. Additionally to monitor xenophagy events, the sensor mCh‐PB1‐AS3_67 revealed a subpopulation of mitochondria that are coated with LC3C during mitophagy, indicating an LC3C/NDP52‐mediated mechanism similar to xenophagy of Salmonella (Verlhac et al, 2015).
The engineered LIR‐ or CLIR‐like motifs of the sensors do not seem to influence autophagy and mitophagy flux indicating that endogenous receptors and effectors specific for LC3C outcompete the sensor and still are able to function. While this is positive on the one side, it also means that the sensor will be unable to monitor events where LC3C is low in concentration and/or tightly associated with effector proteins. Nevertheless, our sensors can be used to study early events in autophagy that precede the recruitment of effector proteins or processes with high local concentration of mATG8s. Similarly to studying xenophagy and mitophagy, mATG8 sensors could be used to investigate cellular processes other than autophagy such as mATG8‐dependent intracellular transport pathways including unconventional secretion and exocytosis (Nowag & Munz, 2015; Stadel et al, 2015), LC3‐associated phagocytosis (Sanjuan et al, 2007), or the role of LC3‐coats on double membranes required for viral infections (Reggiori et al, 2010).
Having established several sensors for individual mATG8 proteins—p62‐derived sensor mCh‐AS3_p62, LC3A and LC3B sensor mCh‐AS3_AB2, LC3B and LC3C sensor mCh‐AS_M67, LC3C sensor mCh‐AS3_67, GABARAPL1 sensor mCh‐AS3_99, and GABARAPL2 sensor mCh‐AS2_10M30—a variety of applications are within reach, including live imaging of dynamic autophagy events as well as simultaneous monitoring of individual mATG8s and their distinct behaviors in cells. Preliminary data on live cell imaging of sensor mCh‐PB1‐AS3_67 (red) decorating MitoTracker Deep Red‐labeled mitochondria (green) upon CCCP treatment support this view (Movie EV1). As final selectivity analysis for sensors in ATG8 KO cell lines remains open, sensors cannot be used as definite proof for localization of a specific ATG8, but they can give important indications, followed by a more detailed analysis of the process. For autophagy‐independent functions of mATG8s, one could imagine further optimization approaches by introduction of other anchor domains customized for the process of interest. Overall, ATG8 sensors provide potent tools for revealing different functions of individual LC3 and GABARAP family members in vivo.
Materials and Methods
Cell culture and generation of stable cell lines
All cells were cultured in DMEM supplemented with 10% (vol/vol) FBS and penicillin and cultured at 37°C in a humidified incubator containing 5% CO2. For transient overexpression, DNA plasmid transfections were performed with GeneJuice (Merck Millipore; HEK293T and HeLa) or TurboFect (Thermo Fisher; U2OS) following the manufacturer's instructions. For the generation of inducible cell lines, respective fusion constructs were cloned into the pcDNA5/FRT/TO vector (Invitrogen) and co‐transfected with Flp recombinase pOG44 into tetracycline transactivator HeLa (T‐REx) cells.
Hygromycin B‐resistant colonies were pooled and expanded. Expression was induced by 1 μg/ml doxycycline for 17–24 h (D9891; Sigma‐Aldrich). HeLa FRT/TO cells for the generation of stable cell lines using the Flp‐In T‐REx system (Invitrogen) were generously provided by S. Taylor and maintained as described (Tighe et al, 2008). Hygromycin B was purchased from Enzo Life Sciences (ALX‐380‐306‐G001) and blasticidin S from Thermo Fisher Scientific (R210‐01). HeLa Kyoto cells were provided by Carsten Schultz, EMBL Heidelberg, Germany. HEK293T and U2OS cells were acquired from ATCC. HeLa cells (mitophagy assay) were acquired from the ATCC and authenticated by the Johns Hopkins GRCF Fragment Analysis Facility using STR profiling. All cell lines used in this study were tested for mycoplasma at least monthly.
Antibodies
Antibodies used for immunofluorescence: anti‐dsRED (rabbit, 632496, Clontech, 1:300), anti‐LC3B (mouse, M152‐3, MBL, 1:300; or rabbit, PM036, MBL, 1:300), anti‐LC3C (rabbit, 1:100; Stadel et al, 2015), and anti‐LAMP1 (mouse, H4A3, DSHB, 1:400).
Antibodies used for Western blot: anti‐GFP (mouse, B‐2, sc‐9996, Santa Cruz Biotechnology, 1:1000), anti‐dsRED (rabbit, 632496, Clontech, 1:1000), anti‐tRFP (rabbit, AB234, BioCat, 1:1000), anti‐LC3B (mouse, 0231‐100/LC3‐5F10, Nanotools, 1:1000), anti‐p62 (mouse, M162‐3, MBL, 1:1000), and anti‐vinculin (mouse, G1160, Sigma, 1:1000).
Phage selections
The p8 peptide library consisted of 1011 unique 16‐mer peptides displayed on phage. The peptide‐coding sequences were assembled from trimeric nucleotide building blocks, so that each naturally occurring amino acid (with the exception of cysteine) was encoded with equal probability at each of the 16 amino acid positions. This approach alleviates the risk of introducing unwanted stop codons.
The p3 peptide library consisted of 1010 unique 12‐mer peptides fused to M13 minor coat protein p3. The peptide‐coding sequence was assembled from 12 consecutive repetitions of “NNK” codons (where N represents all four nucleotides and K represents guanine or thymine).
mATG8 proteins were expressed as GST fusions and purified from Escherichia coli. The phage selections were performed as described (Tonikian et al, 2007). The concentration of the immobilized target protein was gradually decreased from 2 to 0.25 μM in consecutive selection rounds. The phage pool was pre‐adsorbed on immobilized non‐cognate mATG8 proteins (5 μM in total). After the pre‐adsorption, 5 μM (sum) of non‐cognate mATG8 proteins was added to the phage pool and then transferred onto the selection plate. In the initial rounds of selections, input phage pools were additionally pre‐adsorbed on immobilized GST alone (2 μM in coating solution). All pre‐adsorption steps were carried out at 4°C, while binding to the target isoform was performed at room temperature (RT). During selections, we applied increasing number of plate washes (from eight times with cold buffer in the first round to 12 times RT buffer in the last round) and, in the last selection rounds, we diluted the phage input up to 10,000 times.
Individual phage clones were tested for specificity by ELISA on immobilized GST‐mATG8 proteins. Bound phage was detected using HRP‐conjugated mAb against M13 phage. The peptide‐coding sequence of the clones that were specific in the ELISA was determined by DNA sequencing.
GST pull‐down
For GST pull‐down assays using HEK293T cell lysates, cells were transiently transfected with plasmids encoding the indicated proteins using GeneJuice (Merck Millipore) transfection reagent. At 24 h after transfection, cells were harvested, washed with PBS, lysed in lysis buffer (10 mM Tris, pH 7.5; 150 mM NaCl; 0.5 mM EDTA; 0.5% NP‐40) supplemented with protease inhibitors (1 mM PMSF, 1× ROCHE complete), and cleared by centrifugation. For GST pull‐down assays, recombinant GST‐mATG8 on beads in 500 μl pull‐down buffer (10 mM Tris, pH 7.5; 150 mM NaCl; 0.2% NP‐40; 5 mM DTT) was used. Lysates were incubated overnight at 4°C with immobilized GST fusion proteins. Following three washes with the pull‐down buffer, proteins were resolved by SDS–PAGE and analyzed by immunoblotting with the indicated antibodies.
Annexin assay
Annexin assays were performed in HeLa Kyoto cells as described in Piljic and Schultz (2008). Confocal images of the GFP and mCherry channel were taken before and after treatment with ionomycin. Images were acquired by the TCP SP8 laser‐scanning microscope (Leica) using a 63× oil‐immersion lens.
Immunofluorescence
For visualization of ATG8 sensor recruitment to autophagosomes, cells were plated on glass coverslips in a 12‐well plate. Cells were either transfected with 1 μg of DNA, or expression in stable cell lines was induced with 1 μg/ml doxycycline 17–24 h before treatment.
For induction of autophagy, cells were treated with 10 μM KU‐0063784 (autophagy inducer) and 200 ng/ml bafilomycin A1 (deacidification of lysosomes; block of lysosomal degradation). Before induction of mitophagy, MitoTracker Deep Red (Life Technologies) was added to the media for 45 min and removed from the media by one washing step. Cells were subsequently incubated in media containing 20 μM CCCP for 3 h. In Fig 6C, a mCherry‐based marker is used to label mitochondria (Gomes et al, 2011). Salmonella infection was performed as described in Fiskin et al (2016). For fixation, cells were incubated in 4% (wt/vol) paraformaldehyde in PBS for up to 20 min. For staining of endogenous LC3B and staining with anti‐dsRED antibody to enhance sensor signal in MEFs (Fig 3C), cells were permeabilized with 0.1% Triton X‐100 for 3 min, blocked in 5% BSA for 1 h, and incubated with primary and secondary antibodies for 1 h at RT, respectively, each step followed by intensive washing with PBS. For remaining stainings, a saponin‐based method was used: Fixed cells were incubated for > 30 s in PMS (PBS, 5 mM MgCl2, 0.1% saponin). Coverslips were subsequently incubated with primary and secondary antibodies (PBS, 5 mM MgCl2, 0.1% saponin, 1% BSA), respectively, each step followed by intensive washing with PMS. Coverslips were mounted with ProLong Diamond mounting solution with or without DAPI (Molecular Probes) and placed on a glass holder. Images were acquired by the TCP SP8 laser‐scanning microscope (Leica) using a 63× oil‐immersion lens (pinhole setting: ≤ 1.0 Airy). Excitation wavelength, recorded emission spectral range, gain, and laser power were adapted to settings of individual experiments. Image analysis and processing was performed using ImageJ (NIH).
Quantification of sensor recruitment
For semiautomatic quantification of IF images, the Leica Application Suite X 2D Analysis and 2D Analysis Multi Channel Extension software was used. Precise settings can be downloaded via the provided x.cfg file (see Source Data for Expanded View). In brief, the program identifies vesicles/dot‐like structures in two channels (green vesicles = EGFP‐ATG8; red vesicles = mCherry sensor) individually via the following steps: (i) image processing pre‐filter: find white detail—top hat, size 9; (ii) threshold adjustment MaxEntropy; and (iii) binary processing pre‐filter: discard detail, size 1. In case of no recruitment of the sensor, the auto‐setting of the threshold for the red channel is non‐functional. In this case, the threshold has to be adjusted manually. Subsequently, the number of ATG8‐decorated vesicles recognized by the respective sensor was counted by the overlay of the two channels (co‐localization of objects). Performance of the sensor was calculated by percentage of green vesicles recognized by the sensor (number of co‐localization events divided by total number of objects recognized in green channel).
Live cell imaging
Live imaging of U2OS cells, grown in 35‐mm glass‐bottom dishes (No. 0), was performed on Visitron confocal spinning‐disk microscope, equipped with Yokogawa CSU‐X1 scanning head CSU X1‐A1‐5000rpm, single filter sets for GFP, mCherry, EBFP2, and HXP 120 Xenon Lamp for epifluorescence illumination. Still images were processed using ImageJ (NIH).
Peptide array
Lyophilized, biotinylated peptides (approximately 56 nmol; JPT, Germany) were dissolved in 30 μl DMSO and subsequently diluted with 170 μl PBS. Peptides (2 μl/well) were immobilized on 96‐well Nunc immobilizer streptavidin F96 clear plates (#436014, Thermo Scientific) in 100 μl PBS containing 0.1% Tween‐20 (PT) and 5% BSA (PTB) overnight at 8°C. After three washing steps with 200 μl PT, peptides were incubated with 100 μl of 1 μM GST‐mATG8 fusion proteins in PTB, isolated from E. coli, for 1 h at 8°C on a shaker. After three washing steps with 200 μl PT, peptide bound GST‐ATG8 were labeled with anti‐GST‐HRP antibody (A00866, GenScript, 1:5000). After three washing steps with 200 μl PT, 100 μl TMB Substrate Reagent Set (BD OptEIA) was added. The reaction of HRP with the TMB substrate (blue coloration) was stopped by the addition of 1 M H3PO4. Measurements were done on a Synergy H1 ELISA reader from BioTek at 450 nm.
Autophagy flux experiments
Autophagy flux experiments were performed as described earlier (Khaminets et al, 2015). Cells were starved with Earle's balanced salt solution (EBSS) or induced for autophagy by addition of 0.25 μM Torin 1 to the media. About 200 ng/ml bafilomycin A1 (Baf) was added where indicated to block lysosomal degradation. In case of Torin 1 treatment, bafilomycin A1 was added 30 min before the start of the treatment.
Quantification of autophagy flux experiments
Western blots for quantification were detected with the help of a ChemiDoc MP (Bio‐Rad). Images of exposure times adapted to respective signals were exported as tiff, and signal strength was analyzed via ImageJ (NIH). Samples of one experiment (eight samples) were loaded on the same gel. To allow comparison between independent experiments, signals were normalized as follows: For p62 and LC3B I (non‐lipidated LC3B, upper band), the signal strength at the 0 h time point of treatment was set as 100%. For LC3B II, which is barely present at time point 0 h, the sum of the signals of all four time points (0, 1, 2, and 3 h) was set as 100%. As such, distribution of the lipidation progression over time can be displayed.
Mt‐mKeima mitophagy assay
HeLa cells stably expressing mt‐mKeima [a gift from A. Miyawaki, Laboratory for Cell Function and Dynamics, Brain Science Institute (BSI), RIKEN, Japan], HA‐Parkin, and GFP‐variants of peptide binders were generated. For mitophagy, cells were treated with 10 μM antimycin A, 10 μg/ml oligomycin, and 10 μM Q‐VD for up to 6 h. Cells were collected at indicated time points in cell‐sorting media (145 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 10 mM HEPES, 10 mM glucose, 0.1% BSA) containing 0.1 μg/ml DAPI. Mitophagy analysis was performed on a Beckman Coulter MoFlo Astrios cell sorter using Summit software (v6.2.6.16198). Measurements of lysosomal mt‐mKeima were made using dual‐excitation ratiometric pH measurements at 488 (pH 7)‐ and 561 (pH 4)‐nm lasers with 620/29‐nm and 614/20‐nm emission filters, respectively. For each sample, 50,000 events were collected and subsequently gated for YFP/mt‐mKeima double‐positive cells that were DAPI‐negative. Data were analyzed using FlowJo (v10; Tree Star).
ITC and NMR sample preparation
For ITC and NMR studies, the non‐labeled and stable isotope‐labeled LC3 and GABARAP proteins, as well as Ub19‐AS_30 and Ub19‐AS_67 fusion construct, containing ETDSELDWKWEQLGSPLWTWNGGG and ETDSESFTMYEPDQQTIVIESGGG peptides, respectively, under TEV‐cleavable Ub19 leader (Rogov et al, 2012), were obtained according to the protocols described elsewhere (Rozenknop et al, 2011; Rogov et al, 2012; Habisov et al, 2016). AS_67 peptide was purchased from GenScript USA Inc. (NJ, USA). Before the experiments, all proteins and peptides were equilibrated to a buffer containing 50 mM Na2HPO4 and 100 mM NaCl at pH 7.0 and supplied with 5 mM protease inhibitor cocktail.
Isothermal titration calorimetry
All titration experiments were performed at 25°C using a VP‐ITC microcalorimeter (Malvern Instruments, UK). The ITC data were analyzed with the ITC‐Origin 7.0 software with a “one‐site” binding model. The free peptides and fusion construct at concentrations of 0.35–0.60 mM were titrated into 0.010–0.025 mM LC3 and GABARAP proteins in 20–26 steps. The protein and peptide concentrations were calculated from the UV absorption at 280 nm by Nanodrop spectrophotometer (Thermo Fisher Scientific, DE, USA).
Nuclear magnetic resonance spectroscopy
All NMR experiments were performed at 298 K on Bruker Avance spectrometers operating at proton frequencies of 500, 600, and 700 MHz. Titration experiments were performed with 0.1–0.2 mM 15N‐labeled LC3 and GABARAP protein solutions to which the non‐labeled peptides or fusion constructs were added stepwise until 4 times excess.
Author contributions
AS, MP, VD, SSS, AE, and ID designed experiments. AS, MP, IK, and VM performed experiments. JH and VVR designed and performed ITC and NMR experiments. CW and RJY designed and performed the mitophagy flux experiments. AS, MP, AE, and ID jointly wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Movie EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Acknowledgements
We thank Carsten Schultz and Dirk Bumann for the annexin A4 construct and Salmonella strains, respectively, as well as Evgenij Fiskin and Tihana Bionda for support with the Salmonella work. Christian Behrends generously provided us with the LC3C antibody. Natalia Rogova was of great help for cloning, screening, expression, and isolation of proteins for ITC and NMR data. We are grateful to David McEwan for help and advice and Dr. Dragan Maric for FACS analysis at NINDS flow cytometry core facility. This work was supported by grants from the Fritz Thyssen Stiftung (Az. 10.14.2.208) to AS, the Human Frontier Science Program (HFSP RGP55) to SSS and ID, the LOEWE initiative of the State of Hessen to the Research Center for Translational Medicine and Pharmacology (AE and MP), the LOEWE initiative Ub‐Net (AE and ID), the Deutsche Forschungsgemeinschaft (SFB 1177) to VD and ID, and the National Institutes of Health NINDS Intramural Program to RJY. Work of JH, VVR, and VD was further funded by the Center for Biomolecular Magnetic Resonance (BMRZ, Frankfurt), the German Cancer Consortium (DKTK), and the Cluster of Excellence Frankfurt “Macromolecular Complexes”.
The EMBO Journal (2017) 36: 549–564
Contributor Information
Andreas Ernst, Email: ernst@biochem2.de.
Ivan Dikic, Email: dikic@biochem2.uni-frankfurt.de.
References
- Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Overvatn A, Kirkin V, Johansen T (2012) ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3‐interacting region (LIR) motifs. J Biol Chem 287: 39275–39290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beskow A, Grimberg KB, Bott LC, Salomons FA, Dantuma NP, Young P (2009) A conserved unfoldase activity for the p97 AAA‐ATPase in proteasomal degradation. J Mol Biol 394: 732–746 [DOI] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M (2014) The mitochondrial deubiquitinase USP30 opposes parkin‐mediated mitophagy. Nature 510: 370–375 [DOI] [PubMed] [Google Scholar]
- Birgisdottir AB, Lamark T, Johansen T (2013) The LIR motif – crucial for selective autophagy. J Cell Sci 126: 3237–3247 [DOI] [PubMed] [Google Scholar]
- Ernst A, Sazinsky SL, Hui S, Currell B, Dharsee M, Seshagiri S, Bader GD, Sidhu SS (2009) Rapid evolution of functional complexity in a domain family. Sci Signal 2: ra50 [DOI] [PubMed] [Google Scholar]
- Ernst A, Gfeller D, Kan Z, Seshagiri S, Kim PM, Bader GD, Sidhu SS (2010) Coevolution of PDZ domain‐ligand interactions analyzed by high‐throughput phage display and deep sequencing. Mol Biosyst 6: 1782–1790 [DOI] [PubMed] [Google Scholar]
- Farre JC, Burkenroad A, Burnett SF, Subramani S (2013) Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Rep 14: 441–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskin E, Bionda T, Dikic I, Behrends C (2016) Global analysis of host and bacterial ubiquitinome in response to Salmonella typhimurium infection. Mol Cell 62: 967–981 [DOI] [PubMed] [Google Scholar]
- Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, Fields S (2010) High‐resolution mapping of protein sequence‐function relationships. Nat Methods 7: 741–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genau HM, Huber J, Baschieri F, Akutsu M, Dotsch V, Farhan H, Rogov V, Behrends C (2015) CUL3‐KBTBD6/KBTBD7 ubiquitin ligase cooperates with GABARAP proteins to spatially restrict TIAM1‐RAC1 signaling. Mol Cell 57: 995–1010 [DOI] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habisov S, Huber J, Ichimura Y, Akutsu M, Rogova N, Loehr F, McEwan DG, Johansen T, Dikic I, Doetsch V, Komatsu M, Rogov VV, Kirkin V (2016) Structural and functional analysis of a novel interaction motif within UFM1‐activating enzyme 5 (UBA5) required for binding to ubiquitin‐like proteins and ufmylation. J Biol Chem 291: 9025–9041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa A, Hayes SJ, Lawe DC, Sudharshan E, Tuft R, Fogarty K, Lambright D, Corvera S (2004) Structural basis for endosomal targeting by FYVE domains. J Biol Chem 279: 5958–5966 [DOI] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1‐PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60: 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiyama M, Kusakabe KT, Takeshita A, Sugi S, Kuniyoshi N, Imai H, Kano K, Kiso Y (2015) Nutrient starvation affects expression of LC3 family at the feto‐maternal interface during murine placentation. J Vet Med Sci 77: 305–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachim J, Jefferies HB, Razi M, Frith D, Snijders AP, Chakravarty P, Judith D, Tooze SA (2015) Activation of ULK kinase and autophagy by GABARAP trafficking from the centrosome is regulated by WAC and GM130. Mol Cell 60: 899–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaulich M, Lee YJ, Lonn P, Springer AD, Meade BR, Dowdy SF (2015) Efficient CRISPR‐rAAV engineering of endogenous genes to study protein function by allele‐specific RNAi. Nucleic Acids Res 43: e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, Mauthe M, Katona I, Qualmann B, Weis J, Reggiori F, Kurth I, Hubner CA, Dikic I (2015) Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522: 354–358 [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Codogno P (2013) The mechanism and physiological function of macroautophagy. J Innate Immun 5: 427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koukourakis MI, Kalamida D, Giatromanolaki A, Zois CE, Sivridis E, Pouliliou S, Mitrakas A, Gatter KC, Harris AL (2015) Autophagosome proteins LC3A, LC3B and LC3C have distinct subcellular distribution kinetics and expression in cancer cell lines. PLoS One 10: e0137675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40: 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ktistakis NT, Tooze SA (2016) Digesting the expanding mechanisms of autophagy. Trends Cell Biol 26: 624–635 [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036 [DOI] [PubMed] [Google Scholar]
- Landajuela A, Hervas JH, Anton Z, Montes LR, Gil D, Valle M, Rodriguez JF, Goni FM, Alonso A (2016) Lipid geometry and bilayer curvature modulate LC3/GABARAP‐mediated model autophagosomal elongation. Biophys J 110: 411–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand JN, Bon K, Fraichard A, Zhang J, Jouvenot M, Risold PY, Boyer‐Guittaut M, Delage‐Mourroux R (2013) Specific distribution of the autophagic protein GABARAPL1/GEC1 in the developing and adult mouse brain and identification of neuronal populations expressing GABARAPL1/GEC1. PLoS One 8: e63133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leil TA, Chen ZW, Chang CS, Olsen RW (2004) GABAA receptor‐associated protein traffics GABAA receptors to the plasma membrane in neurons. J Neurosci 24: 11429–11438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lystad AH, Ichimura Y, Takagi K, Yang Y, Pankiv S, Kanegae Y, Kageyama S, Suzuki M, Saito I, Mizushima T, Komatsu M, Simonsen A (2014) Structural determinants in GABARAP required for the selective binding and recruitment of ALFY to LC3B‐positive structures. EMBO Rep 15: 557–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P, Dikic I (2015) PLEKHM1 regulates autophagosome‐lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57: 39–54 [DOI] [PubMed] [Google Scholar]
- von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein A, Bloor S, Rutherford TJ, Freund SM, Komander D, Randow F (2012) LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell 48: 329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller JM, Shorter J, Newman R, Deinhardt K, Sagiv Y, Elazar Z, Warren G, Shima DT (2002) Sequential SNARE disassembly and GATE‐16‐GOS‐28 complex assembly mediated by distinct NSF activities drives Golgi membrane fusion. J Cell Biol 157: 1161–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dotsch V, Ney PA, Dikic I (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11: 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowag H, Munz C (2015) Diverting autophagic membranes for exocytosis. Autophagy 11: 425–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsumi Y (2014) Historical landmarks of autophagy research. Cell Res 24: 9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piljic A, Schultz C (2008) Analysis of protein complex hierarchy in living cells. ACS Chem Biol 3: 749–755 [DOI] [PubMed] [Google Scholar]
- Reggiori F, Monastyrska I, Verheije MH, Cali T, Ulasli M, Bianchi S, Bernasconi R, de Haan CA, Molinari M (2010) Coronaviruses Hijack the LC3‐I‐positive EDEMosomes, ER‐derived vesicles exporting short‐lived ERAD regulators, for replication. Cell Host Microbe 7: 500–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, Youle RJ, Dikic I (2016) Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA 113: 4039–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov VV, Rozenknop A, Rogova NY, Lohr F, Tikole S, Jaravine V, Guntert P, Dikic I, Dotsch V (2012) A universal expression tag for structural and functional studies of proteins. Chembiochem 13: 959–963 [DOI] [PubMed] [Google Scholar]
- Rogov VV, Suzuki H, Fiskin E, Wild P, Kniss A, Rozenknop A, Kato R, Kawasaki M, McEwan DG, Lohr F, Guntert P, Dikic I, Wakatsuki S, Dotsch V (2013) Structural basis for phosphorylation‐triggered autophagic clearance of Salmonella . Biochem J 454: 459–466 [DOI] [PubMed] [Google Scholar]
- Rogov V, Dotsch V, Johansen T, Kirkin V (2014) Interactions between autophagy receptors and ubiquitin‐like proteins form the molecular basis for selective autophagy. Mol Cell 53: 167–178 [DOI] [PubMed] [Google Scholar]
- Rozenknop A, Rogov VV, Rogova NY, Lohr F, Guntert P, Dikic I, Dotsch V (2011) Characterization of the interaction of GABARAPL‐1 with the LIR motif of NBR1. J Mol Biol 410: 477–487 [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Shpilka T, Elazar Z (2012) Mechanisms of autophagosome biogenesis. Curr Biol 22: R29–R34 [DOI] [PubMed] [Google Scholar]
- Sagiv Y, Legesse‐Miller A, Porat A, Elazar Z (2000) GATE‐16, a membrane transport modulator, interacts with NSF and the Golgi v‐SNARE GOS‐28. EMBO J 19: 1494–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR (2007) Toll‐like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450: 1253–1257 [DOI] [PubMed] [Google Scholar]
- Sidhu SS, Weiss GA, Wells JA (2000) High copy display of large proteins on phage for functional selections. J Mol Biol 296: 487–495 [DOI] [PubMed] [Google Scholar]
- Sims JJ, Scavone F, Cooper EM, Kane LA, Youle RJ, Boeke JD, Cohen RE (2012) Polyubiquitin‐sensor proteins reveal localization and linkage‐type dependence of cellular ubiquitin signaling. Nat Methods 9: 303–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelton NJ, Koehler MF, Zobel K, Wong WL, Yeh S, Pisabarro MT, Yin JP, Lasky LA, Sidhu SS (2003) Origins of PDZ domain ligand specificity. Structure determination and mutagenesis of the Erbin PDZ domain. J Biol Chem 278: 7645–7654 [DOI] [PubMed] [Google Scholar]
- Stadel D, Millarte V, Tillmann KD, Huber J, Tamin‐Yecheskel BC, Akutsu M, Demishtein A, Ben‐Zeev B, Anikster Y, Perez F, Dotsch V, Elazar Z, Rogov V, Farhan H, Behrends C (2015) TECPR2 cooperates with LC3C to regulate COPII‐dependent ER export. Mol Cell 60: 89–104 [DOI] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501 [DOI] [PubMed] [Google Scholar]
- Tighe A, Staples O, Taylor S (2008) Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J Cell Biol 181: 893–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonikian R, Zhang Y, Boone C, Sidhu SS (2007) Identifying specificity profiles for peptide recognition modules from phage‐displayed peptide libraries. Nat Protoc 2: 1368–1386 [DOI] [PubMed] [Google Scholar]
- Tonikian R, Zhang Y, Sazinsky SL, Currell B, Yeh JH, Reva B, Held HA, Appleton BA, Evangelista M, Wu Y, Xin X, Chan AC, Seshagiri S, Lasky LA, Sander C, Boone C, Bader GD, Sidhu SS (2008) A specificity map for the PDZ domain family. PLoS Biol 6: e239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonikian R, Xin X, Toret CP, Gfeller D, Landgraf C, Panni S, Paoluzi S, Castagnoli L, Currell B, Seshagiri S, Yu H, Winsor B, Vidal M, Gerstein MB, Bader GD, Volkmer R, Cesareni G, Drubin DG, Kim PM, Sidhu SS et al (2009) Bayesian modeling of the yeast SH3 domain interactome predicts spatiotemporal dynamics of endocytosis proteins. PLoS Biol 7: e1000218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoef LG, Heinen C, Selivanova A, Halff EF, Salomons FA, Dantuma NP (2009) Minimal length requirement for proteasomal degradation of ubiquitin‐dependent substrates. FASEB J 23: 123–133 [DOI] [PubMed] [Google Scholar]
- Verlhac P, Gregoire IP, Azocar O, Petkova DS, Baguet J, Viret C, Faure M (2015) Autophagy receptor NDP52 regulates pathogen‐containing autophagosome maturation. Cell Host Microbe 17: 515–525 [DOI] [PubMed] [Google Scholar]
- Wang H, Sun HQ, Zhu X, Zhang L, Albanesi J, Levine B, Yin H (2015) GABARAPs regulate PI4P‐dependent autophagosome: lysosome fusion. Proc Natl Acad Sci USA 112: 7015–7020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z (2010) LC3 and GATE‐16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 29: 1792–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk SJ, Fiskin E, Putyrski M, Pampaloni F, Hou J, Wild P, Kensche T, Grecco HE, Bastiaens P, Dikic I (2012) Fluorescence‐based sensors to monitor localization and functions of linear and K63‐linked ubiquitin chains in cells. Mol Cell 47: 797–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, Martens S (2015) Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. ELife 4: e08941 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Movie EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 4