

Abstract
Targeted detection is one of the most important methods in mass spectrometry (MS)-based metabolomics; however, its major limitation is the reduced metabolome coverage that results from the limited set of targeted metabolites typically used in the analysis. In this study we describe a new approach, globally optimized targeted (GOT)-MS, that combines many of the advantages of targeted detection and global profiling in metabolomics analysis, including the capability to detect unknowns, broad metabolite coverage, and excellent quantitation. The key step in GOT-MS is a global search of precursor and product ions using a single liquid chromatography–triple quadrupole (LC–QQQ) mass spectrometer. Here, focused on measuring serum metabolites, we obtained 595 precursor ions and 1 890 multiple reaction monitoring (MRM) transitions, under positive and negative ionization modes in the mass range of 60–600 Da. For many of the MRMs/metabolites under investigation, the analytical performance of GOT-MS is better than or at least comparable to that obtained by global profiling using a quadrupole-time-of-flight (Q-TOF) instrument of similar vintage. Using a study of serum metabolites in colorectal cancer (CRC) as a representative example, GOT-MS significantly outperformed a large targeted MS assay containing ∼160 biologically important metabolites and provided a complementary approach to traditional global profiling using Q-TOF-MS. GOT-MS thus expands and optimizes the detection capabilities for QQQ-MS through a novel approach and should have the potential to significantly advance both basic and clinical metabolic research.
Graphical abstract
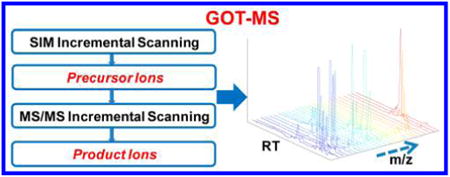
Metabolomics has emerged as a powerful approach for providing detailed and multilayered information about complex biological processes and systems.1–9 Metabolomics studies have resulted in a number of important findings in systems biology and biomarker discovery, including a deeper understanding of cancer metabolism10,11 and drug toxicity,12,13 the potential for improved early disease detection14–16 or therapy monitoring,4,17 as well as applications in environmental science,18 nutrition,19 etc. It is clear from these studies and numerous others that significant potential exists for major breakthroughs in metabolomics that will impact many fields.
Mass spectrometry (MS) based methods, specifically global profiling or targeted detection, play an important role in metabolomics for detecting and quantifying metabolites.20–28 Because of significant advantages of great selectivity and excellent quantitation, targeted MS using a liquid chromatography triple quadrupole (LC–QQQ) mass spectrometer under multiple reaction monitoring (MRM) mode provides an excellent approach for metabolite profiling. Large targeted MS assays, covering ∼200 metabolites in many important metabolic pathways (e.g., glycolysis, the TCA cycle, etc.), can be constructed and used for metabolomics studies.11,15,29–34 However, the major limitation of targeted metabolomics is reduced metabolite coverage, often to a small to moderate number of well-known compounds. Specialized knowledge about metabolism is necessary to make good use of highly targeted assays, especially those with a small number of predefined targets. It is therefore valuable to further develop targeted detection by broadening its coverage, including the detection of unknown metabolites.
In this study, we describe a novel approach, globally optimized targeted (GOT)-MS, which retains the advantages of targeted detection and meanwhile possesses the capacity for broad metabolome coverage. The key step in GOT-MS is a global search of precursor and product ions with a single LC–QQQ mass spectrometer. While there are alternative methods to conduct large-scale MRM experiments,35–39 we expand and optimize the detection capabilities of QQQ-MS through an innovative approach. In this proof-of-principle study, we use aqueous extracts from serum samples to develop the GOT-MS method and also demonstrate the capabilities of GOT-MS in a trial application of metabolomics, namely, to use GOT-MS to differentiate colorectal cancer (CRC) patients from healthy controls. The performance of GOT-MS was compared to two alternative approaches, global and targeted profiling, using the same set of samples, and the results show that GOT-MS compares favorably with these widely used methods.
Experimental Methods
Chemicals
Acetonitrile (LC–MS grade) and methanol (LC–MS grade) were purchased from Sigma-Aldrich (St. Louis, MO). Ammonium acetate (LC–MS grade) and acetic acid (LC–MS grade) were obtained from Fisher Scientific (Pittsburgh, PA). DI water was provided in-house by a Synergy Ultrapure Water System from EMD Millipore (Billerica, MA). The metabolite standards in the Northwest Metabolomics Research Center (NW-MRC) internal database (included in Table S1) were purchased from Sigma-Aldrich or Fisher Scientific. Stable isotope labeled amino acids were purchased from Cambridge Isotope Laboratories (Tewksbury, MA).
Biological Samples
To develop GOT-MS, pooled human serum samples were purchased from Innovative Research, Inc. (Novi, MI). For the CRC metabolomics study, serum samples from 18 CRC patients and 20 healthy controls were selected from those involved in our previous studies.15,25,40 Table S2 shows the demographic and clinical information for these samples. All serum samples were collected in accordance with the protocols approved by the Indiana University School of Medicine and Purdue University Institutional Review Boards. All subjects in the study provided informed consent according to the institutional guidelines.
Aqueous Metabolite Extraction
Frozen human serum samples were thawed at 4 °C, and proteins were precipitated by mixing 50 μL of serum with 250 μL of cold methanol. After 20 min incubation at −20 °C, the mixture was centrifuged at 14 000 RCM for 20 min. The supernatant was transferred into a clean 2.0 mL Eppendorf vial and then dried under vacuum (Eppendorf Vacufuge). The obtained residue was reconstituted in 400 μL of Solvent C (40% Solvent A/60% Solvent B, see details in the LC–MS section) prior to MS analysis. In addition, 5 samples were prepared by reconstituting the metabolite residues from 50 μL of serum containing spiked U–13C15N-amino acids, into 200 μL (1:4 dilution), 400 μL (1:8 dilution), 600 μL (1:12 dilution), 1 200 μL (1:24 dilution), and 2 400 μL (1:48 dilution) of Solvent C. Table S3 shows the concentrations of spiked U–13C15N-amino acids in the 1:4 dilution sample. Intraday coefficients of variation (CVs) were obtained using the data from samples run three times on the same day, and interday CVs were calculated from the samples run three times each day for 3 consecutive days.
LC–MS
Agilent 1260 LC-6410 Triple Quad MS (Agilent Technologies, Inc., Santa Clara, CA)
The LC strategy in Figure S1 is often used in the NW-MRC for both reverse phase (RP) and hydrophilic interaction liquid chromatography (HILIC) separations. After a short (t0) initial isocratic elution (Solvent B, P0), the percentage of Solvent B changes to P1 until t1 and is held at this content until t2. Then the percentage of Solvent B quickly goes back to P0 to prepare for the next injection. In this study, we primarily used HILIC to separate aqueous metabolites in GOT-MS, employing SeQuant ZIC-cHILIC columns (150 mm × 2.1 mm, 3.0 μm, Merck KGaA, Darmstadt, Germany) at 45 °C. Agilent C18 columns (100 mm × 3 mm, 1.8 μm, Agilent Technologies, Inc., Santa Clara, CA) were used for RP separation at 45 °C. In Figure S1, t0 = 1 min, t1 = 6 min, and t2 = 9 min. Solvent A was 5 mM ammonium acetate in 90% H2O/10% acetonitrile/0.2% acetic acid, and Solvent B was 5 mM ammonium acetate in 90% acetonitrile/10% H2O/0.2% acetic acid. For HILIC, P0 = 80%, and P1 = 30%. For RP, P0 = 0%, and P1 = 95%. The flow rate was 0.3 mL/min. Volumes of 10 μL and 5 μL were injected for negative and positive ionization, respectively. The electrospray ionization (ESI) voltage was 3.8 kV. Agilent MassHunter Qualitative Analysis (version B.07.00) and Quantitative Analysis (version B.07.00) software were used to extract MS peak areas.
Agilent 1200 SL LC-6520 Quadrupole-Time of Flight (Q-TOF) MS (Agilent Technologies, Inc., Santa Clara, CA)
The separation conditions for the LC–Q-TOF experiments were the same as those for the LC–QQQ (although the LC hardware was not exactly the same). The ESI voltage was also 3.8 kV, and the m/z scan range was 60–1000. The Q-TOF data were extracted using Agilent MassHunter Qualitative Analysis (version B.07.00), Quantitative Analysis (version B.07.01), and Mass Profiler Professional (MPP, version B.13.00) software. The absolute intensity threshold for the LC–Q-TOF data extraction was 1000, and the mass accuracy limit was set to 10 ppm.
Agilent 1260 LC-AB Sciex QTrap 5500 MS (AB Sciex, Toronto, ON, Canada)
The data of the same 18 CRC samples and 20 healthy controls, collected in our previous study,15 were used to demonstrate the performance of a traditional large targeted MS assay (with ∼160 metabolites in the detection list). Although not exactly the same, the LC conditions of that assay were very similar to those of the Agilent LC–QQQ or LC–Q-TOF in this study. The experimental methods were detailed in our previous paper.15
Multivariate Statistical Analysis
For the CRC study, we performed principal component analysis (PCA) using the PLS toolbox (version 6.2, Eigenvector Research, Inc., Wenatchee, WA) in Matlab (version 7.0.4, Mathworks, Natick, MA). The data were log10 transformed and mean-centered, prior to PCA analysis.
Results
GOT-MS MRMs
Figure 1 shows the flowchart for GOT-MS. As it is very challenging, if not impossible, to develop an optimized separation condition for all metabolites, we chose the LC strategy shown in Figure S1 for both RP and HILIC. HILIC was primarily used to separate aqueous metabolites in the pooled serum samples (1:8 dilution) for GOT-MS development; C18 columns were also used to evaluate whether GOT-MS could be easily adapted to RP separation.
Figure 1.
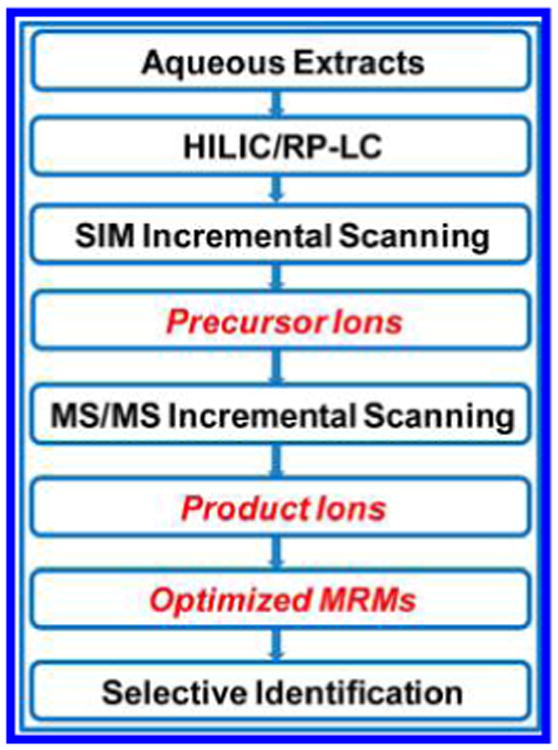
Flowchart for globally optimized targeted mass spectrometry (GOT-MS).
To identify the MRM transitions for GOT-MS, we first performed selected ion monitoring (SIM) incremental scans in the mass range of 60–600 Da in order to cover most of the aqueous metabolites. SIM was employed due to its relatively high sensitivity and good signal-to-noise ratio (S/N, data not shown). The quadrupole has unit mass resolution; therefore, an m/z increment of 0.5 was used (e.g., m/z 60, 60.5, 61, etc.). For each injection, an m/z range of 30 (e.g., 60–89.5 Da) was measured (60 SIMs), using a 10 ms scan time (cycle time ∼0.6 s). We examined each individual SIM, and the m/z values that produced relatively good peak shapes (using manual inspection based on symmetry, peak width, etc.) and S/Ns > 3 were selected as precursor ions. As shown in Figure 2, many HILIC-SIMs were observed in the 100–190 Da mass range from the pooled human serum sample under both positive and negative ion detection. Similarly, Figure S2 shows that GOT-MS can also produce many SIMs from the same sample using C18 columns for RP separation. Similar results from urine were also obtained (data not shown), indicating that GOT-MS can be applied to various kinds of biological samples.
Figure 2.
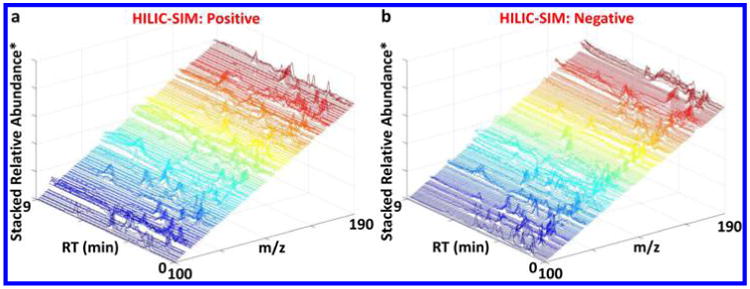
Typical HILIC-SIMs in the m/z range of 100–190 from a pooled serum sample in GOT-MS, under (a) positive and (b) negative ion detection modes. *Each SIM data was first linearly scaled, so that the minimum is 0 and the maximum is 1; an increment of 0.04 was then added to each SIM across different retention time.
We then carried out tandem mass spectrometry (MS/MS) experiments using product ion scans with different values of the collision energy (CE) to profile product ions. While a wider CE range and different increment values were tested, three CE values were selected in this study: 5, 15, and 25 V. Most aqueous metabolites fragmented under these CEs. MS/MS spectra using the CE of 5 V provided more accurate m/z values for precursor ions (e.g., 60 could be updated to 60.1). For many precursor ions, different CE values could produce distinct but somewhat overlapping fragmentation patterns, which was helpful to confirm that the MS/MS spectra were from the same metabolite(s). The low m/z limit for any MS/MS scan was 40. The number of selected precursor ions was decreased in this step, since the ions with similar m/z values (e.g., m/z 60.1 and 60.5) and the same fragmentation patterns were excluded except for the one with the highest signal intensity.
With both precursor and product ions chosen, ions were detected in MRM mode while optimizing the fragmentor voltage, cell accelerator voltage, and CE. The fragmentor voltage was optimized in the range of 40–180 V using an increment of 20 V, and CE was optimized in the range of 5–40 V with an increment of 5 V. The cell accelerator voltage was evaluated at 2, 4, and 6 V. Finally, 595 GOT-MS precursor ions and 1 890 MRMs were determined from the peaks with reasonable peak shapes (manual inspection) and S/Ns > 3. Table S1 summarizes all these MRMs. Figure 3a,b shows the distribution of GOT-MS precursor and product ions in the space of Q1 versus Q3 m/z values in positive and negative ion detection modes, respectively. The resulting distribution was quite dense, especially for positive ion mode, since both precursor and product ions were globally searched. Interestingly, a few fragmentation patterns showed visible trends in Figure 3a,b. For example, many precursor ions lost H2O (M → M – 18), and the fragmentation pattern M → M – 164 was frequently observed under positive ionization. Several product ions (such as those at m/z 104 and 184) were able to be dissociated from many different precursor ions. In contrast, Figure 3c,d shows the distribution of precursor and product ions in a more traditional large targeted MS assay (targeting ∼160 metabolites) that has been used successfully in the NW-MRC.11,15,29,30 It can be clearly seen in Figure 3 that GOT-MS not only increased the number of precursor ions but also expanded the Q3 space. This wide coverage can be very important in metabolomics, since different product ions from the same precursor ion can result from different metabolites in complex biological samples.
Figure 3.
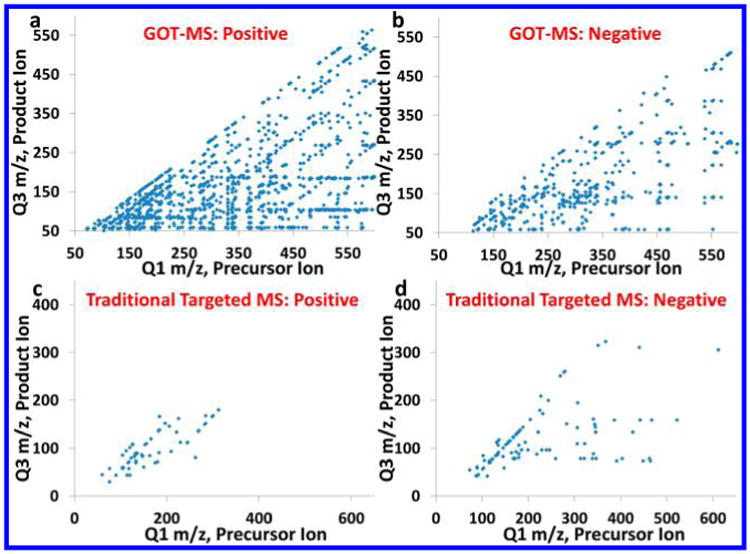
Distribution of precursor and product ions in the space of Q1 versus Q3 m/z values, under (a) GOT-MS positive ionization, (b) GOT-MS negative ionization, (c) positive ionization in a large targeted MS assay,11,15,29,30 and (d) negative ionization in a large targeted MS assay.11,15,29,30
Scheduling MRM transitions based on retention time (“scheduled MRM mode”) can be used to maximize the number of MRMs in each measurement. Using this approach, we were able to measure all 1 890 GOT-MS MRMs (5 ms scan time for each MRM), in 4 injections (three for positive ions and one for negative ions) using a separation time of 9 min each.
Analytical Performance
As anticipated, GOT-MS has good quantitative performance, since it is based on an LC– QQQ platform. We examined the intraday CVs (n = 3) of all the GOT-MS MRMs. As shown in Figure 4a, more than 40% of GOT-MS MRMs had CVs < 5%, and <2% had CVs >30%, indicating excellent reproducibility. Overall, the average CV for GOT-MS detected metabolites was 7.8 ± 7.0%. Figure 4b shows the relationship between the GOT-MS integrated areas and the CV values. The GOT-MS areas extended more than 4 orders of magnitude, and the CVs decreased with increased peak areas.
Figure 4.
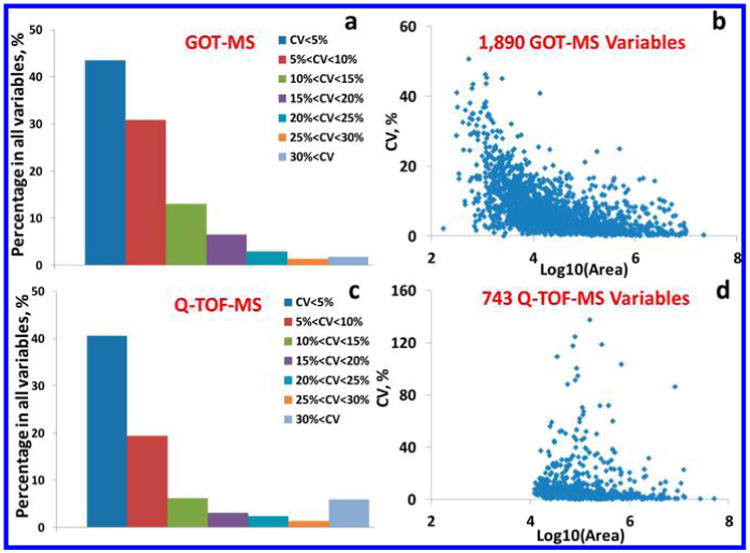
Distribution of intraday (n = 3) CV values (a) for all 1 890 GOT-MS MRMs, (b) against the integrated areas of GOT-MS MRMs, (c) for the 743 Q-TOF variables present in all the data, and (d) against the integrated areas of Q-TOF variables present in all the data.
While it is challenging to carry out a perfect comparison between Q-TOF and QQQ instruments, in this study we ran the same samples (1:8 dilution) using essentially the same LC conditions and the Agilent MS systems of similar generation or vintage. Figure 4c shows the distribution of intraday CV values for 743 Q-TOF variables that were present in all the data (n = 3). The reproducibility in Figure 4c was a little bit worse than that in Figure 4a, with the average CV of 9.2 ± 16.4%. It was confirmed in Figure 4d that most of the Q-TOF variables had small intraday CVs, but the CV distribution was obviously more dispersed than that shown in Figure 4b. The Q-TOF peak areas extended <4 orders of magnitude in Figure 4d (different y scales). Notably, while there were 3 471 detected MS features, we selected these 743 variables because of high reliability (n = 3), although this number decreased to 500 over 3 days (n = 9).
Because analytical detection is compound dependent, we specifically examined amino acids to further compare the performance of GOT-MS and Q-TOF. Figure S3a shows the intraday (n = 3) and interday (n = 3 × 3 consecutive days) CVs of amino acids for GOT-MS. As expected, the interday average CV (8.3 ± 3.4%) was larger than the intraday average CV (3.0 ± 2.2%). Figure S3b shows that normalization to internal standards improved the reproducibility, especially the interday average CV (4.8 ± 4.0%). In addition, GOT-MS had good linearity (R2) for most amino acids, based on the data in Figure S3c collected from the five dilution samples.
Figure S4 shows the analytical performance of the Q-TOF instrument for amino acid detection using the same samples. In Figure S4a, the intraday (9.2 ± 16.6%) and interday (11.7 ± 12.6%) CVs for many amino acids were comparable to those in Figure S3a, especially for the Q-TOF signals well beyond the detection limit. However, the CVs of a few amino acids with low intensities were very large (and cysteine was not detectable in these samples using the Q-TOF). Surprisingly, normalization to isotope labeled internal standards did not improve the reproducibility of Q-TOF data for many amino acids (Figures S3b vs S4b). This is because the concentrations of spiked internal standards (Table S3) were less than the unlabeled amino acids; U–13C15N-cysteine, U–13C15N-serine, and U–13C15N-histidine were not detectable in these samples using the Q-TOF. As anticipated, the detection limit of the Q-TOF was also worse than that of the QQQ, which caused larger variation in the Q-TOF signals (and contributed to the larger CVs during normalization calculations). For example, we measured the limit of detection (LOD, S/N = 3) of valine to be 1 × 10−13 mol and 1 × 10−11 mol using the QQQ and Q-TOF, respectively. Figure S4c shows that the linearity of Q-TOF was good for many amino acids with high intensities. Table 1 summarizes the analytical performance of GOT-MS and Q-TOF-MS.
Table 1. Summary of the Analytical Performance of GOT-MS and Q-TOF in This Study.
| MS features | analytical variables | Q-TOFa | GOT-MSa | |
|---|---|---|---|---|
| intraday (n = 3) | 1 890 GOT-MS MRMs | CVs | 7.8 ± 7.0% | |
| dynamic range | >4 orders | |||
| 743 Q-TOF variables present in all the data | CVs | 9.2 ± 16.4% | ||
| dynamic range | <4 orders | |||
| amino acidsb | CVs | 9.2 ± 16.6%c | 3.0 ± 2.2% | |
| CVs after normalizationd | 14.7 ± 24.6%c,e | 4.1 ± 3.4% | ||
| R2f | 0.76 ± 0.37c | 0.82 ± 0.26 | ||
| interday (n = 9 over 3 consecutive days) | amino acidsb | CVs | 11.7 ± 12.6%c | 8.3 ± 3.4% |
| CVs after normalizationd | 18.5 ± 22.6%c,e | 4.8 ± 4.0% |
GOT-MS is based on QQQ-MS; the samples were run on Agilent MS systems of similar vintage with essentially the same LC conditions.
Ile and Leu were integrated together since they had the same MRMs and baseline separation between them was not observed.
Cysteine was not detectable in these samples using Q-TOF.
Normalization to the corresponding isotope labeled (U–13C15N-) internal standards was performed.
U–13C15N-cysteine, U–13C15N-serine, and U–13C15N-histidine were not detectable in these samples using Q-TOF.
The linearity (R2) of amino acids was obtained from the 5 dilution samples.
GOT-MS-Based Metabolomics: CRC Biomarker Discovery
It is important to improve CRC detection, as CRC is one of the most prevalent and deadly cancers in the U.S. and worldwide.41 To date, many metabolic alterations have been found in CRC tissue,42–45 serum,15,46–49 urine,50 and fecal water.51 In this methodology study, we wanted to evaluate whether GOT-MS has good potential for biomarker discovery (e.g., in CRC). We first examined the performance of our traditional large targeted MS assay for differentiating CRC patients (n = 18) from healthy controls (n = 20).15 This assay included ∼160 metabolites from more than 25 metabolic pathways of great biological significance,11,15,29,30 of which 113 metabolites were detected in these serum samples. Figure 5a shows the PCA score plot (PC1 vs PC2), and no clear separation was observed between CRCs and healthy controls. In the volcano plot (Figure 5b), there was only one variable with fold change (FC) > 2 and P < 0.05. Although previous studies showed that the combination of multiple serum biomarkers can result in differentiation between cancer and normal,15,46–49 our results using 18 CRC and 20 healthy control samples suggest that CRC did not cause many very strong perturbations (using the relatively strict criterion of FC > 2) to major metabolites that are currently often examined.
Figure 5.
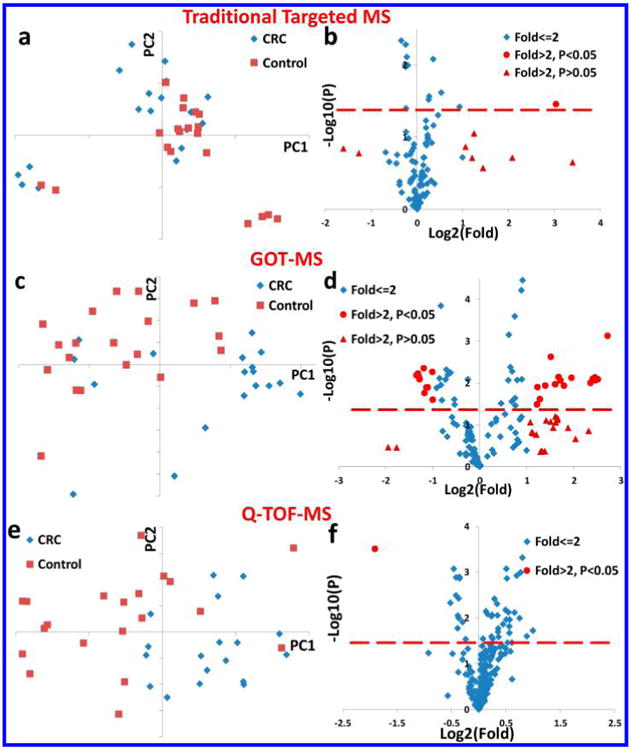
(a) The PCA score plot (PC1 vs PC2) of the data collected from CRC and healthy control samples, using a large targeted MS assay,15 (b) the volcano plot of variables used in part a, (c) the PCA score plot (PC1 vs PC2) of the 26 GOT-MS MRMs with fold changes >2 and P < 0.05, (d) the volcano plot of the 155 GOT-MS variables collected from CRC and healthy control samples, (e) the PCA score plot (PC1 vs PC2) for the 230 Q-TOF variables collected from CRC and healthy control samples, and (f) the volcano plot of variables used in part e. The dashed red-lines show where P = 0.05.
For analysis using GOT-MS, we used the following approach. To save experimental time, we focused on detecting a moderate number of the possible GOT-MS detected metabolites, rather than focusing on all 1 890 GOT-MS MRMs. Briefly, we mixed CRC and healthy control samples separately, to obtain a pooled sample for each group (Pooled-CRC and Pooled-Control). We then examined all the GOT-MS MRMs on these two pooled samples. We selected the top 93 and 85 differential GOT-MS MRMs in positive and negative ionization, respectively, according to fold changes (FC > 2) and pooled sample-CVs (<20%). The 178 selected MRMs were then run on each individual sample (using two separate injections for positive and negative ions), and finally 155 out of 178 (87%) GOT-MS variables were used in data analysis. Figure S5 shows the flowchart of this GOT-MS-based metabolomics method.
In Figure S6, the separation between CRCs and healthy controls (mainly along PC2) was better than that observed in Figure 5a. Figure 5d shows the volcano plot for the 155 GOT-MS variables. We found 26 variables with FC > 2 and P < 0.05. We then performed PCA on these 26 important variables. The separation was further improved in Figure 5c, and the two groups were clearly separated except that 4 healthy samples were mixed with CRC samples.
We then analyzed the same samples using Q-TOF-MS. Traditional global profiling requires sophisticated software for data processing, such as peak picking, peak deconvolution, and peak alignment. Missing values frequently occur in Q-TOF profiling, and 286 (out of 790 in total) variables were present in >80% of the samples in this study. We used the average value of all the samples for data interpolation. Similar to GOT-MS, 230 variables were finally selected for data analysis, after pooled sample-CV (<20%) filtering. In Figure 5e, the separation between the two groups was better than that in Figure 5a, but worse than that in Figure 5c (GOT-MS). Only one variable was found to have FC > 2 and P < 0.05, as seen in Figure 5f.
Preliminary Identification
While a complete study of definitive metabolite identification for GOT-MS is beyond the scope of this methodology paper, we did verify that the identities of ∼80 GOT-MS MRMs were correct using authentic standards. GOT-MS can of course detect metabolites that are well measured by traditional approaches. As an example, Figure S7 (LC retention) and Figure S8 (MS/MS spectra) show the identification of glutamine and lysine (having very similar fragmentation patterns) from the serum GOT-MS results. In addition, we searched each GOT-MS MRM against the Metlin database.52 As shown in Table S1, 770 (out of 1 374) and 183 (out of 516) GOT-MS MRMs could be found in the Metlin database under positive and negative ionization, respectively. We provide up to five possible metabolite identities listed in Table S1 for each GOT-MS MRM, from the Metlin database and our own internal database (only product ions with the highest intensities were recorded).11,15,29,30 These identities are worth pursuing to further confirm metabolite identification. We acknowledge that it is relatively easy to identify potential metabolites that are contributing to MRM peaks; while, due to many possibilities (such as in-source fragments, isomers, and adducts), it is much more difficult to verify that a particular MRM peak results solely from a specific metabolite. Therefore, we have retained all the GOT-MS MRMs in this study. For many metabolomics studies we recommend using GOT-MS to obtain the MRMs of special interest first (using statistical filtering, for example), followed by various steps of metabolite identification thereafter.
GOT-MS is also capable of detecting unknowns. In fact, 937 (604 in positive mode and 333 in negative mode) GOT-MS MRMs were not found in the Metlin database (Table S1). Although the metabolite identities are unknown, these GOT-MS MRMs should be highly reliable, because of the high selectivity of precursor and product ion pairs that were globally searched. This indicates that GOT-MS is well qualified to detect not only well-known metabolites but also unknowns or less-studied small molecules.
Discussion
In this study, we developed a novel approach, GOT-MS, which enables reliable metabolomics analysis with broad coverage. GOT-MS requires only a single LC–QQQ instrument to discover and acquire comprehensive and quantitative MRMs. Data treatment (prior to statistical analysis) is relatively easy, and the data size is small. Although the quadrupole has unit mass resolution, LC retention and fragmentation patterns (MRMs) are useful to resolve metabolites with similar molecular weight. Importantly, the number of precursor ions is limited in a certain mass range by the unit resolution, which is another advantage of GOT-MS.
We note that the GOT-MS approach is different from the newly introduced pseudotargeted metabolomics method that was developed to acquire MRMs without chemical standards.35,36 In pseudotargeted metabolomics, a global profiling mass spectrometer, such as Q-TOF, is used to generate pairs of precursor and product ions, and then these MRMs are measured using a QQQ machine. While pseudotargeted MS has been successfully applied in metabolomics, it is not a global approach, since it uses the auto MS/MS or data- or information-dependent mode to acquire the MRMs, which limits the number of MRMs (generally <5 at each moment). It also requires two (expensive) MS platforms, a Q-TOF-MS for identifying MRMs and a QQQ-MS for detection. It is best if the two instruments come from the same manufacturer. In a separate approach, Nikolskiy et al. developed a computational method that is helpful to conduct large-scale MRM experiments; however, this method is dependent on databases (less specific to samples) and is unable to detect unknowns.37 This approach has not been implemented experimentally to date.
Our results for the many detected MRMs indicate that the analytical performance of GOT-MS is comparable to or is better than that of a Q-TOF instrument for both global profiling and targeted detection (using instruments of similar vintage). This improved performance results primarily from the fact that GOT-MS is based on the QQQ-MS platform, which was also observed in previous studies.35,36 In addition, while our Q-TOF generally has good reproducibility for strong signals, these signals cannot be too large, because signal saturation will deteriorate mass accuracy (as well as linearity). Therefore, the advantages of high resolution and high mass accuracy can be compromised in traditional global profiling.
In the proof-of-principle study focused on biomarker discovery in CRC, GOT-MS provides a significant improvement over a large targeted MS approach and provides supporting information to that of traditional global profiling. Traditional targeted detection is limited by databases or predefined metabolites (that may not be detectable in the samples), which makes it unable to discover unknowns. GOT-MS removes this bottleneck through a global search of precursor and product ions, using the samples under investigation. Meanwhile, GOT-MS can serve as a valuable supplementary approach to traditional global profiling, because GOT-MS is not dependent on mass resolution/accuracy. The improved reliability of GOT-MS is observed in this example of comparing CRC vs normal, where GOT-MS analysis resulted in 26 MRMs/metabolites with fold changes >2 and P < 0.05, compared to only 1 for the Q-TOF and 1 for the traditional large targeted assay. While the multivariate analysis using the Q-TOF data was successful in separating the two classes of samples, this result was dependent on a large number of metabolites that individually had relatively poor performance. This situation is well-known in the metabolomics field and makes the job of choosing metabolites for further validation difficult and often not very successful.
Currently, the major limitation of GOT-MS is the required time to optimize the many MRMs for a particular sample type. Future experiments using a faster and more sensitive instrument will allow the measurement of more GOT-MS MRMs and in principal could reduce the development time to ∼1 week/sample type (using scheduled experiments). In addition, we are making significant efforts to improve metabolite identification in GOT-MS. Finally, we are developing a software package to automate the whole GOT-MS development process, so that GOT-MS will be more user-friendly to the metabolomics community.
Conclusions
In summary, we describe a new MS approach, GOT-MS, which integrates many advantages of traditional global profiling and targeted detection, including the capability to detect unknowns, wide metabolite coverage, and excellent quantitation. GOT-MS was designed to essentially optimize the detection performance of a single LC–QQQ mass spectrometer for broad metabolome coverage, and given the capabilities of QQQ-MS, it is very selective and highly reliable for quantitative analysis. The precursor and product ions are globally searched, resulting in the maximum number of MRMs/metabolites that can be measured by LC–QQQ. GOT-MS has wider metabolite coverage than traditional targeted detection and provides a valuable supplementary approach to traditional global profiling. Faster and more sensitive LC–QQQ instruments will allow further improvements in speed and coverage for GOT-MS.
Supplementary Material
Additional data and spectra (PDF)
List of GOT-MS MRMs (XLSX)
Acknowledgments
This work was supported in part by the NIH (Grants 2R01 GM085291, 2P30 CA015704), AMRMC Grant W81XWH-10-0540, the China Scholarship Council, the Chinese National Instrumentation Program (Grant 2011YQ170067), the PCSIRT program (Grant No. IRT13054), the National Natural Science Foundation of China (Grant No. 21365001), the ITHS Rising Stars Program (Grant UL1TR000423), and the University of Washington. The authors also thank Dr. Xiaoyan Angela Qin (Integrated Brain Imaging Center, Department of Radiology, University of Washington) for her help with data analysis.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.5b03812.
The authors declare the following competing financial interest(s): D.R. serves an executive officer for and holds equity in Matrix-Bio, Inc. The other authors declare that they have no potential conflicts of interest.
References
- 1.Lindon JC, Nicholson JK. Expert Opin Drug Metab Toxicol. 2014;10:915–919. doi: 10.1517/17425255.2014.922954. [DOI] [PubMed] [Google Scholar]
- 2.Griffin JL, Atherton H, Shockcor J, Atzori L. Nat Rev Cardiol. 2011;8:630–643. doi: 10.1038/nrcardio.2011.138. [DOI] [PubMed] [Google Scholar]
- 3.Gowda GAN, Raftery D. Curr Metabolomics. 2013;1:227–240. doi: 10.2174/2213235X113019990005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halama A, Riesen N, Moller G, de Angelis MH, Adamski J. J Intern Med. 2013;274:425–439. doi: 10.1111/joim.12117. [DOI] [PubMed] [Google Scholar]
- 5.Scalbert A, Brennan L, Fiehn O, Hankemeier T, Kristal BS, van Ommen B, Pujos-Guillot E, Verheij E, Wishart D, Wopereis S. Metabolomics. 2009;5:435–458. doi: 10.1007/s11306-009-0168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gu H, Gowda GAN, Raftery D. Future Oncol. 2012;8:1207–1210. doi: 10.2217/fon.12.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunn WB, Broadhurst DI, Atherton HJ, Goodacre R, Griffin JL. Chem Soc Rev. 2011;40:387–426. doi: 10.1039/b906712b. [DOI] [PubMed] [Google Scholar]
- 8.Bain JR, Stevens RD, Wenner BR, Ilkayeva O, Muoio DM, Newgard CB. Diabetes. 2009;58:2429–2443. doi: 10.2337/db09-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiehn O. Plant Mol Biol. 2002;48:155–171. [PubMed] [Google Scholar]
- 10.Van der Heiden MGV, Cantley LC, Thompson CB. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carroll PA, Diolaiti D, McFerrin L, Gu H, Djukovic D, Du J, Cheng PF, Anderson S, Ulrich M, Hurley JB, Raftery D, Ayer DE, Eisenman RN. Cancer Cell. 2015;27:271–285. doi: 10.1016/j.ccell.2014.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griffin JL. Curr Opin Chem Biol. 2003;7:648–654. doi: 10.1016/j.cbpa.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Kumar B, Prakash A, Ruhela RK, Medhi B. Pharmacol Rep. 2014;66:956–963. doi: 10.1016/j.pharep.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 14.Roberts LD, Gerszten RE. Cell Metab. 2013;18:43–50. doi: 10.1016/j.cmet.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu J, Djukovic D, Deng L, Gu H, Himmati F, Chiorean EG, Raftery D. J Proteome Res. 2014;13:4120–4130. doi: 10.1021/pr500494u. [DOI] [PubMed] [Google Scholar]
- 16.Wang ZN, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu XM, Chung YM, Wu YP, Schauer P, Smith JD, Allayee H, Tang WHW, DiDonato JA, Lusis AJ, Hazen SL. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicholson JK, Holmes E, Kinross JM, Darzi AW, Takats Z, Lindon JC. Nature. 2012;491:384–392. doi: 10.1038/nature11708. [DOI] [PubMed] [Google Scholar]
- 18.Bundy JG, Davey MP, Viant MR. Metabolomics. 2009;5:3–21. [Google Scholar]
- 19.Rezzi S, Ramadan Z, Fay LB, Kochhar S. J Proteome Res. 2007;6:513–525. doi: 10.1021/pr060522z. [DOI] [PubMed] [Google Scholar]
- 20.Lei ZT, Huhman DV, Sumner LW. J Biol Chem. 2011;286:25435–25442. doi: 10.1074/jbc.R111.238691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dettmer K, Aronov PA, Hammock BD. Mass Spectrom Rev. 2007;26:51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raftery D, editor. Mass Spectrometry in Metabolomics-Methods and Protocols. Springer; New York: 2014. [Google Scholar]
- 23.Griffiths WJ, Koal T, Wang YQ, Kohl M, Enot DP, Deigner HP. Angew Chem Int Ed. 2010;49:5426–5445. doi: 10.1002/anie.200905579. [DOI] [PubMed] [Google Scholar]
- 24.Theodoridis G, Gika HG, Wilson ID. TrAC, Trends Anal Chem. 2008;27:251–260. [Google Scholar]
- 25.Gu H, Du J, Neto FC, Carroll PA, Turner SJ, Chiorean EG, Eisenman RN, Raftery D. Analyst. 2015;140:2726–2734. doi: 10.1039/c4an02386b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi YP, Gu HW, Song YL, Dong X, Liu AJ, Lou ZY, Fan GR, Chai YF. Evidence-Based Complementary and Alternative Medicine. 2013;2013:1–10. [Google Scholar]
- 27.Yao WF, Gu HW, Zhu JJ, Barding G, Cheng HB, Bao BH, Zhang L, Ding AW, Li W. Anal Bioanal Chem. 2014;406:7367–7378. doi: 10.1007/s00216-014-8169-x. [DOI] [PubMed] [Google Scholar]
- 28.Liu XJ, Ser Z, Locasale JW. Anal Chem. 2014;86:2175–2184. doi: 10.1021/ac403845u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai DF, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ, Chen T, Gu HW, Djukovic D, Raftery D, Beyer RP, MacCoss MJ, Rabinovitch PS. Aging Cell. 2014;13:529–539. doi: 10.1111/acel.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reyes NL, Banks GB, Tsang M, Margineantu D, Gu H, Djukovic D, Chan J, Torres M, Liggitt HD, Hirenallur-S DK, Hockenbery DM, Raftery D, Iritani BM. Proc Natl Acad Sci U S A. 2015;112:424–429. doi: 10.1073/pnas.1413021112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei R, Li GD, Seymour AB. Anal Chem. 2010;82:5527–5533. doi: 10.1021/ac100331b. [DOI] [PubMed] [Google Scholar]
- 32.Yuan M, Breitkopf SB, Yang XM, Asara JM. Nat Protoc. 2012;7:872–881. doi: 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, Yuan C, Bao Y, Townsend MK, Tworoger SS, Davidson SM, Papagiannakopoulos T, Yang A, Dayton TL, Ogino S, Stampfer MJ, Giovannucci EL, Qian ZR, Rubinson DA, Ma J, Sesso HD, Gaziano JM, Cochrane BB, Liu SM, Wactawski-Wende J, Manson JE, Pollak MN, Kimmelman AC, Souza A, Pierce K, Wang TJ, Gerszten RE, Fuchs CS, Vander Heiden MG, Wolpin BM. Nat Med. 2014;20:1193–1198. doi: 10.1038/nm.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen SL, Kong HW, Lu X, Li Y, Yin PY, Zeng ZD, Xu GW. Anal Chem. 2013;85:8326–8333. doi: 10.1021/ac4016787. [DOI] [PubMed] [Google Scholar]
- 36.Shao Y, Zhu B, Zheng R, Zhao X, Yin P, Lu X, Jiao B, Xu G, Yao Z. J Proteome Res. 2015;14:906–916. doi: 10.1021/pr500973d. [DOI] [PubMed] [Google Scholar]
- 37.Nikolskiy I, Siuzdak G, Patti G. J Bioinformatics. 2015;31:2017. doi: 10.1093/bioinformatics/btv085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.O111.016717. O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu XC, Chen YP, Subramanian R. Anal Chem. 2014;86:1202–1209. doi: 10.1021/ac403385y. [DOI] [PubMed] [Google Scholar]
- 40.Chen C, Deng L, Wei S, Gowda GAN, Gu H, Chiorean EG, Zaid MA, Harrison ML, Pekny JF, Loehrer PJ, Zhang D, Zhang M, Raftery D. J Proteome Res. 2015;14:2492–2499. doi: 10.1021/acs.jproteome.5b00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siegel RL, Miller KD, Jemal A. Ca-Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 42.Chan ECY, Koh PK, Mal M, Cheah PY, Eu KW, Backshall A, Cavill R, Nicholson JK, Keun HC. J Proteome Res. 2009;8:352–361. doi: 10.1021/pr8006232. [DOI] [PubMed] [Google Scholar]
- 43.Piotto M, Moussallieh FM, Dillmann B, Imperiale A, Neuville A, Brigand C, Bellocq JP, Elbayed K, Namer I. J Metabolomics. 2009;5:292–301. [Google Scholar]
- 44.Denkert C, Budczies J, Weichert W, Wohlgemuth G, Scholz M, Kind T, Niesporek S, Noske A, Buckendahl A, Dietel M, Fiehn O. Mol Cancer. 2008;7:72. doi: 10.1186/1476-4598-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lean CL, Newland RC, Ende DA, Bokey EL, Smith ICP, Mountford CE. Magn Reson Med. 1993;30:525–533. doi: 10.1002/mrm.1910300502. [DOI] [PubMed] [Google Scholar]
- 46.Qiu YP, Cai GX, Su MM, Chen TL, Zheng XJ, Xu Y, Ni Y, Zhao AH, Xu LX, Cai SJ, Jia W. J Proteome Res. 2009;8:4844–4850. doi: 10.1021/pr9004162. [DOI] [PubMed] [Google Scholar]
- 47.Ritchie SA, Ahiahonu PWK, Jayasinghe D, Heath D, Liu J, Lu YS, Jin W, Kavianpour A, Yamazaki Y, Khan AM, Hossain M, Su-Myat KK, Wood PL, Krenitsky K, Takemasa I, Miyake M, Sekimoto M, Monden M, Matsubara H, Nomura F, Goodenowe DB. BMC Med. 2010;8:13. doi: 10.1186/1741-7015-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leichtle AB, Nuoffer JM, Ceglarek U, Kase J, Conrad T, Witzigmann H, Thiery J, Fiedler GM. Metabolomics. 2012;8:643–653. doi: 10.1007/s11306-011-0357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan BB, Qiu YP, Zou X, Chen TL, Xie GX, Cheng Y, Dong TT, Zhao LJ, Feng B, Hu XF, Xu LX, Zhao AH, Zhang MH, Cai GX, Cai SJ, Zhou ZX, Zheng MH, Zhang Y, Jia W. J Proteome Res. 2013;12:3000–3009. doi: 10.1021/pr400337b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiu YP, Cai GX, Su MM, Chen TL, Liu YM, Xu Y, Ni Y, Zhao AH, Cai SJ, Xu LX, Jia W. J Proteome Res. 2010;9:1627–1634. doi: 10.1021/pr901081y. [DOI] [PubMed] [Google Scholar]
- 51.Monleon D, Morales JM, Barrasa A, Lopez JA, Vazquez C, Celda B. NMR Biomed. 2009;22:342–348. doi: 10.1002/nbm.1345. [DOI] [PubMed] [Google Scholar]
- 52.http://metlin.scripps.edu/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional data and spectra (PDF)
List of GOT-MS MRMs (XLSX)