

Abstract
Metabolic reprogramming of tumor cells toward serine catabolism is now recognized as a hallmark of cancer. Serine hydroxymethyltransferase (SHMT), the enzyme providing one-carbon units by converting serine and tetrahydrofolate (H4PteGlu) to glycine and 5,10-CH2-H4PteGlu, therefore represents a target of interest in developing new chemotherapeutic drugs. In this study, 13 folate analogues under clinical evaluation or in therapeutic use were in silico screened against SHMT, ultimately identifying four antifolate agents worthy of closer evaluation. The interaction mode of SHMT with these four antifolate drugs (lometrexol, nolatrexed, raltitrexed, and methotrexate) was assessed. The mechanism of SHMT inhibition by the selected antifolate agents was investigated in vitro using the human cytosolic isozyme. The results of this study showed that lometrexol competitively inhibits SHMT with inhibition constant (Ki) values in the low micromolar. The binding mode of lometrexol to SHMT was further investigated by molecular docking. These results thus provide insights into the mechanism of action of antifolate drugs and constitute the basis for the rational design of novel and more potent inhibitors of SHMT.
Keywords: antifolates, cancer metabolism, inhibitors, lometrexol, serine hydroxymethyltransferase (SHMT)
Introduction
Proliferating cells require programming of cellular metabolism towards aerobic glycolysis (i.e., Warburg effect, which is recognized as a hallmark of many types of cancer) and the routing of many metabolites into nucleotide metabolism, to increase DNA synthesis.[1] Recent evidence suggests that in human tumors, such as breast cancer and melanoma, and in tumor-initiating cells in non-small-cell lung cancer, the glycolytic carbon is mainly redirected into the synthesis of serine.[2–4] The increase in serine biosynthesis represents a major source of methyl groups for the one-carbon pools required for the de novo biosynthesis of purines and pyrimidines, typical of proliferating cells,[2] and, under hypoxic conditions, contributes to the limiting of reactive oxygen species (ROS) generation by enhancing NADPH production in mitochondria.[5]
Central to the aforementioned series of interconnected metabolic pathways, the ubiquitous pyridoxal 5′-phosphate (PLP)-dependent enzyme serine hydroxymethyltransferase (SHMT; E.C. 2.1.2.1) catalyzes the transfer of the Cβ of serine to 5,6,7,8-tetrahydrofolate (tetrahydropteroylglutamate, H4PteGlu), with formation of glycine and 5,10-CH2-H4PteGlu.
In humans and other higher organisms, two SHMT genes are found (i.e., SHMT1 and SHMT2), encoding the cytoplasmic (hcSHMT) and mitochondrial (hmSHMT) isozymes.[6] Moreover, SHMT2 encodes a second transcript (hcSHMTα), lacking a short exon, that is only required for efficient import into mitochondria. This third isozyme (i.e., hcSHMTα) is identical to hmSHMT but localizes in the cytoplasm together with hcSHMT, and this accounts for the unexpected viability of SHMT1−/− mice.[7, 8]
Based on the observation that Chinese hamster ovary (CHO) cells lacking hmSHMT are auxotrophic for glycine,[9] it has been suggested that hmSHMT is preferentially involved in the synthesis of glycine and mitochondrial deoxythymidine monophosphate (dTMP), while hcSHMT and to a lesser extent (∼25%) hcSHMTα participate in the synthesis of nuclear dTMP, undergoing nuclear import during S-phase and supplying 5,10-CH2-H4PteGlu during the thymidylate cycle, along with thymidylate synthase (TS) and dihydrofolate reductase (DHFR).[7] 5,10-CH2-H4PteGlu (oxidized to 10-CHO-H4PteGlu) is also utilized in the de novo biosynthesis of purines. Nuclear localization of hcSHMT is required to prevent uracil accumulation and maintain DNA integrity;[10] accordingly, knockdown of SHMT1 overexpressed in lung cancer cells leads to p53-dependent apoptosis and cell-cycle arrest.[11]
SHMT therefore occupies a critical position at the convergence of three key pathways for chemotherapeutic intervention: 1) folate metabolism; 2) dTMP biosynthesis; 3) glycine/serine metabolism. Accordingly, since its first isolation, SHMT has been repeatedly hailed as an ideal target for cancer chemotherapy.[12–14] Despite this fact, only a few studies focusing on drug design strategies and discovery of compounds that can inhibit SHMT have been carried out to date. The search for selective serine analogues and amino acid derivatives as SHMT inhibitors has not been very successful.[15] With respect to antifolate agents, the quite toxic sulfonyl fluoride triazine derivative NSC127755 was reported as an irreversible inhibitor of SHMT.[16] Leucovorin (5-formyltetrahydrofolate (fTHF), 5-CHO-H4PteGlu) has also been reported as a potent, low-micromolar inhibitor of both SHMT isoforms;[17,18] the crystal structures of Escherichia coli, Bacillus stearothermophilus and rabbit SHMTs in complex with leucovorin have also been solved, giving detailed structural insights into the binding mode of this inhibitor.[19–21] However, leucovorin cannot be used clinically as an SHMT inhibitor, as it is readily converted to other folic acid derivatives (e.g., H4PteGlu) and thus has vitamin activity equivalent to that of folic acid.
Recently, we reported that (S)-2-[4-[2-(4-amino-2-oxo-3,5,7-triazabicyclo[4.3.0] nona-3,8,10-trien-9-yl) ethyl] benzoyl] aminopentanedioic acid (pemetrexed, brand name Alimta), a multitargeting antifolate drug approved by the US Food and Drug Administration for the treatment of mesothelioma (in combination with cisplatin) and non-small-cell lung cancer, acts as a low micromolar inhibitor of hcSHMT.[22] In the present study, we extend our analysis to investigate the possible inhibitory potency and binding mode of other antifolate agents under clinical evaluation or currently in therapeutic use. Our results show that lometrexol, together with leucovorin and pemetrexed, acts as a low micromolar antifolate inhibitor of hcSHMT.
Results and Discussion
Docking of antifolate agents to hcSHMT
An initial search in the ZINC database[23] and biomedical literature for purchasable folate analogues in clinical trials/use retrieved 13 compounds (Table 1). Leucovorin (5-CHO-H4PteGlu)-based template docking was then used to assess the possible binding mode of the selected antifolates to hcSHMT. The docked compounds were then ranked based on the predicted affinity of binding (Table 1). Through these docking experiments, six compounds were predicted to have no steric clashes with hcSHMT active site residues and bind in a leucovorin-like fashion (lometrexol [LTX], pemetrexed [PTX], methotrexate [MTX], AG2034, nolatrexed [NTX] and raltitrexed [RTX]). In Table 1, this is reflected by the docking scores of the 13 compounds, which shows an energy “gap” between these six antifolate agents (entries 1–6; re-rank score after energy minimization <150.0) and the rest of the 13 compounds evaluated (entries 7–13; re-rank score >90.0).
Table 1.
<w=2>Antifolate agents in clinical trial/use investigated in this study ranked by docking score.
| Entry | Name | 2D-representation | Targets[a] | Score | Poses[b] |
|---|---|---|---|---|---|
| 1 | Lometrexol |
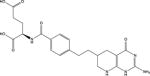
|
GARFT | −173.4 | 10 |
| 2 | Pemetrexed |
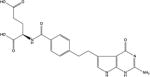
|
TS, DHFR, GARFT | −168.2 | 10 |
| 3 | AG2034 |
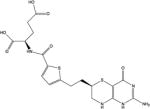
|
GARFT | −166.7 | 9 |
| 4 | Nolatrexed |
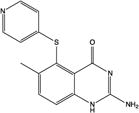
|
TS | −165.6 | 7 |
| 5 | Raltitrexed |
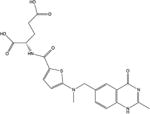
|
TS | −161.4 | 10 |
| 6 | Methotrexate |

|
DHFR, TS, GARFT, AICARF | −152.9 | 10 |
| 7 | Talotrexin |
|
TS, DHFR | −89.3 | 8 |
| 8 | Plevitrexed |
|
TS | −44.7 | 7 |
| 9 | Edatrexate |

|
DHFR | −43.7 | 6 |
| 10 | Pralatrexate |
|
TS, DHFR | −36.2 | 6 |
| 11 | Trimetrexate |
|
DHFR | −22.6 | 7 |
| 12 | Piritrexim |

|
DHFR | −12.7 | 5 |
| 13 | 1843U89 |
|
TS | −5.78 | 6 |
Abbreviations: AICARF: aminoimidazole carboxamide ribonucleotide formyltransferase; DHFR: dihydrofolate reductase; GARFT: glycinamide ribonucleotide formyltransferase; TS: thymidylate synthase.
Number of similar poses (RMSD<1.2 Å) in the top-ten scoring poses.
The interaction between PTX and hcSHMT has been investigated previously,[22] and AG2034 was unavailable for purchase. As such, LTX, MTX, NTX and RTX were used for further studies. The docked conformations of LTX, MTX, NTX and RTX are shown in Figure 1. As expected, the predicted interactions between these compounds and the active site residues of hcSHMT are similar to those observed for leucovorin. For all ligands (with the exception of NTX, which lacks the para-aminobenzoate and glutamate moieties), the α-carboxylate of the glutamate tail is placed at optimum distance to form a hydrogen bond with the side chain of Tyr82, while the γ-carboxylate is further stabilized through a hydrogen-bond interaction with the main chain of Ala395, albeit subtle variations can be noticed by comparing the orientation of the γ-carboxylate of different antifolate agents. Our docking experiments suggest that Tyr82 is also involved in a stacking interaction with the thiophene ring of RTX and the aromatic ring of the benzoic acid moiety of MTX and LTX. In contrast, the 4-pyridinylthio moiety of NLT, while pointing to Tyr82 and predicted to potentially make hydrophobic contacts with the latter residue, is not predicted to be involved in any stacking interaction, since the presence of the thioether bond at the C5 atom provides a different orientation to the aromatic ring.
Figure 1.

Predicted binding poses of antifolate agents in the active site of hcSHMT (PDB code: 1BJ4): A) methotrexate (MTX); B) nolatrexed (NTX); C) raltitrexed (RTX); D) lometrexol (LTX). hcSHMT is shown as grey ribbon. Conserved residues in the active site are indicated; the interactions for each antifolate agent are displayed as grey dashed lines. Pyridoxal 5′-phosphate (PLP) is shown as grey sticks. Leucovorin is shown as grey lines, superposed on the antifolate agent for reference.
Compared with the pterine ring of leucovorin, the bicyclical aromatic moieties of the antifolate agents evaluated are predicted to orient in quite a similar pose; that is, stacked to the external aldimine of the active site PLP–glycine complex. However, these antifolate agents have different substituent groups on their bicyclic cores, and therefore, their binding modes are not identical. In MTX, the Sp2 hybridization of the C6 atom, due to the unsaturated N5–C6 bond, hinders the proper positioning of the bulky diaminopteridine of MTX in the active site of hcSHMT. Also, compared with the physiological co-substrate, MTX displays the substitution of the oxygen in position 4′ with an ammine, and there is the addition of a methyl moiety at the N10 position. These features, which are shared with the lowest ranking compounds (entries 7–13 in Table 1), might account for the lower predicted affinity of MTX for the enzyme. The N10-methyl group is not predicted to hamper MTX binding, as it seems to be accommodated in a cleft formed by the side chains of Tyr82 and Tyr83. However, bulkier substitutions at the N10 atom, as seen in plevitrexed, edatrexate and pralatrexate (entries 8–10, Table 1), are predicted to make steric clashes with the side chains of Tyr82 and Tyr83, and are therefore disfavored.
In the quinazoline moiety of RTX and NLT, the N5 and N8 atoms, which are present in the pterin ring of folate, are replaced by two carbon atoms. Moreover, in RTX, a methyl group replaces the ammine at position 2 of the pterin ring. These substitutions, causing the loss of two hydrogen bonds with Asn387 and with the main chain carbonyl group of Leu143, probably account for the predicted lower affinity towards hcSHMT of RTX compared with the similar antifolates, LTX and AG2034. In contrast, other predicted interactions involving the quinazoline moiety of RTX mimic the binding mode of leucovorin––the N1 atom interacts with the side chain of Asn387, the 3′-NH position is involved in a hydrogen-bond contact with the carbonyl group of the main chain of Gly147, and the oxygen in position 4′ forms a hydrogen-bond interaction with Leu149.
Compared with the other selected antifolate agents, LTX has the highest structural similarity to leucovorin (and H4PteGlu). Therefore, as expected, the predicted binding mode of LTX is almost identical to that of leucovorin, although LTX lacks the nitrogen atom in position 10′, which prevents the interaction with the side chain of Glu75.
Inhibition of SHMT activity by antifolate agents
Inhibition properties of LTX, MTX, NTX and RTX were tested using a competitive binding assay, in which the antifolate agent competes for binding with leucovorin. The assay is based on the spectrophotometric measurement of the quinonoid intermediate that develops when both glycine and leucovorin bind to SHMT, forming an enzyme—glycine–folate ternary complex.[17] Binding of H4PteGlu and 5-CH3-H4PteGlu to the enzyme–glycine complex also yields a quinonoid, however, this is much less stable over time. The quinonoid intermediate, which yields an intense absorption band with a maximum at around 500 nm, derives from deprotonation of glycine (Scheme 1), but it accumulates to a measurable extent only when a folate ligand is also bound to SHMT and a ternary complex is formed.[24] Therefore, absorbance at 500 nm is proportional to the fraction of enzyme present as a ternary complex.
Scheme 1.
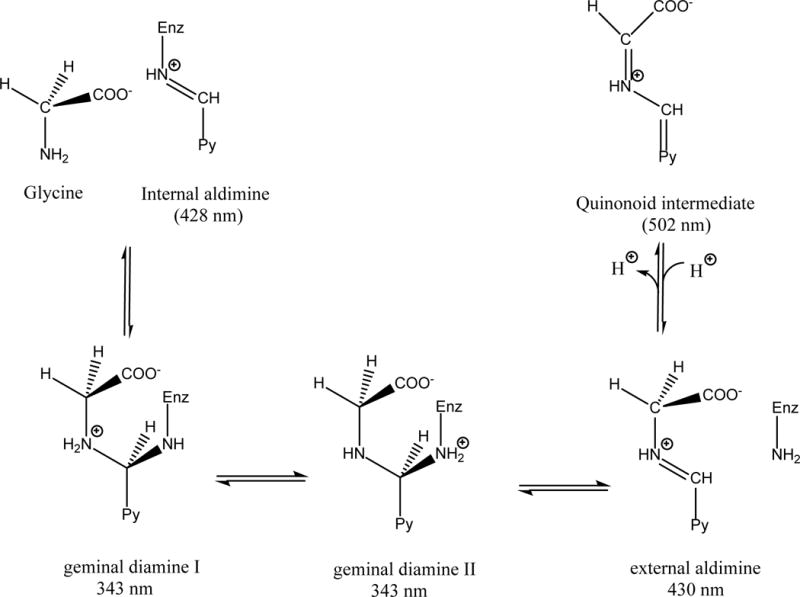
Intermediates formed upon binding of glycine to SHMT; relative absorption maxima are given in parentheses. The quinonoid intermediate accumulates only when a folate ligand is also bound to the enzyme, forming an enzyme–glycine–folate ternary complex. In the scheme, the pyridine ring of the cofactor is indicated by Py.
A pre-requisite for screening the aforementioned antifolate agents was to measure the apparent dissociation constant (Kd) values for leucovorin and glycine, thus validating the experimental set-up for further inhibition studies; the values obtained (Kd=1.5 μM for leucovorin; Kd=30 μM for glycine) were found to be in agreement with literature data,[25] thus confirming the soundness of the protocol design.
Preliminary experiments showed that none of the antifolate compounds tested yield any absorption at 500 nm when added to an enzyme solution saturated with glycine, even at an antifolate concentration equal to 2 mM. Consequently, binding of antifolates to hcSHMT was analyzed by measuring the inhibitory effect that any specific antifolate had on the formation of the quinonoid intermediate in an enzyme solution containing both glycine and leucovorin. Initially, inhibition curves were obtained varying the concentration of antifolate at fixed concentration of glycine (3 mM) and of leucovorin (5 μM). The measured apparent inhibition constant (Ki) values were 23±2 μM for LTX, 122±13 μM for RTX and 201±19 μM for MTX. NTX inhibited hcSHMT only very poorly, with an apparent Ki value higher than 1 mM.
Since LTX showed the most potent inhibitory effect on hcSHMT compared with the other selected antifolate agents tested, the inhibition mechanism of LTX was characterized in more detail. The population of the quinonoid intermediate was measured upon addition of a varying concentration of leucovorin (0.25–100 μM) to solutions containing glycine (3 mM), hcSHMT (5.5 μM) and different concentrations of LTX (0, 7, 30, 60, 125 and 300 μM). Absorbance data at 500 nm were charted in a double reciprocal plot and fitted to straight lines (Figure 2). It is clear that, under this experimental set-up, LTX acts as a competitive inhibitor (straight lines have the same y intercept), consistent with the random Bi-Bi rapid equilibrium system proposed for binding of substrates and release of products by SHMT.[26] A secondary plot of slopes as a function of LTX concentration gave a Ki value of 20±4 μM (data not shown).
Figure 2.
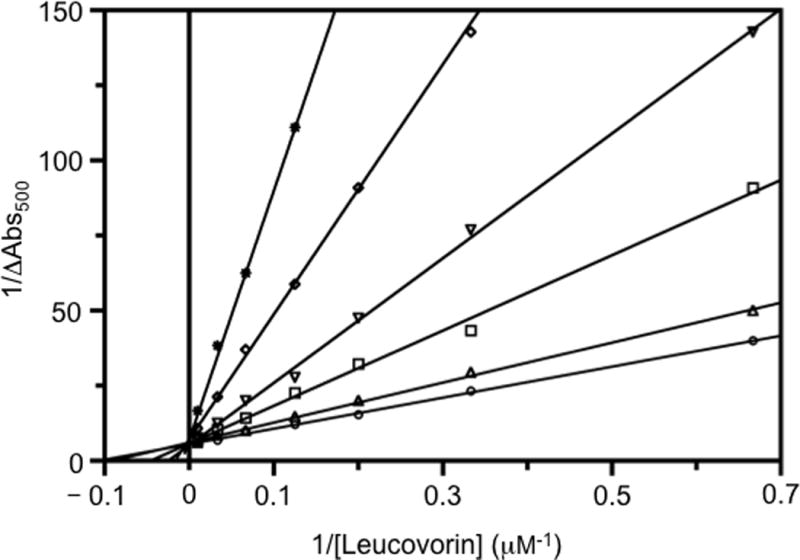
Double reciprocal plots showing lometrexol (LTX) inhibition. Absorbance changes at 500 nm (plotted as 1/ΔAbs500) were measured upon the addition of leucovorin to a solution of 5.5 μM hcSHMT containing glycine and LTX. Assays were carried out varying the concentration of leucovorin at fixed glycine concentration (3 mM) and at various concentrations of LTX (μM): 0 (◦); 7 (▵); 15 (▪); 30 (▫); 60 (▽); 125 (◊); 200 (×); 300 (∗). The continuous straight lines through the experimental points were obtained from the independent linear fitting of data.
To directly estimate the Kd value, we measured the binding constant of LTX to hcSHMT using isothermal titration calorimetry (ITC) (Table 2 and Figure 3A). As a control, the binding of hcSHMT to leucovorin was also determined to validate the experimental set-up (Table 2 and Figure 3B).
Table 2.
<w=1>Thermodynamic parameters of the interactions characterized in this work derived from the curve fit obtained through microcalorimetric titrations of hcSHMT and different ligands.
| Ligand | ΔH[a] [kcal mol−1] |
ΔS [cal mol−1/deg] |
Kd [μM][a] | n[a] |
|---|---|---|---|---|
| LTX | 3.50±0.06 | 38.0 | 1.99±0.99 | 0.53±0.05 |
| Leucovorin | 4.64±0.39 | 42.4 | 1.36±0.49 | 0.51±0.03 |
| Leucovorin[b] | –[c] | –[c] | –[c] | –[c] |
Heat of binding (ΔH), the stoichiometry (n), and the dissociation constant (Kd) were calculated from plots of the heat evolved per mole of ligand injected versus the molar ratio of ligand to protein (for further details see Experimental Section); data represent the mean ±SD of at least two independent experiments.
Experiment carried out in the presence of 200 μM lometrexol (LTX) in both hcSHMT and leucovorin solutions.
No binding observed; the signal measured superposes with that of the dilution of leucovorin into buffer (data not shown).
Figure 3.
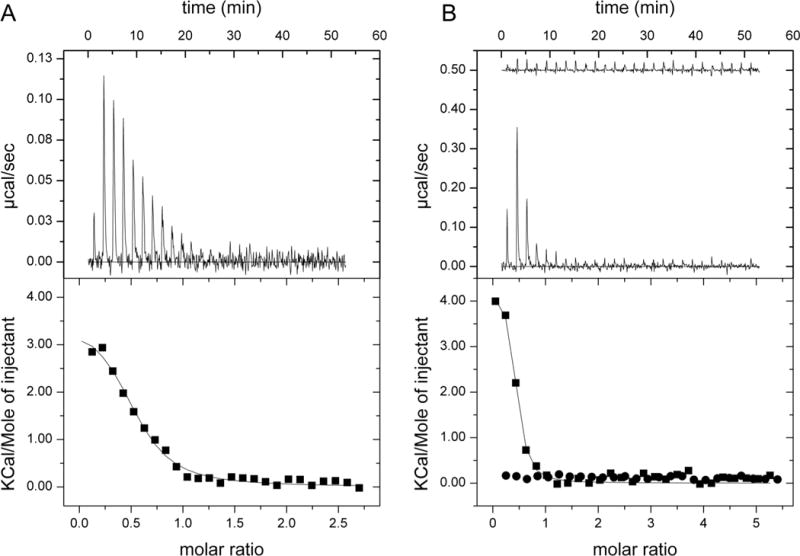
Microcalorimetric titrations of hcSHMT with different ligands. A) Titration of hcSHMT (37 μM) with 0.5 mM lometrexol (LTX); B) Titration of hcSHMT (27 μM) with 1 mM leucovorin with (top trace in upper panel/• in lower panel) or without (lower trace in upper panel/▪ in lower panel) LTX (200 μM) in both protein and ligand solutions. Top panels: Raw isothermal titration calorimetry (ITC) data. Bottom panels: Integrated peak areas and fit with the one-binding-site model of ORIGIN provided by MicroCal (continuous line). Derived thermodynamic parameters are listed in Table 2.
The experiment was carried out by titrating a 37 μM hcSHMT solution with 0.5 mM LTX solution. The titration profile is depicted in Figure 3A (upper panel). As expected for specific binding, integration of the titration peaks produced a sigmoidal enthalpy curve for the interaction between LTX and hcSHMT (Figure 3A, lower panel). The Kd value for LTX, derived from the slope of the binding curve, was 2±1 μM, and the binding stoichiometry (n) was 0.53:1 (±0.05); this value of n is close to that previously found for the co-substrate folate,[21] and for the inhibitors leucovorin (Table 2) and pemetrexed.[22] The ten-fold difference between Ki and Kd values might be explained by the different experimental approach used to determine their values. Here, the Ki value was extrapolated from enzyme activity data, while the Kd value was obtained from direct ITC measurements. The affinity of hcSHMT for LTX indicates that, together with leucovorin, LTX is the most potent antifolate-derived competitive inhibitor of SHMT identified thus far. The ability of LTX to compete with folates for binding to hcSHMT was also demonstrated by titrating hcSHMT with leucovorin in the presence of excess LTX (Figure 3B, upper trace in the upper graph), which completely abolishes binding of leucovorin to hcSHMT, as compared with the control experiment (Figure 3B, lower trace in the upper graph).
Conclusions
Folates play a pivotal role in several steps of the de novo biosynthesis of purines and pyrimidines, serving as substrates in a number of single-carbon-transfer reactions.[27] Accordingly, antifolate agents, drugs that quench the effects of H4PteGlu and its derivatives on cellular processes, are a landmark in anticancer therapy based on antimetabolites, and remain a fruitful source of new drugs, as well as novel therapeutic strategies and important findings in basic cancer research. Drugs targeting TS and DHFR (e.g., 5-fluorouracil and MTX) cause death in highly proliferating cells and remain among the most widely used chemotherapeutic agents in medical oncology.[28] Compared with DHFR and TS, there are only a few studies focusing on drug design strategies for the inhibition of SHMT.
We recently showed that pemetrexed, a chemotherapeutic antimetabolite approved for the treatment of pleural mesothelioma as well as non-small-cell lung cancer, is able to inhibit hcSHMT in addition to TS and DHFR, thus providing initial insights into the rational design of new inhibitors of SHMT.[22] However, the lack of recent studies focusing on the inhibition of human SHMT suggests that this enzyme can still be regarded as an underexplored target in cancer chemotherapy. Thus, in the present study, we extended our previous analysis on the interaction between pemetrexed and hcSHMT to investigate the possible inhibitory potency and binding mode of other antifolates in clinical trials/use. Starting from a dataset of 13 antifolate compounds, the most promising inhibitors of hcSHMT, as assessed by in silico screening, were purchased and tested in vitro. The results of this study showed that LTX competitively inhibits SHMT with respect to leucovorin, with a measured Ki value of 20±4 μM; this value compares with the Kd value of 2±1 μM measured by ITC.
According to modeling studies, several of the acceptor/donor functional groups of LTX are placed in regions already constrained in the unbound state (i.e., the 5-deaza pteridine moiety), and the groups are thus predicted to interact with well-structured regions of the protein (residues Gly147, Leu143, Leu149), minimizing the structuring effect and the loss of conformational entropy. Moreover, according to the docking results, the glutamate moiety of LTX, as in leucovorin and pemetrexed,[22] can bind to the protein in several orientations, thus paying a smaller price in terms of conformational entropy loss.
The information gained from the identified compounds and their predicted binding mode to hcSHMT represent an initial step toward the development of more potent and effective SHMT inhibitors, which could provide a promising new chemotherapeutic strategy against the large variety of cancers overexpressing this enzyme.
Experimental Section
Biological methods
Materials
Ingredients for bacterial growth and chemicals used in protein purification were purchased from Sigma–Aldrich. Chromatography media (CM-Sepharose and Phenyl-Sepharose) were from GE Healthcare. (6S)-5-CHO-H4PteGlu (leucovorin) was a gift from Merck & Co., Schaffhausen, Switzerland. All other reagents were from Sigma–Aldrich. Lometrexol was purchased from Sigma–Aldrich. Nolatrexed, raltitrexed, and methotrexate were purchased from Ambinter (Paris, France). All chemicals were of the highest purity available.
Enzyme purification
Recombinant hcSHMT was purified as previously described.[29] The enzyme activity was measured in a coupled assay, in which the 5,10-CH2-H4PteGlu produced by the reaction was reduced by a methylene-H4PteGlu dehydrogenase.[30]
Inhibition experiments
All assays were carried out at 30°C in 50 mM NaHEPES buffer (pH 7.2) containing 0.2 mM DTT and 0.1 mM EDTA, unless stated otherwise. Initially, formation of the quinonoid intermediate upon addition of a folate ligand to hcSHMT (10 μM) saturated with glycine (10 mM) was analyzed by measuring absorbance at λ 500 nm (Hewlett–Packard 8453 diode-array spectrophotometer) upon addition of either H4PteGlu, 5-CH3-H4PteGlu, or 5-CHO-H4PteGlu (leucovorin) at 10 μM. 5-CH3-H4PteGlu and leucovorin yielded twice as much absorbance as H4PteGlu. Moreover, whereas with H4PteGlu and 5-CH3-H4PteGlu absorbance rapidly decreased with time, the quinonoid developed using leucovorin was stable over a period of 5 min. Therefore, leucovorin was used in all inhibition assays. Dissociation constants of glycine and leucovorin were determined by varying one ligand while keeping the other at a fixed and saturating concentration. When glycine was the varied ligand (from 0 to 20 mM), leucovorin was kept at 200 μM. When varying leucovorin (0–300 μM), glycine was fixed at 20 mM. The dependence of quinonoid formation on pH was also analyzed over a pH range of 6.5–9.5. Buffers were made by a mixture of MES, HEPES, and CHES (50 mM each), brought to pH with NaOH. In these experiments, leucovorin (10 μM) was added to buffer containing glycine (10 mM) and hcSHMT (10 μM). The maximum quinonoid intensity was observed between pH 6.5 and 7, whereas absorbance at λ 500 nm decreased at higher pH values and nearly disappeared at pH 9.5. All antifolate compounds were dissolved in pure DMSO. The effect of DMSO concentration on quinonoid development was analyzed and found to be negligible up to 20% DMSO (v/v). In all inhibition assays, the final DMSO concentration was 5% (v/v), and leucovorin was added as the last component. After a rapid manual mixing, the absorbance change at λ 500 nm was measured. The obtained inhibition curves were fitted to Equation (1) to obtain the observed inhibition constants (Ki).
| (1) |
In this equation, A500 is the absorbance measured at λ 500 nm, A0 is the absorbance measured in the absence of potential inhibitor, and Ki is the observed inhibition constant. Fitting of data was performed with Prism software (version 4.1, GraphPad, La Jolla, CA, USA). Data obtained with LTX, varying leucovorin concentration while keeping glycine fixed at 3 mM, were used to produce a double-reciprocal plot and fitted to linear equations. Slopes and y-axis intercepts of the straight lines so obtained were plotted versus LTX concentration in secondary plots and fitted to a linear equation in order to find the related inhibition constant from x-axis intercepts.
Isothermal titration calorimetry (ITC)
ITC experiments were carried out using an iTC200 microcalorimeter (MicroCal). hcSHMT was equilibrated with binding buffer (50 mM HEPES pH 7.2, 100 μM EDTA), following PD10 gel filtration (GE Healthcare). Ligand stock solution (100 mM) was prepared by dissolving it in 100% DMSO. Titrations were carried out in 92.4% binding buffer, 10 mM glycine, and 1% DMSO. Aliquots (1.5 μL) of 0.5 mM or 0.3 mM LTX solution were injected into a solution of hcSHMT (37 μM) at 25°C. Binding of leucovorin to hcSHMT was assayed by titrating 27 μM hcSHMT with 1.5-μL aliquots of 1 mM leucovorin solution in 94% binding buffer, 10 mM glycine, at 25°C. The same titration was also carried out in the presence of 200 μM LTX in both protein and leucovorin solutions (in 92.4% binding buffer, 10 mM glycine, 1% DMSO). Data were corrected for heat changes from the injection of the ligand into the titration buffer and fitted using the “one-binding-site model” of the MicroCal version of ORIGIN. The heat of binding (ΔH), stoichiometry (n), and the dissociation constant (Kd) were then calculated from plots of the heat evolved per mole of ligand injected versus the molar ratio of ligand to protein using the software Origin provided by the vendor (MicroCal).
Docking of antifolate agents
The crystal structure of hcSHMT in its biologically active tetrameric form, and in complex with PLP as internal aldimine,[12] was used as a starting point to model the “closed” form of the enzyme in complex with PLP-glycine, as previously described.[22] The Dundee PRODRG2 server was used to build the energy-minimized three-dimensional structures of antifolates.[31] Template-based molecular docking was carried out by means of Molegro Virtual Docker (MVD) software (CLCbio, version 5.5). Flexible torsions of antifolates were automatically detected by MVD, and manually checked for consistency. The obtained three-dimensional structure of hcSHMT was prepared by automatically assigning bond orders and hybridization, and adding explicit hydrogens, charges and Tripos atom types. A search space of 15 Å radius, centered on the active site cavity, was used for docking. The aromatic rings of the pteroic acid skeleton of leucovorin were taken as pharmacophoric groups for template-based dockings.[22] In the latter, if an atom of the ligand matches a group definition, it is rewarded by using a weighted score that depends on its distance to the group centers. The grid-based MolDock score with a grid resolution of 0.30 Å was used as scoring function, and MolDock SE was used as docking algorithm.[32] For each ligand, ten runs were defined. Similar poses (RMSD<1.2 Å) were clustered, and the best-scoring one was taken as representative. Other docking parameters were fixed at their default values. After docking, energy optimization of hydrogen bonds was performed.
Acknowledgments
The authors are grateful to Chiara Ruggeri (Sapienza University of Rome, Italy) for help. This work was partially funded by the Italian Association for Cancer Research (AIRC) (AIRC-IG2012 n.13150) and Sapienza University of Rome, Italy (C26A149EC4).
Serine hydroxymethyltransferase (SHMT) has been repeatedly hailed as the missing target for cancer chemotherapy. Here, we identify lometrexol as one of the most potent antifolate inhibitors of human cytosolic SHMT (Kd=2±1 μM) known to date. The results reported represent an initial step toward the development of more potent and effective SHMT inhibitors.
Footnotes
This paper is dedicated to the memory of our friend and colleague Prof. Donatella Barra, prematurely deceased on 28 September 2014.
References
- 1.Weber G. Cancer Res. 1983;43:3466–3492. [PubMed] [Google Scholar]
- 2.Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, Sasaki AT, Anastasiou D, Mullarky E, Vokes NI, Sasaki M, Beroukhim R, Stephanopoulos G, Ligon AH, Meyerson M, Richardson AL, Chin L, Wagner G, Asara JM, Brugge JS, Cantley LC, Vander MG, Heiden P. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, Chen WW, Barrett FG, Stransky N, Tsun ZY, Cowley GS, Barretina J, Kalaany NY, Hsu PP, Ottina K, Chan AM, Yuan B, Garraway LA, Root DE, Mino-Kenudson M, Brachtel EF, Driggers EM, Sabatini DM. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S, Soh BS, Sun LL, Tai BC, Nga ME, Bhakoo KK, Jayapal SR, Nichane M, Yu Q, Ahmed DA, Tan C, Sing WP, Tam J, Thirugananam A, Noghabi MS, Pang YH, Ang HS, Mitchell W, Robson P, Kaldis P, Soo RA, Swarup S, Lim EH, Lim B. Cell. 2012;148:259–272. doi: 10.1016/j.cell.2011.11.050. [DOI] [PubMed] [Google Scholar]
- 5.Ye J, Fan J, Venneti S, Wan WJ, Pawel BR, Zhang J, Finley IW, Lu C, Lindsten T, Cross JR, Qing G, Liu Z, Simon MC, Rabinowitz JD, Thompson CB. Cancer Discovery. 2014;4:1406. doi: 10.1158/2159-8290.CD-14-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garrow TA, Brenner AA, Whitehead WM, Chen XN, Duncan RG, Korenberg JR, Shane B. J Biol Chem. 1993;268:11910–11916. [PubMed] [Google Scholar]
- 7.Anderson DD, Stover PJ. PLoS One. 2009;4:e5839. doi: 10.1371/journal.pone.0005839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacFarlane AJ, Liu X, Perry CA, Flodby P, Allen RH, Stabler SP, Stover PJ. J Biol Chem. 2008;283:25846–25853. doi: 10.1074/jbc.M802671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfendner W, Pizer LI. Arch Biochem Biophys. 1980;200:503–512. doi: 10.1016/0003-9861(80)90382-3. [DOI] [PubMed] [Google Scholar]
- 10.MacFarlane AJ, Anderson DD, Flodby P, Perry CA, Allen RH, Stabler SP, Stover PJ. J Biol Chem. 2011;286:44015–44022. doi: 10.1074/jbc.M111.307629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paone A, Marani M, Fiascarelli A, Rinaldo S, Giardina G, Contestabile R, Paiardini A, Cutruzzolà F. Cell Death Differ. 2014;5:e1525. doi: 10.1038/cddis.2014.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Renwick SB, Snell K, Baumann U. Structure. 1998;6:1105–1116. doi: 10.1016/s0969-2126(98)00112-9. [DOI] [PubMed] [Google Scholar]
- 13.Rao NA, Talwar R, Savithri HS. Int J Biochem Cell Biol. 2000;32:405–416. doi: 10.1016/s1357-2725(99)00126-0. [DOI] [PubMed] [Google Scholar]
- 14.Agrawal S, Kumar A, Srivastava V, Mishra BN. J Mol Microbiol Biotechnol. 2003;6:67–75. doi: 10.1159/000076737. [DOI] [PubMed] [Google Scholar]
- 15.Webb HK, Matthews RG. J Biol Chem. 1995;270:17204–17209. doi: 10.1074/jbc.270.29.17204. [DOI] [PubMed] [Google Scholar]
- 16.Snell K, Riches D. Cancer Lett. 1989;44:217–220. doi: 10.1016/0304-3835(89)90064-5. [DOI] [PubMed] [Google Scholar]
- 17.Stover P, Schirch V. J Biol Chem. 1991;266:1543–1550. [PubMed] [Google Scholar]
- 18.Chang WN, Tsai JN, Chen BH, Huang HS, Fu TF. Drug Metab Dispos. 2007;35:2127–2137. doi: 10.1124/dmd.107.016840. [DOI] [PubMed] [Google Scholar]
- 19.Pai VR, Rajaram V, Bisht S, Bhavani BS, Rao NA, Murthy MR, Savithri HS. Biochem J. 2009;418:635–642.6. doi: 10.1042/BJ20081739. [DOI] [PubMed] [Google Scholar]
- 20.Scarsdale JN, Radaev S, Kazanina G, Schirch V, Wright HT. J Mol Biol. 2000;296:155–168. doi: 10.1006/jmbi.1999.3453. [DOI] [PubMed] [Google Scholar]
- 21.Fu TF, Scarsdale JN, Kazanina G, Schirch V, Wright HT. J Biol Chem. 2003;278:2645–2653. doi: 10.1074/jbc.M210649200. [DOI] [PubMed] [Google Scholar]
- 22.Daidone F, Florio R, Rinaldo S, Contestabile R, Di Salvo ML, Cutruzzolà F, Bossa F, Paiardini A. Eur J Med Chem. 2011;46:1616–1621. doi: 10.1016/j.ejmech.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 23.Irwin JJ, Shoichet BK. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schirch L, Ropp M. Biochemistry. 1967;6:253–257. doi: 10.1021/bi00853a039. [DOI] [PubMed] [Google Scholar]
- 25.Fu TF, Hunt S, Schirch V, Safo MK, Chen BH. Arch Biochem Biophys. 2005;442:92–101. doi: 10.1016/j.abb.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 26.Schirch LV, Tatum CM, Benkovic SJ. Biochemistry. 1977;16:410–419. doi: 10.1021/bi00622a011. [DOI] [PubMed] [Google Scholar]
- 27.Appling DR. FASEB J. 1991;5:2645–2651. doi: 10.1096/fasebj.5.12.1916088. [DOI] [PubMed] [Google Scholar]
- 28.Bertino JR. Best Pract Res Clin Haematol. 2009;22:577–582. doi: 10.1016/j.beha.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Kruschwitz H, Ren S, Di Salvo M, Schirch V. Protein Expression Purif. 1995;6:411–416. doi: 10.1006/prep.1995.1055. [DOI] [PubMed] [Google Scholar]
- 30.Schirch V, Hopkins S, Villar E, Angelaccio S. J Bacteriol. 1985;163:1–7. doi: 10.1128/jb.163.1.1-7.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schüttelkopf AW, van Aalten DMF. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 32.Thomsen R, Christensen MH. J Med Chem. 2006;49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]