

Abstract
Spiroiminodihydantoin (Sp) is a hyperoxidized form of guanine (G) resulting from oxidation by reactive oxygen species (ROS). The lesion is highly mutagenic, and the stereocenter renders the two isomers with distinct behaviors in chemical, spectroscopic, enzymatic, and computational studies. In this work, the α-hemolysin (αHL) latch sensing zone was employed to investigate the base pairing properties of the Sp diastereomers embedded in a double-stranded DNA (dsDNA). Duplexes containing (S)-Sp consistently gave deeper current blockage, and a baseline resolution of ∼0.8 pA was achieved between (S)-Sp:G and (R)-Sp:G base pairs. Ion fluxes were generally more hindered when Sp was placed opposite pyrimidines. Analysis of the current noise of blockade events further provided dynamics information about the Sp-containing base pairs. In general, base pairs comprised of (S)-Sp generated higher current fluctuations than their (R)-Sp counterparts, suggesting enhanced base pairing dynamics. The current noise was also substantially affected by the identity of the base opposite to Sp, increasing in the order of A, G, T, and C. This report provides information about the dynamic structure of Sp in the DNA duplex and therefore has implications for the enzymatic repair of the Sp diastereomers.
Keywords: spiroiminodihydantoin, α-hemolysin, nanopore, DNA repair, NEIL
Graphical abstract

Introduction
DNA is a major target for oxidation by reactive oxygen species (ROS) generated by oxidative and inflammatory stress that leads to genomic alterations, cellular apoptosis, and mutagenesis.1–3 Among the four DNA bases, guanine (G) is the most susceptible to oxidation because it has the lowest redox potential.4 Upon two-electron oxidation of G, the stable product 8-oxo-7,8-dihydroguanine (OG) is formed, which has a much lower redox potential than the parent compound.5,6 Upon two-electron oxidation of OG, the products spiroiminodihydantoin (Sp) and 5-guanidinohydantoin (Gh) are observed; additionally, these products are detected from direct four-electron oxidation of G (e.g., by 1O2).7–13 Yields of Sp dominate over Gh when the reaction proceeds in neutral to alkaline solutions or in a G-quadruplex context, and Gh yields are maximal in reactions conducted at low pH or in double-stranded DNA (dsDNA) contexts.7,12,13 In addition, Sp has been detected in Nei-deficient Escherichia coli upon exposure to chromate14 and in mice with infection-induced colitis.15 In the latter case, the level of Sp was positively correlated with the progression of colon cancer. The Sp lesion is highly mutagenic, and can cause nearly 100% G→T or G→C transversion mutations in vivo if not properly repaired.16–19 These types of mutations are characteristic of cancers resulting from long-term oxidative stress;20,21 however, the source of these mutations is not known. Therefore, further biochemical experiments with Sp are warranted to better understand this lesion and how it impacts genomic structures possibly leading to mutations. Recently, the Núñez and Jamieson groups found that the Sp lesion would have minor effects on the stability and positioning of nucleosomal DNA.22
Interestingly, Sp exists as two stable diastereomers, (+)-(S)-Sp and (−)-(R)-Sp that originate from the stereocenter formed by the junction of the two heterocyclic rings of Sp (Figure 1A). For the sake of brevity, (+)-(S)-Sp and (−)-(R)-Sp are herein abbreviated as (S)-Sp and (R)-Sp, respectively. The absolute configuration of the Sp diastereomers was openly debated in the literature for many years;23,24 recently, the experimental and computational results were reconciled in our laboratory.25 The currently accepted absolute configurations for Sp form the basis for the assignments used in this manuscript. The reason that the stereochemistry of Sp is of interest is because the two diastereomers possess distinct biological properties. For instance, a dependency on Sp stereochemistry of polymerase insertion and bypass efficiency has been demonstrated in vitro and in vivo,7,16–19 showing that templates containing (S)-Sp generally exhibited higher bypass and extension. The stereochemistry of Sp also strongly affects its mutation types in vivo: the mutation profile of (S)-Sp exhibits sequence dependency in the mutations observed; in comparison, (R)-Sp leads to roughly equal amounts of G→T and G→C mutations regardless of sequence context.16–19 Additionally, nuclease P1 was more efficient at digesting dinucleotide substrates bearing (S)-Sp.26 Furthermore, (S)-Sp opposite any of the four canonical nucleotides was more favorably excised by the DNA glycosylase hNEIL1 in comparison to (R)-Sp.27,28 These experimental results were supported by a computational study showing (S)-Sp was better housed in the binding pocket of hNEIL1.29 It is proposed that DNA repair enzymes locate lesion sites through either a slide-and-probe mode30–32 or a passive search mechanism,33 and destabilization of damaged base pairs could facilitate this recognition process.34 Therefore, elucidating the base pairing dynamics of Sp and its stereochemical effects is of critical importance to understand how Sp is found by hNEIL1 or other enzymes.
Figure 1.

Nanopore platform for investigating base pairing properties of the Sp diastereomers. (A) Chemical structure of the Sp diastereomers. Here, the heterocyclic A-ring and B-ring are labeled. (B) Cross-section surface model of the αHL nanopore (pdb 7AHL45). A DNA duplex can be well accommodated within the 2.6 nm-wide αHL latch. (C) Sequence of the DNA duplexes studied. The Sp-containing base pair (X:Y) was synthesized at position 9, placing the base pair at the αHL latch zone. X denotes (S)-Sp, (R)-Sp, OG, G, or T; Y denotes A, G, T, or C.
The α-hemolysin (αHL) nanopore, since its first use with DNA in 1996,35 has developed into a versatile single-molecule analytical platform. Amid various applications,36,37 the αHL protein has demonstrated potential in differentiating chiral molecules.38–40 For example, Kang et al. incorporated a cyclodextrin adapter into the β-barrel of αHL to successfully distinguish enantiomers of ibuprofen and thalidomide, and they monitored the racemization of thalidomide in the protein channel.38 Later, Boersma and coworkers were able to discriminate enantiomers of four amino acids through interactions with an immobilized Cu2+ inside a site-specific mutant of the αHL nanopore39 The Luchian laboratory further observed that mutation of certain histidine residues from the l-form to the d-form in an amyloid peptide fragment would lead to an orders of magnitude decrease in binding affinity with free Cu2+ that was analyzed by the αHL protein nanopore.40
Recently, we discovered the utility of a new sensing region in the wild-type αHL nanopore.41–43 The αHL latch is unique in studying dsDNA, reasoned by the size compatibility between 2.0 nm B-form dsDNA44 and the 2.6 nm latch opening (Figure 1B).45 The size similarity leads to a sensitive response in the ion current to local structural changes of dsDNA within the latch sensing zone. This feature has allowed us to investigate uracil-DNA glycosylase (UDG) activity on a uracil-containing dsDNA,41 recognize A:T vs. G:C variations and cytosine methylation in dsDNA,42 and monitor base flipping of mismatches in dsDNA.43 Inspired by these successful applications of the αHL latch, we set out to utilize this powerful tool to study the base pairing properties of the Sp diastereomers. Use of the latch zone of αHL to monitor Sp base pair flipping as a function of the base opposite provides insight to the dynamics of these base pairs to support conclusions regarding the efficiency of their recognition by DNA repair enzymes. Furthermore, monitoring the individual Sp diastereomers embedded in dsDNA by the latch zone of wild-type αHL adds additional evidence for using the chiral protein context to differentiate stereochemical differences in an analyte. To achieve these goals, we investigated both current amplitude and the noise of the blocking events, and both were found to be strongly affected by the stereochemistry of Sp and the identity of the partner base, i.e., adenine (A), G, thymine (T), or cytosine (C). The results of these experimental studies support a stereochemical dependency of Sp impacting the local duplex structure and dynamics, and this is discussed with respect to enzymatic processing of these diastereomeric lesions.
Experimental Section
Materials
All chemicals were purchased from commercial suppliers and used as received. The phospholipid 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC) was obtained from Avanti Polar Lipids (Alabaster, AL). Wild-type αHL monomers were purchased from Sigma Aldrich (St. Louis, MO).
Oligodeoxynucleotide (ODN) Preparation
The ODNs were synthesized from commercially available phosphoramidites (Glen Research, Sterling, VA) by the DNA-Peptide Core Facility at the University of Utah (see Figure 1C for the sequences). Each strand was then cleaved from the synthetic column and deprotected using the standard protocols. Oligomers containing OG were manually cleaved in 30% NH4OH with 0.25 M β-mercaptoethanol for 24 h at ambient temperature, followed by heating at 55 °C for another 24 h to remove all the protecting groups. The deprotected strands were purified on a semi-preparative ion-exchange HPLC column using a linear gradient of 1–100% B over 35 min while monitoring absorbance at 260 nm (A = 10% CH3CN/90% ddH2O; B = 1.0 M NaCl, 20 mM NaPi, pH 8.0 in 10% CH3CN/90% ddH2O; flow rate = 3 mL/min). Oligomers containing each Sp diastereomer were synthesized by oxidizing OG-containing ODNs (2 nM, 40 μM) with Na2IrCl6 (24 nM, 480 μM) in 20 mM NaPi (pH 8.0) for 20 min at ambient temperature. Purification of the individual diastereomers was achieved on an analytical ion-exchange column (DNAPac PA100 250×4.6 mm) for which the elution order of the Sp diastereomers was previously established.25 The elution method consisted of a flow rate of 1 mL/min and mobile phases of A = 10% CH3CN/90% ddH2O and B = 1.5 M NaOAc, pH 7.0 in 10% CH3CN/90% ddH2O while monitoring the elution profile by the absorbance at 260 nm.
The DNA duplexes were prepared by mixing 100 μM of each corresponding DNA strand (ratio = 1:1) in buffer comprising 1.00 M KCl, 10 mM KPi, pH 7.4 and heating at 90 °C for 10 min followed by slowly cooling to room temperature over ∼4 h.
The melting temperature (Tm) studies were performed on a Shimadzu UV-1800 spectrophotometer coupled with a temperature controller. The data were recorded in the buffer of 1.00 M KCl, 10 mM KPi, pH 7.4, with a heating rate of 1 °C/min. In the Tm measurements, the (CAT)10 tail of the sequences had been removed and concentration of the DNAs used was 0.75 μM. The data were analyzed using the manufacture's software.
Nanopore Measurements
Glass nanopores were prepared by literature methods.46,47 The current–time (i–t) recordings were achieved using a system provided by Electronic BioSciences (EBS, San Diego, CA). After formation of a lipid bilayer across the aperture of the glass nanopore (radius ∼900 nm), wild-type αHL monomers (0.5 μg) were added into the electrolyte buffer solution in the cis chamber. After a reconstituted αHL heptamer nanochannel was obtained,46 DNA samples (25 μM) were added into the cis compartment. Similar to our previous work,48 the DNA duplexes were retained in the αHL channel for ∼1 s under a transmembrane potential of 100 mV (trans vs. cis) to collect the current signatures, followed by transiently reversing the voltage to −100mV for ∼200 ms to remove the molecule. The voltage was then switched back to the original state to capture the next dsDNA. In each experiment, over 200 events were collected for data analysis. All data were collected at a 50 kHz sampling rate with a 10 kHz low-pass filter. Unless otherwise stated, all nanopore experiments were conducted in 1.00 M KCl, 10 mM KPi, pH 7.4 at 100 mV (trans vs. cis) and 22 ± 1 °C.
Data Analysis
Blockade events were extracted using QUB 1.5.0.31. Current histograms and current–noise density plots were constructed with Origin 9.3 and software provided by EBS. Only events with a current drop to less than 20 pA were selected for plotting the current histograms, where the x axis is the current intervals (width = 0.1 pA) and y axis the number of events falling into each interval. Adobe Illustrator CS5 was used to prepare the figures.
Results
The duplexes studied consisted of 20 base pairs (bp) and a single-stranded (CAT)10 overhang at the 3′-end of the strand without the lesion (Figure 1C). The base pair of interest was placed at position 9 of the duplex region (Figure 1C). This DNA position was demonstrated to be in proximity to the latch constriction of the αHL nanopore and offers the best resolution to differentiate subtle base modifications.41 Oligonucleotides containing the Sp diastereomers were prepared from OG-containing strands according to a literature protocol7 and separated by an analytical HPLC ion-exchange column (Figure S1). The Sp diastereomers were synthesized in the shorter strand that allowed purification of the isomers. All nanopore experiments were conducted in 1.00 M KCl, 10 mM KPi, pH 7.4 at 100 mV (trans vs. cis) and 22 ± 1 °C.
The individual Sp diastereomers base paired with A (Figure 2) were studied first. Under the electrophoretic force, the target duplex was preferentially captured by the single-stranded tail and driven into the αHL nanochannel, causing a characteristic current drop to a level of ∼12.0–13.5 pA (Figure 3). When the DNA studied was captured by the blunt-end first, the current drop was to a level >20 pA, and these events were not further analyzed. Initial tests of the (S)-Sp:A-containing duplex revealed that under a voltage of 100 mV, the duplex hybrid was held inside the pore with a lifetime of 12.1 ± 1.2 s before the electrical force induced unzipping of the duplex (Figure S2, the lifetime value was derived from a first order exponential decay fitting). This long lifetime in the nanopore prevented efficient data collection. Therefore, a voltage-reversal method was employed to collect the blockade information for each molecule.48 Briefly, after ∼1 s of event recording, the polarity of the transmembrane potential was transiently reversed to release the trapped duplex, followed by switching back the voltage to capture the next molecule (Figure 3).
Figure 2.

Proposed hydrogen bonding between Sp and the four canonical bases, i.e., A, G, T, or C. The Sp base pairs are drawn according to ref 49.
Figure 3.
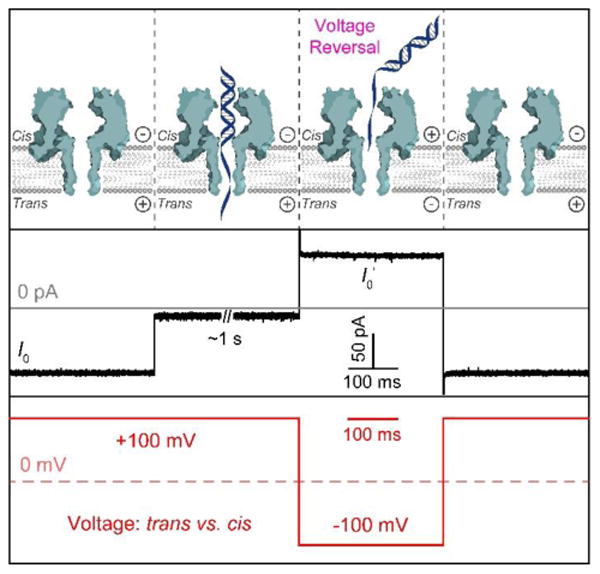
Capturing DNA duplexes with the αHL nanopore for their interrogation in the latch zone. Upper: diagrams showing the molecular mechanism for capturing and releasing a single DNA duplex upon voltage control. Middle: typical current modulations associated with the voltage-reversal process. Lower: illustration of the applied voltage cycle. The trace was recorded in the presence of (S)-Sp:A-containing duplex in 1.00 M KCl, 10 mM KPi, pH 7.4 at 100 mV (trans vs. cis) and 22 ± 1 °C.
The initial results for the Sp isomeric base pairs show that the duplexes with a (S)-Sp:A or (R)-Sp:A base pair each gave a single peak in the blocking current histogram when studied individually (Figure S3, S4), prompting us to investigate the duplexes as a mixture of diastereomers with the latch zone of αHL (Figure 4A, S5). When a mixture was studied, the (S)-Sp:A duplex was added at a twofold greater concentration than the (R)-Sp:A duplex, thus allowing us to assign the two peaks in the current histogram on the basis of the peak area ratio. Figure 4B illustrates a magnified view of a typical tail entrance event. Two parameters were extracted from the blocking events and used for analysis, the average current (I) and current noise (IRMS). The former corresponds to the residual ion fluxes during duplex blockage, while the latter is related to current fluctuations.
Figure 4.

Analysis of the Sp:A base pairs using the αHL latch zone. (A) Representative i–t trace generated by a mixture of (S)-Sp:A- and (R)-Sp:A-containing duplexes. Events with a deep blockage (i) represent tail entrance, whereas events with a shallow blockage (ii) represent blunt-end entrance. The voltage was transiently reversed after ∼1 s of event recording to release the trapped duplex and capture the next. (B) Expanded view of the current trace indicated by a red rectangle in panel A. Herein, I denotes the average residual current of the blockade, and IRMS corresponds to the noise of current. (C) Typical blocking current histogram caused by a mixture of (S)-Sp:A and (R)-Sp:A duplexes. The 2:1 peak ratio reflects the twofold greater concentration of (S)-Sp:A duplex in solution. (D) Corresponding current–noise density plot associated with panel C. Assignment of the two clusters was based on their current blockage. (E) Representative current–noise density plot provided by a mixture of (S)-Sp:A duplex and the T:A-containing duplex utilized as an internal standard. In these cases, the T:A-containing duplex was added at a concentration twofold higher facilitating its recognition. (F) Representative current–noise density plot provided by a mixture of (R)-Sp:A duplex and the T:A internal standard. The traces were recorded in 1.00 M KCl, 10 mM KPi, pH 7.4 at 100 mV (trans vs. cis) and 22 ± 1 °C.
Figure 4C shows the current histogram for the (S)-Sp:A vs. (R)-Sp:A mixture in which two partially overlapped peaks were observed with a peak-to-peak current difference (i.e., ΔIR−S = I(R-Sp − I(S)-Sp) of 0.4 ± 0.1 pA (for current blocks normalized by the open channel current, please refer to Table S1). The reported errors are derived from three independent trials in which each experiment comprised over 200 events. The corresponding current–noise density plot is shown in Figure 4D and yields two clusters with a distinct boundary. By aligning the current–noise density plot to the previously assigned current histogram, the identities of the two clusters were readily obtained. The result shows the (S)-Sp:A duplex induced a higher current noise level than the (R)-Sp:A duplex (Figure 4D).
Next, to gain the intrinsic characteristics of each individual Sp stereoisomer and circumvent interference arising from pore-to-pore variances, each analyte was spiked with an internal standard. The internal standard here refers to a duplex containing a T:A base pair at position 9, and identification of the T:A internal standard is facilitated by adding it at a concentration twofold greater than the Sp analyte. The results (Figure 4E, 4F, S6, S7) show that duplexes containing either (S)-Sp:A or (R)-Sp:A base pairs were substantially less blocking than the T:A internal standard (IT:A = 10.0 ± 0.4 pA). For example, the current difference between analyte and the spiked-in standard (IAnalyte−IT:A) was 2.4 ± 0.1 pA and 2.8 ± 0.1 pA for the (S)-Sp:A and (R)-Sp:A base pair containing duplexes, respectively. This observation identifies the S diastereomer of Sp to block the current more than the R diastereomer when base paired with A.
Encouraged by the success with the study of Sp:A-containing duplexes, we proceeded to explore how the Sp diastereomers would behave when paired opposite the other three canonical DNA bases, i.e., G, T, or C (Figure 2). Interestingly, in all base-pairing combinations the αHL latch could distinguish between duplexes containing (S)-Sp and (R)-Sp as a mixture in the same protein nanopore (Figure 4C, 5A, S8–S10). Moreover, duplexes containing the S conformation were always more blocking than the R isomer. The best separation between the Sp diastereomers was achieved when the base pairing partner was G, which yielded a baseline resolution of ΔIR−S = 0.8 ± 0.1 pA (Figure 5A). All other combinations (i.e., (S)-Sp:A vs. (R)-Sp:A, (S)-Sp:T vs. (R)-Sp:T, or (S)-Sp:C vs. (R)-Sp:C) gave a ΔIR−S of ∼0.3–0.4 pA (Figure 4C, S9, S10). These data demonstrate that regardless of the partner base, the (S)-Sp-containing duplex blocks more current than its R counterpart in the αHL nanopore, and the current difference ΔIR−S is further modulated by the partner base.
Figure 5.

Comparison of the blocking currents recorded between duplexes containing (S)-Sp and (R)-Sp base paired with the canonical bases. (A) Typical current histogram generated by a mixture of (S)-Sp:G and (R)-Sp:G duplexes that yields the largest separation. (B) Bar chart of the blocking current for each combination of the Sp stereoisomer and the partner base. All data are relative to the T:A internal standard (i.e., IAnalyte−IT:A). Data represent the mean (± standard deviation) of three individual experiments, in which each experiment was comprised of more than 200 events. The traces were recorded in 1.00 M KCl, 10 mM KPi, pH 7.4 at 100 mV (trans vs. cis) and 22 ± 1 °C.
In the next set of studies, comparisons between the base pairs with each Sp diastereomer were made against the T:A-containing duplex utilized as an internal standard. Within the T:A internal standard, the relative blocking current for each Sp-containing base pair was obtained (Figure 4, S6, S7, S11–S16), which is summarized in Figure 5B. In the (S)-Sp family, the duplex was the most hindering to the ion current when G or C served as the opposite base, with a current blockage 2.0 ± 0.1 pA smaller than the T:A internal control; in comparison, duplexes containing (S)-Sp:T or (S)-Sp:A bases pairs were ∼0.2 pA and ∼0.5 pA, respectively, less hindering than the former G or C base-paired groups. A slightly more diverse distribution of I was observed in the (R)-Sp family, among which IAnalyte−IT:A was 2.9 ± 0.1 pA for the least blocking (R)-Sp:A duplex and 2.2 ± 0.1 pA for the most blocking (R)-Sp:C duplex. These studies further verify duplexes with (S)-Sp base pairs consistently yield greater blockages (i.e. a lower absolute current level) to the current recorded.
Apart from the blockage amplitude, the noise level IRMS of each signature event was also analyzed, and the results are plotted in Figure 6. It was evident that all lesion base pairs led to noisier events than the natural T:A reference and G:C base pair (IRMSG:C−IRMST:A = 0.10 ± 0.02 pA, Figure S17). The data in Figure 6 further conclusively demonstrated that duplexes containing (S)-Sp caused stronger current fluctuations than their (R)-Sp counterparts, represented by a typical ∼0.08 pA to ∼0.20 pA increase in IRMS. The identity of the partner base contributes another crucial factor in determining the current noise. For both Sp diastereomers, switching the partner from A to C resulted in the most pronounced enhancement of IRMS, reaching ∼0.30 pA.
Figure 6.

Noise analysis of the blocking events generated by the Sp-containing duplexes. The bar chart summarizes the noise levels for all combinations of the Sp stereoisomer and the partner base. All data are relative to the T:A internal standard (i.e., IRMSAnalyte−IRMST:A). Data represent the mean (± standard deviation) of three individual experiments, in which each experiment was comprised of more than 200 events. The traces were recorded in 1.00 M KCl, 10 mM KPi, pH 7.4 at 100 mV (trans vs. cis) and 22 ± 1 °C.
Discussion
As stated above, when keeping the partner base identical, the (S)-Sp-containing base pairs always blocked more current than the (R)-Sp-containing ones when placed at the latch constriction of the αHL nanopore (Figure 5B). Though there are no crystal structures or other direct experimental evidence to explain this phenomenon, a computational study provides some clues. Jia et al. used molecular modeling to extract the structural and thermodynamic features of Sp-incorporated duplexes.49 They found that the propeller-like Sp lesion predominately adopts a syn orientation with the A-ring hydrogen bonding with the opposite base and the perpendicular B-ring accommodated in the major groove.49 In this configuration, the S diastereomer of Sp forms fewer and weaker hydrogen bonds than the R diastereomer and causes more perturbations to adjacent base pairs.49 These two factors synergistically drive the (S)-Sp-containing duplex to be less compact in the local structure when compared to the (R)-Sp-bearing duplexes. Consequently, the less compact duplexes bearing the (S)-Sp diastereomers are anticipated to block the current more than the (R)-Sp diastereomer, consistent with the observations of the present study (Figures 5B).
The effects of the partner base on the current blockage are more complicated. Generally, except for the base pair (S)-Sp:G, the ion fluxes in the αHL nanopore were impeded less when Sp was paired with purines, i.e., A or G (Figure 5B). A probable explanation is that due to the thymine-like feature of Sp, duplexes containing Sp:A or Sp:G base pairs are less strained and bear a tighter architecture, thus, providing less steric hindrance to the nanopore current.49 It is quite interesting that all Sp-containing base pairs blocked the current less than the canonical T:A base pair (Figure 5B), despite the fact T:A is a stable Watson-Crick base pair and planar T causes much less disturbance to neighboring base stacking than the propeller-like Sp. In addition, both (S)-Sp:C and (R)-Sp:C base pairs were less blocking than the undamaged G:C base pair (IG:C−IT:A = 0.6 ± 0.1 pA, Figure S17) or the parent OG:C base pair (IOG:C−IT:A = 0.6 ± 0.1 pA, Figure S18). The mechanism behind these two observations is still not clear, and a possible reason is that distortion of the base pairs in the vicinity of the Sp lesion causes local unwinding and lengthening in the region of the duplex occupying the latch zone,49 thus promoting the passage of more ions leading to the higher current observed.50 Solvation changes can play an important role,51,52 and these are harder to predict and quantify.
In this body of work, with all other conditions identical, alterations of the current noise in the experiments are most likely due to the distinctive dynamics of each Sp base pair. This interpretation is strongly corroborated by various experimental and computational studies in literature reports,27,28,49,53 as is discussed below.
According to the above hypothesis, base pairs containing (S)-Sp that generated noisier current traces (Figure 6) should be more dynamic than their R counterparts. This conclusion is in good accordance with reported enzymatic, NMR, and computational results.27,28,49,53 Enzyme-catalyzed lesion cleavage is an effective method to investigate the dynamics of a damaged base pair, because an increase in base pair dynamics facilitates recognition and excision by the enzyme. In studies of hNEIL1-mediated repair of oxidized guanines,27,28 (S)-Sp was cleaved at least 1.3-fold faster than (R)-Sp that is indicative of higher dynamics of the (S)-Sp-containing base pairs. The Shafirovich laboratory analyzed the NOSEY-NMR signals of the imino–amino protons in base pairs flanking the Sp damaged site.53 The result again supports the S configuration of Sp would cause stronger perturbations to local structure.53 It should be noted that the authors in this report assigned the configuration of the two diastereomers opposite to ours,25,53 and reinterpretation of the data here is based on our more recent re-assignment.25 The computational study by Jia, et al.49 on the other hand, provided the molecular basis for the stereochemical dependency of Sp on base pairing dynamics and associated current noise in the αHL nanopore. First, regardless of the partner base, the (S)-Sp base pairs form weaker hydrogen bonds compared to (R)-Sp, which implies that the corresponding base pair is more likely to temporarily open when being trapped at the αHL latch, and thus adds more to the current fluctuations. Second, the propeller-like architecture of Sp orients the carbonyl oxygen (O6) on the B-ring (Figure 1A) in the opposite direction for the two diastereomers, which results in different disturbances to the adjacent base pairs. Specifically, in the case of (S)-Sp the O6 atom is directed 5′ and positioned in a more crowded context, thus causing greater destabilization to neighboring base pairs; meanwhile, the 3′-oriented O6 atom in the (R)-Sp group generates less distortion of the local structure.
Apart from the evidence outlined above, the correlation between current noise and base pairing dynamics is further supported by the agreement in the partner base dependency between literature reports27,28,54 and the noise analysis here (Figure 6). The hNEIL1 repair experiments disclosed that for either (S)-Sp or (R)-Sp the lesion cleavage was accelerated as the partner changed from A, C, G, or T.27,28 Meanwhile, the IRMS figures followed the pattern A < G < T < C (Figure 6). The two datasets trend with each other quite well in general, with only one exception being the C-paired case in which the cleavage results indicate for either diastereomer, Sp:C is the second-most stable base pair among the four combinations (i.e., Sp:A, Sp:G, Sp:T, or Sp:C) while noise data imply it is the most dynamic. A possible explanation for this discrepancy is that in the hNEIL1 repair studies, a large excess of hNEIL1 (chNEIL1:cdsDNA = 10:1) was used to derive the single-turnover rate constants.27,28 Under these circumstances, the lesion cleavage for the Sp:G and Sp:T base pairs was appreciably enhanced due to the strong hydrogen bonding between the arginine117 residue of hNEIL1 and the corresponding partner base, namely G or T, thus, yielding a higher rate constant than that of the Sp:C base pair that is the most dynamic.29,55,56 Besides the hNEIL1 experiments, our previous study on the structural destabilization effects of oxidized G lesions revealed that the dsDNA containing a Sp:C base pair was unzipped roughly 2-fold faster in the αHL nanopore than the one containing a Sp:A.54 This discrepancy in the unzipping time indicates base pair Sp:C destabilizes the duplex structure to a greater extent than Sp:A, which is consistent with the largest noise being associated with Sp:C (Figure 6).
Measurements of Tm are a common way to infer the stability of base pairs and DNA structures. However, the variation of Tm was small when changing the stereochemistry of Sp (Figure S19), which is in accordance with literature reports.53,57 The αHL latch, on the other hand, demonstrates a high sensitivity in probing the subtle difference in dynamics between the Sp diastereomers. The rationale used in this work could be feasibly extended to the study of other diastereomeric bases, such as the lesion 5-carboxamido-5-formamido-2-iminohydantoin (2Ih). 2Ih was recently discovered to be a major product of guanine oxidation in the attack of hydroxyl radicals under aerobic reducing conditions.58 Similar to Sp, 2Ih also bears a stereocenter for which the absolute configurations are known,59 and the two diastereomers behave differently in enzymatic processes,60 yet their base pairing dynamics are still poorly understood. Additionally, the analysis of noise adds to the capability of the αHL latch to discern variances in base pairs. One instance of applying this capability could be to resolve the equilibrium between Gh and iminoallantoin (Ia), in which Gh gradually isomerizes to Ia with increased pH.61 Future applications of this approach will be helpful in understanding other lesion-containing base pairs in DNA.
Conclusions
Using the αHL nanopore, we were able to discriminate the Sp diastereomers and investigate their base pairing dynamics. All four canonical bases, i.e., A, G, T, or C, were allowed to individually pair with (S)-Sp and (R)-Sp, and confinement of each base pair at the latch zone of the αHL nanopore generated blocking events with characteristic current changes and noise levels. Duplexes containing (S)-Sp consistently gave larger blockage amplitudes, whereas the partner base further modulated the current difference between the two diastereomers with the best separation of ∼0.8 pA achieved when G was the base pairing partner. Furthermore, a general trend was observed that for either (S)-Sp or (R)-Sp, purine partners caused less current blocking than pyrimidine partners. Blockage amplitude is reasoned to inversely correlate with the compactness of the local structure of Sp-containing base pair at the αHL latch. In addition to current blockage level, the noise information was also extracted from the blockade traces to investigate the dynamics of the Sp base pairs. In all base-pairing combinations, (S)-Sp gave a higher current noise than (R)-Sp, indicating enhanced dynamics in the (S)-Sp-containing base pair. The influence of the opposite bases on blockade noise was mapped, which revealed that for each Sp diastereomer the blockade noise successively increased as the partner base varied from A to G, T, or C. The dynamic nature of Sp:C base pairs may have a crucial biological implication: upon ROS attack, hNEIL1 enzyme can quickly locate the Sp lesion site that is inherently paired with C and is very dynamic, and then initiate the repair process to restore the original G:C base pairing, thus preserving the integrity of the genome.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health (R01 GM093099). We also acknowledge EBS for donation of the instrument and software utilized for recording the i–t traces. The oligonucleotides were provided by the DNA/Peptide core facility at the University of Utah which is supported in part by the NCI Cancer Center Support Grant (P30 CA042014).
Footnotes
Conflict of Interest: The authors declare no competing financial interest.
Supporting Information: HPLC chromatograms, i–t traces, current blocking histograms, and current–noise density plots, normalized current blocks, Tm measurements. The Supporting Information is available free of charge on the ACS Publications website at http://pubs.acs.org.
References
- 1.Beckman KB, Ames BN. Oxidative decay of DNA. J Biol Chem. 1997;272:19633–19636. doi: 10.1074/jbc.272.32.19633. [DOI] [PubMed] [Google Scholar]
- 2.Cadet J, Douki T, Ravanat JL. Oxidatively generated base damage to cellular DNA. Free Radical Bio Med. 2010;49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 3.Delaney S, Jarem DA, Volle CB, Yennie CJ. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radical Res. 2012;46:420–441. doi: 10.3109/10715762.2011.653968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steenken S, Jovanovic SV. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 5.Burrows CJ, Muller JG. Oxidative nucleobase modifications leading to strand scission. Chem Rev. 1998;98:1109–1151. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 6.Steenken S, Jovanovic SV, Bietti M, Bernhard K. The trap depth (in DNA) of 8-oxo-7,8-dihydro-2′deoxyguanosine as derived from electron-transfer equilibria in aqueous solution. J Am Chem Soc. 2000;122:2373–2374. [Google Scholar]
- 7.Kornyushyna O, Berges AM, Muller JG, Burrows CJ. In vitro nucleotide misinsertion opposite the oxidized guanosine lesions spiroiminodihydantoin and guanidinohydantoin and DNA synthesis past the lesions using Escherichia coli DNA polymerase I (Klenow fragment) Biochemistry. 2002;41:15304–15314. doi: 10.1021/bi0264925. [DOI] [PubMed] [Google Scholar]
- 8.Ye Y, Muller JG, Luo WC, Mayne CL, Shallop AJ, Jones RA, Burrows CJ. Formation of 13C-, 15N-, and 18O-labeled guanidinohydantoin from guanosine oxidation with singlet oxygen. Implications for structure and mechanism. J Am Chem Soc. 2003;125:13926–13927. doi: 10.1021/ja0378660. [DOI] [PubMed] [Google Scholar]
- 9.Crean C, Geacintov NE, Shafirovich V. Oxidation of guanine and 8-oxo-7,8-dihydroguanine by carbonate radical anions: Insight from oxygen-18 labeling experiments. Angew Chem Int Edit. 2005;44:5057–5060. doi: 10.1002/anie.200500991. [DOI] [PubMed] [Google Scholar]
- 10.Ravanat JL, Martinez GR, Medeiros MHG, Di Mascio P, Cadet J. Singlet oxygen oxidation of 2′-deoxyguanosine. Formation and mechanistic insights, Tetrahedron. 2006;62:10709–10715. [Google Scholar]
- 11.Gremaud JN, Martin BD, Sugden KD. Influence of substrate complexity on the diastereoselective formation of spiroiminodihydantoin and guanidinohydantoin from chromate oxidation. Chem Res Toxicol. 2010;23:379–385. doi: 10.1021/tx900362r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleming AM, Muller JG, Dlouhy AC, Burrows CJ. Structural context effects in the oxidation of 8-oxo-7,8-dihydro-2′-deoxyguanosine to hydantoin products: Electrostatics, base stacking, and base pairing. J Am Chem Soc. 2012;134:15091–15102. doi: 10.1021/ja306077b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleming AM, Burrows CJ. G-quadruplex folds of the human telomere sequence alter the site reactivity and reaction pathway of guanine oxidation compared to duplex DNA. Chem Res Toxicol. 2013;26:593–607. doi: 10.1021/tx400028y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hailer MK, Slade PG, Martin BD, Sugden KD. Nei deficient Escherichia coli are sensitive to chromate and accumulate the oxidized guanine lesion spiroiminodihydantoin. Chem Res Toxicol. 2005;18:1378–1383. doi: 10.1021/tx0501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mangerich A, Knutson CG, Parry NM, Muthupalani S, Ye WJ, Prestwich E, Cui L, McFaline JL, Mobley M, Ge ZM, Taghizadeh K, Wishnok JS, Wogan GN, Fox JG, Tannenbaum SR, Dedon PC. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. P Natl Acad Sci USA. 2012;109:E1820–E1829. doi: 10.1073/pnas.1207829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 17.Delaney S, Delaney JC, Essigmann JM. Chemical–biological fingerprinting: Probing the properties of DNA lesions formed by peroxynitrite. Chem Res Toxicol. 2007;20:1718–1729. doi: 10.1021/tx700273u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delaney S, Neeley WL, Delaney JC, Essigmann JM. The substrate specificity of MutY for hyperoxidized guanine lesions in vivo. Biochemistry. 2007;46:1448–1455. doi: 10.1021/bi061174h. [DOI] [PubMed] [Google Scholar]
- 19.Neeley WL, Delaney S, Alekseyev YO, Jarosz DF, Delaney JC, Walker GC, Essigmann JM. DNA polymerase V allows bypass of toxic guanine oxidation products in vivo. J Biol Chem. 2007;282:12741–12748. doi: 10.1074/jbc.M700575200. [DOI] [PubMed] [Google Scholar]
- 20.Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res–Fund Mol M. 2001;477:7–21. doi: 10.1016/s0027-5107(01)00091-4. [DOI] [PubMed] [Google Scholar]
- 21.Pfeifer GP, Besaratinia A. Mutational spectra of human cancer. Hum Genet. 2009;125:493–506. doi: 10.1007/s00439-009-0657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norabuena EM, Williams SB, Klureza MA, Goehring LJ, Gruessner B, Radhakrishnan ML, Jamieson ER, Nunez ME. Effect of the Spiroiminodihydantoin Lesion on Nucleosome Stability and Positioning. Biochemistry. 2016;55:2411–2421. doi: 10.1021/acs.biochem.6b00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durandin A, Jia L, Crean C, Kolbanovskiy A, Ding S, Shafirovich V, Broyde S, Geacintov NE. Assignment of absolute configurations of the enantiomeric spiroiminodihydantoin nucleobases by experimental and computational optical rotatory dispersion methods. Chem Res Toxicol. 2006;19:908–913. doi: 10.1021/tx060078e. [DOI] [PubMed] [Google Scholar]
- 24.Karwowski B, Dupeyrat F, Bardet M, Ravanat JL, Krajewski P, Cadet J. Nuclear magnetic resonance studies of the 4R and 4S diastereomers of spiroiminodihydantoin 2′-deoxyribonucleosides: Absolute configuration and conformational features. Chem Res Toxicol. 2006;19:1357–1365. doi: 10.1021/tx060088f. [DOI] [PubMed] [Google Scholar]
- 25.Fleming AM, Orendt AM, He YA, Zhu JD, Dukor RK, Burrows CJ. Reconciliation of chemical, enzymatic, spectroscopic and computational data to assign the absolute configuration of the DNA base lesion spiroiminodihydantoin. J Am Chem Soc. 2013;135:18191–18204. doi: 10.1021/ja409254z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X, Fleming AM, Muller JG, Burrows CJ. Endonuclease and exonuclease activities on oligodeoxynucleotides containing spiroiminodihydantoin depend on the sequence context and the lesion stereochemistry. New J Chem. 2013;37:3440–3449. doi: 10.1039/C3NJ00418J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krishnamurthy N, Zhao XB, Burrows CJ, David SS. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry. 2008;47:7137–7146. doi: 10.1021/bi800160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao XB, Krishnamurthy N, Burrows CJ, David SS. Mutation versus repair: NEIL1 removal of hydantoin lesions in single-stranded, bulge, bubble, and duplex DNA contexts. Biochemistry. 2010;49:1658–1666. doi: 10.1021/bi901852q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jia L, Shafirovich V, Geacintov NE, Broyde S. Lesion specificity in the base excision repair enzyme hNeil1: Modeling and dynamics studies. Biochemistry. 2007;46:5305–5314. doi: 10.1021/bi062269m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banerjee A, Santos WL, Verdine GL. Structure of a DNA glycosylase searching for lesions. Science. 2006;311:1153–1157. doi: 10.1126/science.1120288. [DOI] [PubMed] [Google Scholar]
- 31.Blainey PC, van Oijent AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. P Natl Acad Sci USA. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson SR, Dunn AR, Kathe SD, Warshaw DM, Wallace SS. Two glycosylase families diffusively scan DNA using a wedge residue to probe for and identify oxidatively damaged bases. P Natl Acad Sci USA. 2014;111:E2091–E2099. doi: 10.1073/pnas.1400386111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao CY, Jiang YL, Stivers JT, Song FH. Dynamic opening of DNA during the enzymatic search for a damaged base. Nat Struct Mol Biol. 2004;11:1230–1236. doi: 10.1038/nsmb864. [DOI] [PubMed] [Google Scholar]
- 34.Yang W. Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair. 2006;5:654–666. doi: 10.1016/j.dnarep.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Characterization of individual polynucleotide molecules using a membrane channel. P Natl Acad Sci USA. 1996;93:13770–13773. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deamer D, Akeson M, Branton D. Three decades of nanopore sequencing. Nat Biotechnol. 2016;34:518–524. doi: 10.1038/nbt.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howorka S, Siwy Z. Nanopore analytics: Sensing of single molecules. Chem Soc Rev. 2009;38:2360–2384. doi: 10.1039/b813796j. [DOI] [PubMed] [Google Scholar]
- 38.Kang XF, Cheley S, Guan XY, Bayley H. Stochastic detection of enantiomers. J Am Chem Soc. 2006;128:10684–10685. doi: 10.1021/ja063485l. [DOI] [PubMed] [Google Scholar]
- 39.Boersma AJ, Bayley H. Continuous stochastic detection of amino acid enantiomers with a protein nanopore. Angew Chem Int Edit. 2012;51:9606–9609. doi: 10.1002/anie.201205687. [DOI] [PubMed] [Google Scholar]
- 40.Schiopu I, Iftemi S, Luchian T. Nanopore investigation of the stereoselective interactions between Cu2+ and d, l-histidine amino acids engineered into an amyloidic fragment analogue. Langmuir. 2015;31:387–396. doi: 10.1021/la504243r. [DOI] [PubMed] [Google Scholar]
- 41.Jin Q, Fleming AM, Johnson RP, Ding Y, Burrows CJ, White HS. Base-excision repair activity of uracil-DNA glycosylase monitored using the latch zone of α-hemolysin. J Am Chem Soc. 2013;135:19347–19353. doi: 10.1021/ja410615d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.D Ding Y, Fleming AM, White HS, Burrows CJ. Differentiation of G:C vs A:T and G:C vs G:mC base pairs in the latch zone of α-hemolysin. ACS Nano. 2015;9:11325–11332. doi: 10.1021/acsnano.5b05055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson RP, Fleming AM, Beuth LR, Burrows CJ, White HS. Base flipping within the α-hemolysin latch allows single-molecule identification of mismatches in DNA. J Am Chem Soc. 2016;138:594–603. doi: 10.1021/jacs.5b10710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinden RR. DNA Structure and Function. Academic Press; San Diego: 1994. Introduction to the structure, properties, and reactions of DNA; pp. 1–57. [Google Scholar]
- 45.Song LZ, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 46.White RJ, Ervin EN, Yang T, Chen X, Daniel S, Cremer PS, White HS. Single ion-channel recordings using glass nanopore membranes. J Am Chem Soc. 2007;129:11766–11775. doi: 10.1021/ja073174q. [DOI] [PubMed] [Google Scholar]
- 47.Zhang B, Galusha J, Shiozawa PG, Wang GL, Bergren AJ, Jones RM, White RJ, Ervin EN, Cauley CC, White HS. Bench-top method for fabricating glass-sealed nanodisk electrodes, glass nanopore electrodes, and glass nanopore membranes of controlled size. Anal Chem. 2007;79:4778–4787. doi: 10.1021/ac070609j. [DOI] [PubMed] [Google Scholar]
- 48.An N, Fleming AM, White HS, Burrows CJ. Crown ether–electrolyte interactions permit nanopore detection of individual DNA abasic sites in single molecules. P Natl Acad Sci USA. 2012;109:11504–11509. doi: 10.1073/pnas.1201669109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jia L, Shafirovich V, Shapiro R, Geacintov NE, Broyde S. Structural and thermodynamic features of spiroiminodihydantoin damaged DNA duplexes. Biochemistry. 2005;44:13342–13353. doi: 10.1021/bi050790v. [DOI] [PubMed] [Google Scholar]
- 50.Carson S, Wilson J, Aksimentiev A, Weigele PR, Wanunu M. Hydroxymethyluracil modifications enhance the flexibility and hydrophilicity of double-stranded DNA. Nucleic Acids Res. 2016;44:2085–2092. doi: 10.1093/nar/gkv1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolna AH, Fleming AM, An N, He LD, White HS, Burrows CJ. Electrical current signatures of DNA base modifications in single molecules immobilized in the α-Hemolysin Ion Channel. Isr J Chem. 2013;53:417–430. doi: 10.1002/ijch.201300022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhattacharya S, Yoo J, Aksimentiev A. Water mediates recognition of DNA sequence via ionic current blockade in a biological nanopore. ACS Nano. 2016;10:4644–4651. doi: 10.1021/acsnano.6b00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khutsishvili I, Zhang N, Marky LA, Crean C, Patel DJ, Geacintov NE, Shafirovich V. Thermodynamic profiles and nuclear magnetic resonance studies of oligonucleotide duplexes containing single diastereomeric spiroiminodihydantoin lesions. Biochemistry. 2013;52:1354–1363. doi: 10.1021/bi301566v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin Q, Fleming AM, Ding Y, Burrows CJ, White HS. Structural destabilization of DNA duplexes containing single-base lesions investigated by nanopore measurements. Biochemistry. 2013;52:7870–7877. doi: 10.1021/bi4009825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luscombe NM, Laskowski RA, Thornton JM. Amino acid–base interactions: A three-dimensional analysis of protein–DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoffman MM, Khrapov MA, Cox JC, Yao JC, Tong LN, Ellington AD. AANT: The amino acid–nucleotide interaction database. Nucleic Acids Res. 2004;32:D174–D181. doi: 10.1093/nar/gkh128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gruessner B, Dwarakanath M, Stewart E, Bae Y, Jamieson ER. Effect of base-pairing partner on the thermodynamic stability of the diastereomeric spiroiminodihydantoin lesion. Chem Res Toxicol. 2016;29:279–284. doi: 10.1021/acs.chemrestox.5b00453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alshykhly OR, Fleming AM, Burrows CJ. 5-Carboxamido-5-formamido-2-iminohydantoin, in addition to 8-oxo-7,8-dihydroguanine, is the major product of the iron-Fenton or X-ray radiation-induced oxidation of guanine under aerobic reducing conditions in nucleoside and DNA contexts. J Org Chem. 2015;80:6996–7007. doi: 10.1021/acs.joc.5b00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fleming AM, Alshykhly O, Orendt AM, Burrows CJ. Computational studies of electronic circular dichroism spectra predict absolute configuration assignments for the guanine oxidation product 5-carboxamido-5-formamido-2-iminohydantoin. Tetrahedron Lett. 2015;56:3191–3196. doi: 10.1016/j.tetlet.2014.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alshykhly OR, Fleming AM, Burrows CJ. Guanine oxidation product 5-carboxamido-5-formamido-2-iminohydantoin induces mutations when bypassed by DNA polymerases and is a substrate for base excision repair. Chem Res Toxicol. 2015;28:1861–1871. doi: 10.1021/acs.chemrestox.5b00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu J, Fleming AM, Orendt AM, Burrows CJ. pH-dependent equilibrium between 5-guanidinohydantoin and iminoallantoin affects nucleotide insertion opposite the DNA lesion. J Org Chem. 2016;81:351–359. doi: 10.1021/acs.joc.5b02180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.