

Abstract
The use of nanoparticles (NPs) in biomedical applications creates a need for appropriate model systems to systematically investigate NP–membrane interactions under well-defined conditions. Black lipid membranes (BLMs) are free-floating membranes with defined composition that are ideally suited for characterizing NP–membrane interactions free of any potential perturbation through a supporting substrate. Herein, arrays of microfabricated BLMs are integrated into a chip-based platform that is compatible with high-speed optical NP tracking. This system is used to investigate the lateral diffusion of 40 nm gold spheres tethered to biotinylated lipids through antibody-functionalized ligands (single-stranded DNA or polyethylene glycol). Although the NPs show an almost free and ergodic diffusion, their lateral motion is subject to substantial drag at the membrane surface, which leads to systematically smaller diffusion coefficients than those obtained for lipids in the membrane through fluorescence recovery after photobleaching. The lateral mobility of the NPs is influenced by the chemical composition and salt concentration at the NP-membrane interface, but is independent of the ligand density in the membrane. Together with the observation that nanoprisms, which have a larger relative contact area with the membrane than spherical NPs, show an even slower diffusion, these findings indicate that the lateral mobility of NPs tethered in close vicinity to a membrane is significantly reduced by the friction at the NP-membrane interface.
1. Introduction
Nanoparticles (NPs) of various chemical compositions, sizes, and shapes have found important applications in biomedical research, disease diagnostics, and therapeutics.[1] Quantum dots, iron oxide, and noble metal NPs are widely used as imaging reagents;[2,3] gold nanoshells have important emerging applications in photodynamic cancer therapy;[4] and vesicles, dendrimers, polymer nanospheres, etc. are useful carriers for drug and gene delivery.[5] Although all of these applications involve interactions between NPs and membranes, the underlying mechanisms that govern NP–membrane interactions and their dependence on key material properties of the NPs, including density, shape, size, and especially charge and chemical composition of the surface, remain quantitatively insufficiently understood.[6] These parameters determine the number and nature of contacts a NP forms to the plasma membrane and, thus, influences NP uptake and trafficking in cellular systems.[7,8] Observations by several groups that NPs containing specific mixtures of hydrophilic and hydrophobic surface ligands or even oligonucleotides can translocate through membranes in a passive fashion[6,9,10] have spurred additional interest in the NP-membrane interface.[11,12]
A systematic investigation of how NP properties and membrane composition interact requires appropriate model systems in which NP and membrane parameters can be independently varied in a rational fashion. Solid-supported membranes are widely used artificial membrane model systems due to their easy preparation and excellent mechanical stability.[13] One potential concern associated with the supported membrane system is, however, that the contact between the lipid bilayer and the supporting substrate impacts the lipid lateral diffusion in the membrane and leads to a reduction of the diffusion coefficient (D).This problem was in part mitigated through the introduction of hydrophilic cushion-supported lipid bilayers.[14–16] The latter show an improvement in membrane fluidity, but the D values of the lipid lateral diffusion in some of these membranes are still relatively low compared to those of freestanding membranes. Furthermore, the direct contact between the membrane and the supporting cushions can create complications in the interpretation of particle–membrane interactions.[17,18] Black lipid membranes (BLMs) are prepared over an aperture in a hydrophobic material, most commonly a Teflon sheet.[19,20] Consequently, BLMs are freestanding in solution and entirely avoid contact with the underlying substrate. BLMs have characteristic lateral diffusion coefficients (D) in the range of 10–30 μm 2 s−1,[21,22] which compares to approximately 4 μm 2 s−1 for solid-supported membranes.[13] BLMs are, thus, the model system of choice for investigating NP–membrane interactions.
The development of micro-/nanofabrication techniques has led to the realization of “on-chip” integrated BLMs.[23,24] Instead of using an individual Teflon aperture as the base for BLMs, as in the traditional Montal–Mueller approach,[19] entire arrays of apertures can be created in a broad range of materials including silica, polydimethylsiloxane (PDMS), or photoresist with precise control over the size and geometry of the individual apertures.[23–25] Different approaches have been developed to form membranes over the created wells or apertures.[26] Lipid bilayer self-assembly through vesicle deposition is widely used for on-chip BLM formation.[24,27–30] In addition, BLM formation by the contact of two lipid monolayers at the oil/water interface inside a flow chamber has been demonstrated.[31,32]
Most of the BLM systems in use today are, however, optimized for electrical conductance measurements, although optical tracking of NPs can provide detailed insights into NP–membrane interactions as well, especially in the case of noble metal NPs. Due to their large scattering cross sections and extreme photophysical stability,[33–35] gold NPs with diameters larger than 20 nm can be easily detected in a conventional dark-field microscope, which makes them superb probes for optical imaging and particle tracking. Unlike fluorescent labels, gold NPs do not blink or bleach and the size and shape of gold NPs can be well controlled using established synthesis procedures.[36–38] Furthermore, gold NPs can be conveniently functionalized with thiolated polymers (R-SH) of different charge, stiffness, and length through formation of an Au–S-R bond. To take full advantage of the superb optical photo-physical properties of noble metal NPs and their tunability, herein we implement a BLM array system optimized for high-speed imaging of NPs on membranes of defined composition through dark-field microscopy. We then apply it to characterize the lateral diffusion of gold NPs on freestanding 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membranes and to determine the lateral friction associated with the NP diffusion.
2. Results and Discussion
2.1. BLM Array Design, Preparation, and Characterization
Our design of the BLM array was inspired by the work of Ganesan and Boxer who recently introduced an on-chip BLM interferometer based on a shallow well array fabricated through microlithography.[23] They generated the wells through reactive ion etching (RIE) into a SiO2 layer. This procedure is, however, not ideal for dark-field microscopy since the latter will collect signals from all efficient scatterers close to the focal plane under oblique illumination. The roughness introduced to the wafer in the etching process leads to substantial background in dark-field imaging and causes a dramatic increase in the noise level. We, consequently, used an alternative fabrication process (Figure 1a) and created the wells through deposition of SiO2 on lithographically patterned substrates (see the Experimental Section for more details). This approach successfully retained the smoothness of the polished wafer surface and minimized background scattering (Figure 1b). The background in optical tracking was further decreased by generating large wells with a diameter of 100 μm, which provided sufficient membrane area away from the edges of the wells where the scattering background is high. The wells were also made relatively deep (7–9 μm) to minimize the scattering background from dirt on the bottom of the well (Figure 1c).
Figure 1.

BLM array fabrication. a) Schematic illustration of the BLM array fabrication and membrane formation process. b) Dark-field image of a BLM array and c) scanning electron microscopy (SEM) image of a single well in the array. Scale bars in (b) and (c): 100 and 25 μm, respectively. PEG = polyethylene glycol.
The small (1 cm × 1 cm) well array chips were mounted on plastic microscope cover slides. BLMs were then formed by thinning of a decane layer sandwiched between two lipid monolayers at the organic–aqueous interphases.[23] During spontaneous retraction of the decane film across the surface, lipid membranes formed across the fabricated wells (see the Experimental Section for details). After membrane formation, another cover slide connecting the inlet and outlet tubing was placed on top of the first slide supporting the BLM chip, and the two cover slides were sealed by double-sided tape to form a flow chamber as shown in Figure 2a. Figure 2b presents a scheme of our experimental setup with a BLM chip containing the flow chamber integrated into a dark-field microscope. The overall thickness of the chamber was around 1 mm to allow the focusing of both a high numerical aperture (NA) oil dark-field condenser and an imaging objective onto the plane of the BLMs.
Figure 2.

Integration of BLM chips into a dark-field microscope. a) The BLM chip is inserted in a simple flow chamber, which allows addition and removal of antibody-functionalized NPs that bind to biotin–phosphatidylethanolamine (biotin-PE) contained in the POPC membrane. b) Schematic drawing of the optical dark-field setup. c) Trapping of fluorescent dye (carboxyfluorescein) in the fabricated wells by membrane formation enables detection of wells sealed by an intact membrane. d) Fluorescence from Nile red contained in the decane is enriched at the edges of the wells, which indicates the formation of a decane annulus. ssDNA = single-stranded DNA, EMCCD = electron-multiplying charge coupled device.
Although not all of the wells in the array were successfully sealed by membranes in our assembly approach, the addition of a fluorescent dye (carboxyfluorescein, CF) to the aqueous buffer beneath the decane film greatly facilitated the detection of membranes. Only those wells that were sealed by an intact membrane showed strong fluorescence signals (Figure 2c). We dissolved Nile red in the decane to check for the presence of remaining organic solvent enclosed between the two leaflets of the membrane bilayer (Figure 2d). Except for the area in close vicinity of the well edge (Plateau–Gibbs border),[39] in which the two leaflets do not associate due to an enclosed solvent annulus,[40] the Nile red fluorescence signal was negligible, which indicates that the two leaflets form a tight lipid bilayer in the center area of the wells. In some cases we actually observed the process of bilayer formation and the associated “zipping” of the two membrane leaflets in real time. The zipping occurs when the decane gradually retracts and the two lipid monolayers get sufficiently close for thermal fluctuations to induce contact formation. The energetically favored bilayer structure then rapidly expands across the whole membrane area.[41] Figure S1 (Supporting Information) shows this process as observed by dark-field microscopy.
After successful formation of the lipid membranes, we set out to characterize the ensemble diffusion properties of the lipids in the formed bilayer through fluorescence recovery after photobleaching (FRAP). To that end, we integrated CF-labeled phosphatidylethanolamine (CF-PE) into the membrane and determined the diffusion coefficient as D = w2/4t½, where w is the effective radius of the bleaching laser beam and t½ is the time for the bleached spot to recover half of its full recovery intensity. The diffusion coefficient was measured to be D = 25.1 ± 3.4 μm2 s−1 for a membrane containing 10% CF-PE and 90% POPC at 25 °C (Figure S2), which is in very good agreement with the literature value reported for BLMs.[21,22] Overall, the FRAP studies confirmed the formation of well-behaved BLMs.
2.2. Tracking the Lateral Diffusion of NPs on BLMs
We functionalized our 40 nm (diameter) sphere NPs with single-stranded DNAs (ssDNAs). We used a mix of 50-nucleotide-long ssDNAs that were 3′-functionalized with a thiol group and 5′-functionalized with an azide group (HS-DNA- N3) and 30-nucleotide-long ssDNAs that were only 3′-thiolated (HS-DNA) for the assembly of a ssDNA brush on the NP surface. HS-DNA-N3 and HS-DNA were mixed in the ratio of 30:70 mol%. The thiol group efficiently anchored the ssDNAs to the NP surface while the azide group allowed for a convenient cross-linking to alkyne-labeled anti-biotin antibodies through the Cu+-catalyzed azide–alkyne cycloaddition.[42,43] For more details regarding the NP preparation and characterization, please refer to the Experimental Section and the Supporting Information. In the following we will refer to the anti-biotin antibody-functionalized NPs simply as Ab-DNA-NPs.
One of the key advantages of our NP–membrane model system is that it offers both outstanding signal intensity and low background. Figure 3a shows a representative dark-field image of 40 nm Ab-DNA-NPs on BLMs acquired at 500 frames per second (fps). The precise locations of the individual NPs in each frame were determined by fitting their point-spread functions (PSFs) with 3D Gaussians (see Experimental Section for details). The binding of Ab-DNA-NPs in the absence of biotin-PE was very low, thus confirming that nonspecific binding on POPC BLMs was negligible (Figure 3b). By connecting the coordinates of the fitted peak intensities in each frame, the diffusion trajectories of the individual NPs (Figure 3c) were obtained. We tracked individual NPs with frame rates between 100 and 2380 fps. The resulting average signal-to-noise ratios (s/n) showed that the noise scales as the square root of the signal intensity (Figure 3d), which is characteristic of photon noise (shot noise). Due to the low background and the large scattering cross sections of the NPs, an excellent s/n of around 10 (without any image processing) was achieved at 1000 fps. Gold NPs do not blink or bleach, and we routinely tracked individual NPs for several minutes. The maximum observation time in the tracking experiments was not limited by the photophysical properties of the NPs but by the area monitored by our cameras. After some time the NPs simply diffused out of the detection area.
Figure 3.

NP tracking in dark-field microscopy. a)Typical image (31.25 μm × 31.25 μm) of Ab-DNA-NPs on a BLM; individual NPs are clearly identifiable as bright dots. The image was acquired at a frame rate of 500 fps. For each individual NP in each frame, the raw data (upper and middle insets) were fitted with a 3D Gaussian function (bottom inset) to determine the exact coordinates of the NP. b) Number of Ab-DNA-NPs on membranes with different ligand density. c) Representative trajectory of a single particle tracked for 1000 frames at 500 fps (blue spheres). The gray line shows the 2D projection of the trajectory. d) Signal-to-noise ratio (s/n) for the Ab-DNA-NPs as a function of acquisition cycle time.
In the next step, we determined the effect of increasing biotin-PE concentration in the membrane on the lateral diffusion of the Ab-DNA-NPs. The click chemistry applied to bind antibodies to ssDNA-functionalized NPs was anticipated to be highly efficient and to provide gold NPs carrying multiple antibodies. We verified the presence of multiple antibodies on the NP surface by incubating the 40 nm Ab-DNA-NPs with an excess of 10 nm (diameter) biotinylated gold NPs. SEM images of this mix in Figure S4 confirm the association of many 10 nm NPs with each individual 40 nm Ab-DNA-NP. Multivalent Ab-DNA-NPs can form multiple biotin–antibody contacts with the membrane, but at constant antibody/NP ratio the probability of multiple contacts is expected to decrease with decreasing biotin-PE concentration in the membrane. We varied the biotin-PE density in the membrane over two orders of magnitude and assembled membranes containing 0.05, 0.5, and 5 mol% biotin-PE. These concentrations correspond to 1.25, 12.5, and 125 biotins in an area of 40 nm × 40 nm in the upper leaflet of the BLM. As expected, the number of NPs bound to the membrane increased with growing biotin-PE density in the membrane (Figure 3b) due to the increasing number of available binding sites in the membrane.
We performed tracking experiments on membranes containing identical Ab-DNA-NPs in 3-(N-morpholino) propanesulfonic acid (MOPS) buffer (0.5 m NaCl, pH 7). Figure 4a shows the resulting mean-square displacement (MSD) 〈Δr2〉 as a function of time lag (t) for NPs on membranes with different biotin-PE concentrations. The MSD was computed as time and ensemble average as , where N is the total number of trajectories, τ is the acquisition time, and T is the total length of trajectory j. The following N values were used: 81 (0.05 mol% biotin-PE), 394 (0.5 mol% biotin-PE), and 778 (5 mol% biotin-PE). According to the Stokes–Einstein relationship,[44] the MSD 〈Δr2〉 increases linearly with increasing time lag t as described by the relationship 〈Δr2〉 = 4Dtα with α = 1 for Brownian diffusion. In Figure 4a we plot the 〈Δr2〉 as a function of t for BLMs with different biotin-PE concentrations. The plots of 〈Δr2〉 versus t for membranes containing 0.05, 0.5, and 5% biotin-PE show almost ideal linear dependencies. Strikingly, we did not detect any systematic change of the lateral mobility of the NPs for the different biotin-PE concentrations, even though the biotin was varied over two orders of magnitude. We found α ≥0.95 for all biotin-PE concentrations, consistent with an almost ideal Brownian diffusion for all investigated biotin-PE concentrations. We emphasize that we performed the diffusion analysis at different temporal resolutions (100 and 500 fps) and obtained essentially identical NP diffusion behaviors.
Figure 4.

NP diffusion analysis. a) Ensemble-averaged MSD versus time lag plots for NP diffusion on membranes with different ligand density. b) MSS slope and diffusion coefficient distributions of Ab-DNA-nanoprisms on 0.5% biotin-PE containing BLMs (purple triangles), and Ab-DNA-NPs on 0.05% biotin-PE containing supported membranes (green squares) or 5% (orange circles) biotin-PE containing BLMs. Histograms for the diffusion coefficients (D) and SMSS values for Ab-DNA-NPs on 5 mol% biotin-PE BLMs are included in the top (red) and side (blue) panels. c) Ensemble square displacement and temporal square displacement distributions for Ab-DNA-NPs on BLMs containing 5% biotin-PE.
The mode of lateral diffusion can be classified in more detail using the approach of Ferrari et al. who considered moments of displacements, ν = 1 – 6, in their analysis: 〈Δrv〉 ~ Dtγ (v).[45] A plot of γ versus ν is called a moment scaling spectrum (MSS) and its slope (SMSS) facilitates a classification of the translational motion. SMSS values of 0, 0.5, and 1 correspond to immobilized, free (Brownian), and ballistic diffusion, respectively. In Figure 4b, we plotted the obtained SMSS values for individual trajectories of Ab-DNA-NPs on BLMs (with the highest investigated biotin-PE concentration of 5%, recorded at 100 fps) as a function of the fitted diffusion coefficient (ν = 2; orange circles). The SMSS distribution is centered at around SMSS = 0.5 but the distribution is asymmetric with a longer tail on the low SMSS side. Nevertheless, 72% of the trajectories lie within the range of SMSS = 0.5 ± 0.1, and 92% lie within the range of SMSS = 0.5 ± 0.15, thus indicating that the majority of the NPs perform an essentially free diffusion on the free-floating membrane system despite the high concentration of available binding sites in the membrane. This behavior is in stark contrast to the SMSS distribution observed with the same Ab-DNA-NPs on a 0.05% biotin-PE-containing glass-supported membrane, for which the diffusion was overall slower and the contribution from immobile NPs higher (Figure 4b, green squares). The supported membrane was assembled through the Langmuir–Blodgett technique followed by a Langmuir–Schaefer procedure.[46,47] We chose a very low biotin-PE concentration for the supported membrane experiment since for higher ligand concentrations the majority of the NPs were immobile. Even with the 0.05% biotin-PE concentration, about 45% of the observed NPs (32 out of 71) were essentially immobile. The mobile fraction of NPs with SMSS >0.1 have an average D = 0.42 ± 0.41 μm2 s−1, which is systematically slower than the D = 3.69 ± 1.09 μm2 s−1 observed for the BLMs with the same biotin-PE density. This comparison underlines the advantage of the BLM approach, which—by design—avoids perturbation of the membrane or spurious interactions between the NPs and the substrate supporting the membrane.
In the next step we took advantage of the long continuous trajectories facilitated by NPs to test whether the NP diffusion on the implemented BLMs is ergodic.[48] Ergodicity implies that the time average for an individual trajectory is equivalent to the ensemble average at an arbitrary point of time.[49] To test this hypothesis we compared the distributions of temporal square displacement (SD) with the ensemble SD for Ab-DNA-NPs on 5% biotin-PE membranes. The temporal SD distribution was obtained from a single, randomly chosen trajectory (1000 at 100 fps). The ensemble SD distribution was populated by calculating the SD between the 51st and 50th frames in a total of 390 trajectories. A comparison of the resulting distributions in Figure 4c shows that the temporal and ensemble SD distributions superimpose, which implies that the NP diffusion on the BLM system is ergodic.
2.3. Characterizing the Lateral Friction of NPs on BLMs
Although the Ab-DNA-NPs exhibit an almost free and ergodic diffusion on the BLMs, the obtained D values (see Figure 4b) are still quite small (D ≈ 4 μm 2 s−1), especially when compared with the experimental diffusion coefficient of the lipids in the membrane (D = 25.1 ± 3.4 μm2 s−1) and the calculated diffusion coefficient of a 40-nm-diameter NP in water (D = 12.3 μm 2 s−1, ηwater = 8.9 × 10−4 Pa s at 25 °C) from the Stokes–Einstein relationship. Since we can exclude that the relatively slow motion of the NPs arises from spurious interactions with a substrate in the case of the BLMs, we attribute it to direct NP–membrane interactions. One obvious potential reason for a slow lateral diffusion of the NPs on the membrane is the formation of multiple antibody–biotin contacts between individual NPs and the membrane.[50] The fact that we did not observe any systematic change of the lateral mobility of the NPs when the concentration of biotin-PE was varied over two orders of magnitude (Figure 4a) argues against multivalent binding as the main cause for the experimentally observed slow diffusion.
Although the Ab-DNA-NPs show only a negligible unspecific binding to the membrane (Figure 3c), it is unavoidable that they explore interfacial effects after being tethered to the membrane. The resulting short-range interactions between the NPs and the membrane will create forces that act on the membrane and the surrounding fluid. The associated reaction forces will seek to counter these effects and result in an increased effective friction for the lateral translation of the membrane-bound NP.[51] Brenner and Leal have shown that the Brownian diffusion of particles at the inter-phase between two immiscible fluids can be characterized by a diffusion coefficient of the form: where kB is the Boltzmann constant, θs is the temperature, η is the dynamic viscosity, a is the radius of the particle, and f is the lateral friction parameter.[52–53] Assuming ideal Brownian diffusion for the trajectories in Figure 4a, we obtain a friction coefficient of approximately f = 20π for the Ab-DNA-NPs, independent of the biotin concentration in the membrane. This compares with f = 6π for a free NP, and we conclude that the additional frictional drag resulting from the close vicinity of the membrane limits the lateral mobility of the NPs on the membrane.
We reasoned that if nonspecific interactions between the membrane and the tethered NPs are the cause for the lateral friction, these should be sensitive to the composition of the NP-membrane interface. To verify this hypothesis, we performed tracking experiments with a fixed membrane composition (99.5 mol% POPC, 0.5 mol% biotin-PE) but with three different flavors of NPs: 1) Ab-DNA-NPs tracked in buffer containing 0.1 m NaCl, 2) Ab-DNA-NPs tracked in buffer containing 0.5 m NaCl, and 3) Ab-PEG-NPs (PEG=polyethylene glycol) tracked in buffer containing 0.1 m NaCl. For the Ab-PEG-NPs the negatively charged ssDNA ligands were substituted with charge-neutral 3.4-kDa-long thiolated PEGs using similar NP functionalization and antibody cross-linking procedures (see Experimental Section). The persistence lengths/end-to-end distances of the polymers bound to the particles under the different experimental conditions are summarized in Table 1. Although negatively charged NP surfaces have been found to induce membrane gelation, which can lead to a slower lipid lateral mobility,[54] we did not find the negative Ab-DNA-NPs to be associated with systematically lower D values than those observed for the Ab-PEG-NPs. Therefore, the NP surface charge does not seem to be the dominating factor for the measured slow diffusion coefficients. In contrast, the stiffness (quantified by the persistence length) and, thus, the spacing between the gold NP surface and the membrane is correlated with the slope of the MSD (Figure 5). The MSD slope increases with growing stiffness of the polymer layer surrounding the NPs.
Table 1.
Overview of diffusion parameters and surface properties for different NPs and buffer conditions.
| Probe and buffer | Persistence length [Å] a) | End-to-end distances [Å] | Diffusion coefficient [μm 2 s−1] | Power-law dependency α | Friction factor f |
|---|---|---|---|---|---|
| Ab-ssDNA-NPs in 0.1 M NaCl | 12.7[55,56] | 87 | 3.90 | 0.95 | 18.9 |
| Ab-ssDNA-NPs in 0.5 M NaCl | 5.7[55,56] | 70 | 3.60 | 0.95 | 20.5 |
| Ab-PEG-NPs in 0.1 M NaCl | 3.7[57] | 52 | 3.27 | 0.98 | 22.6 |
| Ab-ssDNA-nanoprisms in 0.1 M NaCl | 12.7[55,56] | 87 | 1.94 | 1.01 | 38.0 |
The persistence lengths of single - stranded DNA molecules were calculated using the formula provided in ref. [55]: .
Figure 5.
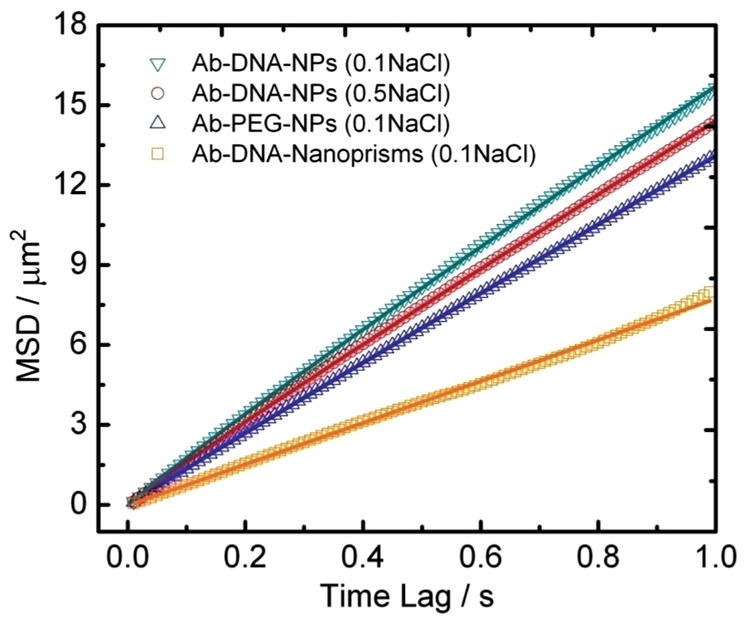
MSD versus time lag for different NP surfaces. The investigated conditions include: 1) Ab-DNA-NPs in buffer with ionic strength of 0.1 mol L−1 (green triangles); 2) Ab-DNA-NPs in buffer with ionic strength of 0.5 mol L−1 (red circles); 3) Ab-PEG-NPs in buffer with ionic strength of 0.1 mol L−1 (blue triangles); 4) Ab-DNA-nanoprisms in buffer with ionic strength of 0.1 mol L−1.
We rationalize this experimental observation in terms of a higher lateral friction for NPs carrying softer polymer brushes due to stronger attractive interactions between the NPs and the membrane. It is worth mentioning that the fitted α values in 〈Δr2〉 = 4Dtα for the different NP surfaces are all very close to 1, which confirms that all of the investigated NPs perform an essential free Brownian diffusion on the BLMs, albeit with different diffusion coefficients due to different degrees of lateral friction.
To further validate the impact of friction on the lateral diffusion of polymer-functionalized NPs tethered to lipid membranes, we analyzed the diffusion of nanoprisms with typical side lengths of 80–100 nm and thickness of 10 nm (Figure S5). Ab-DNA-nanoprisms carrying multiple antibodies are expect to attach with their larger front side to the membrane and, thus, to maximize the NP–membrane contact. The resulting increase in contact area relative to spherical NPs with identical volume translates into an increase in friction for the lateral diffusion. Indeed, the diffusion of the nanoprisms was found to be slower by a factor of 2 when compared with the spherical particles (Figure 5 and Table 1), thus emphasizing the importance of the NP–membrane contact area in determining the diffusion friction and lateral mobility.
3. Conclusion
We have introduced a BLM array compatible with optical dark-field tracking of NPs tethered to membrane lipids and have demonstrated that the low background of the BLM array system, in combination with the large scattering cross sections of the NPs, enables the high-speed tracking of individual NPs on free-floating membranes free of any perturbation through a supporting substrate. The NPs were tethered to the membrane through anti-biotin antibodies binding to biotin-PE contained in the membrane. We systematically investigated the lateral diffusion of NPs that were coated with a brush of a ssDNA mix containing 30- and 50-nt-long ssDNAs or 3.4 kDa PEGs. The NPs exhibited a free and ergodic diffusion on the membrane with typical lateral diffusion coefficients of D ≈ 4 μm 2 s−1, which were dominated by the lateral friction of the polymer-functionalized NPs tethered in close vicinity of the membrane. Our tracking studies indicate that even for NPs that show negligible nonspecific binding to the membrane from solution, a tethering of the NPs in close vicinity to the membrane (<10 nm) results in a substantial lateral frictional drag that reduces their lateral mobility. Nanospheres and nanoprisms functionalized with ssDNAs or PEGs exhibited measurable differences in the lateral diffusion, which suggests that the precise control of the NP-membrane interface represents a potential approach for maximizing the mobility of NPs on the membrane in a rational fashion through variation of the chemical composition, length, and density of the ligands grafted onto the NP surface.
4. Experimental Section
Materials
The following materials were used as obtained from the vendors: octadecanethiol (Sigma); 2-[methoxy(polyethyleneoxy) propyl]trimethoxysilane (PEG-silane) (Gelest); 10X MOPS buffer stock solution (Fisher Scientific); 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (Avanti Polar Lipids, Inc.); 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) sodium salt (biotin-PE) (Avanti Polar Lipids, Inc.); 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-carboxyfluorescein ammonium salt (CF-PE) (Avanti Polar Lipids, Inc.); 40 nm gold colloid (British Biocell International); thiol-30nt DNA (Integrated DNA Technologies); thiol-50ntDNA-azide (HS-50nt DNA-N3), thiol-30ntDNA (HS-30nt DNA) (Fidelity Systems, Inc.); thiol-polyethylene glycol-azide (N3-(CH2CH2O)77-CH2CH2-SH, molecular weight 3400) (NANOCS); propargyl dPEG-NHS ester (Quanta Biodesign); anti-biotin affinity isolated antigen-specific antibody (Sigma); L-ascorbic acid (Aldrich); copper(II) sulfate pentahydrate (Aldrich); carboxyfluores-cein (CF) (Sigma); Nile red (Sigma); tetrachloroauric acid (HAuCl4) and crystalline sodium sulfide (Na2S) (Sigma–Aldrich). We used Zeba spin desalting columns (7k molecular weight cutoff, MWCO) from Thermo Scientific. The reagent solutions were prepared in distilled deionized (DDI) water (double distilled, 18.2 MΩ).
BLM Array Fabrication
Wells were fabricated through standard microlithography followed by thin-film deposition and photoresist lift-off processes (Figure 1a). A 30 μm layer of AZ4620 photoresist spin-coated on a Pyrex wafer was patterned through photolithography (Karl Suss MA6 Aligner). A 7 μm layer of SiO2 was then deposited onto the patterned wafer (Sharon Vacuum, Brockton, MA, USA) followed by 5 nm Cr and 100 nm Au deposition (CHA Industries, Fremont, CA, USA). The photoresist was removed by immersing the substrates in acetone for 10 min followed by 3 min of sonication in an acetone bath.
After drying and oxygen plasma cleaning for 30 min, the substrates were immersed in a 1 mM solution of 1-octadecanethiol in ethanol and subsequently in a 1 mM PEG-silane solution in toluene. Substrates were stored under vacuum until usage to prevent oxidation of the thiol–metal bond.
Membrane Formation and Characterization
A surface-functionalized 1 cm × 1 cm BLM chip was mounted over a central (1.2 cm × 1.2 cm) cutout in a plastic microscope slide (7.6 × 2.5 × 0.4 cm, Fisher Scientific) before membrane formation. The whole slide was then submerged in MOPS buffer (0.5 M NaCl, pH 7) containing CF. The wells on the chip were sealed by dispersing decane on top of the chips with a pipette. Remaining CF-containing buffer in the unsealed wells was then replaced by clear MOPS buffer through extended rinsing. In the subsequent step, preassembled vesicles (formed by lipid rehydration and extrusion) in MOPS buffer were added to deliver lipids into the decane. Upon addition of the lipids, the decane covering the SiO2 substrates retracted in the aqueous buffer to form droplets. During the retraction process a freestanding lipid bilayer was formed over the fabricated wells.
Fluorescence Recovery after Photobleaching
FRAP was performed on membranes assembled from POPC and CF-PE (90:10 mol%) in an Olympus FV1000 scanning confocal microscope. Atto 565 dissolved in MOPS buffer was used to identify those wells that were successfully sealed by a membrane. A 488 nm laser was used to excite the fluorescence from CF-PE and a 35 mW 405 nm laser was used to bleach a hole with diameter around 30 μm.
Antibody Conjugation
Anti-biotin antibody was first cross-linked with propargyl-PEG-NHS ester and then linked to DNA/DNA-N3-coated particles through Cu+-catalyzed azide–alkyne click chemistry following established procedures (Figure S3a).[42] A solution of propargyl-PEG-NHS ester (2 μL, 100 mg mL-1 dissolved in DMSO) was mixed with biotin antibody solution (100 μL, 0.5 mg mL−1) in 1x phosphate-buffered saline (PBS, pH 7.2) in an ice bath for 6 h. The excess propargyl-PEG-NHS ester was then removed by running the solution through a size exclusion column (7k MWCO) twice. The resulting propargyl-PEG-biotin antibody solution (25 μL) was incubated with ssDNA/PEG-modified gold NPs (500 μL) in 0.5× PBS buffer in the presence of a click chemistry catalyst (100 μM ascorbic acid and 20 μM CuSO4) overnight at 4 °C. The resulting antibody-conjugated nanoparticles (Ab-DNA-NPs) were washed three times by centrifugation and then stored at 4 °C for no more than 3 days before use.
Particle Preparation
Commercial citrate-stabilized 40 nm gold NPs (0.15 nM) were concentrated by a factor of 50 by centrifugation. Then Tween 20 solution (8 μL, 2.5% w/w) was added to the NP solution (80 μL, 7.5 nM) and incubated for 30 min. After that, 100 μM HS-DNA-N3/HS-DNA mixture (5 μL, 30:70 mol%) was added and the particle solution was incubated for 2 h. Then x μL of y Msolution of NaCl were added in individual steps of (x, y): (3, 0.4); (10, 0.4); (10, 0.4); (10, 2); (15, 2). At least 2 h of incubation time was applied between each step. After a final incubation overnight, the particles were washed with water four times by centrifugation and resuspension and finally resuspended in MOPS buffer. The formation of a DNA brush surrounding the NPs was monitored by dynamic light scattering (DLS; Figure S3b). PEG-modified NPs were obtained by incubating HS-PEG-N3 aqueous solution (5μL,10 mM) with citrate-stabilized 40 nm gold NPs (1 mL, 0.15 nM) overnight. The NPs were then washed by repeated (3×) centrifugation and resuspension in water and finally resuspended in the buffer of choice.
Nanoprism Synthesis and Functionalization
All glassware was incubated in aqua regia overnight followed by extensive rinsing with soap solution and DDI water before use.
The NPs were prepared in a two-step process by reducing aqueous tetrachloroauric acid (HAuCl4) with sodium sulfide (Na2S). First, aqueous Na2S solution (10 mL, 1 mM) aged for 24 h was added to HAuCl4 solution (10 mL, 2 mM) under vigorous stirring. After 5 min more Na2S solution (3.2 mL) was added and the reaction mix was kept stirring at room temperature. The color of the solution changed from dark yellow to wine red in the first 20 min. After stirring for 4 h the particles were subsequently PEGylated with Acid PEG (HSC11H22(OC2H4)6OCH2COOH). The Acid PEG solution (100 μL, 10 mM) was added to the reaction mixure (23 mL) and incubated overnight. The particles were then loaded into a 1% agarose gel and run at a constant voltage of 170 V for 25 min using 0.5x Tris-borate-EDTA (TBE) as running buffer. The individual bands were cut out of the gels and the particles were recovered from the gel by electro-elution. The functionalization of the isolated nano-prisms with antibodies followed the same procedures as described for the spherical NPs.
Dark-field Microscopy
Dark-field experiments were performed with an Olympus IX71 inverted microscope (Figure 2b). A xenon lamp (λ = 380–720 nm, Agilent Polychrome 3000) was used as illumination source. Incident light was filtered by a long-pass filter (>530 nm) to prevent bleaching of CF. The light scattered from the sample was collected with a 60× air objective lens (Olympus LUCPLFLN, NA = 0.7). The signal was further magnified by a 1.6× lens and collected by electron-multiplying charge coupled devices (EMCCDs). We used Andor IxonEM+ detectors with a maximum detection area of 128 × 128 pixels and a pixel size of 30 μm × 30 μm.
NP Tracking
All data analyses were performed with Matlab software. The particle locations in each frame were obtained by fit-ting a 9 × 9 pixel window around each scatterer with a Gaussian function: , where A is the amplitude of signal intensity and B is the image background. NPs with signal intensities larger than ten times higher than that of the average for single NPs were excluded from the analysis. The peak coordinates of the Gaussian fits in individual frames were generated by linking the NP coordinates in subsequent frames of movies containing at least 100 frames. The diffusion coefficient (D) of each trajectory was calculated using the equation 〈Δr2〉 = 4Dt. The number of mean square displacement (MSD) points (Pmin) required for accurate diffusion coefficient calculation was determined as Pmin = E(2 + 2.7x0.5), , where σ is the localization uncertainty, D is the diffusion coefficient, Δt is the frame duration, and E represents the floor function.[58] Pmin depends on the s/n ratio and, thus, on the temporal resolution. At 100 Hz Pmin = 2 applies to 100% of the trajectories, and at 500 Hz 96.2% still have Pmin = 2, which underlines the superb s/n provided by the gold NPs. For higher frame rates Pmin increased. We used the first ten frames to fit D throughout.
Supplementary Material
Acknowledgments
We thank Yan Hong and Bo Yan for providing the nanoprisms and Paul Mak for his assistance with the BLM array fabrication. We are also grateful to Guoxin Rong for valuable discussions. The work was supported by the National Institutes of Health through grant 5R01CA138509.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.Davis ME, Chen Z, Shin DM. Nat Rev Drug Discov. 2008;7:771. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 2.Gao XH, Cui YY, Levenson RM, Chung LWK, Nie SM. Nat Biotechnol. 2004;22:969. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 3.Jain PK, Huang XH, El-Sayed IH, El-Sayed MA. Acc Chem Res. 2008;41:1578. doi: 10.1021/ar7002804. [DOI] [PubMed] [Google Scholar]
- 4.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Proc Natl Acad Sci USA. 2003;100:13549. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duncan R, Izzo L. Adv Drug Deliv Rev. 2005;57:2215. doi: 10.1016/j.addr.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 6.Verma A, Uzun O, Hu Y, Hu Y, Han HS, Watson N, Chen S, Irvine DJ, Stellacci F. Nat Mater. 2008;7:588. doi: 10.1038/nmat2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao HJ, Shi WD, Freund LB. Proc Natl Acad Sci USA. 2005;102:9469. doi: 10.1073/pnas.0503879102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S. Adv Mater. 2009;21:419. doi: 10.1002/adma.200801393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carney RP, Carney TM, Mueller M, Stellacci F. Biointerphases. 2012;7:17. doi: 10.1007/s13758-011-0017-3. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Li X, Li Z, Gao H. Nanoscale. 2012;4:3768. doi: 10.1039/c2nr30379e. [DOI] [PubMed] [Google Scholar]
- 11.Roiter Y, Ornatska M, Rammohan AR, Balakrishnan J, Heine DR, Minko S. Nano Lett. 2008;8:941. doi: 10.1021/nl080080l. [DOI] [PubMed] [Google Scholar]
- 12.Verma A, Stellacci F. Small. 2010;6:12. doi: 10.1002/smll.200901158. [DOI] [PubMed] [Google Scholar]
- 13.Lin WC, Yu CH, Triffo S, Groves JT. Current Protocols in Chemical Biology. 2010;2:235. doi: 10.1002/9780470559277.ch100131. [DOI] [PubMed] [Google Scholar]
- 14.Kiessling V, Crane JM, Tamm LK. Biophys J. 2007;92:698. doi: 10.1529/biophysj.106.091421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray DH, Tamm LK, Kiessling V. J Struct Biol. 2009;168:183. doi: 10.1016/j.jsb.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson JR, Heron AJ, Santoso Y, Wallace MI. Nano Lett. 2007;7:3875. doi: 10.1021/nl071943y. [DOI] [PubMed] [Google Scholar]
- 17.Jain MK, editor. Introduction to Biological Membranes. John Wiley & Sons; New York: 1988. [Google Scholar]
- 18.Yetukuri L, Ekroos K, Vidal-Puig A, Oresic M. Mol Biosyst. 2008;4:121. doi: 10.1039/b715468b. [DOI] [PubMed] [Google Scholar]
- 19.Montal M, Mueller P. Proc Natl Acad Sci USA. 1972;69:3561. doi: 10.1073/pnas.69.12.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mueller P, Rudin DO, Tien HT, Wescott WC. Nature. 1962;194:979. doi: 10.1038/194979a0. [DOI] [PubMed] [Google Scholar]
- 21.Fahey PF, Webb WW. Biochemistry. 1978;17:3046. doi: 10.1021/bi00608a016. [DOI] [PubMed] [Google Scholar]
- 22.Sonnleitner A, Schutz GJ, Schmidt T. Biophys J. 1999;77:2638. doi: 10.1016/S0006-3495(99)77097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganesan PV, Boxer SG. Proc Natl Acad Sci USA. 2009;106:5627. doi: 10.1073/pnas.0901770106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki H, Le Pioufle B, Takeuhci S. Biomed Microdevices. 2009;11:17. doi: 10.1007/s10544-008-9205-4. [DOI] [PubMed] [Google Scholar]
- 25.Liu B, Rieck D, Van Wie BJ, Cheng GJ, Moffett DF, Kidwell DA. Biosens Bioelectron. 2009;24:1843. doi: 10.1016/j.bios.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirano-Iwata A, Aoto K, Oshima A, Taira T, Yamaguchi R-t, Kimura Y, Niwano M. Langmuir. 2010;26:1949. doi: 10.1021/la902522j. [DOI] [PubMed] [Google Scholar]
- 27.Ota S, Suzuki H, Takeuchi S. Lab Chip. 2011;11:2485. doi: 10.1039/c1lc20334g. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki H, Tabata K, Kato-Yamada Y, Noji H, Takeuchi S. Lab Chip. 2004;4:502. doi: 10.1039/b405967k. [DOI] [PubMed] [Google Scholar]
- 29.Zagnoni M, Sandison ME, Marius P, Lee AG, Morgan H. Lab Chip. 2007;7:1176. doi: 10.1039/b703818f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shao C, Kendall EL, DeVoe DL. Lab Chip. 2012;12:3142. doi: 10.1039/c2lc40077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Funakoshi K, Suzuki H, Takeuchi S. Anal Chem. 2006;78:8169. doi: 10.1021/ac0613479. [DOI] [PubMed] [Google Scholar]
- 32.Malmstadt N, Nash MA, Purnell RF, Schmidt JJ. Nano Lett. 2006;6:1961. doi: 10.1021/nl0611034. [DOI] [PubMed] [Google Scholar]
- 33.Yguerabide J, Yguerabide EE. Anal Biochem. 1998;262:137. doi: 10.1006/abio.1998.2759. [DOI] [PubMed] [Google Scholar]
- 34.Yguerabide J, Yguerabide EE. Anal Biochem. 1998;262:157. doi: 10.1006/abio.1998.2760. [DOI] [PubMed] [Google Scholar]
- 35.Yguerabide J, Yguerabide EE. J Cell Biochem Suppl. 2001;(Suppl 37):71. doi: 10.1002/jcb.10077. [DOI] [PubMed] [Google Scholar]
- 36.Sau TK, Rogach AL. Adv Mater. 2010;22:1781. doi: 10.1002/adma.200901271. [DOI] [PubMed] [Google Scholar]
- 37.Tao AR, Habas S, Yang PD. Small. 2008;4:310. [Google Scholar]
- 38.Ziegler C, Eychmueller A. J Phys Chem C. 2011;115:4502. [Google Scholar]
- 39.Beerlink A, Thutupalli S, Mell M, Bartels M, Cloetens P, Herminghaus S, Salditt T. Soft Matter. 2012;8:4595. [Google Scholar]
- 40.White SH. Biophys J. 1972;12:432. doi: 10.1016/S0006-3495(72)86095-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujiwara H, Fujihara M, Ishiwata T. J Chem Phys. 2003;119:6768. [Google Scholar]
- 42.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Boriskina SV, Wang HY, Reinhard BM. ACS Nano. 2011;5:6619. doi: 10.1021/nn202055b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson P. Biological Physics: Energy, Information Life. Freeman; New York: 2003. [Google Scholar]
- 45.Ferrari R, Manfroi AJ, Young WR. Phys D. 2001;154:111. [Google Scholar]
- 46.Castellana ET, Cremer PS. Surf Sci Rep. 2006;61:429. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamm LK, McConnell HM. Biophys J. 1985;47:105. doi: 10.1016/S0006-3495(85)83882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gray RM. Probability, Random Processes, and Ergodic Properties. Springer; New York: 1988. [Google Scholar]
- 49.Weigel AV, Simon B, Tamkun MM, Krapf D. Proc Natl Acad Sci USA. 2011;108:6438. doi: 10.1073/pnas.1016325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee GM, Ishihara A, Jacobson KA. Proc Natl Acad Sci USA. 1991;88:6274. doi: 10.1073/pnas.88.14.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fischer TM, Dhar P, Heinig P. J Fluid Mech. 2006;558:451. [Google Scholar]
- 52.Brenner H, Leal LG. J Colloid Interface Sci. 1982;88:136. [Google Scholar]
- 53.Brenner H, Leal LG. J Colloid Interface Sci. 1978;65:191. [Google Scholar]
- 54.Wang B, Zhang L, Bae SC, Granick S. Proc Natl Acad Sci USA. 2008;105:18171. doi: 10.1073/pnas.0807296105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tinland B, Pluen A, Sturm J, Weill G. Macromolecules. 1997;30:5763. [Google Scholar]
- 56.Chen H, Meisburger SP, Pabit SA, Sutton JL, Webb WW, Pollack L. Proc Natl Acad Sci USA. 2012;109:799. doi: 10.1073/pnas.1119057109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee H, Venable RM, MacKerell AD, Jr, Pastor RW. Biophys J. 2008;95:1590. doi: 10.1529/biophysj.108.133025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Michalet X. Phys Rev E. 2011;83:059904. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.