

Abstract
A strategy employing a combination of peptide nucleic acid (PNA) probes, an optically amplifying conjugated polymer (CP), and S1 nuclease enzyme is capable of detecting SNPs in a simple, rapid, and sensitive manner. The recognition is accomplished by sequence-specific hybridization between the uncharged, fluorescein-labeled PNA probe and the DNA sequence of interest. After subsequent treatment with S1 nuclease, the cationic water soluble CP electrostatically associates with the remaining anionic PNA/DNA complex, leading to sensitized emission of the labeled PNA probe via FRET from the CP. The generation of fluorescent signal is controlled by strand-specific electrostatic interactions and is governed by the complementarity of the probe/target pair. To assess the method, we compared the ability of the sensor system to detect normal, wild-type human DNA sequences, and those sequences containing a single base mutation. Specifically, we examined a PNA probe complementary to a region of the gene encoding the microtubule associated protein tau. The probe sequence covers a known point mutation implicated in a dominant neurodegenerative dementia known as frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), which has clinical and molecular similarities to Alzheimer's disease. By using an appropriate PNA probe, the conjugated polymer poly[(9,9-bis(6′-N,N,N-trimethylammoniumhexylbromide)fluorene)-co-phenylene] and S1 nuclease, unambiguous FRET signaling is achieved for the wild-type DNA and not the mutant sequence harboring the SNP. Distance relationships in the CP/PNA assay are also discussed to highlight constraints and demonstrate improvements within the system.
Keywords: biosensor, energy transfer, hybridization probes, polyelectrolyte, fluorescence
The largest degree of sequence variation in human DNA is attributed to SNPs, occurring as often as every few hundred to few thousand base pairs in genomic DNA (1–3). The perceived concept of personalized medicine is rooted in the identification of these unique polymorphisms, providing opportunities in both the diagnosis and treatment of disease. Furthermore, SNP's have become an ever increasing concern in the process of drug discovery, forensics, and agriculture.
SNP detection and genotyping assays are also of fundamental importance in the identification of numerous genetic and hereditary diseases. Although these genetic alterations are not necessarily harmful, when such mutations disrupt or alter the coding of amino acids, protein dysfunction can result, often leading to disease. One well known example is the A → T (Glu → Val) point mutation in human β-globin gene leading to sickle cell anemia (4). Another important collection of recently identified SNPs are in the gene encoding the microtubule associated protein tau (5). These SNPs (tauopathies) segregate genetically with frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), a neurodegenerative disease similar in many ways to Alzheimer's disease, characterized by neuronal cell death, abnormal tau pathology, and dementia. Recent studies demonstrate unequivocally that each of the tau SNPs is causative for the disease (6–8).
Automated DNA sequencers provide invaluable information in SNP and genotyping studies; however, these methods are often time and instrumentation intensive More classic methods of detection, such as restriction fragment length polymorphism and mobility shift gel analysis, are limited by cumbersome protocols requiring specific optimization, as well as mutation position and target conformation constraints that may limit the detection range (9–13). Integrating high-throughput screening with standard laboratory techniques for SNP validation testing is highly desirable but logistically and technically challenging (12).
Fluorescently labeled allele- or SNP-specific hybridization probes are common reagents used to satisfy the need for rapid and reliable detection of DNA point mutations. Typical probes comprise a short recognition sequence and two fluorophores whose spatial separation is governed by strand-specific hybridization. One unit serves as the reporting element and is modulated (turned on/off) by the proximity of the second. Examples include stem-loop molecular beacons and TaqMan-type probes, which are of current interest in real-time SNP screening (14, 15). Although probes like molecular beacons offer excellent selectivity (16), their effectiveness is often limited by the sensitivity of the fluorescent reporter. Additionally, the need for multiple probe modifications increases the complexity of probe design and cost.
Recently, a collective response method for DNA detection that provides amplified levels of hybridization probe sensitivity was reported (17, 18). The assay introduced the use of cationic water-soluble conjugated polymers as light-harvesting additions to standard, singly modified hybridization probes [peptide nucleic acid (PNA) or DNA] by means of FRET. The conjugated polymers (CPs) may be envisioned as large, delocalized molecular structures comprised of a series of conjugated segments or optical units held in close proximity by virtue of a polymer backbone. Energy migration within the structure is facile (19–22), allowing photoexcitations to ultimately reside in a small number of low-energy sites (23–25). This collective behavior gives rise to optical enhancement or amplification if the low-energy site comprises a reporter fluorophore. The water solubility of these macromolecules is achieved by attaching ionic side chains pendent to the otherwise hydrophobic main chain, whereas the spectral properties are tuned by altering the actual conjugated backbone structure (26–31).
Scheme 1 illustrates how the CP detection method takes advantage of the optical amplification provided by CPs (32–35) and interactions typical of polyelectrolytes (36, 37). A neutral peptide nucleic acid labeled with a 5′ fluorescein (FL) acceptor (PNA-FL) is used to probe an unknown DNA target. The probe acquires negative charges (from DNA) only upon strand-specific PNA/DNA hybridization. The result is an attractive electrostatic force between the positively charged CP and the complementary PNA/DNA duplex. This electrostatic force brings the two within close proximity and satisfies the distance requirement for FRET (38, 39) from the CP to the PNA-FL probe. Noncomplementary DNA targets will not form the negatively charged duplex and little attraction is expected between the neutral PNA-FL and cationic CP antenna. Overall, the system provides elevated fluorescent signals and eliminates the need for dual probe modification while maintaining high sequence specificity (17, 18).
Scheme 1.

Conjugated polymer (CP) and PNA-FL probe assay for sequence specific DNA detection.
Materials and Methods
Testing Materials. Reagents. PAGE-purified DNA oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA), and the concentrations were determined by using 260-nm absorbance measurements in 200-μl quartz cells in a Beckman DU800 spectrophotometer. Concentrations of the larger DNA sequences obtained by PCR and gel purification were measured by fluorescence using an OliGreen single stranded DNA (ssDNA) Quantitation kit (Molecular Probes). The HPLC-purified PNA-FL (5′-FL-OO-TCCACGGCATCTCA-EE) was purchased from Applied Biosystems (Foster City, CA) and used as received (mismatch site is shown in bold italics). Water used was distilled and deionized by using a Millipore filtration system. The S1 enzyme was obtained from Promega, and all reactions were carried out in Promega S1 reaction buffer.
PCRs. All PCRs were done in a PerkinElmer Cetus DNA Thermal Cycler. The wild-type and mutant (R406W) sequences were generated by using forward (5′-GTCGCGGGATCCAATAAAAAGATTGAAACCCACAAGCTGA-3′) and reverse (5′-GATCCGGA AT TCTCACA A ACCCTGCT TGGCCA-3′) primers at the C terminus, and a sequence noncomplementary to the PNA probe was generated by using forward (5′-GCCTGGAAGACGAAGCTGCTGGTCACG-3′) and reverse (5′-GCTGCGATCCCCTGATTTTGGAGGTTCACCAGAG-3′) primers near the N terminus of the tau DNA sequence. The one-base discrepancy between C and N terminus PCR products was a result of primer optimization. High-fidelity PfuTurbo DNA Polymerase (Stratagene) was used to minimize PCR-induced mutations. Reactions (100 μl) were carried out by using 2.5 units of Pfu and 200 μM dNTPs. Thermocycling parameters and primer, template, and dNTP concentrations were all adjusted and optimized by analyzing reaction products by gel electrophoresis.
dsDNA templates. The human wild-type tau cDNA sequence encoding the longest four-repeat (4R2N wt; 441 aa) isoform in the prK expression vector was a gift from Ken Kosik (University of California, Santa Barbara). The R406W mutation was introduced by site-directed mutagenesis (Stratagene). All constructs were sequenced before use (Iowa State Sequencing Facility, Ames). Standard (symmetric) PCR was performed to generate dsDNA templates for subsequent asymmetric PCRs. The resulting amplicons were excised from a 1.2% agarose gel and purified by using a Qiagen QIAquick Gel Extraction kit (Valencia, CA). Purity was confirmed on a 1.2% agarose gel stained with ethidium bromide and imaged with an Ultraviolet Products EPI Chemi Darkroom and UV transilluminator.
ssDNA targets. Asymmetric PCR was done by using a 1:25 ratio of forward (0.02 μM) to reverse (0.5 μM) primer and 10 ng of dsDNA template for 40 cycles. The asymmetric PCR products were loaded on a 3% MetaPhor agarose gel run at constant voltage (110 V) for 1.5 h and stained with ethidium bromide. ssDNA was extracted and purified by using a Qiagen QIAquick Gel Extraction kit. Samples were eluted from the purification columns in 1 mM Tris·Cl buffer (pH 8.5), and the purity was confirmed by using 1-μl samples on an Agilent (Palo Alto, CA) 2100 Bioanalyzer and RNA 6000 Nano LabChip kit.
Annealing Procedures. Annealing of DNA targets was performed by mixing equal molar quantities (moles of chains) of target DNA with the FL-labeled PNA probe sequence (PNA-FL). Oligomer and larger DNA targets were heated at 70°C and 90°C, respectively, for 10 min and slowly cooled to room temperature. All hybridization and S1 enzyme reactions were done in a 1 mM sodium acetate buffer, pH 6.5.
S1 Reactions and Denaturing Gels. To all S1 reactions, 1.25× Promega S1 reaction buffer (500 mM sodium acetate/2.8 M sodium chloride/45 mM zinc sulfate, pH 4.5) was added to the annealed PNA/DNA sample mixtures. Digestion reactions were carried out at [ssDNA/PNA-FL] = 2 × 10-6 M for oligomers and [ssDNA/PNA-FL] = 1 × 10-6 M for the longer DNA targets and at [S1] = 0.75 and 2.5 units/μl, respectively. After 60 min at 37°C, all reactions were stopped by addition of 10 mM EDTA (pH 8) and placed on ice. Samples for gel electrophoresis were diluted 1:1 with TBE–urea sample buffer (Bio-Rad; 89 mM Tris·HCl/89 mM boric acid/2 mM EDTA/7 M urea/12% Ficoll/0.01% Bromophenol Blue/0.02% Xylene Cyanol FF) and loaded on a 20% acrylamide urea denaturing gel. Gels were run at a constant power of 30 W for 60 min in TBE buffer (pH 8.3). Samples were heated to 90°C for 10 min before loading on a warmed gel that had been prerun for 30 min. Gels were stained with Sybr Gold (Molecular Probes) for 45 min and imaged directly out of the stain by using either an Ultraviolet Products EPI Chemi Darkroom and UV transilluminator or a Bio-Rad Molecular Imager FX. Single-stranded 14-base DNA samples were observable by using 300-nm excitation at loading levels as low as 50 ng.
Fluorescence Experiments. Fluorescence was measured by using a PTI (South Brunswick, NJ) Quantum Master fluorometer equipped with a xenon lamp excitation source and a Hamamatsu (Japan) 928 PMT and 3 ml plastic cuvettes for all DNA targets at a fixed concentration. Oligomer DNA was tested with 2-ml sample volumes and longer sequences with 1- to 0.725-ml sample volumes. Successive additions of CP were done to determine optimum ratios of FRET. Samples were gently agitated for 2–3 s between each fluorescence measurement (after each addition of polymer). Fluorescence intensities were determined as the integrated area of the emission spectra and FRET ratios were calculated as the area of the acceptor (FL) over the area of the donor (CP). The data were normalized with respect to the CP emission to highlight differences in energy transfer as a function of target sequence. Experimentally, this is done by monitoring the maximum intensity of both the donor and acceptor at the corresponding wavelengths (420 nm and 525 nm, respectively). Representative raw spectra are given in supporting information, which is published on the PNAS web site. The buffer used for all spectroscopy was 30 mM potassium phosphate, pH 7.4.
Results and Discussion
As an initial test of the ability of the CP/PNA assay to distinguish between normal nucleic acid sequences (wild type) and those harboring SNPs, we screened sequences encoding fragments of either wild-type tau or the identical sequences containing an SNP near the C terminus of the protein. This mutation results in an arginine to tryptophan substitution at amino acid position 406 (R406W or RW) in the microtubule-associated protein tau. The mutant version of the protein, known to cause frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), displays a diminished ability to assemble microtubules and is characterized by abnormal levels of phosphorylation (6, 40).
Double-stranded test sequences were generated by amplifying a region of tau DNA spanning the desired mutation site. Plasmids containing both the wild-type and mutant (RW) tau DNA were used as templates for generating 249-bp amplicons by PCR. A 248-bp sequence from the N terminus of the tau DNA was also generated as a noncomplementary, negative control target. Asymmetric PCRs were performed by using the wild-type, RW, and N-terminal double-stranded templates to generate single-stranded targets for evaluation. Optimized reaction products for all three sequences were purified by extraction from a 3% MetaPhor agarose gel where distinct single- and double-stranded bands were clearly resolved and confirmed by capillary gel electrophoresis. A 14-base PNA probe sequence was designed to test for the R406W SNP mismatch. The probe contained a 5′ FL modification and two 3′ solubility enhancing groups (Scheme 2).
Scheme 2.

PNA-FL probe, CP (poly[(9,9-bis(6′-N,N,N-trimethylammoniumhexylbromide)fluorene)-co-phenylene]) and 28-base DNA oligomer targets used (D0–D3), with mismatches relative to the PNA highlighted in tan. The R406W mutation position within the PNA probe is shown in green. The D0 sequence represents the perfect probe complement, D1 sequence represents the single R406W point mutation, the D2 sequence represents a 2-base mutation, and the D3 sequence represents a random noncomplementary sequence.
An equimolar quantity of the PNA probe (Scheme 2) was mixed with each of the three ssDNA sequences in 1 mM sodium acetate, and these solutions were annealed at 90°C. As the degree of complementarity between the PNA probe and DNA target is reduced, so is the stability of the resulting duplex. The mutated (mismatched) DNA targets are expected to display a diminished FL signal response according to the sensor system depicted in Scheme 1, because the resulting anionic PNA/DNA duplexes are less stable. Furthermore, it should be noted that PNA oligonucleotides are excellent candidates for SNP detection probes because the PNA/DNA duplexes formed can be nearly twice as destabilized by mismatches, relative to natural DNA/DNA complexes (41). The PNA/DNA duplexes are also much less sensitive to the ionic strength of the solution (42).
The annealed samples were diluted to [PNA/ssDNA] = 1 × 10-8 M in a phosphate buffer and treated with 4 × 10-7 M (in monomer repeat units) of the CP (poly[(9,9-bis(6′-N,N,N-trimethylammoniumhexylbromide)f luorene)-co-phenylene], Scheme 2). Photoexcitation of the CP at room temperature produces a FRET response sensitive to the complementarity of the PNA probe and the target DNA sequence. The results, normalized to the CP emission (400–500 nm), are shown in Fig. 1. One can discriminate between the wild-type and SNP-containing mutant DNA based on the relative FL emission intensity (500–600 nm). That the noncomplementary (N-terminal) sequence shows no evidence of FRET is consistent with the action described in Scheme 1 and highlights the assay's ability to discern matching from nonmatching target sequences (17).
Fig. 1.

Fluorescence analysis of full-length ssDNA targets. (A) PNA-FL probe (red) annealed to asymmetric PCR products, with mutations indicated as tan segments in the black DNA sequence, for fluorescence analysis according to Scheme 1. (B) Normalized fluorescence of PNA-FL/ssDNA upon addition and excitation (λex = 380 nm) of the CP ([CP] = 4 × 10-7 M and [PNA/ssDNA] = 1 × 10-8 M) in 30 mM potassium phosphate buffer, pH 7.4, at room temperature. The black curve corresponds to the complementary (wild type) DNA, and the red and blue curves correspond to the single mismatch (RW) and noncomplementary (N-term) DNA targets, respectively.
Fig. 2 demonstrates the amplification provided by the light harvesting conjugated polymer. Comparing the emission intensity upon direct FL excitation (λex = 480 nm) to the sensitized FL emission by excitation of the CP in the energy transfer complex (DNA/PNA-FL/CP) reveals an ≈1 order of magnitude increase in optical signature.
Fig. 2.

Signal amplification of the PNA-FL hybridization probe. The signal intensity of FL generated from direct excitation (λex = 480 nm), shown in red, compared to the signal generated from the same FL reporter in the CP energy transfer complex (λex = 380 nm), shown in black ([CP] = 1 × 10-6 M and [PNA/WT ssDNA] = 1 × 10-8 M). The test was performed in 30 mM potassium phosphate buffer, pH 7.4.
Analogous experiments were conducted by using shorter 28-base tau DNA oligonucleotide targets (Scheme 2). The selected targets include the R406W mutation site (D0 and D1), a two-base mismatch (D2) sequence, and a randomly generated noncomplementary sequence (D3). After annealing the four targets with the PNA probe, a concentrated CP solution was added and the FRET signatures were evaluated (Fig. 3B). The resulting spectra allow one to distinguish the four different DNA targets based on PNA probe complementarity.
Fig. 3.

Fluorescence analysis of ssDNA oligomer targets. (A) PNA-FL probe (red) annealed to 28-base oligomers (black) for fluorescence analysis according to Scheme 1 with mutations shown as tan segments/loops in the DNA targets. Sequences D0–D3 range from perfectly complementary, one base mismatch, two base mismatch, and noncomplementary, respectively. (B) Normalized fluorescence of PNA-FL/ssDNA (D0–D3, black, red, blue, and green, respectively) upon addition and excitation (λex = 380 nm) of the CP ([CP] = 2.5 × 10-7 M and [PNA/ssDNA] = 1 × 10-8 M) in 30 mM potassium phosphate buffer, pH 7.4.
Comparison of the 28- and 249-base DNA targets, in terms of the ratio of FL signal relative to the residual CP emission, reveals a loss in signal to background (500–600 nm) for the larger target (Fig. 4). This disparity is related to distance relationships in the Förster energy transfer process resulting from size differences between the target DNA and CP.
Fig. 4.

Fluorescence of PNA-FL annealed to complementary 28 (black, D0) and 249 (red, WT) base ssDNA targets ([PNA/ssDNA] = 1 × 10-8 M) upon addition and excitation (λex = 380 nm) of the CP ([CP] = 2 × 10-7 M) in 30 mM potassium phosphate buffer, pH 7.4. The noncomplementary (28 base, D3) DNA target is shown in blue for reference. The arrow indicates the drop in the relative FL-to-CP emission peak ratio between the two different DNA target lengths.
From the Förster equation (Eq. 1), the donor/acceptor separation distance (r) at which the rate of energy transfer, kT(r), equals the rate of donor emission, 1/τD, is referred to as the Förster radius or Ro (38).
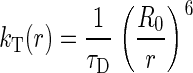 |
[1] |
A recent report from Heeger et al. (43) estimated this distance for the same CP structure and a similar PNA-FL/DNA acceptor complex by using the steady state fluorescence and absorption spectra of the donor–acceptor pair. Experimental separation distances were reported for time-resolved spectroscopy measurements and provided values within the estimated Ro of 37.2 Å. These measurements confirm that for short, oligomeric DNA targets, the rate of transfer is faster than polymer emission and, thus, effective energy transfer from donor to acceptor is observed (17, 18).
Because the energy transfer process shows strong distance dependence, it is important to consider the relative length scales of the target DNA and cationic CP. Based on the molecular weight of the polymer (Mw ≈ 13,000 g/mole, ≈13 Å per repeat unit), it is evident that the CP is longer than the oligonucleotide (15–28 base) target sequences tested previously (27–31, 44). Complex formation yields an acceptor (PNA-FL/DNA duplex) with a strong contact along the longer CP chain and a small donor/acceptor separation distance (Scheme 3A). Structural relationships change when the DNA target length exceeds the length of the CP. The nonspecific electrostatic interactions responsible for the polymer/DNA complex will favor a random distribution of polymer chains along the longer DNA target (Scheme 3B) and thus a mixture of different donor/acceptor distances. Any polymer chains that associate outside of the Ro distance will be more likely to emit absorbed photons by their normal radiative pathway leading to a higher CP-to-FL intensity ratio and thus a stronger background signal. Ultimately, when comparing different target lengths, the signal-to-background is reduced as the length of DNA increases.
Scheme 3.

Cationic CPs (blue) have the ability to associate nonspecifically along any region of a targeted ssDNA sequence (black). (A) Association of a short DNA target with the CP. (B) The probability of CP association along a longer DNA target. CPs complexed at distances greater than Ro from the FL acceptor will be less efficient donors to the PNA-FL probe (red and green).
In response to the assay's limitation toward longer DNA sequences and in an effort to improve SNP selectivity, we sought to specifically reduce DNA target lengths by using common nuclease enzymes. Given that PNA molecules are inherently resistant to nucleases and proteases (45), the protocol was modified to include an S1 nuclease digestion step after the PNA/DNA annealing reaction. In principle, this commonly used single strand-specific nuclease should digest all nonannealed ssDNA, overlapping ssDNA segments that extend beyond the region of the PNA/DNA duplex, and any unbound mismatches within a duplex (46–49).
The use of the S1 nuclease step was first examined by using the oligonucleotide sequences in Scheme 2. The PNA probe was annealed to all four targets in 1 mM sodium acetate, pH 6.5. The enzyme and appropriate buffer were then added to the annealed PNA/DNA solutions, incubated at 37°C, and eventually quenched with 10 mM EDTA. The overall process is shown in Scheme 4 with the spectroscopic and gel analysis provided in Fig. 5.
Scheme 4.

Long ssDNA target sequences (black) are digested by S1 nuclease, leaving intact only those regions bound to the PNA probe (red). CPs (blue) added directly to the resulting solutions can only associate with the remaining PNA-FL/DNA duplex. Any PNA/DNA mismatches will result in complete DNA digestion; therefore, energy transfer from the CP occurs only for the perfect PNA-FL/DNA complement.
Fig. 5.

Fluorescence analysis of ssDNA oligomer targets after enzymatic digestion. (A) PNA-FL was hybridized to oligomer sequences D0–D3, and then digested with S1 nuclease. The 28- and 14-base size standards (S) correspond to the starting D0 sequence and the predicted 14-base product, respectively. The denaturing polyacrylamide gel was visualized via Sybr Gold staining (λex = 365 nm) and the band of interest is indicated by the red asterisk. (B) Normalized fluorescence of S1 treated hybridization products (D0–D3). Spectra were obtained (λex = 380 nm) by adding CP directly to the quenched reaction mixtures diluted ([CP] = 1.5 × 10-7 M and [PNA/ssDNA] = 1 × 10-8 M) in 30 mM potassium phosphate buffer, pH 7.4.
The denaturing polyacrylamide gel in Fig. 5A, visualized with Sybr Gold stain (50), shows complete digestion of both mismatched sequences (D1 and D2) and the noncomplementary sequence (D3) relative to 14- and 28-base ssDNA standards. The lane corresponding to the perfect DNA complement (D0) indicates that the original 28-base DNA target has been reduced to the length of the PNA probe.
The action described in Scheme 4 was tested by direct addition of CP to the PNA/DNA solutions after enzymatic reaction (diluted to 10 nanomolar in a phosphate buffer at pH 7.4). Excitation of the CP (λex = 380 nm) results in selective FRET for only the perfectly complementary pair (Fig. 5B). Samples containing even one mismatch (R406W mutation) showed no evidence of energy transfer from cationic CP to neutral PNA probe, consistent with the loss of electrostatic attraction afforded by the negatively charged DNA target. Emission spectra such as those in Fig. 5B provide for straightforward discrimination of strand-specific SNP sites on short DNA target sequences.
The importance of the enzyme-resistant PNA probe was confirmed by performing the digestion step with an analogous DNA probe (less the FL label). Under the same conditions, no DNA (target or probe) was visible on the denaturing gel even for the perfect duplex pair (46). Furthermore, less enzyme, shorter times, and lower incubation temperatures are required for samples with more than a single PNA/DNA mismatch (i.e., D2 and D3).
S1 reactions were then performed with the longer PCR products (which include the actual genetic tau mutation) under the same conditions as above, except with slightly higher enzyme concentrations to account for the increased target size. Comparison of the 249-base wild-type and R406W mutant DNA on the denaturing gel shows a probe length DNA product for only the perfect complement (Fig. 6A), indicating single base resolution for the S1 enzymatic reaction on longer DNA sequences. Addition of the CP to the diluted reaction mixtures was done for fluorescence evaluation. Energy transfer resulting in intense FL emission was only observed for the wild-type DNA (Fig. 6B), providing improved signal response over those samples not treated with the S1 enzyme. The substantial increase in selectivity is clearly apparent when one compares Figs. 1 and 6B. These spectra provide unambiguous evidence of SNP identification by using the CP/PNA based detection assay with the aid of S1 nuclease.
Fig. 6.

Fluorescence analysis of full-length ssDNA targets after enzymatic digestion. (A) PNA-FL was hybridized to 249-base WT and R406W sequences, and then digested with S1 nuclease. The 28- and 14-base size standards (S) correspond to the D0 sequence and the predicted 14-base product, respectively. The denaturing polyacrylamide gel was visualized via Sybr Gold staining (λex = 488 nm), and the band of interest is indicated by the red asterisk. (B) Normalized fluorescence of S1-treated hybridization products (WT, black; R406W, red). Spectra were obtained (λex = 380 nm) by adding CP directly to the quenched reaction mixtures diluted ([CP] = 1.4 × 10-7 M and [PNA/ssDNA] = 1 × 10-8 M) in 30 mM potassium phosphate buffer, pH 7.4.
Conclusions
A general method for optically enhanced DNA detection employing conjugated polymers and fluorescently labeled PNA probe molecules has been demonstrated and applied to SNP identification. The two recognition events responsible for signal transduction are the specific base pair recognition of a target/probe pair in combination with the electrostatic interactions between the cationic light-harvesting conjugated polymer and the PNA-FL/DNA duplex. The action of a nonspecific enzyme before addition of the conjugated polymer increases both the selectivity of the overall assaying process and the range of DNA target lengths. Fluorescence tests on both DNA oligomers and fragments of human DNA generated by PCR clearly reflect the loss in duplex stability by the introduction of mismatches in the probe/target pairs. The inherent selectivity of the PNA/enzyme combination allows for unambiguous single base mismatch identification. The use of S1 enzyme also lowers the assay's background signal by reducing size discrepancies between the target DNA and CP.
Compared to other fluorescent methods, the assay in Scheme 4 eliminates the need for two signaling elements (dual modification) on the probe molecule, thereby simplifying the design and overall complexity of the fluorescent reagents. The background and sensitivity levels of such reagents can also be limited by the probe purity and by the physical properties of the appended fluorophore. It is also noteworthy that the PNA/CP system provides added sensitivity to standard single fluorophore-labeled probes without the need for complex/expensive optical setups.
Supplementary Material
Acknowledgments
We thank Dr. Monte Radeke, Michelle Staples, Dr. Pat Johnson, and Prof. Lincoln Johnson (Center for Macular Degeneration, University of California, Santa Barbara) for use of instrumentation and useful discussions. This work was supported by the Materials Research Laboratory Program of the National Science Foundation (DMR00-80034), National Institutes of Health Grants NS35010 and GM62958-01, and the Institute for Collaborative Biotechnologies.
Author contributions: B.S.G., M.R.M., and G.C.B. designed research; B.S.G. and M.R.M. performed research; M.R.M. contributed new reagents/analytic tools; B.S.G., M.R.M., S.C.F., and G.C.B. analyzed data; and B.S.G., S.C.F., and G.C.B. wrote the paper.
Abbreviations: PNA, peptide nucleic acid; CP, conjugated polymer; FL, fluorescein; ssDNA, single-stranded DNA.
References
- 1.Cooper, D. N., Smith, B. A., Cooke, H. J., Niemann, S. & Schmidtke, J. (1985) Hum. Genet. 69, 201-205. [DOI] [PubMed] [Google Scholar]
- 2.Wang, D. G., Fan, J. B., Siao, C. J., Berno, A., Young, P., Sapolsky, R., Ghandour, G., Perkins, N., Wnchester, E., Spencer, J., et al. (1998) Science 280, 1077-1082. [DOI] [PubMed] [Google Scholar]
- 3.Halushka, M. K., Fan, J. B., Bentley, K., Hsie, L., Shen, N., Weder, A., Cooper, R., Lipshutz, R. & Chakravarti, A. (1999) Nat. Genet. 22, 239-247. [DOI] [PubMed] [Google Scholar]
- 4.Wu, D. Y., Ugozzoli, L., Bal, B. K. & Wallace, R. B. (1989) Proc. Natl. Acad. Sci. USA 86, 2757-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee, V. M-Y., Goedert, M. & Trojanowski, J. Q. (2001) Annu. Rev. Neurosci. 24, 1121-1159. [DOI] [PubMed] [Google Scholar]
- 6.Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., Pickering-Brown, S., Chakraverty, S., Isaacs, A., Grover, A., et al. (1998) Nature 393, 702-705. [DOI] [PubMed] [Google Scholar]
- 7.Spillantini, M. G., Murrell J. R., Goedert, M., Farlow, M. R., Klung, A. & Ghetti, B. (1998) Proc. Natl. Acad. Sci. USA 95, 7737-7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark, L. N., Poorkaj, P., Wszolek, Z., Geschwind, D. H., Nasreddine, Z. S., Miller, B., Li, D., Payami, H., Awert, F. & Markopoulou, K. (1998) Proc. Natl. Acad. Sci. USA 95, 13103-13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Botstein, D., White, R. L., Skolnick, M. & Davis R. W. (1980) Am. J. Hum. Genet. 32, 314-331. [PMC free article] [PubMed] [Google Scholar]
- 10.Orita, M., Iwahana, H., Kanazawa, H., Hayashi, K. & Sekiya, T. (1989) Proc. Natl. Acad. Sci. USA 86, 2766-2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maxam, A. M. & Gilbert, W. (1980) Methods Enzymol. 65, 499-560. [DOI] [PubMed] [Google Scholar]
- 12.Kanazawa, H., Noumi, T. & Futai, M. (1986) Methods Enzymol. 126, 595-603. [DOI] [PubMed] [Google Scholar]
- 13.Carlson, C. S., Newman, T. L. & Nickerson, D. A. (2001) Curr. Opin. Chem. Biol. 5, 78-85. [DOI] [PubMed] [Google Scholar]
- 14.Marras, S. A., Kramer, F. R. & Tyagi, S. (1999) Genet. Anal. 14, 151-156. [DOI] [PubMed] [Google Scholar]
- 15.Livak, K .J. (1999) Genet. Anal. 14, 143-149. [DOI] [PubMed] [Google Scholar]
- 16.Bonnet, G., Tyagi, S., Libchaber, A. & Kramer, F. R. (1999) Proc. Natl. Acad. Sci. USA 96, 6171-6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaylord, B. S., Heeger, A. J. & Bazan, G. C. (2002) Proc. Natl. Acad. Sci. USA 99, 10954-10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaylord, B. S., Heeger, A. J. & Bazan, G. C. (2003) J. Am. Chem. Soc. 125, 896-900. [DOI] [PubMed] [Google Scholar]
- 19.Guillet, J. E. (1985) in Polymer Photophysics and Photochemistry (Cambridge Univ. Press, Cambridge, U.K.).
- 20.Webber, S. E. (1990) Chem. Rev. 90, 1469-1482. [Google Scholar]
- 21.Levitsky, I. A., Kim, J. & Swager, T. M. (1999) J. Am. Chem. Soc. 121, 1466-1472. [Google Scholar]
- 22.Miao, Y.-J., Herkstroeter, W. G., Sun, B. J., Wong-Foy, A. G. & Bazan, G. C. (1995) J. Am. Chem. Soc. 117, 11407-11420. [Google Scholar]
- 23.Sariciftci, N. S.; Heeger, A. J. (1997) in Handbook of Conductive Molecules and Polymers, ed. Nalwa, H. S. (Wiley, Chichester, U.K.), Vol. 1, pp. 413-455. [Google Scholar]
- 24.Wang, J., Wang, D., Miller, E. K., Moses, D., Bazan, G. C. & Heeger, A. J. (2000) Macromolecules 33, 5153-5158. [Google Scholar]
- 25.Gaylord, B. S., Wang, S., Heeger, A. J. & Bazan, G. C. (2001) J. Am. Chem. Soc. 123, 6417-6418. [DOI] [PubMed] [Google Scholar]
- 26.Shi, S. Q. & Wudl, F. (1990) Macromolecules 23, 2119-2124. [Google Scholar]
- 27.Stork, M., Gaylord, B. S., Heeger, A. J. & Bazan, G. C. (2002) Adv. Mater. 14, 361-366. [Google Scholar]
- 28.Wang, S., Liu, B., Gaylord, B. S. & Bazan, G. C. (2003) Adv. Funct. Mat. 13, 463-467. [Google Scholar]
- 29.Liu, B., Gaylord, B. S., Wang S. & Bazan, G. C. (2003) J. Am. Chem. Soc. 125, 6705-6714. [DOI] [PubMed] [Google Scholar]
- 30.Liu, B., Wang, S., Bazan, G. C. & Mikhailovsky, A. (2003) J. Am. Chem. Soc. 125, 13306-13307. [DOI] [PubMed] [Google Scholar]
- 31.Liu, B. & Bazan, G. C. (2004) J. Am. Chem. Soc. 126, 1942-1943. [DOI] [PubMed] [Google Scholar]
- 32.McQuade, D. T., Pullen. A. E. & Swager, T. M. (2000) Chem. Rev. 100, 2537-2574. [DOI] [PubMed] [Google Scholar]
- 33.McQuade, D. T., Hegedus, A. H. & Swager, T. M. (2000) J. Am. Chem. Soc. 122, 12389-12390. [Google Scholar]
- 34.Yang, J. S. & Swager, T. M. (1998) J. Am. Chem. Soc. 120, 11864-11873. [Google Scholar]
- 35.Harrison, B. S., Ramey, M. B. & Reynolds, J. R. (2000) J. Am. Chem. Soc. 122, 8561-8563. [Google Scholar]
- 36.Bronich, T. K., Nguyen, H. K., Eisenberg, A. & Kabanov, A. V. (2000) J. Am. Chem. Soc. 122, 8339-8343. [Google Scholar]
- 37.Kabanov, A. V., Felgner, P. & Seymour, L. W., eds. (1998) in Self-Assembling Complexes for Gene Delivery: From Laboratory to Clinical Trial (Wiley, Chichester, U.K.).
- 38.Lakowicz, J. R. (1999) in Principles of Fluorescence Spectroscopy (Kluwer Academic/Plenum, New York), pp. 368-394.
- 39.Förster, T. (1948) Ann. Phys. 2, 55-75. [Google Scholar]
- 40.Sahara, N., Tomiyama, T. & Mori, H. (2000) J. Neurosci. Res. 60, 380-387. [DOI] [PubMed] [Google Scholar]
- 41.Egholm, M., Buchardt, O., Christensen, L., Behrens, C., Freier, S. M., Driver, D. A., Berg, R. H., Kim S. K., Norden, B. & Nielsen, P. (1993) Nature 365, 566-568. [DOI] [PubMed] [Google Scholar]
- 42.Tomac, S., Sarkar, M., Ratilainen, T., Wittung, P., Nielsen, P. E., Nordén, B. & Gräslund, A. (1996) J. Am. Chem. Soc. 118, 5544-5552. [Google Scholar]
- 43.Xu, Q.-H., Gaylord, B. S., Wang, S., Bazan, G. C., Moses, D. & Heeger, A. J. (2004) Proc. Natl. Acad. Sci. USA 101, 11634-11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stryer, L. (1995) in Biochemistry (Freeman, New York), 4th ed., pp. 75-94.
- 45.Nielson, P. E. & Egholm, M. (1999) in Peptide Nucleic Acids: Protocols and Applications (Horizon Scientific Press, Portland, OR), pp. 1-19.
- 46.Komiyama, M., Ye, S., Liang, X., Yamamoto, Y., Tomita, T., Zhou, J.-M. & Aburatani, H. (2003) J. Am. Chem. Soc. 125, 3758-3762. [DOI] [PubMed] [Google Scholar]
- 47.Ren, B., Zhou, J.-M. & Komiyama, M. (2004) Nucleic Acids Res. 32, e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Demidov, V., Frank-Kamenetskii, M. D., Egholm, M., Buchardt, O. & Nielsen, P. E. (1993) Nucleic Acids Res. 21, 2103-2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sambrook, J. & Russell, D. (2001) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Plainview, NY), 3rd Ed., pp. A4.1-A4.52.
- 50.Tuma, R. S., Beaudet, M. P., Jin, X., Jones, L. J., Cheung, C.-Y., Yue, S. & Singer, V. L. (1999) Anal. Biochem. 268, 278-288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.