

ABSTRACT
The enrichment of H2-oxidizing bacteria (HOB) by H2 generated by nitrogen-fixing nodules has been shown to have a fertilization effect on several different crops. The benefit of HOB is attributed to their production of plant growth-promoting factors, yet their interactions with other members of soil microbial communities have received little attention. Here we report that the energy potential of H2, when supplied to soil, alters ecological niche partitioning of bacteria and fungi, with multifaceted consequences for both generalist and specialist microbial functions. We used dynamic microcosms to expose soil to the typical atmospheric H2 mixing ratio (0.5 ppmv) permeating soils, as well as mixing ratios comparable to those found at the soil-nodule interface (10,000 ppmv). Elevated H2 exposure exerted direct effects on two HOB subpopulations distinguished by their affinity for H2 while enhancing community level carbon substrate utilization potential and lowering CH4 uptake activity in soil. We found that H2 triggered changes in the abundance of microorganisms that were reproducible yet inconsistent across soils at the taxonomic level and even among HOB. Overall, H2 exposure altered microbial process rates at an intensity that depends upon soil abiotic and biotic features. We argue that further examination of direct and indirect effects of H2 on soil microbial communities will lead to a better understanding of the H2 fertilization effect and soil biogeochemical processes.
IMPORTANCE An innovative dynamic microcosm chamber system was used to demonstrate that H2 diffusing in soil triggers changes in the distribution of HOB and non-HOB. Although the response was uneven at the taxonomic level, an unexpected coordinated response of microbial functions was observed, including abatement of CH4 oxidation activity and stimulation of carbon turnover. Our work suggests that elevated H2 rewires soil biogeochemical structure through a combination of direct effects on the growth and persistence of HOB and indirect effects on a variety of microbial processes involving HOB and non-HOB.
KEYWORDS: biogeochemistry, microbial ecology, soil microbiology, trace gas
INTRODUCTION
Microorganisms are the metabolic engine of essential biogeochemical processes supplying food and natural resources to mankind, all the while playing a pivotal role in energy balance and plant, animal, and human health. Despite the fact that microbial communities often consist of microbial “dark matter” eluding cultivation efforts (1), advances in high-throughput sequencing technologies provide new means to obtain a reasonable portrait of their taxonomic composition and functional potential. Such progress has led to a very promising approach in molecular biogeochemistry, i.e., using molecular indicators in combination with environmental variables as parameters for process rate predictive models (2–5). The inclusion of microbe-microbe interactions to improve these models and harness ecosystem services provided by microbial communities is of paramount importance (6, 7).
As a ubiquitous energy source found in all compartments of the biosphere, molecular hydrogen (H2) has been selected as a case study to infer potential outcomes of stimulating a specialized guild of microbes for other processes in soil. An H2 mixing ratio of 0.53 ppmv is typically found in the global atmosphere (8), which then diffuses into surface soils (9), whereas hot spots can be found in hypersaline cyanobacterial mats, with H2 concentrations between 16,000 and 90,000 ppmv (10), and inside N2-fixing legume nodules, with concentrations ranging between 9,000 and 27,000 ppmv (11–14). Indeed, H2 is produced at a rate of 240,000 liters/hectare per growing season in legume fields as an obligate by-product of biological nitrogen fixation (15). It is then quickly scavenged by aerobic H2-oxidizing bacteria (HOB) within the first few centimeters surrounding nitrogen-fixing nodules (16), after which its remaining concentration is slightly lower than atmospheric mixing ratios. It is assumed that a succession of HOB use this trace gas as an energy source for lithoautotrophic growth or mixotrophic growth and survival according to their affinity for H2 and regulation of hydrogenase gene expression (17). Under aerobic conditions, H2 oxidation is carried out by [NiFe]-hydrogenases that are unevenly distributed among microbial taxa. So far, Bradyrhizobium japonicum (Alphaproteobacteria) and Cupriavidus necator (Betaproteobacteria; previously Ralstonia eutropha) are the best-characterized low-affinity soil-dwelling HOB (18), while the Actinobacteria Mycobacterium smegmatis (19, 20), Rhodococcus equi (21), and Streptomyces spp. (22, 23), as well as the Acidobacteria Pyrinomonas methylaliphatogenes (24), are the best-studied high-affinity H2 oxidizers. H2 evolved from nitrogen-fixing nodules was shown to exert a fertilization effect in soil (25), likely caused by enrichment of HOB displaying plant growth-promoting effects (26).
Subsequent studies focusing on HOB have shown that soil H2 exposure shapes the taxonomic fingerprint of microbial communities (27–29). However, neither the conservation of these changes in different soils nor their consequences for microbial community functioning, other than H2 oxidation, were addressed. The present study tested the hypothesis that the response of soil microbial community structure to H2 exposure is idiosyncratic, here defined as an inconsistent response across land use types, thus under the influence of soil biodiversity and physicochemical properties. On top of that, changes in community structure induced by the activation of HOB exposed to H2 were expected to covary with alterations of soil microbial processes mediated by HOB and non-HOB. To address this prime matter, we have exposed soils embodying three land use types to different levels of H2 and investigated the impact of the treatment on soil microbial communities by using both marker gene PCR amplicon sequencing and shotgun metagenomics. Soil H2 and CH4 uptake, involving specialized bacterial guilds of microbes, and community level carbon substrate utilization profiles, involving generalist bacterial and fungal guilds, were also monitored. In this report, a “direct effect” of soil H2 exposure is defined as an alteration of the H2 oxidation rate of aerobic HOB and “indirect effects” refer to the responses of other microbe-mediated processes (i.e., CH4 uptake and the carbon utilization profile) involving HOB and/or non-HOB.
RESULTS
Soil physicochemical properties and microbial metabolism.
Soils were incubated in microcosm chambers under a dynamic headspace simulating H2 concentrations found in the atmosphere (aH2; 0.53 ppmv) or at the soil-nodule interface (eH2; 10,000 ppmv). Soils were acidic (pH 4.7 to 5.2) and encompassed a wide range of carbon (2.4 to 7.9%) and nitrogen (0.24 to 0.66%) contents (Table 1). H2 exposure did not alter measured soil physicochemical properties, with the exception of lower C/N ratios and pH values in larch and poplar soils exposed to eH2, respectively.
TABLE 1.
Soil physicochemical parameters and process rates measured in soil microcosmsa
| Variableb | Poplar monoculture |
Farmland |
Larch monoculture |
|||
|---|---|---|---|---|---|---|
| eH2-P | aH2-P | eH2-F | aH2-F | eH2-L | aH2-L | |
| pH | 5.11 (0.02)c | 5.21 (0.02)c | 5.25 (0.02) | 5.20 (0.05) | 4.81 (0.01) | 4.77 (0.01) |
| % C | 7.6 (0.4) | 7.9 (0.1) | 2.4 (0.1) | 2.5 (0.3) | 4.3 (0.4) | 4.7 (0.2) |
| % N | 0.64 (0.03) | 0.66 (0.01) | 0.24 (0.01) | 0.24 (0.03) | 0.36 (0.02) | 0.37 (0.01) |
| C/N ratio | 11.8 (0.1) | 11.8 (0.1) | 10.1 (0.2) | 10.3 (0.1) | 11.9 (0.2)c | 12.7 (0.31)c |
| % H2O | 0.46 (0.01) | 0.51 (0.01) | 0.19 (0.01)c | 0.25 (0.01)c | 0.34 (0.02) | 0.31 (0.01) |
| High-affinity H2 oxidatione | 59 (0)c | 1003 (45)c | 22 (2)c | 361 (87)c | 47 (18)c | 1045 (250)c |
| Low-affinity H2 oxidationf | 264 (32)c | 43 (2)c | 61 (1)c | 19 (3)c | 59 (18)c | 29 (2)c |
| CH4 oxidatione | 45 (12)c | 198 (2)c | 29 (7)c | 59 (0)c | 16 (0)c | 45 (9)c |
| Net CO2 productionf | 82 (6)c | 178 (20)c | 9 (1)c | 37 (1)c | 18 (2)c | 58 (3)c |
| EcoPlatesg | 3.20 (0.01)c | 3.06 (0.01)c | 3.22 (0.04)c | 3.05 (0.02)c | 3.11 (0.01)c | 3.00 (0.02)c |
| Bacterial species richnessg | 6.39 (0.02)c | 5.85 (0.08)c,d | 5.83 (0.09)c | 6.26 (0.03)c | 6.02 (0.06) | 6.16 (0.01) |
| Fungal species richnessg | 4.11 (0.05) | NA | 4.33 (0.03)d | 4.50 (0.05) | 4.23 (0.04) | 3.98 (0.18) |
| Bacterial species evennessh | 0.85 (0.00)c | 0.78 (0.01)c,d | 0.78 (0.01)c | 0.84 (0.00)c | 0.81 (0.01) | 0.82 (0.00) |
| Fungal species evennessh | 0.66 (0.01) | NA | 0.72 (0.01)d | 0.73 (0.01) | 0.69 (0.01) | 0.65 (0.03) |
Shown are the mean (standard deviation) for triplicate microcosms exposed to aH2 and eH2 treatments. NA, not available.
The variables water content, EcoPlates, bacterial species richness (Shannon index), fungal species richness (Shannon index), bacterial species evenness (Pielou index), and fungal species evenness (Pielou index) followed a normal distribution (Shapiro-Wilk test). Transformations were applied to obtain normal distribution of the other variables. The variable net CO2 production was square root transformed, low-affinity H2 oxidation was log transformed, C was cosine transformed, and C/N was Box-Cox transformed. The variables pH, CH4 oxidation, and high-affinity H2 oxidation could not be transformed to obtain normal distribution. dw, dry weight.
Significant difference (α = 0.05) between the two H2 treatments (one-way analysis of variance for normally distributed variables and Wilcoxon-Mann-Whitney test for nonnormally distributed variables).
Calculated from duplicate instead of triplicate results.
Expressed in picomoles per gram of soil (dry weight) per hour.
Expressed in nanomoles per gram of soil (dry weight) per hour.
Shannon index.
Pielou index.
All measured metabolic processes were significantly altered by H2 treatment of the three soils, albeit to various extents at the functional and land use levels (Table 1). The direct effects of H2 exposure included a 1.2- to 7-fold higher activity of low-affinity HOB and a 15- to 23-fold lower activity of high-affinity HOB in soils exposed to eH2. Stimulation of low-affinity HOB using CO2 and H2 as carbon and energy sources was linked to the lower net CO2 production measured in eH2-treated microcosms than in those incubated under aH2. Both carbon utilization profiling and CH4 oxidation rate analyses demonstrated the indirect effect of H2 on soil microbial functions. The highest carbon substrate utilization by bacteria and fungi was observed in soils exposed to eH2, as shown by average well color development (see Fig. S1 in the supplemental material) and the Shannon index (Table 1). Furthermore, CH4 oxidation rates in soil microcosms exposed to eH2 were 2 ± 0.2, 3 ± 0.2, and 4 ± 0.3 times lower in farmland, larch, and poplar soils than in their aH2-exposed counterparts (Table 1). As soil-air CH4 exchanges might reflect concomitant production and oxidation processes in aerobic soils (30), microcosms were also examined for potential methanogenic activity. No CH4 emission was detected in soil, indicating that the CH4 oxidation activity of methane-oxidizing bacteria (MOB), rather than CH4 production by methanogenic archaea, was influenced by H2 (see Table S1).
Taxonomic profile of soil microbial communities.
The three soils were dominated by bacteria encompassing the Proteobacteria, Actinobacteria, and Acidobacteria phyla and fungi affiliated with the Ascomycota phylum (see Fig. S2A). Bacterial and fungal operational taxonomic units (OTUs) detected in all three land use types represented 60 and 18% of the retrieved OTUs (see Fig. S2B). The species richness (Shannon index) and evenness (Pielou index) of bacterial communities were significantly influenced by H2 exposure, although no significant effect was recorded for fungi (Table 1). The impact of H2 exposure on bacterial richness was idiosyncratic in that eH2 treatments led to higher species richness in poplar soil, while farmland soil followed the opposite trend and larch soil did not express any significant response compared to microcosms exposed to aH2. The idiosyncratic response of soil microbial communities was illustrated with β diversity analyses of bacterial (Fig. 1A) and fungal (Fig. 1B) communities. The land use type contributed to 74 and 84% of the variation in bacterial and fungal community profiles, while the contribution of H2 treatment was approximately 5% (Fig. 1D). Poplar soil was the land use type showing the strongest response to H2 exposure. Indeed, the Euclidean distances between replicated bacterial profiles in microcosms exposed to aH2 and those exposed to eH2 were 11.2, 7.2, and 5.1 for poplar, farmland, and larch soils, respectively, with the relative abundance of 857 bacterial and 63 fungal OTUs influenced by H2 treatment (see Data Set S1). These responsive OTUs were grouped taxonomically to ascertain whether OTUs within higher-level taxa respond similarly to H2 treatments. No reproducible distribution pattern was found at the family level (Table 2). While many individual taxa responded to the treatment in one or two soil land use types, only a few OTUs representative of the rare biosphere (<0.01% relative abundance) were characterized by a reproducible response to H2 exposure in the three soils (see Data Set S1).
FIG 1.
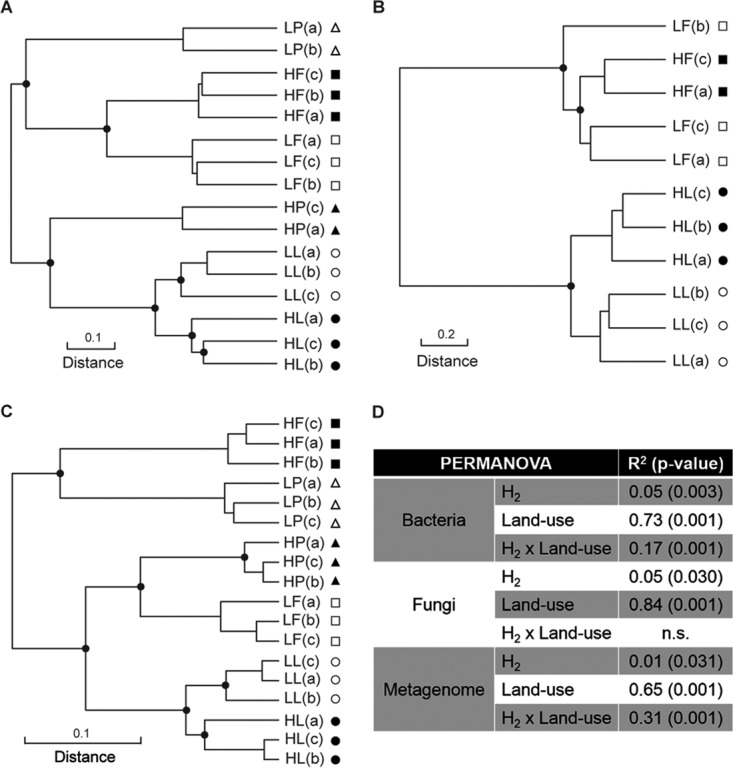
UPGMA agglomerative clustering of soil microcosms according to a Euclidean distance matrix calculated with Hellinger-transformed bacterial (A) and fungal (B) ribotyping profiles and a Bray-Curtis distance matrix of genome bin profiles (C). (D) PERMANOVA displaying the proportion of total β diversity variance explained by soil land use type and H2 treatment. Land use types are represented by the following symbols: squares, farmland; circles, larch; triangles, poplar. Black symbols indicate soil microcosms exposed to eH2 treatment, and white symbols indicate soil microcosms exposed to aH2 treatment. 16S rRNA gene PCR amplicon sequencing failures for samples LP(c) and HP(b) and ITS2 PCR amplicon sequencing failures for six poplar samples and HF(b) impaired their inclusion in the analysis. The term n.s. represents nonsignificant relationships.
TABLE 2.
Uneven responses of different taxonomic groups (family level) of bacteria and fungi to H2 exposure as a function of land use typea
| Taxonomic group | % more abundant in: |
|||||
|---|---|---|---|---|---|---|
| Poplar |
Farmland |
Larch |
||||
| eH2 | aH2 | eH2 | aH2 | eH2 | aH2 | |
| Bacteria | ||||||
| Nocardioidaceae (10, 9, 8)b | 0 | 100 | 70 | 0 | 56 | 0 |
| Xanthomonadaceae (32, 29, 33) | 0 | 61 | 69 | 0 | 69 | 0 |
| Intrasporangiaceae (7, 6, 7) | 0 | 43 | 57 | 0 | 100 | 0 |
| Sphingomonadaceae (13, 11, 12) | 0 | 75 | 54 | 0 | 27 | 18 |
| Hyphomonadaceae (6, 6, 7) | 0 | 0 | 50 | 0 | 33 | 0 |
| Sphingobacteriaceae (8, 8, 8) | 0 | 63 | 13 | 13 | 25 | 0 |
| Gemmatimonadaceae (30, 26, 28) | 11 | 50 | 7 | 13 | 19 | 0 |
| S47 (6, 6, 6) | 0 | 50 | 0 | 0 | 0 | 17 |
| Frankiaceae (6, 6, 6) | 0 | 17 | 0 | 0 | 50 | 0 |
| 0319-7L14OR (18, 14, 18) | 72 | 0 | 0 | 11 | 0 | 0 |
| RB25CL (9, 7, 9) | 67 | 0 | 33 | 0 | 14 | 0 |
| Flexibacteriaceae (19, 22, 20) | 60 | 15 | 16 | 0 | 5 | 18 |
| OM190OR (6, 7, 7) | 57 | 0 | 0 | 17 | 0 | 14 |
| Bdellovibrionaceae (13, 9, 8) | 0 | 0 | 0 | 54 | 0 | 0 |
| Rhodobacteraceae (10, 8, 9) | 0 | 11 | 0 | 10 | 13 | 50 |
| Fungi | ||||||
| Cortinariaceae (6, 14, 0) | NAc | NA | 0 | 17 | 21 | 64 |
| Helotiaceae (8, 22, 0) | NA | NA | 25 | 0 | 0 | 50 |
The percentages shown are the proportions of OTUs that were more abundant in the aH2 or eH2 treatment for each land use type. This reduced data set consists of microbial families containing at least six OTUs and of which >50% of the OTUs responded to the treatment in at least one land use type.
The values in parentheses are the numbers of OTUs found in the respective land use types.
NA, not available.
Metagenomic profile of soil microbial communities.
PCR-amplified rRNA marker gene profiles provided evidence that the bacterial and fungal response to H2 exposure is idiosyncratic. Nonetheless, uneven distribution of hydrogenases among taxonomic groups impaired the identification of direct and indirect impacts of H2 exposure on soil microbial communities. A metagenomic binning procedure proved efficient at pinpointing microorganisms responding to H2 exposure, searching for hydrogenase genes to outline direct and indirect responses from those genome bins, and identifying the underlying environmental factors influencing their distribution. Clustering analysis showed that genome bin distribution in soils was mainly driven by the land use type, with a modest influence of H2 treatment (Fig. 1C and D). The metagenomic profiles of larch soil were the most resistant to H2 treatment, followed by those of farmland and poplar soils. The same pattern was observed when the whole metagenomic database was considered in the analysis (see Fig. S3). A principal-component analysis (PCA) was used to identify the genome bins that contributed the most to distinguish the metagenomic profiles and their relationship with underlying biotic and abiotic environmental factors (Fig. 2A). The first principal component was correlated with soil nutrients, with C/N having negative loading. The second principal component reflected microbial metabolism, with a negative loading exerted by soil net CO2 production. One genome bin (bin 1) affiliated with the order Rhizobiales and three genome bins affiliated with Xanthomonadales bacteria (bins 2, 3, and 6) exerted more weight than average to define the ordination in the reduced space. The affiliation of these genome bins inferred by gene annotation was supported by a correlation network analysis of the rRNA marker gene amplicon sequencing profile that led to the identification of Rhizobiales and Xanthomonadales OTUs whose elevated relative abundance (up to 7%) and distribution profile corresponded to the four genome bins (see Fig. S4). Network topological properties were analyzed, and it has been determined that the bin 1 representative OTU displayed a very high connectivity (top 10%), bin 2 had average connectivity, and bins 3 and 6 had lower-than-average connectivity, indicating different contributions in shaping soil microbial community structure.
FIG 2.

(A) PCA showing the distribution of soil microcosms in a reduced space defined by the relative abundances of the 93 genome bins. Land use types are represented by the following symbols: squares, farmland; circles, larch; triangles, poplar. Black symbols indicate soil microcosms exposed to eH2 treatment, and white symbols indicate soil microcosms exposed to aH2 treatment. (B) Relative abundances of the four genome bins outlined by the PCA. Asterisks denote genome bins whose relative abundance was significantly different (Wilcoxon-Mann-Whitney test) between treatments. The term n.s. represents nonsignificant relationships. CPM, counts per million.
In accordance with the rRNA marker gene survey, none of the 93 genome bin distribution profiles displayed a reproducible response to H2 exposure in the three soils (see Table S2). For instance, Xanthomonadales genome bins were significantly more abundant under eH2 conditions (than under aH2 conditions) in larch and farmland soils, while they showed the reverse trend in poplar soil (Fig. 2B). This idiosyncratic response was attributed to soil factors influencing the growth and distribution of bacteria. In fact, multiple regression analyses unveiled that, of the independent variables measured, C/N, pH, and the interaction between CO2 respiration and H2 treatment best explained the distribution of the four genome bins (see Table S3A). Parameters of the multiple regression were consistent to explain the variation of OTU candidate representatives of the four genome bins (see Table S3B).
The application of a hydrogenase-based hidden Markov model (HMM) to all of the assembled contigs in this project led to the identification of 122 putative gene fragments encoding the structural subunits of [NiFe]-hydrogenases (see Data Set S2). Among them, 29 sequenced hydrogenase genes were validated by examination of the canonical L1 or L2 cysteine motifs binding the metal ions of the catalytic site in [NiFe]-hydrogenases (31, 32). More than 67% of the sequences retrieved belonged to group 3 [NiFe]-hydrogenases, followed by representatives of groups 1 (15 to 23%) and 2 (10 to 20%). Of those 29 partial genes, 2 were found in assembled genome bins. These putative HOB genome bins included bin 1 (Rhizobiales), possessing the gene encoding the small subunit of a group 1 [NiFe]-hydrogenase. The abundance of most potential HOB genome bins did not change according to H2 treatment, indicating that not all HOB take advantage of H2 availability for lithoautotrophic growth (see Table S4). On the other hand, the higher relative abundance of Xanthomonadales genome bins in eH2 reflects the effect of H2 on non-HOB. Indeed, none of these three bins (genome sequence completeness estimated at 84, 57, and 40% for genome bins 2, 3, and 6, respectively) displayed genes encoding hydrogenase structural subunits or auxiliary components necessary to harness energy from H2 in their respective genomes based on gene annotation and HMM (see Data Set S3).
Disentangling the idiosyncratic impact of H2 on microbial communities.
A structural equation model (SEM) was developed to test the hypothesis that the idiosyncratic impact of H2 exposure observed on soil microbial community structure and function is explained by soil biotic and abiotic features acting as ecological filters for microbial species and functional groups. The hypothetical model comprised four independent variables selected on the basis of a number of assumptions supported by the literature (see Method S1) to predict trace gas turnover and carbon utilization profiles (Fig. 3). Dependent variables included the H2 exposure level and soil water content, as well as two composite variables representing soil abiotic features (Soil) and microbial diversity (Bio). Parameterization of the model with experimental data indicated that the idiosyncratic response of microbial processes upon H2 exposure is process specific, owing to the different degree of dependence of each function on physicochemical and biological properties related to their environment. First, loss of high-affinity H2 oxidation activity in soils exposed to eH2 was attenuated by soil biodiversity and higher soil moisture content (Fig. 3A). The absence of a significant path between the composite variable Soil and the high-affinity H2 oxidation rate was unique for this functional guild. Second, stimulation of low-affinity HOB by eH2 was partly explained by soil physicochemical properties and microbial diversity (Fig. 3B). Third, the indirect impact of eH2 on CH4 oxidation activity was impaired by elevated species richness and soil moisture content and to a lesser extent by soil physicochemical properties (Fig. 3C). Finally, carbon substrate utilization was mainly driven by H2 exposure and soil abiotic properties and the absence of a significant path from the composite variable Bio was unique to this microbial function (Fig. 3D). An elevated path coefficient linking H2 treatment to the high-affinity H2 oxidation rate (−0.94) and carbon substrate utilization (1.0) indicated that soil abiotic and biotic features exerted less influence than H2 treatment on the inhibition and stimulation of these processes.
FIG 3.

SEMs to test causal assumptions of soil physicochemical properties, biodiversity, moisture, and H2 exposure on the high-affinity H2 oxidation rates (A), low-affinity H2 oxidation rates (B), high-affinity CH4 oxidation rates (C), and carbon utilization profiles (D) measured in soil microcosms. Variables shown in hexagons are statistical composites estimated by partial least-squares linear regression analysis of observed variables. The R2 values are presented for dependent variables. Path coefficients were standardized and were significant at a P value of <0.05. The term n.s. represents nonsignificant relationships.
DISCUSSION
The structure of microbial communities is driven by a combination of biotic and abiotic environmental features, including soil physicochemical properties, climate, and vegetation cover. Integration of these microbial community dynamics into an ecological theory framework is necessary to generate predictive models of practical value to mitigate the influence of anthropogenic activities on global biogeochemical cycles (33). For instance, a relationship between energy supply to the ecosystem through primary production and taxonomic diversity was proposed to predict ecological patterns of microbial communities in aquatic ecosystems (34). As observed with animals and plants, this relationship is characterized by a variety of patterns, ranging from insignificant trends to humped patterns where microbial diversity exhibits a maximum at intermediate productivity levels (35). Our findings broaden these classical ecological patterns with inorganic energy inputs through H2 addition exerting a positive, negative, or insignificant impact on soil microbial diversity (Table 1). Hence, we can conclude with confidence that both soil biotic and abiotic properties alter the diversity-energy relationship driven by H2. Furthermore, H2 energy potential influenced the ecological niche of several members of the soil microbial community, which is in contrast to the traditional knowledge that only a few HOB benefit from H2 sources in soil (27, 28). This impact on microbial communities is expected to be proportional to the H2 concentration, since this actual concentration limits the maximum energy potential accessible to HOB. Indeed, in a previous study analyzing the impact of H2 exposure on soil microbial communities, the relative abundance of 958 taxonomically diverse OTUs, representing 0.001 to 1.8% of the community, was altered in a farmland soil exposed to 500 ppmv H2, despite having no significant impact on bacterial species richness overall (29). However, the higher level of H2 used in this study resulted in a more dramatic alteration of microbial community composition, considering that both rare and abundant taxa (up to 7.5% abundance) responded to H2 treatment. Soil biotic and abiotic features have proven to be influential in the responses of individual taxa to H2 exposure, as only a few bacterial or fungal OTUs were shown to display the same response in all three soils. This outcome contrasts with observations made in soil amended with nutrients wherein a convergent response of taxa was observed in grasslands displaying a broad range of biotic and abiotic properties (36). This discrepancy is likely explained by a wider range of microbes benefiting from nitrogen fertilizers compared to those able to use H2.
The combination of rRNA marker gene amplicon sequencing, metagenomics, and process rate measurements in the present investigation led to the first evidence of a combination of direct and indirect impacts of H2 on soil microorganisms. This direct impact consists of inhibition of high-affinity H2 oxidation activity and enrichment of low-affinity HOB in soil exposed to eH2 (16, 37). These alterations of HOB led to a shift in kinetic parameters governing soil H2 uptake activity, with an increase of both (app)Vmax and (app)Km values (29, 38). In contrast, potential indirect side effects of the inhibition and stimulation of HOB on the distribution and metabolism of other members of a microbial community have received no attention. Here we show that H2 can significantly affect the community as a whole, not only HOB. The first evidence was the response of certain fungal OTUs to H2 exposure, despite recent genome database mining unveiling that no fungus species possesses genes encoding motifs associated with H2-oxidizing enzymatic machinery (31, 32). A second piece of evidence was the observation that the relative abundance of three Xanthomonadales genome bins was higher in soil microcosms exposed to eH2, without possessing the genetic potential to oxidize H2. However, considering the incomplete genome coverage of these three candidates (40 to 84% completeness), their ability to oxidize H2 cannot be completely ruled out at this stage. A third piece of evidence of an indirect effect of H2 on soil microorganisms was the lower CH4 oxidation rate and higher diversification of carbon substrate utilization potential in soils exposed to eH2. None of these processes are known to be affected by H2, but future studies shedding light upon those underlying mechanisms are crucial, considering the importance of these processes, with MOB contributing to 15% of the global losses of atmospheric CH4 (39) and carbon turnover being an indicator of the microbial community's physiological state (40). Although CH4 uptake activity measured in this study involves unknown high-affinity MOB and/or conventional MOB under starvation (41), their ability to use H2 as an energy source cannot be excluded. In fact, hydrogenases are widespread in MOB (C. R. Carere et al., submitted for publication) and the ability to oxidize H2 for energy generation has been reported in certain conventional MOB, such as Methylococcus capsulatus (Bath) and Methylosinus trichosporium OB3b (42, 43). Our data may imply the activation and enrichment of antagonistic bacteria and fungi to MOB under eH2 treatment, but a mixotrophic energy metabolism of MOB with preferential use of H2 under CH4 starvation in eH2 microcosms cannot be completely ruled out at this stage. Similarly, the higher community level carbon substrate utilization potential in soils exposed to eH2 could be partly explained by the mixotrophic metabolism of HOB, combining both organic carbon and H2 as energy sources. Regardless of the underlying mechanisms, these observations are in sharp contrast to the uneven responses of taxonomic groups and the energy-biodiversity relationships reported before, with a convergent response of microbial functions to H2 exposure.
SEM revealed that the magnitude of the microbial activity gains and losses observed under eH2 exposure fluctuated according to both soil physicochemical properties and microbial diversity. The relative contribution of biodiversity was more important for the metabolic activity of specialized HOB and MOB than the widely distributed metabolism of organic carbon substrates. This is in accordance with previous investigations performed across a land use gradient where the biodiversity of soil microbial communities represented a poorer predictor of CO2 soil respiration than the more specialized metabolism of CH4 (44). In this work, the contribution of species richness was positive in supporting microbial processes, which is in line with the relationship between biodiversity and ecosystem functioning (45). A combination of a “selection effect,” by which the dominance of certain species with a particular trait influences ecosystem processes, and a “complementary effect,” where the level of biodiversity influences the use of available resources, is expected to explain the observed relationship between biodiversity and measured functions (46, 47). In the presence of high concentrations of CH4, interactions between MOB and heterotrophic bacteria were shown to promote CH4 oxidation activity (48, 49). The SEM provides a framework for future investigations aimed at testing how soil microbial diversity and physicochemical properties mitigate alterations of biogeochemical processes induced by H2. The high-affinity H2 oxidation rate and community level carbon substrate utilization were the processes most sensitive to H2 exposure, and their responses are expected to be reproducible in other soil types. In contrast, the fate of high-affinity CH4 and low-affinity H2 oxidation activities in other soil types exposed to H2 is expected to be more variable, relying on soil biotic and abiotic features.
In conclusion, we validated our hypothesis that the taxonomic response of soil microbial community composition to H2 exposure is inconsistent across land use types. Surprisingly, these changes in community structure occurred along with a common metabolic response mediated by HOB and non-HOB. A few studies have shown the beneficial effect of H2 exposure on plant growth, presumably due to a phytohormonal control (e.g., via 1-aminocyclopropane-1-carboxylate deaminase) by HOB (25). Considering both direct and indirect effects on microbial community structure and functions that have been attributed to H2 exposure in this study, the so-called H2 fertilization effect could be attributed to the alteration of biogeochemical cycles through complex microbe-microbe interactions, as well as other synergistic factors. However, this work is a proof-of-concept experiment performed under artificial, controlled incubation conditions. The elevated H2 exposure treatment represented extreme conditions in soil, as steep H2 concentration gradients occur in the area surrounding nitrogen-fixing nodules. As a result, the impact of H2 on microbial community structure and function observed here might be overrepresented compared to microbial succession along H2 concentration gradients found in natural environments. For instance, other variables, such as root exudates, gas diffusion limitation, and weather conditions, may exert a dominant control of soil microbial communities. Detection of the minimal H2 threshold concentration necessary to induce changes in microbial communities, as well as field investigations, will be important to assess the environmental impacts of H2 in soil. Although preliminary, this work provides the first evidence that H2 exposure disrupts community level carbon substrate and CH4 utilization profiles predetermined by biotic and abiotic features of soils. This study has focused on aerated upland soils, and it remains to be tested whether observed effects of H2 hold true for other soils, including anoxic waterlogged soils exposed to elevated H2 produced during organic matter fermentation and N2 fixation.
MATERIALS AND METHODS
Soil samples.
Soil samples were collected in a tree nursery managed by the Quebec Minister of Forests, Wildlife, and Parks located in the municipality of St. Claude, Quebec, Canada. A 12-year-old larch plantation (45.68114°N, 071.99876°W), a 10-year-old poplar plantation (45.68024°N, 071.99823°W), and a farmland consisting of a potato-and-maize rotation (45.67688°N, 071.99814°W) were visited in June 2014. The litter, fermentation, and humus soil horizon (upper 2- to 5-cm layer) was removed before the upper layer (first 10 cm of B horizon) was collected from tree plantations. Soil samples were transferred into 37.9-liter plastic boxes and stored at 4°C until processing (within 2 months). Soil was first air dried for 60 to 72 h at room temperature (approximately 22°C), homogenized (2-mm sieve) with a vibratory sieve shaker (AS 200; Retsch GmbH, Haan, Germany), and then used for physicochemical analyses and microcosm incubations. Soil texture, pH (1:2.5 [wt/vol] soil suspension in 0.01 M CaCl2), and total carbon, nitrogen, and water contents were determined as described previously (4, 29). All three soils were classified as loamy sand according to particle size distribution. Raw pH values and C, N, and water contents are provided in Data Set S1.
Soil microcosm incubations.
Soil samples were incubated in a dynamic microcosm chamber unit comprising a gas mixing station and a gas distribution network (see Method S2). Dilutions made in the gas blending station were controlled by two mass flow controllers, achieving precise stoichiometric ratios. The gas mixture was distributed into each individual microcosm at 40 ml min−1 through seven mass flow controllers. Outlet tubes were vented to the atmosphere. The gas blending station was parameterized to test two H2 exposure treatments, a high concentration (10,000 ppmv H2) and a low concentration (0.5 ppmv H2). Our setup was an artificial design used in a proof-of-concept experiment to explore the potential impact of the elevated H2 concentrations found in certain ecosystems comprising H2 hot spots, including the N2-fixing nodule-soil interface, hydrothermal vents, hypersaline cyanobacterial mats, and hot springs, as opposed to the low concentrations (0.5 ppmv H2) found in the global atmosphere and surface soils (8). Both H2 exposure treatments were tested by using three independent replicates (three land use types times two treatments times three replicates for a total of 18 microcosms). Microcosms were designated according to H2 treatment (H and L for elevated and ambient H2 exposures, respectively) and soil type (F, farmland; L, larch; P, poplar), followed by a letter separating replicates (a, b, or c). For instance, the first replicate of farmland soil exposed to H2 at 10,000 ppmv was named HF(a). Each microcosm consisted of a 0.9-liter polystyrene cell culture flask (Corning, Tewksbury, MA) holding 200 g of homogenized soil. The soil water content was adjusted to a 30% water-holding capacity with sterile ultrapure water to promote gas diffusion in soils and provide an aerobic ecosystem. Soil was incubated in the dynamic microcosm chamber unit for 15 days at 22°C in the dark. Soil water content was monitored on a regular basis by a standard gravimetric method. Sterile water was added to keep the soil moisture level steady.
Measurement of gas exchanges.
Low-affinity H2 oxidation, net CO2 production, and high-affinity CH4 oxidation rates were measured at the end of the incubation, after soil subsamples were taken for molecular analysis, by a gas chromatographic assay. Microcosms were flushed with a gas mixture comprising 10,000 ppmv H2 and 3 ppmv CH4 in synthetic air (certified standard mixture; Praxair Distribution Inc., PA, USA) for 30 min. These initial H2 and CH4 concentrations can be used to measure the activity of low-affinity HOB and high-affinity MOB. Variations in H2, CH4, and CO2 mixing ratios were monitored over an 8-h time period by analyzing aliquots (10 ml) of the headspace with a gas chromatograph (gas chromatography [GC] system SP1 7890-0504; Agilent Technologies, CA, USA) comprising an electron capture detector (ECD), a thermal conductivity detector (TCD) for H2 analysis and a flame ionization detector (FID) for CH4 and CO2 detection. Agilent Greenhouse Gas Checkout Sample (5 ppmv CH4 and 600 ppmv CO2, balance air, ±2% analytical accuracy) was used for CH4 and CO2 calibration, while the 10,000 ppmv H2 gas mixture (Praxair Distribution Inc., PA, USA) was used for H2 calibration. The ECD used a mixture of 4.95% CH4 in argon (Praxair Distribution Inc., PA, USA) as makeup gas. High-purity H2 (99.999% H2; Praxair Distribution Inc., PA, USA) and hydrocarbon-free synthetic air (zero air; Praxair Distribution Inc., PA, USA) were used to ignite the flame, and 99.9995% N2 was supplied by a nitrogen generator (Parker Hannifin Corp., QC, Canada) as makeup gas for the FID. The makeup gas of the TCD was high-purity argon (99.999% argon; Praxair Distribution Inc., PA, USA). The GC system consists of two separate channels with 1/8-in. stainless-steel-packed columns (HayeSep Q 80/100). The oven temperature was set to 60°C, while the FID, TCD, and ECD temperatures were maintained at 250°C, 200°C, and 350°C, respectively. GC signal acquisition and peak quantification were performed with Agilent OpenLab CDS software. Each analytical run lasted 8 min. The high-affinity H2 oxidation rate was measured at the end of the incubation period by a gas chromatographic assay as described by Khdhiri et al. (4). Briefly, microcosms were flushed with synthetic air for 30 min before the injection of 3 ml of a certified gas mixture containing 500 ppmv H2 (Praxair Distribution Inc., PA, USA). As the initial H2 level in the headspace was approximately 3 ppmv, high-affinity HOB were responsible for the loss of H2 in the static headspace. Raw values of gaseous exchanges are provided in Data Set S1.
Carbon metabolism.
Community level carbon utilization profiles were calculated by EcoPlates assay (Biolog, Hayward, CA, USA) at the end of the incubation period. Soil suspensions were prepared by diluting soil (1:9, wt/vol) in 0.1 NaH2PO4 (pH 6) and then further diluting it in 0.15 M NaCl (1:10, vol/vol) after mixing it with a vortex mixer. A 100-μl volume of the soil suspensions was inoculated into each well of the EcoPlates. Each plate was then incubated for 4 days at 22°C in the dark. After incubation, a color change from clear to purple (reduction of tetrazolium dye) was monitored with an Infinite M1000pro plate reader (Tecan Group Ltd., Männedorf, Switzerland) at 590 nm, proportionally reflecting the microbial use of each substrate. Profiles were standardized by subtracting blank controls from each well (inoculated wells without substrate). Raw values of EcoPlates are provided in Data Set S1.
Nucleic acid extraction and purification.
Soil subsamples were collected in each microcosm at the end of the incubation period for genomic DNA extraction. DNA extraction was performed through a sequence of mechanical lysis (bead beating), phenol-chloroform extraction, ethanol precipitation, and purification with acid-washed polyvinylpolypyrrolidone columns (29). DNA was quantified with the QuantiFluor dsDNA System (Promega, Fitchburg, WI, USA) and a Rotor-Gene 6000 thermocycler (Qiagen, NRW, Germany).
Bacterial, archaeal, and fungal communities.
The V4 region of bacterial and archaeal 16S rRNA genes and the ITS2 region of fungal rRNA were PCR amplified (see Data Set S4). Paired-end sequencing was performed at the Joint Genome Institute (Walnut Creek, CA, USA) with the Illumina MiSeq system (2-by-250 configuration). Our sequencing analysis pipeline using QIIME 1.8.0 software (50) was based on the Itagger pipeline (29, 51). In total, 8,436,319 bacterial and archaeal and 3,391,264 fungal high-quality clustered reads remained after quality control. To avoid bias caused by unequal sequencing effort between samples, the amount of sequences of each type of library was subsampled to the library with the least sequences. This standardization resulted in 77,884 bacterial and archaeal sequences (1,925 OTUs) and 53,498 fungal sequences (817 OTUs) per sample (see Data Set S1).
Metagenomic analysis.
Metagenomic libraries were prepared and sequenced on an Illumina HiSeq 2000 system with a 2-by-150 configuration. Raw sequencing data (256 Gb) were processed through our metagenomic bioinformatic pipeline (see Method S3 and Data Set S5), which includes metagenome binning with Metabat v0.26.1 (52). Bins obtained from Metabat were further processed/decontaminated by splitting each bin into three subbins on the basis of the assigned taxonomic lineage at the order level, as each bin typically had a significant amount of contigs associated with the same-order taxon. The genome sequence completeness of sorted bins was estimated to vary between <0.01 and 89% (see Data Set S5D). To increase the detection power when searching for hydrogenase genes, we made multiple alignments of all amino acid sequences comprised in an extensive database of the [NiFe]-hydrogenase large subunit (31) with MUSCLE v.3.8.31 (53). HMMs (named groups 1, 2, 3, and 4) were generated from these alignments with Hmmer v3.1b1 (54), and gene sequences from metagenome assembly were compared against these training sets with hmmscan (Hmmer v3.1b1). Hits having an E value of ≤1e−10, a query length of ≥100 amino acid residues, and an alignment length of ≥100 were kept, for a total of 122 putative hydrogenase genes. Only sequences containing the canonical L1 or L2 cysteine motif binding the metal ions of the catalytic site in [NiFe]-hydrogenases (31, 32) were kept. This step was necessary to remove nonspecific sequences encoding energy-converting hydrogenase-related complexes.
Statistical analyses.
Statistical analyses were performed with R software v3.1.1 (55). The impact of H2 treatments on soil physicochemical properties, microbial activities, and microbial diversity was tested for each land use type by one-way analysis of variance for normally distributed variables and a Wilcoxon-Mann-Whitney test for nonnormally distributed variables with the stats package (55). Pairwise comparison of the relative abundances of all of the OTUs in soil microcosms incubated under different H2 exposure treatments was done by using the likelihood ratio test implemented in the edgeR package (56). Agglomerative clustering of molecular profiles by the unweighted pair group method using average linkages (UPGMA) was computed with the stats package (55) based on a Euclidean matrix of Hellinger-transformed relative abundance data for ribotyping analysis and a Bray-Curtis distance matrix on nontransformed relative abundance (counts per million) for genome bins. Significantly distinct clusters were identified with the Similarity Profile Tool (SIMPROF) implemented in the clustsig package (57) with 999 permutations. The contributions of the land use type and H2 treatments to partition distance matrices were computed by permutational multivariate analysis of variance (PERMANOVA) with the vegan package (58). A PCA was used to explore soil microcosm partitioning in a reduced space defined by the genome bin profile. An equilibrium circle of descriptors with the radius (where d is the number of dimensions of the reduced space [2] and p is the total space [93]) was computed to identify variables significantly contributing to the axes defining the positions of soil microcosms. The WGCNA package (59) was used to analyze covariation among ribotypes and subsequently to provide a potential taxonomic affiliation with genome bins assembled in the metagenomic analysis. All ribotyping libraries were used to generate a single weighted correlation network containing bacterial, archaeal, and fungal data. The network comprised 16 samples, since HP(b) and LP(c) were removed because of low sequencing effort compared to all of the other microcosms. Network construction was done as described by Piché-Choquette et al. (29). The soft-thresholding power used was 18, and each pair of modules with <0.40 dissimilarity was merged together as a single module. To identify the most probable taxonomic affiliation of genome bins, eigengenes were first computed for all modules. Spearman correlations between module eigengenes and genome bins were computed for taxonomic identification. Modules with the highest module membership for each genome bin were considered to retrieve OTUs with the highest correlation coefficient for each genome bin. The taxonomic affiliation, the relative abundance, and the distribution profile of genome bins and OTUs were compared to infer the most probable affiliation of each bin. Multiple regression analyses (stepwise forward selection of independent variables) were computed with the mfp package (55) to identify the best predictors for genome bin and OTU relative abundance in soil microcosms. A SEM was computed with the Lavaan package (60). Missing Shannon diversity indices for bacterial and fungal communities in SEM due to PCR amplicon sequencing failures were estimated by linear regression with unpublished RNA-based profiling as independent variables (see Method S1).
Accession number(s).
Raw sequence reads of rRNA marker genes from PCR amplicons and shotgun metagenomics were deposited in the Sequence Read Archive of the National Center for Biotechnology Information under BioProject numbers PRJNA329645 and PRJNA343121, respectively. Metagenome data sets can be found on the Integrated Microbial Genome and Microbiome Sample (IMG/M) website (https://img.jgi.doe.gov/cgi-bin/m/main.cgi) under the IMG study name “Soil and rhizosphere microbial communities from Centre INRS–Institut Armand-Frappier, Laval, Canada.”
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Natural Sciences and Engineering Research Council of Canada Discovery grant to P.C. and by the Community Science Program of the Joint Genome Institute (U.S. Department of Energy) to P.C. and J.T. The work conducted by the U.S. Department of Energy Joint Genome Institute, a U.S. Department of Energy Office of Science user facility, is supported by the Office of Science of the U.S. Department of Energy under contract DE-AC02-05CH11231. M.K. is grateful to the Ministry of Higher Education and Scientific Research of Tunisia for his Ph.D. scholarship.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00275-17.
REFERENCES
- 1.Bai Y, Müller DB, Srinivas G, Garrido-Oter R, Potthoff E, Rott M, Dombrowski N, Münch PC, Spaepen S, Remus-Emsermann M, Hüttel B, McHardy AC, Vorholt JA, Schulze-Lefert P. 2015. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528:364–369. doi: 10.1038/nature16192. [DOI] [PubMed] [Google Scholar]
- 2.Lammel DR, Feigl B, Cerri CC, Nüsslein K. 2015. Specific microbial gene abundances and soil parameters contribute to C, N, and greenhouse gas process rates after land use change in Southern Amazonian soils. Front Microbiol 6:1057. doi: 10.3389/fmicb.2015.01057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zak DR, Blackwood CB, Waldrop MP. 2006. A molecular dawn for biogeochemistry. Trends Ecol Evol 21:288–295. doi: 10.1016/j.tree.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Khdhiri M, Hesse L, Popa ME, Quiza L, Lalonde I, Meredith LK, Röckmann T, Constant P. 2015. Soil carbon content and relative abundance of high affinity H2-oxidizing bacteria predict atmospheric H2 soil uptake activity better than soil microbial community composition. Soil Biol Biochem 85:1–9. doi: 10.1016/j.soilbio.2015.02.030. [DOI] [Google Scholar]
- 5.Freitag TE, Prosser JI. 2009. Correlation of methane production and functional gene transcriptional activity in a peat soil. Appl Environ Microbiol 75:6679–6687. doi: 10.1128/AEM.01021-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaser MJ, Cardon ZG, Cho MK, Dangl JL, Donohue TJ, Green JL, Knight R, Maxon ME, Northen TR, Pollard KS, Brodie EL. 2016. Toward a predictive understanding of Earth's microbiomes to address 21st century challenges mBio 7:e00714-16. doi: 10.1128/mBio.00714-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daebeler A, Bodelier PLE, Yan Z, Hefting MM, Jia Z, Laanbroek HJ. 2014. Interactions between Thaumarchaea, Nitrospira and methanotrophs modulate autotrophic nitrification in volcanic grassland soil. ISME J 8:2397–2410. doi: 10.1038/ismej.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Novelli PC, Lang PM, Masarie KA, Hurst DF, Myers R, Elkins JW. 1999. Molecular hydrogen in the troposphere: global distribution and budget. J Geophys Res 104:30427–30444. doi: 10.1029/1999JD900788. [DOI] [Google Scholar]
- 9.Smith-Downey NV, Randerson JT, Eiler JM. 2008. Molecular hydrogen uptake by soils in forest, desert, and marsh ecosystems in California. J Geophys Res 113:G03037. doi: 10.1029/2008JG000701. [DOI] [Google Scholar]
- 10.Nielsen M, Revsbech NP, Kühl M. 2015. Microsensor measurements of hydrogen gas dynamics in cyanobacterial microbial mats. Front Microbiol 6:726. doi: 10.3389/fmicb.2015.00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witty JF. 1991. Microelectrode measurements of hydrogen concentrations and gradients in legume nodules. J Exp Bot 42:765–771. doi: 10.1093/jxb/42.6.765. [DOI] [Google Scholar]
- 12.Hunt S, Gaito S, Layzell D. 1988. Model of gas exchange and diffusion in legume nodules. Planta 173:128–141. doi: 10.1007/BF00394497. [DOI] [PubMed] [Google Scholar]
- 13.Witty JF, Minchin FR. 1998. Hydrogen measurements provide direct evidence for a variable physical barrier to gas diffusion in legume nodules. J Exp Bot 49:1015–1020. doi: 10.1093/jxb/49.323.1015. [DOI] [Google Scholar]
- 14.Rasche ME, Arp DJ. 1989. Hydrogen inhibition of nitrogen reduction by nitrogenase in isolated soybean nodule bacteroids. Plant Physiol 91:663–668. doi: 10.1104/pp.91.2.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong Z, Wu L, Kettlewell B, Caldwell CD, Layzell DB. 2003. Hydrogen fertilization of soils—is this a benefit of legumes in rotation? Plant Cell Environ 26:1875–1879. doi: 10.1046/j.1365-3040.2003.01103.x. [DOI] [Google Scholar]
- 16.La Favre JS, Focht DD. 1983. Conservation in soil of H2 liberated from N2 fixation by Hup− nodules. Appl Environ Microbiol 46:304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liot Q, Constant P. 2016. Breathing air to save energy—new insights into the ecophysiological role of high-affinity [NiFe]-hydrogenase in Streptomyces avermitilis. Microbiologyopen 5:47–59. doi: 10.1002/mbo3.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartz E, Friedrich B. 2006. The H2-metabolizing prokaryotes, p 496–563. In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (ed), The prokaryotes. Springer, New York, NY. [Google Scholar]
- 19.Greening C, Berney M, Hards K, Cook GM, Conrad R. 2014. A soil actinobacterium scavenges atmospheric H2 using two membrane-associated, oxygen-dependent [NiFe] hydrogenases. Proc Natl Acad Sci U S A 111:4257–4261. doi: 10.1073/pnas.1320586111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greening C, Villas-Bôas SG, Robson JR, Berney M, Cook GM. 2014. The growth and survival of Mycobacterium smegmatis is [sic] enhanced by co-metabolism of atmospheric H2. PLoS One 9:e103034. doi: 10.1371/journal.pone.0103034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meredith LK, Rao D, Bosak T, Klepac-Ceraj V, Tada KR, Hansel CM, Ono S, Prinn RG. 2014. Consumption of atmospheric hydrogen during the life cycle of soil-dwelling actinobacteria. Environ Microbiol Rep 6:226–238. doi: 10.1111/1758-2229.12116. [DOI] [PubMed] [Google Scholar]
- 22.Constant P, Chowdhury SP, Pratscher J, Conrad R. 2010. Streptomycetes contributing to atmospheric molecular hydrogen soil uptake are widespread and encode a putative high-affinity [NiFe]-hydrogenase. Environ Microbiol 12:821–829. doi: 10.1111/j.1462-2920.2009.02130.x. [DOI] [PubMed] [Google Scholar]
- 23.Constant P, Poissant L, Villemur R. 2008. Isolation of Streptomyces sp. PCB7, the first microorganism demonstrating high-affinity uptake of tropospheric H2. ISME J 2:1066–1076. doi: 10.1038/ismej.2008.59. [DOI] [PubMed] [Google Scholar]
- 24.Greening C, Carere CR, Rushton-Green R, Harold LK, Hards K, Taylor MC, Morales SE, Stott MB, Cook GM. 2015. Persistence of the dominant soil phylum Acidobacteria by trace gas scavenging. Proc Natl Acad Sci USA 112:10497–10502. doi: 10.1073/pnas.1508385112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Golding A-L, Dong Z. 2010. Hydrogen production by nitrogenase as a potential crop rotation benefit. Environ Chem Lett 8:101–121. doi: 10.1007/s10311-010-0278-y. [DOI] [Google Scholar]
- 26.Maimaiti J, Zhang Y, Yang J, Cen Y-P, Layzell DB, Peoples M, Dong Z. 2007. Isolation and characterization of hydrogen-oxidizing bacteria induced following exposure of soil to hydrogen gas and their impact on plant growth. Environ Microbiol 9:435–444. doi: 10.1111/j.1462-2920.2006.01155.x. [DOI] [PubMed] [Google Scholar]
- 27.Osborne CA, Peoples MB, Janssen PH. 2010. Detection of a reproducible, single-member shift in soil bacterial communities exposed to low levels of hydrogen. Appl Environ Microbiol 76:1471–1479. doi: 10.1128/AEM.02072-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, He X, Dong Z. 2009. Effect of hydrogen on soil bacterial community structure in two soils as determined by terminal restriction fragment length polymorphism. Plant Soil 320:295–305. doi: 10.1007/s11104-009-9894-3. [DOI] [Google Scholar]
- 29.Piché-Choquette S, Tremblay J, Tringe SG, Constant P. 2016. H2-saturation of high affinity H2-oxidizing bacteria alters the ecological niche of soil microorganisms unevenly among taxonomic groups. PeerJ 4:e1782. doi: 10.7717/peerj.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Angel R, Matthies D, Conrad R. 2011. Activation of methanogenesis in arid biological soil crusts despite the presence of oxygen. PLoS One 6:e20453. doi: 10.1371/journal.pone.0020453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greening C, Biswas A, Carere CR, Jackson CJ, Taylor MC, Stott MB, Cook GM, Morales SE. 2016. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J 10:761–777. doi: 10.1038/ismej.2015.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vignais PM, Billoud B. 2007. Occurrence, classification, and biological function of hydrogenases: an overview. Chem Rev 107:4206–4272. doi: 10.1021/cr050196r. [DOI] [PubMed] [Google Scholar]
- 33.Prosser JI, Bohannan BJM, Curtis TP, Ellis RJ, Firestone MK, Freckleton RP, Green JL, Green LE, Killham K, Lennon JJ, Osborn AM, Solan M, van der Gast CJ, Young JPW. 2007. The role of ecological theory in microbial ecology. Nat Rev Microbiol 5:384–392. doi: 10.1038/nrmicro1643. [DOI] [PubMed] [Google Scholar]
- 34.Horner-Devine CM, Leibold MA, Smith VH, Bohannan BJ. 2003. Bacterial diversity patterns along a gradient of primary productivity. Ecol Lett 6:613–622. doi: 10.1046/j.1461-0248.2003.00472.x. [DOI] [Google Scholar]
- 35.Smith VH. 2007. Microbial diversity-productivity relationships in aquatic ecosystems. FEMS Microbiol Ecol 62:181–186. doi: 10.1111/j.1574-6941.2007.00381.x. [DOI] [PubMed] [Google Scholar]
- 36.Leff JW, Jones SE, Prober SM, Barberán A, Borer ET, Firn JL, Harpole WS, Hobbie SE, Hofmockel KS, Knops JMH, McCulley RL, La Pierre K, Risch AC, Seabloom EW, Schütz M, Steenbock C, Stevens CJ, Fierer N. 2015. Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc Natl Acad Sci U S A 112:10967–10972. doi: 10.1073/pnas.1508382112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conrad R, Weber M, Seiler W. 1983. Kinetics and electron transport of soil hydrogenases catalyzing the oxidation of atmospheric hydrogen. Soil Biol Biochem 15:167–173. doi: 10.1016/0038-0717(83)90098-6. [DOI] [Google Scholar]
- 38.Dong Z, Layzell DB. 2001. H2 oxidation, O2 uptake and CO2 fixation in hydrogen treated soils. Plant Soil 229:1–12. doi: 10.1023/A:1004810017490. [DOI] [Google Scholar]
- 39.Conrad R. 2009. The global methane cycle: recent advances in understanding the microbial processes involved. Environ Microbiol Rep 1:285–292. doi: 10.1111/j.1758-2229.2009.00038.x. [DOI] [PubMed] [Google Scholar]
- 40.Garland JL, Mills AL. 1991. Classification and characterization of heterotrophic microbial communities on the basis of patterns of community-level sole-carbon-source utilization. Appl Environ Microbiol 57:2351–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai Y, Zheng Y, Bodelier PLE, Conrad R, Jia Z. 2016. Conventional methanotrophs are responsible for atmospheric methane oxidation in paddy soils. Nat Commun 7:11728. doi: 10.1038/ncomms11728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanczár T, Csáki R, Bodrossy L, Murrell CJ, Kovács KL. 2002. Detection and localization of two hydrogenases in Methylococcus capsulatus (Bath) and their potential role in methane metabolism. Arch Microbiol 177:167–172. doi: 10.1007/s00203-001-0372-4. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, Yoch D. 1987. Regulation of two nickel-requiring (inducible and constitutive) hydrogenases and their coupling to nitrogenase in Methylosinus trichosporium OB3b. J Bacteriol 169:4778–4783. doi: 10.1128/jb.169.10.4778-4783.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levine UY, Teal TK, Robertson GP, Schmidt TM. 2011. Agriculture's impact on microbial diversity and associated fluxes of carbon dioxide and methane. ISME J 5:1683–1691. doi: 10.1038/ismej.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bell T, Newman JA, Silverman BW, Turner SL, Lilley AK. 2005. The contribution of species richness and composition to bacterial services. Nature 436:1157–1160. doi: 10.1038/nature03891. [DOI] [PubMed] [Google Scholar]
- 46.Peter H, Beier S, Bertilsson S, Lindstrom ES, Langenheder S, Tranvik LJ. 2011. Function-specific response to depletion of microbial diversity. ISME J 5:351–361. doi: 10.1038/ismej.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loreau M, Hector A. 2001. Partitioning selection and complementarity in biodiversity experiments. Nature 412:72–76. doi: 10.1038/35083573. [DOI] [PubMed] [Google Scholar]
- 48.Stock M, Hoefman S, Kerckhof F-M, Boon N, De Vos P, De Baets B, Heylen K, Waegeman W. 2013. Exploration and prediction of interactions between methanotrophs and heterotrophs. Res Microbiol 164:1045–1054. doi: 10.1016/j.resmic.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 49.Ho A, de Roy K, Thas O, De Neve J, Hoefman S, Vandamme P, Heylen K, Boon N. 2014. The more, the merrier: heterotroph richness stimulates methanotrophic activity. ISME J 8:1945–1948. doi: 10.1038/ismej.2014.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuczynski J, Stombaugh J, Walters WA, González A, Caporaso JG, Knight R. 2011. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr Protoc Bioinformatics Chapter 10:Unit 10.7. doi: 10.1002/0471250953.bi1007s36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tremblay J, Singh K, Fern A, Kirton ES, He S, Woyke T, Lee J, Chen F, Dangl JL, Tringe SG. 2015. Primer and platform effects on 16S rRNA tag sequencing. Front Microbiol 6:771. doi: 10.3389/fmicb.2015.00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kang DD, Froula J, Egan R, Wang Z. 2015. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eddy SR. 1998. Profile hidden Markov models. Bioinformatics 14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 55.R Development Core Team. 2008. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org. [Google Scholar]
- 56.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clarke KR, Somerfield PJ, Gorley RN. 2008. Testing of null hypotheses in exploratory community analyses: similarity profiles and biota-environment linkage. J Exp Mar Biol Ecol 366:56–69. doi: 10.1016/j.jembe.2008.07.009. [DOI] [Google Scholar]
- 58.Oksanen J, Blanchet F, Kindt R, Legendre P, Minchin P, O'Hara R, Simpson G, Solymos P, Henry M, Stevens H, Wagner H. 2012. vegan: community ecology package. R package version 2.0-4 http://cran.r-project.org/package=vegan.
- 59.Langfelder P, Horvath S. 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosseel Y. 2012. lavaan: an R package for structural equation modeling. J Stat Softw 48:1–36. doi: 10.18637/jss.v048.i02. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.