

Abstract
The Embden–Meyerhof–Parnas (EMP) pathway is generally considered to be the biochemical standard for glucose catabolism. Alas, its native genomic organization and the control of gene expression in Escherichia coli are both very intricate, which limits the portability of the EMP pathway to other biotechnologically important bacterial hosts that lack the route. In this work, the genes encoding all the enzymes of the linear EMP route have been individually recruited from the genome of E. coli K-12, edited in silico to remove their endogenous regulatory signals, and synthesized de novo following a standard (GlucoBrick) that enables their grouping in the form of functional modules at the user’s will. After verifying their activity in several glycolytic mutants of E. coli, the versatility of these GlucoBricks was demonstrated in quantitative physiology tests and biochemical assays carried out in Pseudomonas putida KT2440 and P. aeruginosa PAO1 as the heterologous hosts. Specific configurations of GlucoBricks were also adopted to streamline the downward circulation of carbon from hexoses to pyruvate in E. coli recombinants, thereby resulting in a 3-fold increase of poly(3-hydroxybutyrate) synthesis from glucose. Refactoring whole metabolic blocks in the fashion described in this work thus eases the engineering of biochemical processes where the optimization of carbon traffic is facilitated by the operation of the EMP pathway—which yields more ATP than other glycolytic routes such as the Entner–Doudoroff pathway.
Keywords: Escherichia coli, Pseudomonas putida, standardization, glycolysis, metabolic engineering, PHB
The past few years have witnessed an increase in the number of microorganisms that can be metabolically engineered within the conceptual frame of Systems Biology and the molecular tools of contemporary Synthetic Biology.1−5 Longstanding platforms (e.g., Escherichia coli, Bacillus, Corynebacterium, and yeast) are increasingly accompanied by others (e.g., Pseudomonas species) that, due to their indigenous endurance to environmental stresses, have an improved ability to execute harsh biochemical transformations.6 Whether well-established or emerging, such platforms are empowered by a complex central carbon metabolic network where heterologous pathways must nest in any given host. Despite this fact, the majority of ongoing Metabolic Engineering efforts are preoccupied with what one could call peripheral aspects,7−10e.g., the elimination of competing endogenous pathways, the fine-tuning of gene expression and substrate transport, and the rerouting of small molecules at given nodes of the network. In contrast, the biochemical core that fuels the microbial cell factory is generally taken for granted and hardly ever touched; i.e., few efforts have tried to reshape central enzymatic processes in its entirety. Relevant examples of this sort include auxotrophic CO2 fixation11 and CH3OH assimilation12 by engineered strains of E. coli. Such efforts suffer from the difficulties of re-engineering some of the most extremely interconnected and fine-tuned components of any biological system as central metabolism is.13,14 Against this background, we wondered whether the most archetypal metabolic pathway (i.e., a linear glycolysis) could be reshaped in a way that, while maintaining its biochemical role and identity, could be freed of its native regulatory complexity and made portable either as a whole or as a functional subset of portable elements for specific Metabolic Engineering needs.
In its simplest definition, glycolysis is the metabolic breakdown of glucose into pyruvate (Pyr). Thought to have originated from simple chemical constraints in a prebiotic environment, glycolysis is one of the most widespread and conserved metabolic blocks in nature.15 Several biochemical sequences lead to the formation of Pyr from hexoses. The most studied example is the Embden–Meyerhof–Parnas (EMP) pathway, composed by the sequential activity of ten individual enzymes. The first five enzymes are involved in the so-called preparatory phase, which uses ATP to convert hexoses into trioses phosphate [i.e., glucose → glyceraldehyde-3-P (GA3P)]. The pay-off phase comprises the second half of the EMP enzymes, and it yields 2 NADH molecules and 2 ATP molecules per each processed glucose molecule by converting trioses phosphate into Pyr [i.e., GA3P → Pyr]. Therefore, the overall stoichiometry of the EMP pathway is glucose + 2NAD+ + 2ADP + 2Pi → 2Pyr + 2NADH + 2H+ + 2ATP. Another glycolytic route is the Entner–Doudoroff (ED) pathway, widely distributed in prokaryotes (even more so than the EMP route). The overall stoichiometry of the ED pathway is glucose + NAD+ + NADP+ + ADP + Pi → 2Pyr + NADH + NADPH + 2H+ + ATP. Although this biochemical sequence yields half the amount of ATP as compared to that of the EMP pathway,16 the ED route has been demonstrated to be a key source of reducing power in many environmental microorganisms.17,18 Because of a lifestyle of constant exposure to physicochemical insults often found in natural scenarios,19 such bacteria have evolutionarily favored the formation of reducing power20 to counteract oxidative stress.21 This is in contrast with the generation of ATP and the sheer buildup of biomass that appears to be the main metabolic driving force in Enterobacteria. Besides these highly conserved biochemical sequences, variants of carbohydrate breakdown processes are also present on other prokaryotes. Archaea, for instance, are characterized by the presence of unique, modified versions of the EMP and ED pathways which often include nonphosphorylating enzymes.22,23
The research below was prompted by our ongoing efforts to establish the soil-dweller microorganism Pseudomonas putida as a platform for hosting strong redox reactions.24−27 This bacterium lacks a 6-phosphofructo-1-kinase (Pfk) activity, and therefore the EMP pathway is not functional. The mere addition of Pfk to the biochemical network of P. putida was not only insufficient to activate an EMP-based hexose catabolism, but it also resulted in deleterious effects on growth and limited resistance to oxidative stress.17 Thus, the channeling of glucose toward Pyr in P. putida, while delivering enough NAD(P)H and yielding the highest possible amount of ATP, demands a multitiered approach involving more glycolytic genes or specific combinations thereof. To this end, in our present contribution we describe the reformatting of the entire EMP route of E. coli in a layout that we have called GlucoBricks, that allows complete or partial implantation of the glycolytic route in a variety of Gram-negative bacteria including (but not limited to) P. putida. This work finds inspiration in the attempts to decompress the regulatory density of the E. coli T7 bacteriophage28 and the deconstruction/reconstruction of the whole N2-fixation system of Klebsiella oxytoca(29) in an easy-to-manipulate setup. This time, however, the objective was the implantation of central metabolic functions in different Gram-negative hosts by means of portable, standardized modules assembled in promiscuous plasmids belonging to the SEVA (Standard European Vector Architecture) collection.30 The data discussed below not only demonstrates the versatility of the thereby reported device in various bacterial hosts, but it also expands the toolbox for deep engineering of central biochemical tasks in a microbial cell factory.
Results and Discussion
Functional Elements, Layout, and Design of Glycolytic Modules I and II
The starting point of this research was to design a streamlined set of genes to aid implanting (or increasing) glycolytic capacities in the microbial cell. To this end, we first pinpointed each of the ten enzymes carrying out the complete transformation of glucose into Pyr through the EMP pathway in the extant metabolic network of E. coli. In its naturally occurring configuration the corresponding genes are either scattered throughout the genome or arranged in two operons (e.g., fbaA and pgk) under the transcriptional control of at least five regulators (Cra, SoxS, Crp, Fur, and FnrS) as well as a number of post-transcriptional control devices.31 Our first task was to exclusively capture the enzymatic complement of the system while releasing the corresponding genes (either separately or as a whole) from any host-specific regulatory connection. Against this background, the basic organization of what we call a GlucoBrick is sketched in Figure 1. When more than one gene encoded a given EMP reaction (i.e., pfkA/pfkB, fbaA/fbaB, gpmA/gpmM, and pykA/pykF), the one bringing about the highest activity was selected according to the information available in the literature. The structural sequence of each glycolytic gene starts with a leading ATG and ends up with a STOP codon. The corresponding DNA was edited to eliminate restriction sites incompatible with the assembly standard discussed below, and any internal transcriptional signal was erased. Such ORFs are preceded by a standardized Shine-Dalgarno sequence and a DNA spacer, and the whole segment is flanked upstream and downstream by restriction sites that match given positions of the default multiple cloning site of the SEVA format. SEVA vectors include a choice of plasmids with four compatible broad-host range origins of replication (i.e., RK2, pBBR1, pRO1600/ColE1, and RSF1010) and six independent antibiotic markers [i.e., ampicillin, kanamycin (Km), cloramphenicol, streptomycin, tetracyclin, and gentamicin]. This standard allows for the effective propagation and maintenance of up to four plasmids in any given Gram-negative host, and it also enables the simultaneous expression of several genes. Since each glycolytic gene was preceded by a synthetic ribosome binding site (RBS), and bracketed by two directional SEVA restriction enzymes (i.e., following the standard format Restriction Enzyme 1–RBS–glycolytic gene–Restriction Enzyme 2), the assembly standard allows to directly subclone or swap any gene combination by a simple digestion and ligation step. Moreover, the order of insertion of each of the ten GlucoBricks in the SEVA’s multiple cloning site reflects the biochemical sequence of operation in the EMP pathway, thereby allowing for whatever the combination of the parts within the route at stake.
Figure 1.

Schematic representation of the GlucoBrick platform layout and the cognate glycolytic reactions. (a) The minimal set of genes from Escherichia coli K-12 needed for the activation of a functional and linear Embden–Meyerhof–Parnas pathway were edited according to the Standard European Architecture Vector rules and assembled into two synthetic operons. The first operon, termed Module I, encodes all the reactions within the upper catabolic block of the pathway (i.e., bioreactions of the preparatory phase of glycolysis). The second operon, termed Module II, spans the reactions of the lower catabolic block of the pathway (i.e., bioreactions of the pay-off phase of glycolysis). All the glycolytic reactions are shown below the gene encoding them. Note that each gene is preceded by a synthetic regulatory element, indicated by a purple circle, composed of a ribosome binding site (sequence underlined) and a short spacer sequence. (b) Linear glycolytic pathway encoded by the GlucoBrick platform, transforming glucose into glyceraldehyde-3-P (GA3P) by means of the activities of Module I; and GA3P into pyruvate (Pyr) by means of the activities of Module II. The two sets of glycolytic transformations are indicated with blue and red arrows, representing the genes within Modules I and II, respectively. Other abbreviations used in this outline are as follows: G6P, glucose-6-P; F6P, fructose-6-P; FBP, fructose-1,6-P2; DHAP, dihydroxyacetone-P; BPG, glycerate-1,3-P2; 3PG, glycerate-3-P; 2PG, glycerate-2-P; and PEP, phosphoenolpyruvate.
Building on this concept, we designed two DNA modules that encode all the necessary enzymes for the activation of a flawless, linear EMP-based glycolytic route. The EMP pathway was purposely split into two catabolic blocks: the upper catabolic block, termed Module I, comprises the activities of the preparatory phase; and the lower catabolic block, dubbed Module II, spans the activities of the pay-off phase (Figure 1a). Module I thus encodes all the enzymes needed for glucose phosphorylation and conversion into trioses phosphate, whereas activities originating from Module II use GA3P, the final product of Module I, as the precursor to form Pyr (Figure 1b). A detailed map of the DNA architecture of this platform (Figure 2a) indicates that the two DNA blocks can be also combined sequentially in a single SEVA vector if desired (and even in transposon vectors designed for chromosomal integration of large DNA segments32,33). Note that adopting the SEVA standard also enables the user to express any group of genes under the control of the large number of constitutive and effector-responsive, broad-host-range transcriptional devices available in the database.34−36
Figure 2.

Genetic architecture of the GlucoBrick platform. (a) Physical map of Modules I and II, indicating restriction enzymes bracketing individual glycolytic genes. The enzyme targets are colored in the sequence of the multiple cloning site of all the plasmids belonging to the Standard European Architecture Vector to identify the DNA block they belong to (i.e., blue, Module I; and red, Module II). Other restriction targets that could be used to add different regulatory or structural elements and thereby expand the usability of this platform are shown in gray. The abbreviations used in this outline are GA3P, glyceraldehyde-3-P; and Pyr, pyruvate. (b) Restriction analysis of Module I and II. Plasmids pS224·GBI (upper panel) and pS224·GBII (lower panel) were digested with the appropriate enzymes as indicated and the products were separated by electrophoresis in a 0.7% (w/v) agarose gel. Plasmid pS224·GBI was digested with AvrII-BamHI (i, releases the whole Module I segment); AvrII-EcoRI (ii, releases glk); EcoRI-SacI (iii, releases pgi); SacI-KpnI (iv, releases pfkA); KpnI-SmaI (v, releases fbaA); and SmaI-BamHI (vi, releases tpiA). Plasmid pS224·GBII was digested with BamHI-HindIII (i, releases the whole Module II segment); BamHI-XbaI (ii, releases gapA); XbaI-SalI (iii, releases pgk); SalI-PstI (iv, releases gpmA); PstI-SphI (v, releases eno); and SphI-HindIII (vi, releases pykF).
As a first step in the characterization of this tool in E. coli strains, Modules I and II were separately cloned in vector pSEVA224 (RK2, KmR), giving rise to pS224·GBI and pS224·GBII, respectively (Tables S1 and S2 in the Supporting Information). The expression of the glycolytic genes in these low-copy-number plasmids is under control of the LacIQ/Ptrc expression system, inducible by addition of isopropyl-β-d-1-thiogalactopyranoside (IPTG) to the culture medium. As an additional test of the structural versatility of this plasmid-based GlucoBrick platform, a restriction analysis of both pS224·GBI and pS224·GBII was carried out (Figure 2b). All the glycolytic genes could be separately recovered upon digestion with the appropriate pair of restriction enzymes–thereby facilitating direct subcloning into suitable SEVA vectors whenever needed as indicated above.
The Activities Encoded in the GlucoBricks Restore or Enhance the Growth of Glycolytic Escherichia coli Mutants
The functional characterization of the GlucoBrick system was carried out by assessing their phenotypic impact when the corresponding plasmids were introduced in glycolytic E. coli mutants lacking either single or several combined activities of the EMP pathway (Table 1). E. coli BW25113, a wild-type K-12 strain,37 was used as a positive control in all these growth experiments, in which the final cell density and the specific growth rate was recorded for each strain in cultures containing both Km and IPTG. First, the initial steps in glucose processing were targeted at the level of Glk (i.e., glucokinase) and PtsI [i.e., the EI component of the phosphoenolpyruvate (PEP):carbohydrate phosphotransferase (PTS) system]. No glucose phosphorylation is possible in such mutant as Glk and the PTS-dependent transport of the hexose coupled to phosphorylation are both blocked.38 Note, however, that glucose transport should not be significantly affected, since alternative transporters (e.g., the low-affinity galactose:H+ GalP symporter and the ATP-dependent MglABC system) internalize the nonphosphorylated hexose when the PTS system is not active.39 Accordingly, the Δglk ΔptsI strain grew in M9GCM semisynthetic medium (formulated in such a way that all the glycolytic mutants tested could grow, albeit some of them did so poorly) but not in M9 minimal medium with glucose as the only carbon source. The presence of Module I, however, restored the growth of the double mutant on the hexose back to the levels observed in the semisynthetic medium. When the reaction catalyzed by Pfk (which mediates the glycolytic formation of fructose-1,6-P2 from fructose-6-P) was eliminated by deleting both pfkA and pfkB, the growth of the resulting strain was severely compromised even in M9GCM medium—and no growth at all was observed in minimal medium with glucose. The Pfk activity brought about by Module I was, however, enough to restore this growth deficiency in both culture media, and it also lead to a 2.3-fold increase in the specific growth rate of the recombinant in M9GCM medium. Another critical step of the EMP pathways is TpiA (i.e., triose phosphate isomerase), which plays a central role not only in downward glycolysis but also in gluconeogenesis.40 Expectedly, the growth of a ΔtpiA mutant was impaired among all the culture conditions tested, probably because of the buildup of methylglyoxal as a toxic intermediate.41 Expression of tpiA within Module I alleviated this metabolic situation, even restoring the growth of the mutant from glucose and significantly enhancing it in the semisynthetic medium.
Table 1. Functional Validation of Module I and II in Glycolytic Mutants of Escherichia coli BW25113.
| growth
parametersc in |
|||||
|---|---|---|---|---|---|
| M9GCM
semisynthetic medium |
M9 minimal
medium +20 mM glucose |
||||
| E. coli straina | plasmidb | CDW (g L–1) | growth coefficient | CDW (g L–1) | growth coefficient |
| BW25113 (wild-type strain) | None | 2.7 ± 0.3 | – | 1.7 ± 0.1 | – |
| Δglk ΔptsI | pSEVA224 | 1.1 ± 0.2 | 1.2 ± 0.1 | N.G. | G.R. |
| pS224·GBI | 1.3 ± 0.3 | 1.3 ± 0.2 | |||
| ΔpfkA ΔpfkB | pSEVA224 | 0.4 ± 0.1 | 2.3 ± 0.3 | N.G. | G.R. |
| pS224·GBI | 1.4 ± 0.2 | 1.9 ± 0.1 | |||
| ΔtpiA | pSEVA224 | 0.6 ± 0.1 | 1.4 ± 0.2 | N.G. | G.R. |
| pS224·GBI | 1.7 ± 0.3 | 1.2 ± 0.3 | |||
| ΔgapA Δepd ΔptsI | pSEVA224 | 0.5 ± 0.1 | 6.1 ± 0.5 | N.G. | G.R. |
| pS224·GBII | 1.2 ± 0.1 | 0.9 ± 0.1 | |||
| Δpgk | pSEVA224 | 0.3 ± 0.1 | 2.9 ± 0.1 | N.G. | G.R. |
| pS224·GBII | 1.4 ± 0.5 | 1.6 ± 0.4 | |||
| Δeno | pSEVA224 | 0.8 ± 0.2 | 3.1 ± 0.4 | N.G. | G.R. |
| pS224·GBII | 1.5 ± 0.2 | 0.9 ± 0.1 | |||
The detailed genotype of the strains is given in Table S1 in the Supporting Information.
Plasmid pSEVA224 was used as the control vector; plasmids pS224·GBI and pS224·GBII are pSEVA224 derivatives carrying either Module I or II, respectively. IPTG was added to all cultures at 1 mM at the onset of the cultivation.
M9GCM semisynthetic medium contains the same salts as M9 minimal medium, casein hydrolyzate, glycerol, and sodium malate. CDW, cell dry weight after 48 h of aerobic incubation at 37 °C. N.G., no growth (defined as a change in the CDW < 0.05 g L–1). The growth coefficient is the ratio between the specific growth rate of a strain carrying either Module I or II and the specific growth rate of the same strain carrying pSEVA224; the cases in which the mutant did not grow (and hence, the growth coefficient could not be calculated) are indicated as G.R., growth restored. Results represent the mean value ± standard deviation from triplicate measurements in at least two independent experiments. All the differences in growth coefficients for the strains grown in M9GCM semisynthetic medium were significant (P < 0.05, as evaluated by means of the Student’s t test) in the pairwise comparison of a given recombinant to the control strain carrying the empty pSEVA224 vector.
The capability of Module II to mediate EMP activities in the pay-off phase was analyzed in another set of E. coli glycolytic mutants. The conversion of GA3P into PEP (and, consequently, Pyr) was targeted to test Module II. Note that the mere elimination of GA3P dehydrogenase in E. coli (represented by GapA, and possibly the GapB isozyme42) does not block Pyr formation, as this metabolite could be also produced by the PTS system from PEP. Most of the PEP pool comes from the EMP pathway, yet anaplerotic reactions could also generate this intermediate. Considering this complex metabolic scenario (and in order to ensure that there is no Pyr formation), a triple E. coli mutant was constructed by eliminating gapA (encoding a NAD+-dependent GA3P dehydrogenase, and also the major GA3P dehydrogenase prevalent in Enterobacteria), epd (also known as gapB, encoding a NAD+–erythrose-4-P dehydrogenase), and also ptsI. Glucose-dependent growth was completely abolished in this mutant, and M9GCM medium cultures only attained a modest cell density. Once again, the presence of Module II enhanced or even restored the growth of the triple mutant by means of the GapA activity encoded therein. Interestingly, the specific growth rate of the recombinant cells in semisynthetic medium increased >6-fold when Module II was transformed in the ΔgapA Δepd ΔptsI mutant—the highest among the conditions and strains analyzed in this study. Finally, the Δpgk and Δeno mutants had a similar qualitative behavior: they grew very poorly in semisynthetic medium and could not grow at all in M9 minimal medium with glucose. The corresponding activities encoded in Module II sufficed to restore the growth of the corresponding recombinants on the hexose, while the specific growth rate in M9GCM medium increased by ca. 3-fold in either case (Table 1). Note that, besides the functional complementation of individual enzyme activities missing in the single or multiple mutant strains studied herein, the GlucoBrick blocks can also amplify glycolytic activities that are already in place in E. coli, thus boosting the channeling of hexoses through the linear EMP pathway. In all the cases studied here, the final cell densities attained by the recombinants carrying GlucoBricks in M9 minimal medium with glucose was comparable to that of wild-type E. coli BW25113.
In order to evaluate if these growth phenotypes correlate with higher specific activities of some of the enzymes involved in glucose processing, some key steps in the EMP pathway were evaluated in cell-free extracts in vitro. The activity of Glk in the glucose-grown Δglk ΔptsI mutant expressing Module I was 1,370 ± 120 nmol min–1 mg protein–1 upon induction of gene expression with IPTG, representing a ca. 23- and 10-fold increase as compared with the same strain carrying the empty vector and wild-type BW25113 carrying the empty vector, respectively (note that some residual Glk activity was observed in the knockout strain, possibly arising from some other nonspecific hexose kinases43). Similarly, introduction and induction of the genes in Module I in the ΔpfkA ΔpfkB strain resulted in 10-fold increase in the Pfk activity (1,080 ± 70 nmol min–1 mg protein–1) as compared with the same strain carrying the empty pSEVA224 vector. In contrast, the Pfk activity in wild-type BW25113 was 751 ± 36 nmol min–1 mg protein–1. The residual in vitro phosphofructokinase activity observed in the E. coli ΔpfkA ΔpfkB mutant (<100 nmol min–1 mg protein–1) could arise from a side activity of an enzyme belonging to the PfkB family of phosphosugar kinases, which form a subset of the large ribokinase superfamily.44 Possibly, ribokinase (or another enzyme belonging to this family) phosphorylates fructose-6-P to fructose-1,6-P2in vitro, which in turn may be due to the dilution of an allosteric inhibitor in the assay as previously described by Chin and Cirino.45
Taken together, these results indicate that the efficiency of either Glk or Pfk in E. coli can be appropriately complemented by means of the activities borne by the GlucoBrick platform. The evidence gathered so far highlight the versatility of the GlucoBrick system in bestowing or even enhancing glycolytic activities in E. coli mutants—the next relevant question being whether this tool can also be used in other, unrelated bacterial species.
The Activities Encoded by GlucoBricks Are Instrumental for Engineering Glycolysis in Two Pseudomonas Species
Pseudomonads are a wide group of aerobic Gram-negative γ-proteobacteria characterized by their remarkable metabolic versatility and ubiquitous presence in many environmental niches.46−48P. putida and P. aeruginosa are representative members of the genus, which prevalently use the ED pathway for hexoses breakdown.25,49P. putida KT2440, a current platform for Synthetic Biology and Metabolic Engineering,6,21 uses a cyclic combination of enzymes of the EMP pathway, the ED pathway, and the pentose phosphate pathway to catabolize glucose via a metabolic array termed as EDEMP cycle(26) (Figure 3a)—probably operative in other members of the Pseudomonas group as well, such as P. aeruginosa. The intrinsic metabolic complexity of glycolysis in these bacteria makes the targeted Metabolic Engineering of primary metabolism particularly difficult, and we thus decided to adopt both P. putida KT2440 and P. aeruginosa PAO1 as the heterologous hosts for testing the GlucoBrick platform by assessing physiological and growth parameters as well as the in vitro activity of key glycolytic enzymes.
Figure 3.

Characterization of physiological parameters in recombinant Pseudomonas putida and P. aeruginosa strains carrying Module I. (a) Schematic representation of central carbon metabolism in Pseudomonas species. Glucose catabolism occurs mainly through the activity of the Entner–Doudoroff (ED) pathway, but part of the trioses-P thereby generated are recycled back to hexoses-P by means of the EDEMP cycle, that also encompasses activities from the Embden–Meyerhof–Parnas (EMP) and the pentose phosphate (PP) pathways. Note that a set of peripheral reactions can also oxidize glucose to gluconate and/or 2-ketogluconate (2KG) before any phosphorylation of the intermediates occurs. Each metabolic block is indicated with a different color along with the relevant enzymes catalyzing each step, and the EDEMP cycle is shaded in blue in this diagram. Note that the 6-phosphofructo-1-kinase activity, missing in most Pseudomonas species, is highlighted with a dashed gray arrow. The abbreviations used for the metabolic intermediates are as indicated in the legend to Figure 1; other abbreviations are as follows: 6PG, 6-phosphogluconate; KDPG, 2-keto-3-deoxy-6-phosphogluconate; acetyl-CoA, acetyl-coenzyme A; 2KG, 2-ketogluconate; and 2K6PG, 2-keto-6-phosphogluconate. (b) Glucose consumption profile and (c) growth curves of P. putida KT2440, its Δglk derivative, and P. aeruginosa PAO1, carrying either the control vector (pSEVA224) or pS224·GBI (Module I). Glucose consumption is reported as the mean value ± standard deviation from duplicate measurements in at least three independent experiments. CDW, cell dry weight. Significant differences (P < 0.05, as evaluated by means of the Student’s t test) in the pairwise comparison of a given recombinant to the control strain, carrying the empty pSEVA224 vector, are indicated by an asterisk. In the growth curves, each data point represents the mean value of the optical density measured at 600 nm (OD600) of quadruplicate measurements from at least three independent experiments. The specific growth rates were calculated from these data during exponential growth, and the inset shows the mean values ± standard deviations for each strain.
Since the expression of the glycolytic modules is expected to boost the activity of the extant EMP pathway enzymes in Pseudomonas and to provide, at the same time, the missing step catalyzed by Pfk, we evaluated the overall glucose consumption in recombinant P. putida and P. aeruginosa strains carrying either an empty pSEVA224 vector or the pS224·GBI plasmid, bearing Module I (Figure 3b). In both cases, the overall rate of glucose consumption was boosted by expressing the genes of Module I (i.e., 2.5- and 1.5-fold in P. putida KT2440 and P. aeruginosa PAO1, respectively, as compared to the strains bearing the pSEVA224 vector). Interestingly, no major growth deficiencies (both in terms of final biomass densities and specific growth rates) were observed in the strains carrying Module I (Figure 3c). P. aeruginosa PAO1 had a slight reduction in the specific growth rate when expressing Module I, but the difference in this parameter as compared to that in the same strain carrying an empty vector was not significant. In any case, this result is somewhat in contrast with the observed deleterious effect of separately overexpressing pfkA from E. coli in P. putida KT2440.17 It is plausible that the differences arise from expressing the kinase gene alone versus introducing the whole upper metabolic block from E. coli in P. putida—thereby enabling circulation of carbon from glucose down to GA3P by connecting the corresponding reactions. The consequences of blocking this situation, broadly known as metabolic channeling, have been recently described in a Δpfk mutant of E. coli.50 The authors indicate that intermediates of the EMP pathway are passed from one enzyme to the next one without equilibration within the cellular medium. As such, all the enzymes in the biochemical sequence are needed to reach an efficient traffic through the whole pathway—a finding that helps explaining why the concerted expression of all the EMP genes in P. putida KT2440 does not impact the cell physiology significantly.
If the hypothesis of metabolic channeling advanced above holds true, one might expect that an increase in the activities of the whole EMP pathway is a conditio sine qua non for activating a linear glycolysis in Pseudomonas species. We therefore decided to evaluate if the glycolytic activities implanted could actually account for a functional EMP pathway by evaluating the in vitro activities of enzymes of the preparatory phase of this pathway in different Pseudomonas recombinants.
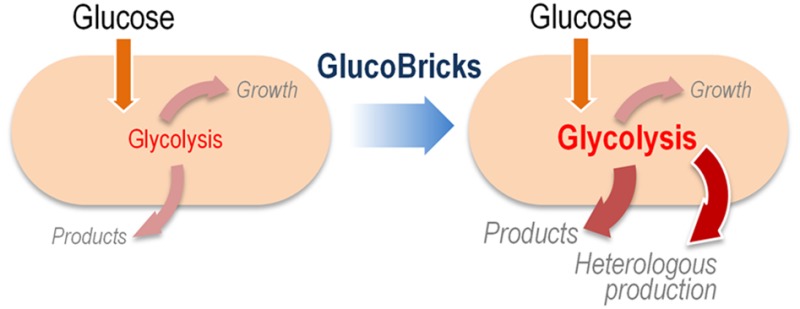
The activity of Glk was first assayed in strains KT2440 and PAO1 carrying pS224·GBI (Module I) and growing on glucose as the sole carbon source, and added with Km and IPTG as needed (Figure 4a). Unlike E. coli, Pseudomonas species do not have a devoted PTS system for glucose uptake, and direct phosphorylation of the hexose is catalyzed by Glk (i.e., PP_1011 in P. putida and PA_3193 in P. aeruginosa) in the cytoplasm. The native Glk activity in both Pseudomonas species ranged from ca. 89 to 184 nmol min–1 mg protein–1 in P. aeruginosa PAO1 and P. putida KT2440, respectively, and this kinase activity was boosted by expression of E. coli glk from plasmid pS224·GBI (i.e., a 1.4-fold increase in strain KT2440 and a remarkable 6.2-fold increase in strain PAO1). A P. putida Δglk strain was also constructed, in which the in vitro Glk activity resulted negligible. Transformation of this glk mutant strain with plasmid pS224·GBI and induction of gene expression by IPTG addition augmented the kinase activity up to 45 nmol min–1 mg protein–1 (i.e., a 100-fold increase in Glk activity).
Figure 4.
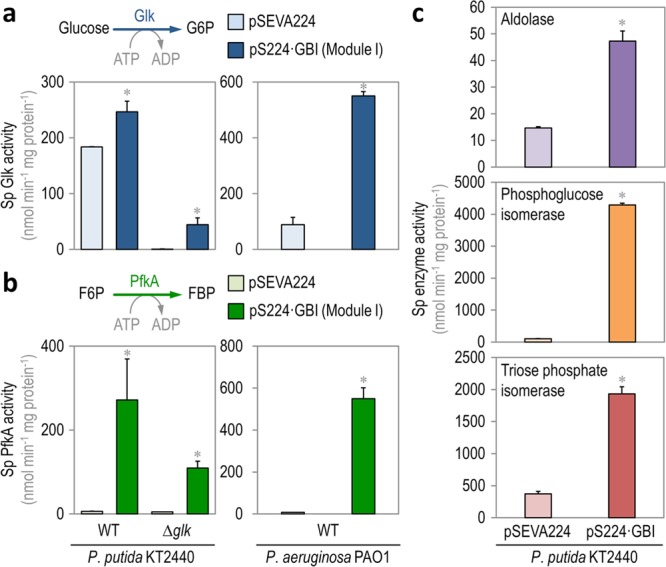
Biochemical characterization of native and implanted enzyme activities in Pseudomonas species. (a) In vitro quantification of the specific (Sp) glucokinase (Glk) activity, which phosphorylates glucose to glucose-6-P (G6P) in wild-type (WT) P. putida KT2440 and its Δglk derivative (left panel), and WT P. aeruginosa PAO1 (right panel) carrying either the empty pSEVA224 vector or Module I. (b) In vitro quantification of the specific (Sp) 6-phosphofructo-1-kinase (PfkA) activity, which converts fructose-6-P (F6P) into fructose-1,6-P2 (FBP) in WT P. putida KT2440 and its Δglk derivative (left panel), and WT P. aeruginosa PAO1 (right panel) carrying either the empty pSEVA224 vector or Module I. (c) In vitro quantification of the specific (Sp) activities of aldolase, phosphoglucoisomerase, and triose phosphate isomerase in P. putida KT2440 carrying either the empty pSEVA224 vector or Module I. These three activities, combined with Glk and PfkA, constitute the preparatory phase of the Embden–Meyerhof–Parnas pathway (i.e., from glucose to glyceraldehyde-3-P). All the strains tested were grown on M9 minimal medium added with glucose at 20 mM and cells were harvested in midexponential phase for these in vitro enzymatic assays. Each bar represents the mean value of the corresponding enzymatic activity ± standard deviation of quadruplicate measurements from at least two independent experiments. Significant differences (P < 0.05, as evaluated by means of the Student’s t test) in the pairwise comparison of a given recombinant to the control strain, carrying the empty pSEVA224 vector, are indicated by an asterisk.
As mentioned before, a Pfk activity is altogether missing in several Pseudomonas species, strains KT2440 and PAO1 being prime examples of the absence of such glycolytic step. Thus, the introduction of Module I in these strains would fill the gap between fructose-6-P and fructose-1,6-P2 by grafting the PfkA activity from E. coli. This in vitro kinase activity was analyzed on both Pseudomonas species transformed with plasmid pSEVA224·GBI and grown in M9 minimal medium containing glucose, Km, and IPTG (Figure 4b). Expectedly, almost no Pfk activity was detected neither in P. putida nor in P. aeruginosa. Expression of the genes borne by plasmid pS224·GBI resulted in increased levels of Pfk activity, which were similarly high in both P. putida KT2440 and P. aeruginosa PAO1 (i.e., ca. 272 and 550 nmol min–1 mg protein–1, respectively). The change in the kinase activity brought about by heterologous expression of pfkA in P. putida Δglk was somewhat low, but it still represents a 23-fold increase as compared to the control strain transformed with an empty vector. The differences in the activities detected in the wild-type strain and its Δglk derivative likely arise from alterations in the intracellular metabolite pools, which may determine a different pattern of regulation on the PfkA enzyme.
The three remaining activities within the preparatory phase (i.e., aldolase, phosphoglucose isomerase, and triose phosphate isomerase) of the EMP pathway were also evaluated in vitro in glucose-grown P. putida KT2440 carrying either pSEVA224 or pS224·GBI (Figure 4c). All three enzyme activities had a significant increase in the recombinant carrying Module I as compared to the same strain transformed with the empty pSEVA224 vector. In particular, the total aldolase and triose phosphate isomerase activities had a 3- and 5-fold increase, respectively, in P. putida KT2440 expressing Module I, whereas the activity of triose phosphate isomerase augmented a surprising 43-fold. Taken together, these in vitro results indicate that all the enzymes within the preparatory phase of the EMP pathway are active upon introduction of Module I in P. putida, thereby accounting for a complete linear glycolysis. Yet, if this is the case, one would also expect an impact of these manipulations on the intracellular metabolome in the engineered strain–an issue that was investigated as disclosed below.
Impact of the GlucoBrick Platform in the Intracellular Metabolome of P. putida KT2440
The next step was to evaluate the intracellular concentration of some critical metabolic intermediates in P. putida KT2440 carrying either pSEVA224 or pS224·GBI. The first intermediate after glucose phosphorylation (i.e., glucose-6-P), and the end product of the preparatory phase of the EMP pathway (i.e., GA3P) were targeted and their concentration was measured by means of liquid chromatography coupled to mass spectrometry (Table 2). Both glycolytic building blocks had an increased abundance in the strain expressing the genes of Module I (i.e., 1.7-fold in the case of the hexose-P and 3.2-fold in the case of the triose-P), a further experimental indication that the EMP pathway is active in this recombinant strain. Moreover, if glucose gets channeled into a linear EMP route instead of the native EDEMP cycle, a reduction in the NADPH availability (that would be otherwise generated through the activity of Zwf, see Figure 3a) is to be expected. The direct measurement of this redox cofactor confirms that is actually the case: the P. putida KT2440 derivative expressing Module I showed a 26% reduction in the intracellular content of NADPH (Table 2). Taken together, these targeted metabolomic determinations (along with the measurement of EMP enzyme activities) indicate that glucose is channeled into a linear glycolysis of sorts in P. putida KT2440 when expressing the genes within Module I.
Table 2. Metabolomic Determinationsa in Glucose-Grown Pseudomonas putida KT2440 Carrying Glycolytic Genes Borne by Module I.
| intracellular
content (nmol mg CDW–1) of |
|||
|---|---|---|---|
| P. putida KT2440 carrying plasmid | glucose-6-P | glyceraldehyde-3-P | NADPH |
| pSEVA224 (empty vector) | 52 ± 9 | 0.39 ± 0.08 | 43 ± 5 |
| pS224·GBI (Module I) | 89 ± 4 | 1.23 ± 0.07 | 32 ± 9 |
Cells were grown aerobically in M9 minimal medium added with glucose at 20 mM, harvested during exponential growth, and rapidly quenched with liquid N2. The intracellular metabolites were extracted and their concentration determined by means of liquid chromatography coupled to mass spectrometry. Each parameter is reported as the mean value ± standard deviation from duplicate measurements in at least two independent experiments.
The results of the two preceding sections highlight the versatility of the GlucoBrick platform to boost existing glycolytic activities or to introduce new metabolic steps that are alien to the biochemical network of Pseudomonas, thereby facilitating the engineering of primary metabolism not only in E. coli but also in unrelated bacterial species. Against this background, the next step was the functional evaluation of this Synthetic Biology tool in the context of the ongoing efforts aimed at manipulating the production of added-value metabolites.
Enhancing Heterologous PHB Production by Boosting Glycolytic Activity in Recombinant E. coli
Polyhydroxyalkanoates are a complex family of bacterial biopolymers.51−53 PHB is an isotactic polyester composed by 3-hydroxybutyrate units.54 The PHB synthesis pathway in Cupriavidus necator (formerly known as Ralstonia eutropha) comprises three enzymes (Figure 5a). PhaA, a 3-ketoacyl-CoA thiolase, condenses two acetyl-CoA moieties, yielding 3-acetoacetyl-CoA. This intermediate is the substrate for PhaB, a NADPH-dependent 3-acetoacetyl-CoA reductase (encoded by phaB1). In the final step of this biosynthetic pathway, (R)-(−)-3-hydroxybutyryl-CoA units are polymerized to PHB by PhaC, a PHA synthase (encoded by phaC1). The idea of a thermoplastic and biocompatible material which is also readily biodegraded by a number of bacteria has become very attractive in an era of increasing environmental concern and shortage of oil supply.55 A number of different recombinant E. coli strains have been constructed thus far by outsourcing structural and regulatory pha genes from several bacterial species,56 given that E. coli does not possess the metabolic machinery needed for the synthesis or the degradation of polyhydroxyalkanoates. A problem recursively encountered when attempting to improve the yield and productivity of biopolymer in recombinant E. coli strains is that the PHB biosynthetic pathway is nested in the core biochemical network of this bacterium,57 not only drawing acetyl-CoA coming from the EMP pathway, but also using NADPH as a cofactor58 and competing with the native fermentation pathways, e.g., acetate formation from acetyl-CoA59 (Figure 5a). Therefore, the availability of biosynthetic precursors constitutes a potential bottleneck compromising not only PHB accumulation but also bacterial growth.
Figure 5.
Enhanced poly(3-hydroxybutyrate) synthesis in recombinant Escherichia coli carrying Modules I and II. (a) Three enzymes are necessary for de novo synthesis of poly(3-hydroxybutyrate) (PHB). In Cupriavidus necator, from which the cognate genes were harnessed, PHB accumulation depends on the sequential activity of a 3-ketoacyl-coenzyme A (CoA) thiolase (PhaA), a NADPH-dependent 3-acetoacetyl-CoA reductase (PhaB1), and a PHB synthase (PhaC1). PhaA and PhaB1 catalyze the condensation of two molecules of acetyl-CoA to 3-acetoacetyl-CoA and the reduction of this intermediate to R-(−)-3-hydroxybutyryl-CoA (3-HB-CoA), respectively. PhaC1 polymerizes 3-HB-CoA monomers to PHB by releasing one CoA-SH molecule per monomer added. Note that acetyl-CoA can also be used in the major fermentation pathway of E. coli, that produces acetate. The main metabolic blocks within the biochemical network are identified with different colors in the outline: the Embden–Meyerhof–Parnas (EMP) pathway, red; the pentose phosphate (PP) pathway, purple; and the tricarboxylic acid (TCA) cycle and gluconeogenesis, green. Abbreviations of metabolic intermediates are as shown in the caption to Figure 1; other abbreviations are as follows: 6PG, 6-phosphogluconate; acetyl-CoA, acetyl-coenzyme A; OAA, oxaloacetate; and 2-OX, 2-oxoglutarate. (b) Glucose consumption profile and (c) PHB accumulation by E. coli BW25113 carrying plasmid pAeT41 (i.e., constitutively expressing the phaC1AB1 gene cluster from C. necator) transformed with plasmids carrying the genes or modules indicated (see Table 2 for further details). Cells were grown aerobically in LB medium added with glucose at 10 g L–1 for 24 h. Each parameter is reported as the mean value ± standard deviation from duplicate measurements in at least three independent experiments. CDW, cell dry weight. Significant differences (P < 0.05, as evaluated by means of the Student’s t test) in the pairwise comparison of a given recombinant to the control strain, carrying the empty pSEVA224 vector, are indicated by an asterisk.
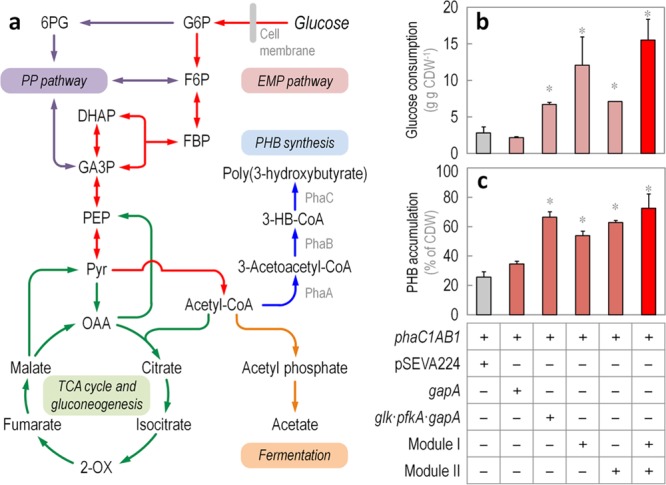
A recombinant, PHB-producing E. coli strain was constructed by transforming plasmid pAeT41 (carrying the phaC1AB1 gene cluster from C. necator) into wild-type E. coli BW25113. Plasmids pSEVA224 (KmR, empty vector), pS438·GBI (SmR, carrying Module I), and/or pS224·GBII (KmR, carrying Module II) were introduced in this strain, and growth, polymer accumulation, and acetate excretion were evaluated in LB medium cultures added with glucose and the corresponding antibiotics and inducers after 24 h of aerobic growth (Table 3 and Figure 5b and c). The separate introduction of Module I and Module II stimulated both glucose consumption and polymer accumulation with respect to the control strain. Glucose consumption significantly increased by 4- and 3-fold upon induction of the genes encoded by Module I and II, respectively (Figure 5b). Furthermore, the simultaneous introduction of the two GlucoBrick clusters resulted in a 6-fold increment of the specific glucose consumption with respect to the control strain, indicating a synergistic effect of each of them on the uptake of the carbon substrate. The increase in the total glucose consumption in this strain, accompanied by a decrease in residual biomass formation, indicates that the metabolic intermediates generated through the EMP pathway are channeled into PHB accumulation (rather than biomass formation).
Table 3. Growth Parameters and Polymer Synthesis of a PHB-Accumulating Escherichia coli BW25113a Strain Carrying Different Combinations of Glycolytic Genes.
| plasmidb | CDWRc (g L–1) | PHB (g L–1) | μ (h–1) | specific rate of acetate formation (mmol g CDW–1 h–1) | acetyl-coenzyme A content (nmol mg CDWR–1) |
|---|---|---|---|---|---|
| pSEVA224 (empty vector) | 0.54 ± 0.06 | 0.21 ± 0.04 | 0.46 ± 0.03 | 3.3 ± 0.4 | 0.43 ± 0.09 |
| pS438·GBI (Module I) | 0.35 ± 0.02 | 0.43 ± 0.02 | 0.29 ± 0.03 | 1.1 ± 0.2 | 0.75 ± 0.03 |
| pS224·GBII (Module II) | 0.37 ± 0.01 | 0.61 ± 0.01 | 0.29 ± 0.01 | 1.8 ± 0.2 | 0.59 ± 0.04 |
| pS438·GBI + pS224·GBII | 0.11 ± 0.01 | 0.35 ± 0.01 | 0.21 ± 0.02 | 1.3 ± 0.1 | N.D. |
| pS424·gapA | 0.45 ± 0.02 | 0.24 ± 0.02 | 0.44 ± 0.01 | 2.8 ± 0.3 | N.D. |
| pS224·GPG (glk·pfkA·gapA) | 0.32 ± 0.01 | 0.72 ± 0.01 | 0.42 ± 0.02 | 1.5 ± 0.1 | N.D. |
E. coli BW25113 carrying plasmid pAeT41 (i.e., constitutively expressing the phaC1AB1 gene cluster from C. necator) was transformed with the plasmids indicated, and grown aerobically in LB medium added with glucose at 10 g L–1 for 24 h. Each parameter is reported as the mean value ± standard deviation from duplicate measurements in at least three independent experiments. N.D., not determined.
The full description of each plasmid can be found in Table S2 in the Supporting Information.
The residual cell dry weight (CDWR) was calculated as the difference between the total CDW and the PHB concentration.
Interestingly, and while the overexpression of either gapA or glk·pfkA·gapA did not affect the specific growth rate of the recombinants significantly, Modules I and II reduced this parameter by ca. 30%, perhaps indicating some sort of metabolic burden due to plasmid maintenance.60 Overflow metabolism was also evaluated by measuring the specific rate of acetate formation (Table 3), since this metabolite is known to be the main byproduct of aerobic glucose catabolism.61E. coli BW25113 carrying pSEVA224 excreted acetate at a rate of 4.8 ± 0.2 mmol g CDW–1 h–1 when cultured in LB medium containing glucose. All the strains expressing the phaC1AB1 gene cluster had a reduced rate of acetate formation (e.g., the strain transformed with both Module I and II had a 73% reduction in this parameter); a finding in line with previous observations indicating that the synthesis of PHB overcomes acetate formation in recombinant E. coli.59 When the intracellular concentration of acetyl-CoA was determined in these strains (Table 3), a correlation between the concentration of this metabolite and PHB formation was clearly observed. The expression of the glycolytic genes borne by Module I or Module II significantly boosted the availability of this metabolic precursor of 1.7- and 1.4-fold, respectively, as compared to E. coli BW25113/pAeT41 + pSEVA224 (P < 0.05, as evaluated by means of the Student’s t test). Considering that acetyl-CoA is the substrate for the PHB synthesis pathway, it was postulated that polymer accumulation should also increase as the availability of this precursor increases. This was actually the case: PHB accumulation in the control strain reached 25.6 ± 3.7% of the cell dry weight (CDW), while the same strain transformed with either Module I or Module II attained 53.9 ± 3.1% and 62.9 ± 1.3% of the CDW, respectively (Figure 5c). The residual CDW of either strain under PHB accumulation conditions was similar (Table 3), indicating that neither Module I nor II affected biomass formation. Expectedly, the simultaneous expression of all GlucoBricks from two independent plasmids further boosted polymer accumulation up to 72.5 ± 9.8% of the CDW—thus representing an increase of ca. 3-fold as compared to E. coli BW25113/pAeT41 + pSEVA224. Interestingly, the overexpression of the glycolytic modules did not lead to an increase in overflow metabolism since acetyl-CoA was rerouted into PHB accumulation rather than acetate formation. This result further illustrates how the availability of key precursors in central metabolism can be channeled into the formation of an added-value product while minimally affecting the physiology of the bacterial host.
In an attempt to determine which glycolytic steps were determinant in increasing PHB accumulation by these recombinants, two different SEVA plasmids were constructed, in which individual GlucoBricks belonging to either Module I or II are expressed under the transcriptional control of an LacIQ/Ptrc regulatory element. Plasmids pS424·gapA and pS224·GPG, carrying glk, pfkA, and gapA (Table S2 in the Supporting Information) were separately introduced into E. coli BW25113/pAeT41, and growth, glucose consumption, and PHB accumulation were tested under the same culture conditions indicated above. While an increase in the traffic through GA3P dehydrogenase (i.e., GapA) did not result in a significant increase of either glucose consumption or polymer synthesis, the expression of the glk·pfkA·gapA cluster from plasmid pS224·GPG significantly enhanced both parameters with respect to the control strain (Figure 5b and c). PHB accumulation in this recombinant peaked at 66.4 ± 3.7% of the CDW (although there was no differences in the actual PHB concentration, see Table 3). Yet, boosting the glycolytic traffic by means of Glk, Pfk and GapA alone was not enough to reach the highest accumulation of PHB, as observed in the strain bearing both Modules I and II. This result may in turn suggest that an appropriate channeling of metabolic intermediates from glucose all the way down to acetyl-CoA is only possible if an adequate balance of all the individual enzymatic activities involved is met. Such scenario also supports the notion that all the EMP enzymes are need in a Pseudomonas strain lacking Pfk in order to activate a linear glycolysis.
Conclusion
The work above shows that the ten genes that shape the EMP route in E. coli can be excised from its native context and still deliver their biochemical input in a fashion that equals (and, in some cases, improves) the natural physiology and metabolic wiring of the microbial cells. This enhancement happens despite the scattered genomic organization, the regulatory complexity, and the high biochemical centrality and connectivity of the extant EMP pathway in Enterobacteria. Such situation is in contrast with previous efforts to decompress the regulatory complexity of the lytic bacteriophage T7 (resulting in a decrease of ca. 20% of infectivity with respect to the wild-type virus28), or the nif genes of K. oxytoca (resulting in ca. 57% of N2 fixation as compared to the naturally occurring system29). In our case, the versatility of the refactored EMP genes enables rational rerouting of the overall glycolytic carbon to meet specific demands for given metabolic precursors. To meet this end, tuning primary metabolism is in principle an effective strategy to foster availability of key carbon intermediates. Yet, the only attempts for, e.g., increase the glycolytic activities of E. coli have thus far focused on individual genes.62 For instance, Solomon et al. designed a recombinant E. coli improved for gluconate production by endowing it with different (and adjustable) levels of Glk activity.63 However, the biological activities of the synthetic EMP route presented here demonstrate the portability and efficiency of some block metabolic pathways (or subsets of them) formerly thought to be short of untouchable and/or difficult to handle as a multienzyme whole.
Besides validating the autonomy of the implanted EMP route, the data above show the utility of a flexible toolbox for boosting the efficiency of carbon consumption and distribution in a variety of cell factories of biotechnological interest. Some of the engineered modules were able to recover glucose-dependent growth of E. coli mutants (deficient in either individual or several glycolytic steps) to an extent beyond the wild-type situation. Increasing glycolytic activities in PHB-accumulating recombinant E. coli resulted in polymer contents (up to ca. 73% of the CDW) that rank among the highest reported in the literature for batch cultures using glucose as the carbon source.56,64 From a different perspective, and owing to the broad-host-range vectors and expression systems available, the enzymatic activities endowed by the platform could be promiscuously transferred to other species (e.g., Pseudomonas, as illustrated in the present study) or even to complete microbial communities (a development of the platform currently under development in our laboratory). Finally, the modular arrangement of the GlucoBricks and the possibility to assemble them as promoter-less gene arrays in SEVA-compatible transposon vectors allows exploration of the right level of expression that makes them more effective when knocked-in in different heterologous hosts32 (which would also reduce any possible metabolic burden related to plasmid maintenance and/or addition of inducers). Further improvements are expected as long as the expression of the genes within each module is appropriately adjusted, and a minimal subset of glycolytic genes, promoting an efficient EMP-based catabolism, is identified. The specific impact of the GlucoBricks on the metabolic networks of different recipients is likely to differ depending on the bacterial species, an issue that can be addressed wherever necessary by measuring fluxes with 13C-labeled substrates. In sum, we believe that the GlucoBricks concept herein presented provide a valuable example of how rationally formatting biological constituents eases the engineering of new-to-nature properties with a reasonable degree of efficiency and predictability.
Materials and Methods
Bacterial Strains, Plasmids, and Culture Conditions
The bacterial strains and plasmids used in this study are listed in Table S1 and Table S2, respectively, in the Supporting Information. E. coli and P. aeruginosa were grown at 37 °C; P. putida cultures were incubated at 30 °C. For regular growth and for the propagation and construction of plasmids, E. coli strains CC118 and DH5α λpir were cultured in LB medium.33,65 For physiology experiments and in order to obtain cell-free extracts for enzyme assays, bacterial cells were grown with rotary agitation at 170 r.p.m. in 250-mL Erlenmeyer flasks filled with 50 mL of M9 minimal medium, containing 6 g L–1 Na2HPO4, 3 g L–1 KH2PO4, 1.4 g L–1 (NH4)2SO4, 0.5 g L–1 NaCl, 0.2 g L–1 MgSO4·7H2O, and 2.5 mL l–1 of a trace elements solution.27 When culturing E. coli strains in minimal media, CaCl2 was added at 0.1 mM and vitamin B1 was added at 0.05% (w/v). Unless indicated otherwise, minimal medium cultures were added with glucose at 20 mM and IPTG at 1 mM as explained in the text. The growth of some glycolytic mutants of E. coli is known to be impaired even in rich LB medium;66 these mutants were grown in M9GCM semisynthetic medium. This culture medium contains the same salts as M9 minimal medium, but also 0.75% (w/v) amino acids from casein hydrolyzate (Becton-Dickinson Diagnostics Co., Sparks, MD, USA), 10 mM glucose, 20 mM glycerol, 15 mM sodium malate, and 0.05% (w/v) vitamin B1. In the case of E. coli ΔgapA Δepd and ΔgapA Δepd ΔptsI mutants (i.e., deficient in the epd-encoded erythrose-4-P dehydrogenase), pyridoxine hydrochloride was added to the culture medium at 5 μM.67 For the purpose of adapting the cells to growth on glucose from rich LB medium, preinocula, prepared with a few isolated colonies from LB medium plates, were grown overnight in 10 mL of M9 minimal medium with glucose or M9GCM semisynthetic medium with the corresponding antibiotics in 100-mL Erlenmeyer flasks. Solid media used to streak cells contained 15 g L–1 agar. The antibiotics employed for selection were added to the media when needed at the following final concentrations: ampicillin, 150 μg mL–1 for E. coli strains or 500 μg mL–1 for P. putida strains; and Km, 50 μg mL–1 with the exception of P. aeruginosa, for which 300 μg mL–1 Km was needed to select colonies after transformation of plasmids by electroporation. For the construction of P. putida mutants, 3-methylbenzoate (3-mB) was used at 15 mM to induce the XylS-dependent Pm promoter. Experiments for PHB accumulation were carried out in LB medium added with glucose at 10 g L–1 and the antibiotics and inducers described in the text (in these experiments, 3-mB was used at 0.5 mM to induce the expression of the genes in Module I).
DNA Manipulation and Sequencing, and Construction of Mutant Strains
DNA manipulations followed routine laboratory techniques.65 Plasmid DNA was obtained with the QIAprep Spin Miniprep kit (Qiagen Inc., Valencia, CA, USA) according to the manufacturer’s instructions. Oligonucleotides were ordered from Sigma-Aldrich Co. (St. Louis, MO, USA). Restriction and DNA modification enzymes were purchased from New England Biolabs (Ipswich, MA, USA). Isolated colonies from fresh LB medium plates were used as the starting material in colony PCR amplifications for checking gene deletions or the presence of plasmids. PCR products were purified with the NucleoSpin Extract II kit (Macherey-Nagel, Düren, Germany). Agarose gel visualization was carried out using a Molecular Imager VersaDoc apparatus (Bio-Rad Corp., Hercules, CA, USA). Electroporation of plasmid DNA in P. putida and P. aeruginosa was carried out as indicated by Choi et al.,68 and in E. coli as described by Datsenko and Wanner.37 The accuracy of all the DNA constructs was confirmed by Sanger sequencing (Secugen SL, Madrid, Spain). A clean P. putida glk knockout mutant was obtained following the protocol described by Martínez-García and de Lorenzo.69 The detailed protocol for the construction of this Δglk mutant is described in the Supporting Information. Mutations in glycolytic genes were accumulated in E. coli derivatives via sequential transduction of individual alleles with bacteriophage P138,70 using individual mutants from the KEIO collection71 as donors, followed by elimination of the antibiotic resistance marker using plasmid pCP20.72
Design and Assembly of the GlucoBrick Modules
DNA sequences of the glycolytic genes of E. coli strain K-12 substrain MG1655 were obtained from the web-based EcoCyc database collection (GenBank accession number U00096.2). The genes were assembled in two blocks, the first of them, termed GlucoBrick Module I, spanning glk (b2388), pgi (b4025), pfkA (b3916), fbaA (b2925), and tpiA (b3919); and the second one, termed GlucoBrick Module II, comprising gapA (b1779), pgk (b2926), gpmA (b0755), eno (b2779), and pykF (b1676). Each gene was preceded by a synthetic RBS (5′-AGG AGG AAA AAC AT-3′), and the structural coding sequences were manually edited to erase targets for restriction enzymes present in SEVA plasmids (i.e., AvrII, EcoRI, SacI, KpnI, SmaI, BamHI, XbaI, SalI, PstI, SphI, and HindIII) while conserving the amino acid sequence of the encoded polypeptides. The two blocks were synthesized de novo by GeneCust Europe (GeneCust Laboratoire de Biotechnologie du Luxembourg S.A.; Dudelange, Luxembourg), and checked for accuracy by Sanger sequencing. The two DNA modules were subcloned in pSEVA vectors to obtain the plasmids indicated in Table S2 in the Supporting Information. Rapid screening of recombinants carrying Module I was carried out by colony PCR using oligonucleotides pfkA·fbaA-Check-F and pfkA·fbaA-Check-R (Table S3 in the Supporting Information). In the case of Module II, oligonucleotides gpmA·eno-Check-F and gpmA·eno-Check-R were used (Table S3 in the Supporting Information).
Preparation of Cell-Free Extracts and In Vitro Enzymatic Assays
Cell-free extracts of E. coli, P. putida, and P. aeruginosa were obtained by following published protocols.73−78 A detailed description of the procedure and the specific methods used for in vitro assays of Glk, Pfk, and GA3P dehydrogenase are given in the Supporting Information. The limit of detection for all the enzymatic assays was consistently below 2–5 nmol min–1 mg protein–1.
Analytical Determinations
PHB quantification was carried out by flow cytometry after staining the cells with Nile Red as indicated by Martínez-García et al.,32 and by means of gas chromatography as described elsewhere.79 Acetate concentration in culture supernatants of selected samples was determined by using a commercially available enzymatic kit essentially as indicated by Nikel et al.(80) with the modifications described by Nikel and de Lorenzo.81 The intracellular concentration of acetyl-CoA was determined as described by Pflüger-Grau et al.,82 with the adjustments specified by Martínez-García et al.(24) An adapted protocol based on the glucose assay kit of Sigma-Aldrich Co. was used to quantify residual glucose in culture supernatants in 96-well microtiter plates (NunclonΔSurface; Nunc A/S, Roskilde, Denmark). The assay reagent was prepared as indicated in the technical bulletin; the final mix per well contained 80 μL of the assay reagent, 40 μL of the sample (diluted with water to approximately 20–80 μg glucose mL–1), and 80 μL of 12 N H2SO4. The amount of the final pink-colored product (oxidized o-dianisidine) was quantified at 540 nm using a SpectraMax M2e plate reader (Molecular Devices, LLC., Sunnyvale, CA, USA). The supernatants for these determinations were obtained by centrifugation of 50 mL cultures (grown for 24 h) at 4000 r.p.m. for 15 min at 4 °C.
Determination of Intracellular Metabolite Concentrations
P. putida cultures carrying either the empty vector pSEVA224 or pS224·GBI were grown in M9 minimal medium containing glucose at 20 mM as explained above until they reached the midexponential phase (i.e., optical density measured at 600 nm of ca. 0.5), at which point the biomass corresponding to 0.5–0.6 mg of CDW was collected in duplicates by fast centrifugation (13 000 r.p.m., 30 s, −4 °C). Bacterial pellets were immediately frozen by immersing the cell sediment in liquid N2. Samples were then extracted three times with 0.5 mL of 60% (v/v) ethanol buffered with 10 mM ammonium acetate (pH = 7.2) at 78 °C for 1 min. After each extraction step, the biomass was separated by centrifugation at 13 000 r.p.m. for 1 min. The three liquid extracts were pooled in a new tube and dried at 120 μbar, and finally stored at −80 °C. Samples were resuspended in 20 μL of Milli-Q water and injected into a Waters Acquity UPLC system (Waters Corp., Milford, MA, USA) with a Waters Acquity T3 column (150 mm × 2.1 mm × 1.8 μm, Waters Corp.) coupled to a Thermo TSQ Quantum Ultra triple quadrupole instrument (Thermo Fisher Scientific Inc., Waltham, MA, USA) with electrospray ionization. The quantitative analysis of raw metabolomic data and the normalization procedure were conducted as explained by Nikel et al.(26) and van der Werf et al.(83)
Statistical Analysis
All the experiments reported were independently repeated at least twice (as indicated in the corresponding figure or table legend), and the mean value of the corresponding parameter ± standard deviation is presented. In some cases, the level of significance of the differences when comparing results was evaluated by means of the Student’s t test with α = 0.05.
Nucleotide Sequence Accession Numbers
The sequences of the GlucoBrick modules were deposited in the GenBank database with the GenBank accession numbers KU886714 (GlucoBrick Module I) and KU886715 (GlucoBrick Module II).
Acknowledgments
The authors are indebted to M. Chavarría (University of Costa Rica, Costa Rica), F. Corona-Pajares and J. L. Martínez (CNB-CSIC, Spain), and A. Sinskey (Massachusetts Institute of Technology, USA) for helpful discussions and for sharing materials. The skilful advice of F. Jovelin (GeneCust Europe) for in silico edition of DNA sequences is gratefully acknowledged. This work was supported by the CAMBIOS Project of the Spanish Ministry of Economy and Competitiveness (RTC-2014-1777-3), EVOPROG (FP7-ICT-610730), ARISYS (ERC-2012-ADG-322797), and EmPowerPutida (EU-H2020-BIOTEC-2014-2015-6335536) Contracts of the European Union, and the PROMPT Project of the Autonomous Community of Madrid (CAM-S2010/BMD-2414). A.S.P. was supported by a predoctoral fellowship from the JAE Programme (JAEpre CP) from CSIC (Spain).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.6b00230.
Full list of bacterial strains (Table S1), plasmids (Table S2), and oligonucleotides (Table S3) used in this study; detailed methods applied to construct mutant strains and the protocols followed for the in vitro enzymatic assays (PDF)
Author Present Address
† The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, 2800 Kongens Lyngby, Denmark.
Author Contributions
A.S.P., V.D.L., and P.I.N. designed the experiments. V.D.L. and P.I.N. conceived the whole study and wrote the article. A.S.P. and P.I.N. carried out the genetic manipulations, quantitative physiology experiments, and in vitro enzyme assays.
The authors declare no competing financial interest.
Supplementary Material
References
- Chubukov V.; Mukhopadhyay A.; Petzold C. J.; Keasling J. D.; García-Martín H. (2016) Synthetic and systems biology for microbial production of commodity chemicals. Syst. Biol. Appl. 2, 16009. 10.1038/npjsba.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. Y.; Kim H. U. (2015) Systems strategies for developing industrial microbial strains. Nat. Biotechnol. 33, 1061–1072. 10.1038/nbt.3365. [DOI] [PubMed] [Google Scholar]
- Smanski M. J.; Zhou H.; Claesen J.; Shen B.; Fischbach M. A.; Voigt C. A. (2016) Synthetic biology to access and expand Nature’s chemical diversity. Nat. Rev. Microbiol. 14, 135–149. 10.1038/nrmicro.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh C. T.; Liu Y.; Conner D. J. (2015) Rational design of efficient modular cells. Metab. Eng. 32, 220–231. 10.1016/j.ymben.2015.10.005. [DOI] [PubMed] [Google Scholar]
- Jullesson D.; David F.; Pfleger B.; Nielsen J. (2015) Impact of synthetic biology and metabolic engineering on industrial production of fine chemicals. Biotechnol. Adv. 33, 1395–1402. 10.1016/j.biotechadv.2015.02.011. [DOI] [PubMed] [Google Scholar]
- Nikel P. I.; Chavarría M.; Danchin A.; de Lorenzo V. (2016) From dirt to industrial applications: Pseudomonas putida as a Synthetic Biology chassis for hosting harsh biochemical reactions. Curr. Opin. Chem. Biol. 34, 20–29. 10.1016/j.cbpa.2016.05.011. [DOI] [PubMed] [Google Scholar]
- Wu G.; Yan Q.; Jones J. A.; Tang Y. J.; Fong S. S.; Koffas M. A. G. (2016) Metabolic burden: cornerstones in synthetic biology and metabolic engineering applications. Trends Biotechnol. 34, 652–664. 10.1016/j.tibtech.2016.02.010. [DOI] [PubMed] [Google Scholar]
- Ajikumar P. K.; Xiao W. H.; Tyo K. E. J.; Wang Y.; Simeon F.; Leonard E.; Mucha O.; Phon T. H.; Pfeifer B.; Stephanopoulos G. (2010) Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science 330, 70–74. 10.1126/science.1191652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephanopoulos G. (2012) Synthetic biology and metabolic engineering. ACS Synth. Biol. 1, 514–525. 10.1021/sb300094q. [DOI] [PubMed] [Google Scholar]
- Raab R. M.; Tyo K. E. J.; Stephanopoulos G. (2005) Metabolic engineering. Adv. Biochem. Eng. Biotechnol. 100, 1–17. 10.1007/b136411. [DOI] [PubMed] [Google Scholar]
- Antonovsky N.; Gleizer S.; Noor E.; Zohar Y.; Herz E.; Barenholz U.; Zelcbuch L.; Amram S.; Wides A.; Tepper N.; Davidi D.; Bar-On Y.; Bareia T.; Wernick D. G.; Shani I.; Malitsky S.; Jona G.; Bar-Even A.; Milo R. (2016) Sugar synthesis from CO2 in Escherichia coli. Cell 166, 115–125. 10.1016/j.cell.2016.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J. E. N.; Meyer F.; Litsanov B.; Kiefer P.; Potthoff E.; Heux S.; Quax W. J.; Wendisch V. F.; Brautaset T.; Portais J. C.; Vorholt J. A. (2015) Engineering Escherichia coli for methanol conversion. Metab. Eng. 28, 190–201. 10.1016/j.ymben.2014.12.008. [DOI] [PubMed] [Google Scholar]
- Chubukov V.; Gerosa L.; Kochanowski K.; Sauer U. (2014) Coordination of microbial metabolism. Nat. Rev. Microbiol. 12, 327–340. 10.1038/nrmicro3238. [DOI] [PubMed] [Google Scholar]
- Noor E.; Eden E.; Milo R.; Alon U. (2010) Central carbon metabolism as a minimal biochemical walk between precursors for biomass and energy. Mol. Cell 39, 809–820. 10.1016/j.molcel.2010.08.031. [DOI] [PubMed] [Google Scholar]
- Bar-Even A.; Flamholz A.; Noor E.; Milo R. (2012) Rethinking glycolysis: on the biochemical logic of metabolic pathways. Nat. Chem. Biol. 8, 509–517. 10.1038/nchembio.971. [DOI] [PubMed] [Google Scholar]
- Peekhaus N.; Conway T. (1998) What’s for dinner?: Entner–Doudoroff metabolism in Escherichia coli. J. Bacteriol. 180, 3495–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavarría M.; Nikel P. I.; Pérez-Pantoja D.; de Lorenzo V. (2013) The Entner–Doudoroff pathway empowers Pseudomonas putida KT2440 with a high tolerance to oxidative stress. Environ. Microbiol. 15, 1772–1785. 10.1111/1462-2920.12069. [DOI] [PubMed] [Google Scholar]
- Klingner A.; Bartsch A.; Dogs M.; Wagner-Döbler I.; Jahn D.; Simon M.; Brinkhoff T.; Becker J.; Wittmann C. (2015) Large-scale 13C flux profiling reveals conservation of the Entner–Doudoroff pathway as a glycolytic strategy among marine bacteria that use glucose. Appl. Environ. Microbiol. 81, 2408–2422. 10.1128/AEM.03157-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins dos Santos V. A. P.; Heim S.; Moore E. R.; Strätz M.; Timmis K. N. (2004) Insights into the genomic basis of niche specificity of Pseudomonas putida KT2440. Environ. Microbiol. 6, 1264–1286. 10.1111/j.1462-2920.2004.00734.x. [DOI] [PubMed] [Google Scholar]
- Kuepper J.; Zobel S.; Wierckx N.; Blank L. M. (2016) A rapid method to estimate NADH regeneration rates in living cells. J. Microbiol. Methods 130, 92–94. 10.1016/j.mimet.2016.09.001. [DOI] [PubMed] [Google Scholar]
- Nikel P. I.; Martínez-García E.; de Lorenzo V. (2014) Biotechnological domestication of pseudomonads using synthetic biology. Nat. Rev. Microbiol. 12, 368–379. 10.1038/nrmicro3253. [DOI] [PubMed] [Google Scholar]
- Bräsen C.; Esser D.; Rauch B.; Siebers B. (2014) Carbohydrate metabolism in Archaea: current insights into unusual enzymes and pathways and their regulation. Microbiol. Mol. Biol. Rev. 78, 89–175. 10.1128/MMBR.00041-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhees C. H.; Kengen S. W.; Tuininga J. E.; Schut G. J.; Adams M. W.; de Vos W. M.; van der Oost J. (2003) The unique features of glycolytic pathways in Archaea. Biochem. J. 375, 231–246. 10.1042/bj20021472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-García E.; Nikel P. I.; Aparicio T.; de Lorenzo V. (2014) Pseudomonas 2.0: genetic upgrading of P. putida KT2440 as an enhanced host for heterologous gene expression. Microb. Cell Fact. 13, 159. 10.1186/s12934-014-0159-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belda E.; van Heck R. G. A.; López-Sánchez M. J.; Cruveiller S.; Barbe V.; Fraser C.; Klenk H. P.; Petersen J.; Morgat A.; Nikel P. I.; Vallenet D.; Rouy Z.; Sekowska A.; Martins dos Santos V. A. P.; de Lorenzo V.; Danchin A.; Médigue C. (2016) The revisited genome of Pseudomonas putida KT2440 enlightens its value as a robust metabolic chassis. Environ. Microbiol. 18, 3403–3424. 10.1111/1462-2920.13230. [DOI] [PubMed] [Google Scholar]
- Nikel P. I.; Chavarría M.; Fuhrer T.; Sauer U.; de Lorenzo V. (2015) Pseudomonas putida KT2440 strain metabolizes glucose through a cycle formed by enzymes of the Entner–Doudoroff, Embden–Meyerhof–Parnas, and pentose phosphate pathways. J. Biol. Chem. 290, 25920–25932. 10.1074/jbc.M115.687749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel P. I.; de Lorenzo V. (2013) Engineering an anaerobic metabolic regime in Pseudomonas putida KT2440 for the anoxic biodegradation of 1,3-dichloroprop-1-ene. Metab. Eng. 15, 98–112. 10.1016/j.ymben.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Chan L. Y.; Kosuri S.; Endy D. (2005) Refactoring bacteriophage T7. Mol. Syst. Biol. 1, 2005.0018. 10.1038/msb4100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme K.; Zhao D.; Voigt C. A. (2012) Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. Proc. Natl. Acad. Sci. U. S. A. 109, 7085–7090. 10.1073/pnas.1120788109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Rocha R.; Martínez-García E.; Calles B.; Chavarría M.; Arce-Rodríguez A.; de las Heras A.; Páez-Espino A. D.; Durante-Rodríguez G.; Kim J.; Nikel P. I.; Platero R.; de Lorenzo V. (2013) The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 41, D666–D675. 10.1093/nar/gks1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K.; Sun Y.; Miyakoshi M.; Vanderpool C. K.; Vogel J. (2013) Small RNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell 153, 426–437. 10.1016/j.cell.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-García E.; Aparicio T.; de Lorenzo V.; Nikel P. I. (2014) New transposon tools tailored for metabolic engineering of Gram-negative microbial cell factories. Front. Bioeng. Biotechnol. 2, 46. 10.3389/fbioe.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-García E.; Aparicio T.; de Lorenzo V.; Nikel P. I. (2017) Engineering Gram-negative microbial cell factories using transposon vectors. Methods Mol. Biol. 1498, 273–293. 10.1007/978-1-4939-6472-7_18. [DOI] [PubMed] [Google Scholar]
- Calero P.; Jensen S. I.; Nielsen A. T. (2016) Broad-host-range ProUSER vectors enable fast characterization of inducible promoters and optimization of p-coumaric acid production in Pseudomonas putida KT2440. ACS Synth. Biol. 5, 741–753. 10.1021/acssynbio.6b00081. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Cavaleiro A. M.; Rennig M.; Nørholm M. H. (2016) SEVA Linkers: a versatile and automatable DNA backbone exchange standard for Synthetic Biology. ACS Synth. Biol. 5, 1177–1181. 10.1021/acssynbio.5b00257. [DOI] [PubMed] [Google Scholar]
- Martínez-García E.; Aparicio T.; Goñi-Moreno A.; Fraile S.; de Lorenzo V. (2015) SEVA 2.0: an update of the Standard European Vector Architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res. 43, D1183–D1189. 10.1093/nar/gku1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K. A.; Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97, 6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel P. I.; de Lorenzo V. (2013) Implantation of unmarked regulatory and metabolic modules in Gram-negative bacteria with specialised mini-transposon delivery vectors. J. Biotechnol. 163, 143–154. 10.1016/j.jbiotec.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Jahreis K.; Pimentel-Schmitt E. F.; Brückner R.; Titgemeyer F. (2008) Ins and outs of glucose transport systems in Eubacteria. FEMS Microbiol. Rev. 32, 891–907. 10.1111/j.1574-6976.2008.00125.x. [DOI] [PubMed] [Google Scholar]
- Fraenkel D. G. (1986) Mutants in glucose metabolism. Annu. Rev. Biochem. 55, 317–337. 10.1146/annurev.biochem.55.1.317. [DOI] [PubMed] [Google Scholar]
- Velur Selvamani R. S.; Telaar M.; Friehs K.; Flaschel E. (2014) Antibiotic-free segregational plasmid stabilization in Escherichia coli owing to the knockout of triosephosphate isomerase (tpiA). Microb. Cell Fact. 13, 58. 10.1186/1475-2859-13-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschi-Muller S.; Azza S.; Pollastro D.; Corbier C.; Branlant G. (1997) Comparative enzymatic properties of GapB-encoded erythrose-4-phosphate dehydrogenase of Escherichia coli and phosphorylating glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 272, 15106–15112. 10.1074/jbc.272.24.15106. [DOI] [PubMed] [Google Scholar]
- Meyer D.; Schneider-Fresenius C.; Horlacher R.; Peist R.; Boos W. (1997) Molecular characterization of glucokinase from Escherichia coli K-12. J. Bacteriol. 179, 1298–1306. 10.1128/jb.179.4.1298-1306.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Gupta R. S. (2008) Adenosine kinase and ribokinase–the RK family of proteins. Cell. Mol. Life Sci. 65, 2875–2896. 10.1007/s00018-008-8123-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J. W.; Cirino P. C. (2011) Improved NADPH supply for xylitol production by engineered Escherichia coli with glycolytic mutations. Biotechnol. Prog. 27, 333–341. 10.1002/btpr.559. [DOI] [PubMed] [Google Scholar]
- Clarke P. H. (1982) The metabolic versatility of pseudomonads. Antonie van Leeuwenhoek 48, 105–130. 10.1007/BF00405197. [DOI] [PubMed] [Google Scholar]
- Sudarsan S.; Dethlefsen S.; Blank L. M.; Siemann-Herzberg M.; Schmid A. (2014) The functional structure of central carbon metabolism in Pseudomonas putida KT2440. Appl. Environ. Microbiol. 80, 5292–5303. 10.1128/AEM.01643-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Oliveros J. C.; Nikel P. I.; de Lorenzo V.; Silva-Rocha R. (2013) Transcriptomic fingerprinting of Pseudomonas putida under alternative physiological regimes. Environ. Microbiol. Rep. 5, 883–891. 10.1111/1758-2229.12090. [DOI] [PubMed] [Google Scholar]
- Berger A.; Dohnt K.; Tielen P.; Jahn D.; Becker J.; Wittmann C. (2014) Robustness and plasticity of metabolic pathway flux among uropathogenic isolates of Pseudomonas aeruginosa. PLoS One 9, e88368. 10.1371/journal.pone.0088368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollinshead W. D.; Rodriguez S.; García-Martín H.; Wang G.; Baidoo E. E. K.; Sale K. L.; Keasling J. D.; Mukhopadhyay A.; Tang Y. J. (2016) Examining Escherichia coli glycolytic pathways, catabolite repression, and metabolite channeling using Δpfk mutants. Biotechnol. Biofuels 9, 212. 10.1186/s13068-016-0630-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez J. G. C., Méndez B. S., Nikel P. I., Pettinari M. J., Prieto M. A., and Silva L. F. (2012) Making green polymers even greener: towards sustainable production of polyhydroxyalkanoates from agroindustrial by-products, In Advances in Applied Biotechnology (Petre M., Ed.), pp 41–62, InTech, Rijeka, Croatia. [Google Scholar]
- López N. I.; Pettinari M. J.; Nikel P. I.; Méndez B. S. (2015) Polyhydroxyalkanoates: much more than biodegradable plastics. Adv. Appl. Microbiol. 93, 93–106. 10.1016/bs.aambs.2015.06.001. [DOI] [PubMed] [Google Scholar]
- Ruiz J. A.; Fernández R. O.; Nikel P. I.; Méndez B. S.; Pettinari M. J. (2006) dye (arc) Mutants: insights into an unexplained phenotype and its suppression by the synthesis of poly(3-hydroxybutyrate) in Escherichia coli recombinants. FEMS Microbiol. Lett. 258, 55–60. 10.1111/j.1574-6968.2006.00196.x. [DOI] [PubMed] [Google Scholar]
- Anderson A. J.; Dawes E. A. (1990) Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol. Rev. 54, 450–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suriyamongkol P.; Weselake R.; Narine S.; Moloney M.; Shah S. (2007) Biotechnological approaches for the production of polyhydroxyalkanoates in microorganisms and plants–A review. Biotechnol. Adv. 25, 148–175. 10.1016/j.biotechadv.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Leong Y. K.; Show P. L.; Ooi C. W.; Ling T. C.; Lan J. C. (2014) Current trends in polyhydroxyalkanoates (PHAs) biosynthesis: insights from the recombinant Escherichia coli. J. Biotechnol. 180, 52–65. 10.1016/j.jbiotec.2014.03.020. [DOI] [PubMed] [Google Scholar]
- Nikel P. I.; Giordano A. M.; de Almeida A.; Godoy M. S.; Pettinari M. J. (2010) Elimination of D-lactate synthesis increases poly(3-hydroxybutyrate) and ethanol synthesis from glycerol and affects cofactor distribution in recombinant Escherichia coli. Appl. Environ. Microbiol. 76, 7400–7406. 10.1128/AEM.02067-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz J. A.; de Almeida A.; Godoy M. S.; Mezzina M. P.; Bidart G. N.; Méndez B. S.; Pettinari M. J.; Nikel P. I. (2012) Escherichia coli redox mutants as microbial cell factories for the synthesis of reduced biochemicals. Comput. Struct. Biotechnol. J. 3, e201210019. 10.5936/csbj.201210019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D. E.; Shin S.; Rhee J. S.; Pan J. G. (1999) Acetate metabolism in a pta mutant of Escherichia coli W3110: importance of maintaining acetyl coenzyme A flux for growth and survival. J. Bacteriol. 181, 6656–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvořák P.; Chrást L.; Nikel P. I.; Fedr R.; Soucek K.; Sedlacková M.; Chaloupková R.; de Lorenzo V.; Prokop Z.; Damborský J. (2015) Exacerbation of substrate toxicity by IPTG in Escherichia coli BL21(DE3) carrying a synthetic metabolic pathway. Microb. Cell Fact. 14, 201. 10.1186/s12934-015-0393-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal V.; Castaño-Cerezo S.; Cánovas M. (2016) Acetate metabolism regulation in Escherichia coli: carbon overflow, pathogenicity, and beyond. Appl. Microbiol. Biotechnol. 100, 8985–9001. 10.1007/s00253-016-7832-x. [DOI] [PubMed] [Google Scholar]
- Emmerling M.; Bailey J. E.; Sauer U. (1999) Glucose catabolism of Escherichia coli strains with increased activity and altered regulation of key glycolytic enzymes. Metab. Eng. 1, 117–127. 10.1006/mben.1998.0109. [DOI] [PubMed] [Google Scholar]
- Solomon K. V.; Moon T. S.; Ma B.; Sanders T. M.; Prather K. L. (2013) Tuning primary metabolism for heterologous pathway productivity. ACS Synth. Biol. 2, 126–135. 10.1021/sb300055e. [DOI] [PubMed] [Google Scholar]
- Li R.; Zhang H.; Qi Q. (2007) The production of polyhydroxyalkanoates in recombinant Escherichia coli. Bioresour. Technol. 98, 2313–2320. 10.1016/j.biortech.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Green M. R., and Sambrook J. (2012) Molecular Cloning: A Laboratory Manual, 4th ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Thomson J.; Gerstenberger P. D.; Goldberg D. E.; Gociar E.; Orozco de Silva A.; Fraenkel D. G. (1979) ColE1 hybrid plasmids for Escherichia coli genes of glycolysis and the hexose monophosphate shunt. J. Bacteriol. 137, 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Zhao G.; Man T. K.; Winkler M. E. (1998) Involvement of the gapA- and epd (gapB)-encoded dehydrogenases in pyridoxal 5′-phosphate coenzyme biosynthesis in Escherichia coli K-12. J. Bacteriol. 180, 4294–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K. H.; Kumar A.; Schweizer H. P. (2006) A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64, 391–397. 10.1016/j.mimet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Martínez-García E.; de Lorenzo V. (2011) Engineering multiple genomic deletions in Gram-negative bacteria: analysis of the multi-resistant antibiotic profile of Pseudomonas putida KT2440. Environ. Microbiol. 13, 2702–2716. 10.1111/j.1462-2920.2011.02538.x. [DOI] [PubMed] [Google Scholar]
- Godoy M. S.; Nikel P. I.; Cabrera Gomez J. G.; Pettinari M. J. (2016) The CreC regulator of Escherichia coli, a new target for metabolic manipulations. Appl. Environ. Microbiol. 82, 244–254. 10.1128/AEM.02984-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T.; Ara T.; Hasegawa M.; Takai Y.; Okumura Y.; Baba M.; Datsenko K. A.; Tomita M.; Wanner B. L.; Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008. 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P. P.; Wackernagel W. (1995) Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14. 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- Nikel P. I.; Kim J.; de Lorenzo V. (2014) Metabolic and regulatory rearrangements underlying glycerol metabolism in Pseudomonas putida KT2440. Environ. Microbiol. 16, 239–254. 10.1111/1462-2920.12224. [DOI] [PubMed] [Google Scholar]
- Nikel P. I.; Pérez-Pantoja D.; de Lorenzo V. (2013) Why are chlorinated pollutants so difficult to degrade aerobically? Redox stress limits 1,3-dichloroprop-1-ene metabolism by Pseudomonas pavonaceae. Philos. Trans. R. Soc. B 368, 20120377. 10.1098/rstb.2012.0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel P. I.; Romero-Campero F. J.; Zeidman J. A.; Goñi-Moreno A.; de Lorenzo V. (2015) The glycerol-dependent metabolic persistence of Pseudomonas putida KT2440 reflects the regulatory logic of the GlpR repressor. mBio 6, e00340–00315. 10.1128/mBio.00340-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel P. I.; Zhu J.; San K. Y.; Méndez B. S.; Bennett G. N. (2009) Metabolic flux analysis of Escherichia coli creB and arcA mutants reveals shared control of carbon catabolism under microaerobic growth conditions. J. Bacteriol. 191, 5538–5548. 10.1128/JB.00174-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel P. I., and Chavarría M. (2016) Quantitative physiology approaches to understand and optimize reducing power availability in environmental bacteria, In Hydrocarbon and Lipid Microbiology Protocols—Synthetic and Systems Biology—Tools (McGenity T. J., Timmis K. N., and Nogales-Fernández B., Eds.), pp 39–70, Humana Press, Heidelberg, Germany. [Google Scholar]
- Chavarría M.; Goñi-Moreno A.; de Lorenzo V.; Nikel P. I. (2016) A metabolic widget adjusts the phosphoenolpyruvate-dependent fructose influx in Pseudomonas putida. mSystems 1, e00154–00116. 10.1128/mSystems.00154-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel P. I.; Pettinari M. J.; Galvagno M. A.; Méndez B. S. (2006) Poly(3-hydroxybutyrate) synthesis by recombinant Escherichia coli arcA mutants in microaerobiosis. Appl. Environ. Microbiol. 72, 2614–2620. 10.1128/AEM.72.4.2614-2620.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel P. I.; Ramirez M. C.; Pettinari M. J.; Méndez B. S.; Galvagno M. A. (2010) Ethanol synthesis from glycerol by Escherichia coli redox mutants expressing adhE from Leuconostoc mesenteroides. J. Appl. Microbiol. 109, 492–504. 10.1111/j.1365-2672.2010.04668.x. [DOI] [PubMed] [Google Scholar]
- Nikel P. I.; de Lorenzo V. (2014) Robustness of Pseudomonas putida KT2440 as a host for ethanol biosynthesis. New Biotechnol. 31, 562–571. 10.1016/j.nbt.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Pflüger-Grau K.; Chavarría M.; de Lorenzo V. (2011) The interplay of the EIIANtr component of the nitrogen-related phosphotransferase system (PTSNtr) of Pseudomonas putida with pyruvate dehydrogenase. Biochim. Biophys. Acta. 1810, 995–1005. 10.1016/j.bbagen.2011.01.002. [DOI] [PubMed] [Google Scholar]
- van der Werf M. J.; Overkamp K. M.; Muilwijk B.; Koek M. M.; van der Werff-van der Vat B. J.; Jellema R. H.; Coulier L.; Hankemeier T. (2008) Comprehensive analysis of the metabolome of Pseudomonas putida S12 grown on different carbon sources. Mol. BioSyst. 4, 315–327. 10.1039/b717340g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.