

Abstract
West Nile virus (WNV) is a human pathogen that can cause neurological disorders, including meningoencephalitis. Experiments with mice and mammalian cell cultures revealed that WNV exhibited resistance to the innate immune program induced by alpha interferon (IFN-α). We have investigated the nature of this inhibition and have found that WNV replication inhibited the activation of many known IFN-inducible genes, because it prevented the phosphorylation and activation of the Janus kinases JAK1 and Tyk2. As a consequence, activation of the transcription factors STAT1 and STAT2 did not occur in WNV-infected cells. Moreover, we demonstrated that the viral nonstructural proteins are responsible for this effect. Thus, our results provided an explanation for the observed resistance of WNV to IFN-α in cells of vertebrate origin.
West Nile virus (WNV) is an enveloped positive-strand RNA virus which, along with other mosquito-borne human pathogenic viruses, including Yellow fever virus and dengue viruses, belongs to the genus Flavivirus in the family Flaviviridae (3). WNV infects migratory and other birds, which produce high virus titers in their blood and thereby permit transmission of the virus to mosquitoes and, eventually, to humans. Although WNV was isolated from an individual in Uganda more than 6 decades ago, it has been recognized as a major human pathogen only recently, when outbreaks of human encephalitis were reported in Romania, Russia, Israel, and, in particular, in New York City in 1999 (1, 19, 31). Subsequently, the virus has spread throughout the continental United States. Whether recent disease outbreaks were a consequence of the emergence of new, pathogenic WNV strains or reflected a lack of immunity in the population is not yet known. Phylogenetic analyses based on the nucleotide sequence of a segment of the envelope gene led to the classification of WNV isolates into two lineages (19). This analysis also revealed a close relationship among the WNV isolates involved in the recent outbreaks in the western hemisphere.
Infectious cDNA clones were first reported for Kunjin virus, a subtype of WNV belonging to lineage 1, and later for two WNV isolates representing both lineages (16, 32, 36). The genomes are approximately 11 kb long and contain a large open reading frame that is flanked by noncoding regions containing the promoters for RNA-dependent RNA synthesis (3, 35). The polyprotein is processed into 10 polypeptides by cellular and viral proteases. Three of these products are structural proteins required for capsid formation (capsid protein) and assembly into enveloped viral particles (premembrane and envelope proteins). The nonstructural (NS) proteins comprise a serine protease and ATP-dependent helicase (NS3), a RNA-dependent RNA polymerase (NS5), and a cofactor of the NS3 protease (NS2B). The functions of the remaining four NS proteins, NS1, NS2A, NS4A, and NS4B, are not yet known. Khromykh and Westaway demonstrated that subgenomic replicons of Kunjin virus expressing the NS proteins were competent for RNA replication (17). Their studies set the path for the development of similar replicon systems with other members of the Flaviviridae, including Hepatitis C virus (HCV) (23).
Like other arboviruses, WNV has the remarkable ability to replicate and assemble virus particles in insect and mammalian cells; hence, it can complete its life cycle under very different environmental conditions. Both invertebrates and vertebrates rely on cellular antiviral programs, the innate immune response, to regulate amplification of viral genomes and to protect cells from infection. Darnell et al. discovered that WNV, compared with other viruses, including vesicular stomatitis virus (VSV) and Sindbis virus, exhibited a marked resistance to the innate immune response elicited by alpha interferon (IFN-α) (4, 5). Moreover, additional studies with WNV and Dengue virus revealed that IFN-α did not inhibit viral replication following the establishment of an infection, suggesting that expression of one or several viral proteins may inhibit the IFN response (7, 24, 27). These observations stand in marked contrast with results reported with HCV, demonstrating that this virus is very sensitive to the antiviral program induced by IFN-α in tissue culture cells (2, 12, 15).
IFNs mediate their biological functions by binding to their cognate receptors on target cells, which induces a signal transduction pathway leading to the induction of tens or even hundreds of genes (6, 21). Besides the IFN receptors, the principal components include the Janus tyrosine kinases JAK1, JAK2, and Tyk2 and the latent transcription factors STAT1, STAT2, and IRF9 (p48). The IFN-induced signals are transduced through sequential phosphorylations of tyrosine residues on the Janus kinases, the IFN receptors, and the STAT proteins. The induction of gene expression is mediated by the formation of multimeric transcription factors that bind to their cognate DNA sequence motifs located in the promoters of IFN-induced genes (21, 33).
The purpose of this study was to investigate the basis for the observed resistance of WNV to the antiviral program induced by IFN-α. The results showed that the expression of one or more of the WNV NS proteins interfered with the activation of an early step in the JAK-STAT signal transduction pathway.
MATERIALS AND METHODS
Cells, viruses, and reagents.
HeLa, Vero, and BHK cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin G, streptomycin, nonessential amino acids, and l-glutamine. SL1 is a HeLa cell line expressing an HCV subgenomic replicon (40). The KUNCD20 cells were derived from a pool of approximately 200 colonies of G418-resistant HeLa cells obtained after transfection with the Kunjin virus subgenomic replicon C20DxrepNeo RNA (14, 17). The cell lines were cultured in medium containing 500 μg of G418/ml. WNV (lineage 2) was produced in BHK cells that were electroporated with 1 μg of synthetic RNA transcribed from plasmid pSP6WNrep/Xba as described by Yamshchikov et al. (36) (accession number NC_001563).
Recombinant IFN-α2b (Intron A) and IFN-γ were obtained from Schering-Plough and Pierce, respectively. Antibodies against STAT1, STAT2, JAK1, Tyk2, IFN-α/β receptor chain 1 (IFNAR1), and β-actin were obtained from Santa Cruz Biotechnology. Antibodies against p-STAT1, p-JAK1, and p-Tyk2 were obtained from Cell Signaling Technologies. Antibodies against p-STAT2 and IFNAR2 were obtained from Upstate Biotechnology and Pierce, respectively.
RNA extraction and Northern blot hybridization.
SL1 and KUNCD20 cells were left untreated or were treated for 3 days with the indicated concentrations of recombinant IFN-α2b (Intron A; Schering-Plough) and IFN-γ (Pierce), respectively. Total cellular RNA was extracted with TRIzol reagent (Invitrogen) following the manufacturer's directions. Five micrograms of total RNA was fractionated on a 1% agarose gel containing 2.2 M formaldehyde and transferred onto nylon membranes. Membranes were hybridized with a riboprobe specific for the neomycin phosphotransferase II (NPT II) gene.
Western blot analysis.
For the analysis of STAT1 and STAT2 phosphorylation, cells were left untreated or were treated for 30 min with 1,000 IU of IFN-α and IFN-γ/ml, respectively. For the analysis of IFN receptor and its associated JAK family of protein tyrosine kinases, cells were cultured in Dulbecco's modified Eagle's medium containing 1% fetal bovine serum for 24 h and then mock treated or treated for 15 min with 1,000 IU of IFN-α and IFN-γ/ml, respectively. Upon the completion of treatment, cells in 100-mm-diameter petri dishes were washed once with ice-cold phosphate-buffered saline (PBS) and subsequently lysed with 800 μl of lysis buffer (20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 1% NP-40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate [SDS]) supplemented with protease inhibitor cocktail (Roche) and 0.1 mM sodium orthovanadate. Lysates were centrifuged for 5 min at 10,000 × g at 4°C. Equal amounts of cell lysates were separated by SDS-polyacrylamide gel electrophoresis, transferred on Immuno-P membrane (Millipore), probed with antibodies for STAT1α p91 (sc-417; Santa Cruz Biotechnology) and STAT2 (sc-1668; Santa Cruz Biotechnology), phosphotyrosine 701-STAT1 (9171; Cell Signaling Technologies), phosphotyrosine 689-STAT2 (07-224; Upstate Technology), IFNAR1 (sc-7391; Santa Cruz Biotechnology), JAK1 (sc-277; Santa Cruz Biotechnology), Tyk2 (sc-169; Santa Cruz Biotechnology), phosphotyrosine 1022/1023-JAK1 (3331; Cell Signaling Technologies), phosphotyrosine 1054/1055-Tyk2 (9321; Cell Signaling Technologies), or β-actin (sc-1616; Santa Cruz Biotechnology), and visualized with Super-Signal chemiluminescence reagents (Pierce).
DNA microarray analysis.
Normal HeLa and KUNCD20 cells were mock treated or treated with 100 IU of IFN-α or IFN-γ/ml for 6 h, and total cellular RNAs were extracted with TRIzol reagent (Invitrogen) and further purified with a RNeasy kit (QIAGEN). A total of 25 μg of each RNA sample was used as a template to make double-stranded cDNA by reverse transcription with oligo(dT)12-18 primer and deoxynucleoside triphosphate mixed with amino allyl-dUTP by using a cDNA indirect labeling kit essentially as described previously (13). The cDNA was then purified and labeled with N-hydroxysuccinimide ester containing Cy3 or Cy5 dye in coupling reactions (Amersham Biosciences). The labeled probes were purified by using a QIAquick PCR purification kit (QIAGEN) by following the manufacturer's directions and hybridized to a microarray containing 15,552 human oligonucleotide sets obtained from MWG Biotech (High Point, N.C.) in 38 μl of hybridization buffer (5× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 0.1% SDS, and 50% formamide) at 42°C for 16 h. Arrays were then washed and analyzed with an Affymetrix 428 scanner. The raw data were analyzed with Imagene 5.6 and Genesight 4 software (Biodiscovery). The complete data set of the microarray experiments can be viewed at the following URL: http://www.fccc.edu/research/labs/seeger/docs/.
CPE assay.
Cells were seeded in 24-well tissue culture dishes with 5 × 104 cells per well and were incubated for 24 h. The cells were treated with the indicated concentrations of IFN-α or IFN-γ for 24 h and then infected with VSV strain Indiana at a multiplicity of infection of 1. Twenty-four hours postinfection, the cells were washed once with PBS, fixed with 10% formaldehyde, and then stained with crystal violet to examine the cytopathic effect (CPE).
Immunofluorescence.
Cells cultured on glass coverslips in six-well plates were mock-treated or treated with 1,000 IU of IFN-α or IFN-γ/ml for 30 min and fixed with 4% of paraformaldehyde in PBS for 10 min. After three washes with PBS, cells were permeabilized with 0.5% Triton X-100 in a blocker solution (2% bovine serum albumin and 10% fetal bovine serum in PBS) for 30 min. Samples were then incubated with mouse monoclonal antibodies for STAT1 and STAT2 for 1 h. Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse immunoglobulin G (IgG; Jackson Laboratories) was used to visualize STAT1 and STAT2 proteins. For the double immunostaining of WNV-infected HeLa cells, cells cultured on glass coverslips in six-well plates were infected with WNV. Twenty-four hours postinfection, cells were treated with IFNs, fixed with 4% of paraformaldehyde, and permeabilized with 0.5% Triton X-100 in the blocker solution as described above. After each subsequent antibody exposure, samples were washed with PBS and blocked with blocker solution. Antibody staining was performed sequentially, starting with STAT1 or STAT2 antibody at a 1:100 dilution, followed by FITC-conjugated goat anti-mouse IgG at a 1:200 dilution and then West Nile hyperimmune ascitic fluid (V554-701-562; American Type Culture Collection) at a dilution of 1:1,000 and rhodamine-conjugated goat anti-mouse IgG (Jackson Laboratories) at a dilution of 1:1,000. Cell nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole). The slides were examined with a Nikon fluorescence microscope and photographed with a charge-coupled device camera.
RESULTS
Attenuated IFN responses in cells expressing Kunjin but not HCV subgenomes.
Our investigations were prompted by the observation that the replication of subgenomic replicons expressing the NS proteins of Kunjin virus was much more resistant than the replication of similar replicons derived from HCV to the antiviral response induced by both IFN-α and IFN-γ (Fig. 1) (14). The 50% inhibitory concentration of IFN-α against Kunjin virus was approximately 100 times that observed with HCV replicons; with IFN-γ, the 50% inhibitory concentration differed about 1,000-fold. Although Kunjin virus and WNV belong to the same lineage of known WNV isolates, sharing more than 95% amino acid homology, we will refer to the subgenomic replicon as Kunjin, which is consistent with the original nomenclature of this isolate (17).
FIG. 1.

Antiviral activities of IFN-α and IFN-γ against HCV and Kunjin replicons in HeLa cell lines. (A) Viral RNA levels present in SL1 and KUNCD20 cells incubated with increasing concentrations of IFN-α (0 and 10−4 to 100 IU/ml in increments of 10; lanes 1 to 8 and 10 to 17) and IFN-γ (lanes 18 to 25 and 27 to 34) for 72 h were determined by Northern blot analysis. rRNA (28S rRNA) served as a control for the amount of RNA loaded per lane. (B) The amounts of HCV RNA were determined with a Fuji phosphoimager. The values plotted as □, ♦, ○, and ▵ are the percentages of the values obtained with the cells in lanes 1, 18, 10, and 27, respectively, of panel A. KUN, KUNCD20 cells.
To determine whether the replication of Kunjin replicons led to a general inhibition of the IFN response or whether the replicons were otherwise protected from the antiviral program induced by the cytokines, we compared the protective effects of IFN-α and IFN-γ on VSV infection in HeLa cells, in HeLa cells expressing HCV (termed SL1), and in HeLa cells expressing Kunjin virus subgenomes (termed KUNCD20). The results showed that IFN-α did not protect KUNCD20 cells from the cytopathic effects caused by VSV and that IFN-γ had only a modest effect (Fig. 2). In contrast, the cytokines were effective in HeLa and HCV-replicating SL1 cells. As reported previously, a fraction of SL1 cells underwent apoptosis in the presence of the cytokine (14). Hence, these results indicated that the resistance of Kunjin replicons to IFN was caused by a more general inhibition of the IFN response in KUNCD20 cells rather than by the absence of a viral target for the IFN-induced innate immune response.
FIG. 2.

Replication of Kunjin replicons in HeLa cells blocks the antiviral response of IFN-α and IFN-γ against VSV. Normal HeLa, SL1, and KUNCD20 (KUN) cells were incubated for 24 h with the indicated concentrations of IFN-α and IFN-γ and then infected with VSV at a multiplicity of infection of 1. Twenty-four hours later, cells were stained with crystal violet to determine the cytopathic effects. All cell lines were assayed in duplicate. The cells in columns C were not infected with VSV.
Replication of Kunjin subgenomic replicons in HeLa cells inhibited IFN-induced gene expression.
To gain a better understanding of the mechanism responsible for the observed inhibition of the IFN response in KUNCD20 cells, we performed DNA microarray analyses to compare the induction of IFN-stimulated genes (ISG) in parental HeLa and KUNCD20 cells treated with IFN-α. The results revealed that after IFN-α treatment, many of the known ISG were expressed at reduced levels in KUNCD20 cells compared with HeLa cells (Table 1). Among 41 genes that were induced more than 2-fold in HeLa cells, the levels of inhibition in KUNCD20 cells varied from more than 10-fold (ifit1, p56) to 2- to 3-fold depending on the gene. Notably, the induction of at least one gene [NM_003641_1; interferon-induced transmembrane protein 1 (9-27); ifitm1] was not inhibited by Kunjin replication, and five other genes differed less than twofold, suggesting that the observed inhibition was not absolute. Nevertheless, these results invoked the hypothesis that one or several NS proteins interfered with the activation of the IFN-induced antiviral program. Consistent with the CPE results (Fig. 2), IFN-α treatment of SL1 cells resulted in the induction of ISG essentially as observed with HeLa cells (J. P. Hayashi, J.-T. Guo, and C. Seeger, unpublished results).
TABLE 1.
IFN-α-inducible gene expression is inhibited in KUNCD20 cells
| Gene identification no. and description | Mean induction level fora
|
Fold change | |
|---|---|---|---|
| HeLa cells | KUNCD20 cells | ||
| NM_001548_1; IFN-induced protein with tetratricopeptide repeats 1; ifit1 | 34.28* | 2.75 | 12.5 |
| XM_038988_1; similar to unknown (protein for image: 3155889) | 16.54 | 2.34 | 7.1 |
| NM_005101_1; IFN-stimulated protein, 15 kDa; isg15 | 12.81 | 2.01 | 6.4 |
| NM_006417_1; HCV-associated microtubular aggregate; mtap44 | 8.27 | 1.56 | 5.3 |
| AL160008.3.1.85671.1; Ensembl Genscan prediction | 7.62 | 1.62 | 4.7 |
| NM_002053_1; guanylate binding protein 1, IFN inducible, 67 kDa; gbp1 | 7.82 | 1.68 | 4.7 |
| NM_004223_1; ubiquitin-conjugating enzyme E2l6; ube2l6 | 7.99 | 1.85 | 4.3 |
| AF086924_1; protein phosphatase 2al b gamma subunit; imypnol | 2.73 | 0.66 | 4.2 |
| NM_017654_1; hypothetical protein Flj20073; flj20073 | 9.04 | 2.19 | 4.1 |
| NM_016323_1; cyclin-e binding protein 1; loc51191 | 5.14 | 1.31 | 3.9 |
| BC005986_1; unknown (protein for image 3685398) | 4.06 | 1.08 | 3.8 |
| NM_002463_1; myxovirus (influenza) resistance 2; mx2 | 4.72 | 1.26 | 3.7 |
| NM_032036_1; Tlh29 protein precursor; tlh29 | 5.17 | 1.44 | 3.6 |
| NM_005531_1; IFN-γ-inducible protein 16; ifi16 | 7.06 | 2.02 | 3.5 |
| AB025254_1; tudor repeat associator with PCTAIRE 2 | 7.09 | 2.13 | 3.3 |
| NM_001572_1; IFN regulatory factor 7 isoform a; irf7 | 4.49 | 1.39 | 3.2 |
| AB006746_1; hmmtra1b | 10.37 | 3.32 | 3.1 |
| NM_016817_1; 2′, 5′-oligoadenylate synthetase 2, isoform p71; oas2 | 4.45 | 1.48 | 3 |
| NM_012420_1; retinoic acid- and IFN-inducible protein (58kDa), ri58 | 4.59 | 1.63 | 2.8 |
| NM_002534_1; 2′, 5′-oligoadenylate synthetase 1, isoform e16, oas1 | 4.56 | 1.65 | 2.8 |
| NM_006187_1; 2′, 5′-oligoadenylate synthetase 3; oas3 | 3.93 | 1.45 | 2.7 |
| NM_007315_1; signal transducer and activator of transcription 1, 91 kDa; stat1 | 5.09 | 1.88 | 2.7 |
| NM_015474_1; Dkfzp564a032 protein, samhd1 | 3.18 | 1.26 | 2.5 |
| NM_022750_1; hypothetical protein Flj22693; flj22693 | 4.91 | 1.98 | 2.5 |
| NM_006820_1; hypothetical protein, expressed in osteoblast; gs3686 | 7.61 | 3.12 | 2.4 |
| NM_002675_1; promyelocytic leukemia protein, isoform 6; pml | 2.81 | 1.21 | 2.3 |
| NM_002462_1; myxovirus (influenza) resistance 1, mx1 | 11.09 | 4.87 | 2.3 |
| AF159441_1; phospholipid scramblase 2 | 7.02 | 3.06 | 2.3 |
| BC017212_1; unknown (protein for mgc 10254) | 2.59 | 1.15 | 2.3 |
| NM_014314_1; RNA helicase, rig-1 | 11.99 | 5.41 | 2.2 |
| AF297093_5; UDP glucuronosyltransferase 1a6; ugt1a6 | 4.72 | 2.17 | 2.2 |
| NM_024293; hypothetical protein Mgc3035, mgc3035 | 2.26 | 1.05 | 2.2 |
| NM_007346_1; 7-60 protein; ogfr | 3.23 | 1.57 | 2.1 |
| NM_017414_1; ubiquitin specific protease 18; usp18 | 4.89 | 2.46 | 2 |
| NM_003733_1; 2′, 5′-oligoadenylate synthetase-like; oas1 | 3.66 | 1.89 | 2 |
| NM_001549_1; IFN-induced protein with tetratricopeptide repeats 4; ifit4 | 2.88 | 1.55 | 1.9 |
| NM_006084_1; IFN stimulated transcription factor 3, gamma (48 kDa) | 2.52 | 1.41 | 1.8 |
| AF167472_1; double-stranded RNA-activated protein kinase; pkr | 2.49 | 1.44 | 1.7 |
| AF307338_1; b aggressive lymphoma long isoform; bal | 8.99 | 6.45 | 1.4 |
| NM_016582_1; peptide transporter 3; pht2 | 3.31 | 2.62 | 1.3 |
| NM_003641_1; IFN-induced transmembrane protein 1 (9-27); ifitm1 | 8.01 | 14.05 | 0.6 |
Mean induction levels for two experiments are shown.
WNV infection prevented phosphorylation of STAT1 and STAT2.
To test our hypothesis, we investigated the levels of tyrosine phosphorylation and nuclear translocation of STAT1 and STAT2 in IFN-treated cells. Both events are required for the induction of IFN-induced genes. Analyses of total STAT1 and STAT2 proteins in cell extracts prepared from IFN-α- and IFN-γ-treated cells showed that they were present at similar levels in all three cell lines analyzed (Fig. 3A). In contrast, the levels of tyrosine-phosphorylated STAT1 and STAT2 were reduced in IFN-α-treated KUNCD20 cells compared with HeLa or SL1 cells. In IFN-γ-treated KUNCD20 cells, some phosphorylated STAT1 could still be detected, which could explain the attenuated antiviral response observed with VSV-infected cells (Fig. 2). However, the amounts of phosphorylated STAT proteins in HeLa and SL1 cells were relatively low; therefore, we could not determine whether the levels of phosphorylated STAT1 in KUNCD20 cells were reduced more than two- to threefold compared with the other HeLa cells.
FIG. 3.

WNV infection prevents the activation of STAT1 and STAT2. (A) Normal HeLa, SL1, and KUNCD20 cells were left untreated or were treated with 1,000 IU of IFN-α or IFN-γ/ml for 30 min., and equal amounts of cell lysates were separated by SDS-polyacrylamide gel electrophoresis and transferred onto Immobilon-P membranes. The expression of STAT and tyrosine-phosphorylated STAT proteins were detected by Western blot analysis with the corresponding specific antibodies. β-Actin levels served as a loading control. (B) Vero cells were infected with WNV, and at 24 h postinfection, the cells were left untreated or treated for 30 min. with 1,000 IU of IFN-α or IFN-γ/ml. Western blot analysis was performed as described above. The figure shows results from one of three experiments that yielded consistent results.
To validate our observations made with HeLa cells replicating subgenomic Kunjin replicons, we performed additional experiments with Vero cells that were infected with WNV released from BHK cells transfected with an infectious full-length cDNA clone (see Materials and Methods). The Vero cells were harvested 24 h after infection, when almost all of the cells were infected as determined by immunofluorescence but before the appearance of CPE (results not shown). While the accumulation of total STAT1 and STAT2 proteins was elevated in WNV-infected Vero cells, IFN-α-induced phosphorylation of both STAT proteins was significantly inhibited compared with control cells (Fig. 3B). As with KUNCD20 cells, IFN-γ-induced STAT1 phosphorylation could be detected in WNV-infected Vero cells, but at a significantly reduced level compared with uninfected cells.
To further confirm our results, we determined the cellular distribution of STAT1 and STAT2 in IFN-treated HeLa cells. As expected, IFN-α induced the nuclear translocation of both proteins in HeLa and SL1 cells. In contrast, in IFN-α-treated KUNCD20 cells both STAT proteins remained in the cytoplasm (Fig. 4A and B). STAT1 could be detected in the nuclei of IFN-γ-treated KUNCD20 cells, which was consistent with the observed accumulation of tyrosine-phosphorylated STAT1 in these cells (Fig. 3A and 4A). Similar observations were made with WNV-infected HeLa cells that were used in lieu of Vero cells for these experiments. Under our experimental conditions, approximately 40% of HeLa cells were infected by WNV. IFN-α-induced nuclear translocation of both STAT proteins was completely blocked only in cells that expressed WNV proteins (Fig. 5A and B). In contrast, in IFN-γ-treated cells STAT1 appeared to accumulate in both the cytoplasmic and nuclear compartments.
FIG. 4.

Nuclear accumulation of STAT1 and STAT2 in the presence of IFN-α and IFN-γ. Normal HeLa, SL1, and KUNCD20 cells were seeded on glass coverslips and left untreated (control) or were treated with 1,000 IU of IFN-α or IFN-γ/ml for 30 min. The cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and incubated first with STAT1 (A) and STAT2 (B) monoclonal antibodies and then with FITC-conjugated goat anti-mouse IgG. The cell nuclei were stained with DAPI. The slides were examined with a Nikon fluorescence microscope and photographed with a charge-coupled device camera.
FIG. 5.
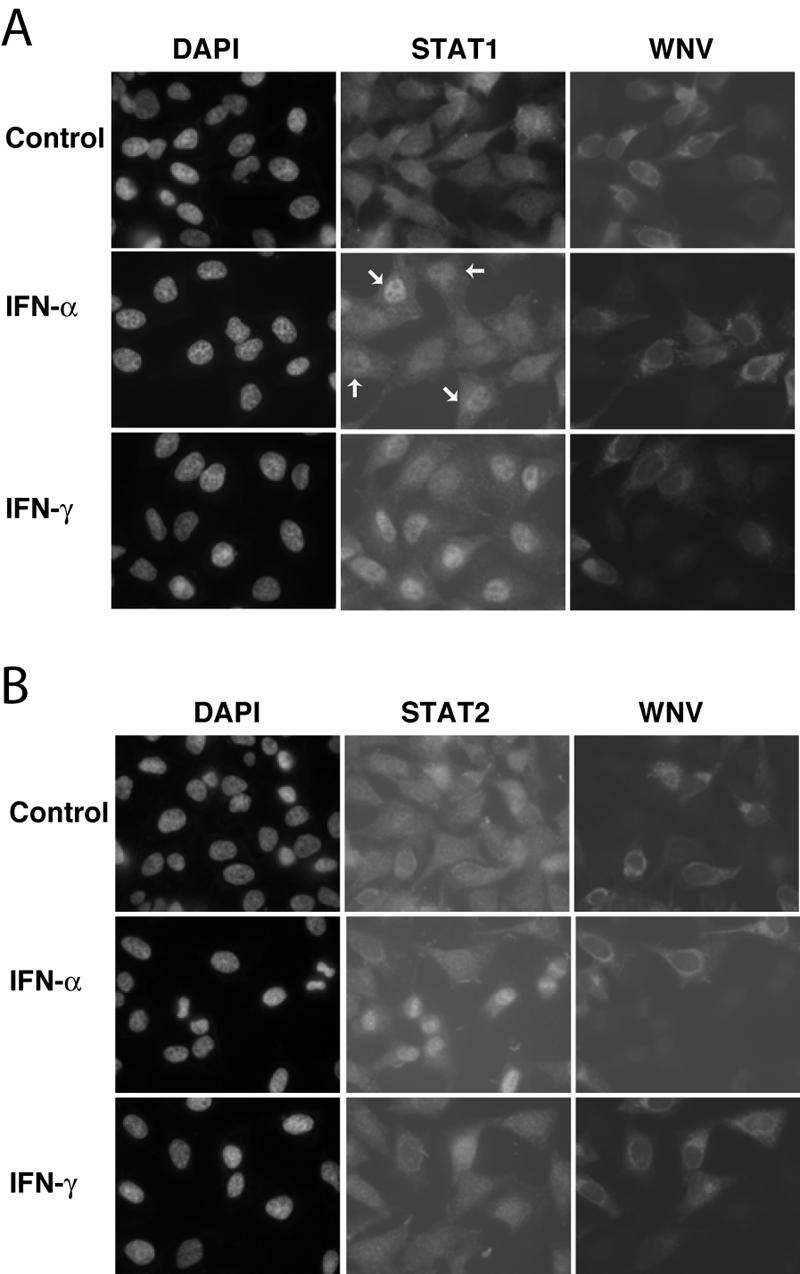
Localization of STAT1 and STAT2 in WNV-infected HeLa cells. HeLa cells were seeded on glass coverslips and infected with WNV for 24 h. The cells were then left untreated (control) or were treated with 1,000 IU of IFN-α or IFN-γ/ml for 30 min, fixed with 4% paraformaldehyde, and permeabilized with 0.5% Triton X-100. STAT1, STAT2, and WNV proteins were visualized by sequential staining of the samples with STAT1 (A) or STAT2 (B) antibodies and WNV-specific antibodies. The cell nuclei were stained with DAPI. The slides were examined with a Nikon fluorescence microscope and photographed with a charge-coupled device camera. Arrows mark uninfected cells with nuclear accumulation of STAT1.
Taken together, our results showed that WNV and Kunjin subgenomes did not inhibit the accumulation of total STAT proteins, but either reduced the steady-state levels of tyrosine phosphorylated STAT proteins, inhibited their phosphorylation, or both. Hence, our results were consistent with the hypothesis predicting that replication of WNV blocked the signal transduction pathways responsible for the activation of IFN-α-and IFN-γ-induced genes.
WNV reduces the tyrosine phosphorylation of JAK1 and Tyk2.
A first step in the activation of the IFN signal transduction cascade is the autophosphorylation of the tyrosine kinases JAK1 and Tyk2 in response to a ligand-induced conformational change of the IFN-α/β receptors. Expression of Kunjin replicons in HeLa cells did not significantly alter the steady-state levels of JAK1 and Tyk2, whereas WNV infection of Vero cells slightly down-regulated the expression of both kinases (Fig. 6). However, IFN-α-induced phosphorylation of both JAK1 and Tyk2 was efficiently blocked in KUNCD20 and in WNV-infected Vero cells (Fig. 6). Moreover, reduced levels of JAK1 phosphorylation were also observed with IFN-γ-treated KUNCD20 and WNV-infected cells. As expected, IFN-γ treatment did not induce Tyk2 phosphorylation in any of the cell lines examined during this study.
FIG. 6.

WNV inhibits the tyrosine phosphorylation of JAK1 and Tyk2. (A) Normal HeLa, SL1, and KUNCD20 cells were left untreated or were treated with 1,000 IU of IFN-α or IFN-γ/ml for 15 min. Proteins from total cellular extracts were analyzed by Western blot analysis. The blots were probed with antibodies against IFNAR1, JAK1, phosphotyrosine 1022/1023-JAK1, Tyk2, phosphotyrosine 1054/1055-Tyk2, and β-actin. (B) Vero cells were infected with WNV, and at 24 h postinfection, the cells were left untreated or were treated with 1,000 IU of IFN-α or IFN-γ/ml for 15 min. Western blot analysis was performed as described above. Results shown are representative of those obtained from three independent experiments.
Because JAK1 and Tyk2 phosphorylation is the first event in the IFN-induced phosphorylation cascade, our results suggested that the observed inhibition could occur at the level of the IFN-α/β receptor. Therefore, we sought to compare the levels of IFN-α/β receptor in normal and WNV-expressing cells. The results showed that the total levels of IFNAR1 as well as the surface expression of IFNAR2 did not vary in a significant way among normal HeLa, SL1, and KUNCD20 cells and between normal and WNV-infected Vero cells (Fig. 6 and results not shown). We concluded that WNV likely interferes with signaling between the IFN receptor and JAK1/Tyk2.
DISCUSSION
Our results demonstrated that WNV inhibits one of the first steps of the IFN signal transduction pathway and, hence, provided an explanation for previous observations made with WNV-infected mice and tissue culture cells (5, 27). Interactions between viral proteins and components of the innate immune response are major determinants in viral pathogenesis. This fact is best exemplified with genetically modified mice that are deficient in their response to IFN-α due to the lack of functional IFN-α/β receptors or STAT proteins and, as a consequence, rapidly succumb to viral infections (9, 26, 28). On the other hand, most, if not all, viruses depend on mechanisms to attenuate the IFN response for their survival (22). For example, certain parainfluenza viruses can inhibit the IFN pathway in human cells with the help of the accessory protein V that induces the proteasome-dependent degradation of STAT proteins (8, 30).
The exact mechanism by which WNV inhibits the IFN response is not yet known. Our results showed that the structural proteins are not required and, hence, by inference, have invoked a role for one or several NS proteins in blocking the activation of the innate immune response. However, it is also possible that the mechanism of inhibition was indirect. For example, it could have been caused by changes in the cytoplasm of infected cells that are known to occur as a consequence of viral replication. These changes include the induction and rearrangement of intracellular membranes (25, 34). Because HCV replication induces similar changes (10) and yet is very sensitive to IFNs (Fig. 1), we do not favor this possibility. Instead, we propose the following scenarios to explain our results. First, NS proteins may directly bind to the IFN receptor and block the activation of the Janus kinases. This model would imply that the NS viral proteins bind to both IFN receptors, because both IFN responses were inhibited albeit with slightly different efficiencies (Fig. 2, 3, and 6). Alternatively, NS proteins could directly interact with JAK1 and Tyk2 and inhibit their tyrosine kinase activities, perhaps similar to the V proteins of human parainfluenza viruses as described above. In fact, we observed a more profound inhibition of Tyk2 than of JAK1, which could be caused by differences in the affinity of one or more NS proteins for the two kinases. Second, WNV replication may lead to the activation of negative feedback loops that could inhibit or reverse JAK1 and Tyk2 phosphorylation. The suppressor of cytokine signaling-3 (SOCS3) or the protein tyrosine phophatases, such as the Src homology 2 (SH2) domain-containing protein tyrosine phophatases (SHP-2), are known members of such pathways (18, 39). For example, herpes simplex virus type I (HSV1) was recently shown to induce SOCS3 expression leading to the inhibition of the IFN response (37). Finally, inhibition may occur through a reduction in the surface expression of IFN receptors. Although a comparison of IFNAR2 present on the surface of parental HeLa and KUNCD20 cells by flow cytometry analysis revealed a slightly lower expression in KUNCD20 cells, similar comparison with normal and WNV-infected Vero cells did not reveal any substantial differences (results not shown). Moreover, a Western blot analysis of IFNRA1 expression did not reveal any significant alteration in KUNCD20 and WNV-infected Vero cells compared with normal HeLa and Vero cells (Fig. 6 and results not shown). Hence, based on these results, we consider this possibility unlikely.
While this study was in progress, Muñoz-Jordán et al. (29) reported that the Dengue virus replication could inhibit the phosphorylation of STAT1 and, hence, explain the previously observed inhibition of the IFN response. Moreover, these authors provided evidence for a role of the NS protein 4B in the modulation of Stat1 phosphorylation. However, this activity was relatively weak compared with that of other known inhibitors of the IFN response and was amplified when NS4B was coexpressed with other NS proteins. To identify the WNV protein(s) responsible for the observed inhibition of the IFN signaling pathway, we used a similar approach and transfected HeLa cells with plasmids encoding individual NS proteins. Unfortunately, our efforts have so far not yielded any positive results (results not shown). It is possible that under our selected conditions, the expression levels of individual NS proteins were lower than those obtained with Dengue NS proteins and those present in KUNCD20 or WNV-infected cells. Alternatively, it is conceivable that during natural replication viral proteins are sequestered in a manner that was not reproduced under our experimental conditions.
The physiological relevance of the well-documented resistance of WNV to IFN treatment can so far only be explained by inference based on other systems, because viral mutants that have lost the ability to block the JAK-STAT pathway are not yet available. One of the best examples is herpes simplex virus, where a functional relationship between the viral infected cell protein 34.5 (ICP34.5) and the IFN-inducible double-stranded RNA-dependent protein kinase R exists that controls viral replication (20). Interestingly, Gale and colleagues have just reported that WNV could delay the expression of IFN-β (11). The expression of this gene is controlled by a pathway that depends on its activation by proteins that can sense virus particles or products of replication, such as certain viral RNA structures or viral proteins. An example of a sensor would be the recently identified retinoic acid inducible gene 1 (RIG-1) (38). Hence, under physiological conditions, WNV might inhibit more than one cellular pathway of the innate immune response.
In summary, our study revealed the mechanism by which WNV inhibits the cellular innate immune response elicited by IFN-α and IFN-γ and provided an explanation for the previously observed resistance of this virus to IFN treatment in animals and cell cultures. These findings set the stage for the identification of the viral proteins and cellular factors that control the innate immune reaction and, hence, will provide opportunities to better understand novel virus-host interactions.
Acknowledgments
We thank Bill Mason and Rich Katz for their helpful comments on the manuscript, and we acknowledge the services provided by the FCCC tissue culture and cell imaging facilities and DNA microarray facilities. Alex Khromykh (SASVRZ, Brisbane, Australia), Vladimir Yamshchikov (University of Kansas), and Ron Harty (University of Pennsylvania) provided reagents that were essential to conduct this study. We acknowledge Margo Brinton for valuable information about WNV biology and Qing Zhu and Frank Puig for their help with flow cytometry and the construction of plasmids.
This work was supported by grants from the National Institutes of Health and by an appropriation from the Commonwealth of Pennsylvania.
REFERENCES
- 1.Anderson, J. F., T. G. Andreadis, C. R. Vossbrinck, S. Tirrell, E. M. Wakem, R. A. French, A. E. Garmendia, and H. J. Van Kruiningen. 1999. Isolation of West Nile virus from mosquitoes, crows, and a Cooper's hawk in Connecticut. Science 286:2331-2333. [DOI] [PubMed] [Google Scholar]
- 2.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972-1974. [DOI] [PubMed] [Google Scholar]
- 3.Brinton, M. A. 2002. The molecular biology of West Nile virus: a new invader of the western hemisphere. Annu. Rev. Microbiol. 56:371-402. [DOI] [PubMed] [Google Scholar]
- 4.Darnell, M. B., and H. Koprowski. 1974. Genetically determined resistance to infection with group B arboviruses. II. Increased production of interfering particles in cell cultures from resistant mice. J. Infect. Dis. 129:248-256. [DOI] [PubMed] [Google Scholar]
- 5.Darnell, M. B., H. Koprowski, and K. Lagerspetz. 1974. Genetically determined resistance to infection with group B arboviruses. I. Distribution of the resistance gene among various mouse populations and characteristics of gene expression in vivo. J. Infect. Dis. 129:240-247. [DOI] [PubMed] [Google Scholar]
- 6.Der, S. D., A. Zhou, B. R. Williams, and R. H. Silverman. 1998. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 95:15623-15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diamond, M. S., T. G. Roberts, D. Edgil, B. Lu, J. Ernst, and E. Harris. 2000. Modulation of Dengue virus infection in human cells by alpha, beta, and gamma interferons. J. Virol. 74:4957-4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Didcock, L., D. F. Young, S. Goodbourn, and R. E. Randall. 1999. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol. 73:9928-9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durbin, J. E., R. Hackenmiller, M. C. Simon, and D. E. Levy. 1996. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84:443-450. [DOI] [PubMed] [Google Scholar]
- 10.Egger, D., B. Wolk, R. Gosert, L. Bianchi, H. E. Blum, D. Moradpour, and K. Bienz. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76:5974-5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fredericksen, B. L., M. Smith, M. G. Katze, P. Y. Shi, and M. Gale, Jr. 2004. The host response to West Nile virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway. J. Virol. 78:7737-7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frese, M., T. Pietschmann, D. Moradpour, O. Haller, and R. Bartenschlager. 2001. Interferon-alpha inhibits hepatitis C virus subgenomic RNA replication by an MxA-independent pathway. J. Gen. Virol. 82:723-733. [DOI] [PubMed] [Google Scholar]
- 13.Frolov, A., S. Chahwan, M. Ochs, J. P. Arnoletti, Z.-Z. Pan, O. Favorova, J. Fletcher, M. von Mehren, B. Eisenberg, and A. K. Godwin. 2003. Response markers and the molecular mechanisms of action of Gleevec in gastrointestinal stromal tumors. Mol. Cancer Ther. 2:699-709. [PubMed] [Google Scholar]
- 14.Guo, J.-T., Q. Zhu, and C. Seeger. 2003. Cytopathic and noncytopathic interferon responses in cells expressing hepatitis C virus subgenomic replicons. J. Virol. 77:10769-10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo, J.-T., V. V. Bichko, and C. Seeger. 2001. Effect of alpha interferon on the hepatitis C virus replicon. J. Virol. 75:8516-8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khromykh, A. A., and E. G. Westaway. 1994. Completion of Kunjin virus RNA sequence and recovery of an infectious RNA transcribed from stably cloned full-length cDNA. J. Virol. 68:4580-4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khromykh, A. A., and E. G. Westaway. 1997. Subgenomic replicons of the flavivirus Kunjin: construction and applications. J. Virol. 71:1497-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krebs, D. L., and D. J. Hilton. 2001. SOCS proteins: negative regulators of cytokine signaling. Stem Cells 19:378-387. [DOI] [PubMed] [Google Scholar]
- 19.Lanciotti, R. S., J. T. Roehrig, V. Deubel, J. Smith, M. Parker, K. Steele, B. Crise, K. E. Volpe, M. B. Crabtree, J. H. Scherret, R. A. Hall, J. S. MacKenzie, C. B. Cropp, B. Panigrahy, E. Ostlund, B. Schmitt, M. Malkinson, C. Banet, J. Weissman, N. Komar, H. M. Savage, W. Stone, T. McNamara, and D. J. Gubler. 1999. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 286:2333-2337. [DOI] [PubMed] [Google Scholar]
- 20.Leib, D. A., M. A. Machalek, B. R. Williams, R. H. Silverman, and H. W. Virgin. 2000. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. USA 97:6097-6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy, D. E., and J. E. Darnell, Jr. 2002. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3:651-662. [DOI] [PubMed] [Google Scholar]
- 22.Levy, D. E., and A. García-Sastre. 2001. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 12:143-156. [DOI] [PubMed] [Google Scholar]
- 23.Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110-113. [DOI] [PubMed] [Google Scholar]
- 24.Lucas, M., T. Mashimo, M. P. Frenkiel, D. Simon-Chazottes, X. Montagutelli, P.-E. Ceccaldi, J. L. Guénet, and P. Desprès. 2003. Infection of mouse neurones by West Nile virus is modulated by the interferon-inducible 2′-5′ oligoadenylate synthetase 1b protein. Immunol. Cell Biol. 81:230-236. [DOI] [PubMed] [Google Scholar]
- 25.Mackenzie, J. M., M. K. Jones, and E. G. Westaway. 1999. Markers for trans-Golgi membranes and the intermediate compartment localize to induced membranes with distinct replication functions in flavivirus-infected cells. J. Virol. 73:9555-9567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meraz, M. A., J. M. White, K. C. Sheehan, E. A. Bach, S. J. Rodig, A. S. Dighe, D. H. Kaplan, J. K. Riley, A. C. Greenlund, D. Campbell, K. Carver-Moore, R. N. DuBois, R. Clark, M. Aguet, and R. D. Schreiber. 1996. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 84:431-442. [DOI] [PubMed] [Google Scholar]
- 27.Morrey, J. D., C. W. Day, J. G. Julander, L. M. Blatt, D. F. Smee, and R. W. Sidwell. 2004. Effect of interferon-alpha and interferon-inducers on West Nile virus in mouse and hamster animal models. Antivir. Chem. Chemother. 15:101-109. [DOI] [PubMed] [Google Scholar]
- 28.Muller, U., U. Steinhoff, L. F. Reis, S. Hemmi, J. Pavlovic, R. M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918-1921. [DOI] [PubMed] [Google Scholar]
- 29.Muñoz-Jordán, J. L., G. G. Sánchez-Burgos, M. Laurent-Rolle, and A. García-Sastre. 2003. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 100:14333-14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parisien, J. P., J. F. Lau, J. J. Rodriguez, B. M. Sullivan, A. Moscona, G. D. Parks, R. A. Lamb, and C. M. Horvath. 2001. The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology 283:230-239. [DOI] [PubMed] [Google Scholar]
- 31.Petersen, L. R., A. A. Marfin, and D. J. Gubler. 2003. West Nile virus. JAMA 290:524-528. [DOI] [PubMed] [Google Scholar]
- 32.Shi, P. Y., M. Tilgner, M. K. Lo, K. A. Kent, and K. A. Bernard. 2002. Infectious cDNA clone of the epidemic West Nile virus from New York City. J. Virol. 76:5847-5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227-264. [DOI] [PubMed] [Google Scholar]
- 34.Westaway, E. G., J. M. Mackenzie, M. T. Kenney, M. K. Jones, and A. A. Khromykh. 1997. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 71:6650-6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Westaway, E. G., J. M. Mackenzie, and A. A. Khromykh. 2002. Replication and gene function in Kunjin virus. Curr. Top. Microbiol. Immunol. 267:323-351. [DOI] [PubMed] [Google Scholar]
- 36.Yamshchikov, V. F., G. Wengler, A. A. Perelygin, M. A. Brinton, and R. W. Compans. 2001. An infectious clone of the West Nile flavivirus. Virology 281:294-304. [DOI] [PubMed] [Google Scholar]
- 37.Yokota, S., N. Yokosawa, T. Okabayashi, T. Suzutani, S. Miura, K. Jimbow, and N. Fujii. 2004. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 78:6282-6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi, M. Miyagishi, K. Taira, S. Akira, and T. Fujita. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730-737. [DOI] [PubMed] [Google Scholar]
- 39.You, M., D. H. Yu, and G. S. Feng. 1999. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol. Cell. Biol. 19:2416-2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu, Q., J.-T. Guo, and C. Seeger. 2003. Replication of hepatitis C virus subgenomes in nonhepatic epithelial and mouse hepatoma cells. J. Virol. 77:9204-9210. [DOI] [PMC free article] [PubMed] [Google Scholar]