

ABSTRACT
Group A Streptococcus (GAS) is a human-only pathogen that causes a spectrum of disease conditions. Given its survival in inflamed lesions, the ability to sense and overcome oxidative stress is critical for GAS pathogenesis. PerR senses oxidative stress and coordinates the regulation of genes involved in GAS antioxidant defenses. In this study, we investigated the role of PerR-controlled metal transporter A (PmtA) in GAS pathogenesis. Previously, PmtA was implicated in GAS antioxidant defenses and suggested to protect against zinc toxicity. Here, we report that PmtA is a P1B4-type ATPase that functions as an Fe(II) exporter and aids GAS defenses against iron intoxication and oxidative stress. The expression of pmtA is specifically induced by excess iron, and this induction requires PerR. Furthermore, a pmtA mutant exhibited increased sensitivity to iron toxicity and oxidative stress due to an elevated intracellular accumulation of iron. RNA-sequencing analysis revealed that GAS undergoes significant alterations in gene expression to adapt to iron toxicity. Finally, using two mouse models of invasive infection, we demonstrated that iron efflux by PmtA is critical for bacterial survival during infection and GAS virulence. Together, these data demonstrate that PmtA is a key component of GAS antioxidant defenses and contributes significantly to GAS virulence.
KEYWORDS: bacterial pathogenesis, gene regulation, iron efflux, metal homeostasis, oxidative stress
INTRODUCTION
Iron is a critical micronutrient required for bacterial survival and proliferation due to its role as a cofactor in cellular macromolecules involved in metabolism and electron transport. Given the bacterial requirement for iron, hosts limit iron availability to pathogens by using a variety of extracellular and intracellular mechanisms (1, 2). The eukaryotic host recruits an array of extracellular antimicrobial factors to chelate iron present at the microbial colonization surfaces to retard bacterial growth (2–4). As a countermeasure, pathogens employ high-affinity iron importers and iron-scavenging siderophores to acquire metal ions from nutrient-sparse infection sites (5). Consistent with this, the inactivation of iron importers resulted in an attenuated virulence of several pathogens, underscoring their importance to bacterial pathogenesis (5–7). Emerging evidence indicates that the host also employs metal toxicity at microbial colonization surfaces, predominantly within phagosomes, to mediate microbial killing and control bacterial infection (8, 9). Host innate immune cells, such as neutrophils and macrophages, accumulate copper and zinc around phagocytosed bacteria and impose metal toxicity (8–12). Conversely, bacteria employ metal efflux pumps to reduce the intracellular metal concentration and negate host-induced metal toxicity (8, 11, 12). In contrast with copper and zinc, relatively little is known regarding the possible role of iron toxicity as a host nutritional immune mechanism during infection. Recent data, however, suggest that iron efflux contributes to microbial pathogenesis in some systems, although it is presently unclear whether this is a stress actively imposed by the host (13).
Group A Streptococcus (GAS), also known as Streptococcus pyogenes, is a human-only pathogen that causes a variety of inflammatory disease conditions, including pharyngitis, impetigo, and necrotizing fasciitis (14, 15). Given its survival in inflamed tissues and the relative abundance of reactive oxygen species (ROS)-producing neutrophils at infection sites, it is likely that GAS encounters oxidative stress during infection. Oxidative stress occurs upon exposure to highly toxic ROS, such as hydroxyl radicals and superoxide anions, as they cause deleterious oxidative damage to cellular proteins, DNA, and membrane lipids that can lead to cell death (16–18). ROS are a key component of the host innate defense against invading pathogens. Superoxide anions interact with Fe(II) ions at the catalytic centers of iron-sulfur-containing enzymes and render the proteins inactive by causing the dissociation of iron (16, 19). The released free pool of iron reacts with hydrogen peroxide to produce highly reactive hydroxyl radicals that cause oxidative assault on bacterial macromolecules. Thus, the availability of free iron in the cytosol is a critical determinant in potentiating ROS toxicity.
Bacterial pathogens have evolved regulatory circuits coupling oxidative stress sensing by cytosolic regulators with the regulation of antioxidant adaptive responses that are involved in ROS detoxification, macromolecule damage repair, and cytosolic metal homeostasis (16). The inducible oxidative stress responses in GAS are predominantly controlled by the peroxide-sensing regulator PerR (20). PerR is a 155-amino-acid protein that belongs to the ferric uptake regulator (Fur) family of regulators. Members of this family are homodimeric metalloregulators that control the expression of genes involved in metal homeostasis (21). PerR forms the subfamily of Fur regulators that mediate gene regulation in response to peroxide stress (22). Structurally, PerR exists as a homodimer with two functional domains, an N-terminal DNA-binding domain and a C-terminal dimerization domain (20, 23–25). Each subunit of PerR has a structural Zn-binding site in its dimerization domain and a regulatory metal-binding site in the interdomain region (20, 23–25). Under physiological conditions, apo-PerR (PerR:Zn) binds to either manganese (PerR:Zn,Mn) or iron (PerR:Zn,Fe). The regulatory metal-bound holorepressor binds to a highly conserved binding motif referred to as the per box, in target promoters, to negatively regulate transcription (20, 26). However, only the PerR:Zn,Fe form, and not the PerR:Zn,Mn form, is responsive to peroxide stress (20, 26, 27). The iron at the regulatory site reacts with hydrogen peroxide to generate a localized hydroxyl radical that oxidizes one of two regulatory metal-coordinating histidines (25, 27). Conformational changes induced by the oxidation of PerR, and the resulting loss of regulatory metal, lead to dissociation from DNA and the derepression of target genes (20, 25, 27).
PerR-mediated oxidative stress sensing and gene regulation are critical for resistance against ROS-mediated killing by phagocytes, GAS survival in blood, and successful pharyngeal colonization (28–32). Consistent with this, dysregulation of the PerR regulon in an isogenic perR mutant significantly attenuated GAS virulence (20, 33, 34). Depending on the strain serotype and growth conditions employed, the GAS peroxide regulon varies significantly in the composition and number of regulated genes (28, 30–32). However, the transcription of PerR-regulated metal transporter A (PmtA) was controlled by PerR in all tested serotypes and under all tested growth conditions. In accordance with this, the promoter sequence of pmtA has a per box motif, indicating that pmtA is directly regulated by the peroxide stress sensor PerR. Subsequent characterization of PmtA revealed that it belongs to P-type ATPases, and derepression of PmtA is associated with the induction of genes regulated by AdcR that are regulated by the depletion of intracellular Zn(II) (28). Although this observation suggests that PmtA may function in Zn(II) efflux, the relationship between PmtA-mediated Zn(II) efflux, GAS antioxidant defenses, and bacterial pathogenesis remains unknown. In this study, we report that PmtA is a PerR-controlled iron efflux pump that contributes significantly to GAS virulence by promoting bacterial survival during oxidative stress.
RESULTS
PmtA belongs to the P1B-4 subfamily of P-type ATPases.
Previous studies suggested that PmtA is a P-type ATPase that aids GAS antioxidant defense by conferring resistance against Zn(II) toxicity (28). The members of P1-type ATPases display a broad spectrum of metal selectivity and transport different metals. Based on domain architecture, transmembrane metal-binding motifs, and metal specificity, they can be classified into several subgroups (35, 36). Amino acid sequence analysis of PmtA revealed that it contains an actuator (A) domain, a C-terminal ATPase domain, and 6 transmembrane helices but lacks the N- and C-terminal metal-binding domains (Fig. 1A). Furthermore, it has the signature metal-binding motifs Ser-Pro-Cys and His-Glu-Gly-Ser-Thr from transmembrane helices M4 and M6, respectively, which are characteristic of the P1B-4 subgroup of P1-type ATPases (Fig. 1A and B) (35, 36). Bioinformatics analyses of streptococcal genomes revealed that PmtA homologs are conserved among pathogenic streptococci, including several human pathogens (see Fig. S2 in the supplemental material). Although the subgroup of P1B-4 ATPases was originally considered to comprise Co(II) efflux pumps, recent characterizations of PmtA orthologs, including PfeT from Bacillus subtilis and FrvA from Listeria monocytogenes, identified them as Fe(II) exporters (Fig. 1B) (13, 37). These observations led us to hypothesize that PmtA is a P1B-4 ATPase that is primarily involved in Fe(II) efflux.
FIG 1.
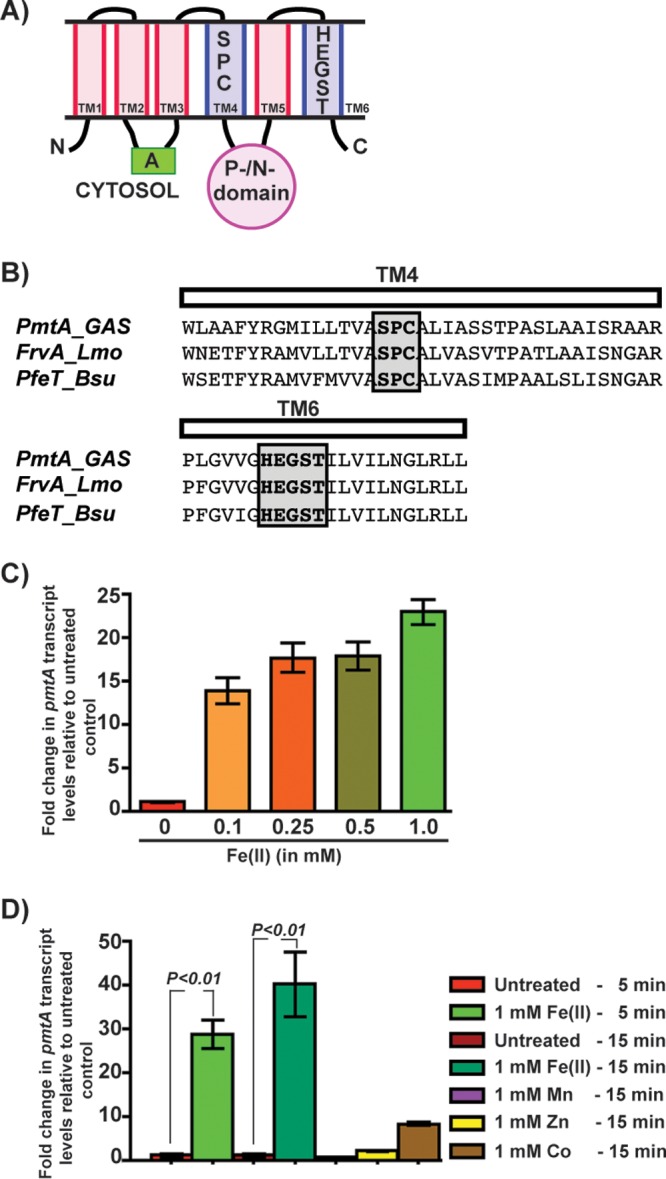
Expression of the pmtA gene, encoding a P1B-4-type ATPase, is upregulated during GAS growth in the presence of excess iron. (A) Schematic representation of the topology and domain architecture of the P1B-4-type ATPase. The predicted six transmembrane helices are labeled. The amino and carboxy termini are marked with N and C, respectively. The cytosolic actuator (A) and the phosphorylation/nucleotide-binding (P-/N-) domains are indicated. The transmembrane metal-binding residues within the transmembrane 4 (TM4) and TM6 helices that are conserved among members of the P1B-4 subgroup of the P1 family of ATPases are shown. (B) Amino acid sequence alignment of the predicted TM4 and TM6 helices from PmtA of GAS and its paralogs FrvA of L. monocytogenes (Lmo) and PfeT of B. subtilis (Bsu). The conserved metal-binding residues are shaded and boxed. (C) Transcript levels of pmtA in GAS grown in THY-C medium supplemented with increasing concentrations of Fe(II) compared to GAS grown in unsupplemented medium measured by qRT-PCR. (D) Transcript levels of pmtA in GAS grown in THY-C medium supplemented with different metals compared to the untreated sample measured by qRT-PCR. Three biological replicates were performed and analyzed in triplicate. Data graphed are means ± standard deviations. Average values for unsupplemented samples were used as a reference, and fold changes in transcript levels of the indicated strains relative to the reference sample are shown.
GAS upregulates pmtA expression in response to increasing Fe(II) concentrations.
Typically, bacterial adaptive responses are induced in response to specific environmental stimuli to negate the appropriate stress conditions. Thus, to determine the metal ligand exported by PmtA, we tested whether pmtA expression is induced in GAS grown in the presence of increasing concentrations of Fe(II). Transcript levels of pmtA increased by 14-fold at 100 μM Fe(II), and induction levels reached up to 22-fold at 1 mM Fe(II) compared to the growth of untreated GAS (Fig. 1C). Next, to determine whether the upregulation of pmtA is specific for Fe(II), we performed similar experiments in the presence of Fe(II), Zn(II), Mn(II), and Co(II). Although low-level induction of pmtA expression was observed in the presence of Zn(II) and Co(II), growth in the presence of Fe(II) caused robust induction (Fig. 1D). Transcript levels of pmtA increased by 28- and 40-fold after incubation with 1 mM Fe(II) for 5 and 15 min, respectively (Fig. 1D). Together, these results indicate that GAS upregulates pmtA expression in response to elevated iron concentrations, consistent with a model in which PmtA facilitates GAS survival during iron toxicity.
PmtA is critical for GAS growth during iron toxicity.
To determine whether PmtA is required for GAS growth in the presence of excess iron, we constructed an isogenic pmtA mutant (ΔpmtA) and a trans-complementation plasmid (pDC-pmtA) that has the coding region of pmtA fused with its native promoter. Complementation of the ΔpmtA mutant by pDC-pmtA at the level of transcription was verified by reverse transcription-quantitative PCR (qRT-PCR) (Fig. 2A). In the absence of iron, all 3 strains exhibited comparable growth kinetics (Fig. 2B). However, when supplemented with various concentrations of Fe(II), the growth of the ΔpmtA mutant was impaired in concert with increasing Fe(II) concentrations (Fig. 2B). The iron sensitivity of the ΔpmtA mutant was reversed to the wild-type (WT)-like phenotype in the trans-complemented strain, indicating that the growth defect of the mutant strain is due to PmtA inactivation (Fig. 2B). In a second line of investigation, we compared the abilities of the above-mentioned strains to negate transient iron toxicity by CFU analysis. Cells were grown to the mid-exponential growth phase, exposed to 4 mM FeSO4 for 15 min, and allowed to recover in Fe(II)-free medium for 3 h. As shown in Fig. 2C, WT GAS incubated with iron restored growth similarly to untreated GAS, whereas the ΔpmtA strain displayed a drastic reduction (>50-fold) in viability relative to the WT, suggesting that pmtA is crucial for GAS survival during iron toxicity (Fig. 2C).
FIG 2.

PmtA is critical for GAS growth under iron toxicity. (A) Transcript levels of pmtA in the indicated strains as measured by qRT-PCR. (B) Growth kinetics of the indicated strains in THY-C medium supplemented with increasing concentrations of FeSO4. Three biological replicates were performed, and the graph represents means ± standard deviations. Compl, complemented strain. (C) GAS strains were grown to mid-exponential phase in THY-C broth, incubated with 4 mM FeSO4 for 15 min, and grown for an additional 3 h in THY broth. The mean CFU recovered after a 3-h recovery period are shown, with P values as determined by a t test. Triplicate cultures were grown under the indicated conditions, and each biological replicate was assessed in duplicate. *, P < 0.0001.
Given that PmtA is implicated in resistance to Zn(II) toxicity (28) and the P1B-4 subgroup of ATPases is implicated in Co efflux, we also carried out growth studies in the presence of toxic Zn(II), Mn(II), and Co(II) concentrations. No differential inhibition was observed for the ΔpmtA mutant strain in the presence of Zn(II) or Mn(II) (see Fig. S3 in the supplemental material). However, when grown in the presence of Co(II), the growth of the ΔpmtA mutant was significantly impaired compared to the WT (Fig. S3). Collectively, these results suggest that PmtA is an iron efflux transporter that aids GAS survival under conditions of Fe(II) toxicity and additionally may protect against Co(II) toxicity.
Defective efflux in a ΔpmtA mutant leads to intracellular accumulation of iron.
To test whether the observed growth defect of the ΔpmtA mutant with excess Fe(II) is correlated with an increased cytosolic accumulation of iron, we compared the intracellular metal contents of the indicated strains by inductively coupled plasma mass spectrometry (ICP-MS). The WT, ΔpmtA mutant, and trans-complemented strains were grown to the mid-exponential phase of growth in the presence or absence of 1 mM FeSO4, and the intracellular metal content was measured. As expected, intracellular iron levels remained similar in cells of the 3 strains grown in unsupplemented Todd-Hewitt broth containing 0.2% (wt/vol) yeast extract (THY broth) supplemented with 1% citrate trisodium dehydrate (THY-C medium). However, when grown in iron-supplemented medium, the ΔpmtA mutant strain had significantly elevated intracellular iron levels compared to the WT and trans-complemented strains (Fig. 3A). The cytosolic concentrations of Zn(II) and Mn(II) remained similar among the strains (Fig. 3A). These results suggest that the growth phenotype of the ΔpmtA mutant under conditions of Fe(II) toxicity is due to defective iron efflux and that PmtA is an iron efflux transporter.
FIG 3.

The ΔpmtA mutant is defective in iron efflux and sensitive to oxidative stress. (A) The WT, ΔpmtA, and trans-complemented strains were grown in THY-C broth with (+) or without (−) 1 mM FeSO4 to the mid-exponential phase of growth, and the intracellular metal content was measured by ICP-MS. n.s., not significant. (B) GAS strains were grown to mid-exponential phase in THY-C broth, incubated with 4 mM FeSO4 for 15 min, and grown in THY broth in the presence or absence of 1 mM H2O2. Mean CFU recovered after the 6-h recovery period are shown, with P values (*, P < 0.0001) as determined by a t test. Triplicate cultures were grown under the indicated conditions, and each biological replicate was assessed in duplicate.
Iron efflux by PmtA is critical for GAS oxidative stress resistance.
The cytosolic labile pool of Fe(II) potentiates the toxicity of peroxide by its participation in the Fenton reaction and the consequent generation of toxic hydroxyl radicals, which affects bacterial survival by causing oxidative damage to cellular macromolecules. Thus, we investigated whether the increased intracellular accumulation of Fe(II) in the ΔpmtA mutant increases GAS sensitivity to peroxide stress. Cells were grown to mid-exponential phase, exposed to 4 mM FeSO4 for 15 min, and washed and resuspended in fresh THY broth supplemented with or without 1 mM H2O2. Cells were allowed to recover for 6 h at 37°C, and viability was assessed by CFU analysis. Interestingly, the isogenic ΔpmtA mutant strain was able to recover from transient Fe(II) toxicity, as it displayed a WT-like growth phenotype after 6 h of incubation (Fig. 3B). Similarly, all 3 strains exhibited comparable sensitivities to peroxide stress in the absence of iron intoxication (Fig. 3B). However, when pretreated with excess iron, peroxide stress caused a drastic reduction in the survival of the ΔpmtA mutant compared to the WT and trans-complemented strains (Fig. 3B). Together, these data suggest that defective iron efflux in the ΔpmtA mutant increases GAS sensitivity to oxidative stress.
GAS adaptive responses to iron toxicity.
To elucidate the molecular components of GAS adaptive responses to iron toxicity, we compared the transcription profiles of WT GAS cells grown in the presence and absence of 1 mM FeSO4 by RNA sequencing (RNA-seq). Briefly, cells were grown to mid-exponential phase and exposed to 1 mM FeSO4 for 15 min, and RNA isolation and cDNA library preparation were performed by using commercial protocols. For the purpose of statistical analysis, two biological replicates were used under each growth condition. Transcript levels of GAS genes with ≥2-fold changes with statistical significance (P < 0.05) are summarized in Tables S1 and S2 in the supplemental material. A total of 82 genes were differentially regulated (∼4% of the 1,951 predicted genes), with 26 genes being upregulated and 56 genes being downregulated in response to iron toxicity. Consistent with our qRT-PCR analysis, the expression of pmtA was highly upregulated (∼18-fold) in the presence of excess iron (Table S1). Additional categories of genes that were upregulated (≥2-fold) with excess iron belong to alternate carbohydrate utilization systems (man and fru operons) and arginine and serine catabolism (arcBCD and sdhBA) (Table S1). The upregulation of alternate sugar transport and utilization as well as arginine and serine catabolic pathways likely ensures continuous sugar import into the cytosol and energy generation during oxidative stress. GAS also downregulates several genes under conditions of excess iron to minimize oxidative damage and reduce the de novo synthesis of macromolecules. Major categories of genes that were downregulated include genes involved in the fatty acid biosynthesis (FAS) pathway (fabDFGHKTZ and accABCD), the iron-sulfur cluster assembly pathway (suf operon), amino acid transport (gln and atm operons), metal transport, and DNA metabolism (nrd operon) (Table S2). Modulation of fatty acid synthesis by the downregulation of the FAS pathway may result in altered fatty acid and phospholipid compositions of the bacterial cell membrane, which can lead to changes in membrane permeability. Such alterations in membrane permeability have been demonstrated to modulate bacterial adaptation to hydrogen peroxide and oxidative stress resistance (38, 39). Similarly, downregulation of iron-sulfur assembly, the primary targets of ROS toxicity, may contribute to the mitigation of ROS toxicity by preventing oxidative damage to bacterial macromolecules. Collectively, these data suggest that GAS undergoes significant remodeling of transcriptional programs to adapt to and survive iron toxicity.
PmtA expression is differentially controlled by the metallated state of PerR.
Since PerR is the oxidative stress sensor in GAS and PerR directly regulates pmtA expression, we tested whether the Fe-dependent upregulation of pmtA requires PerR. The WT, isogenic ΔperR mutant, and trans-complemented (ΔperR::pDC-perR) strains were grown to mid-exponential phase and exposed to 1 mM FeSO4 for 15 min, and pmtA transcript levels were measured by qRT-PCR. Consistent with our above-described results, WT GAS displayed an Fe-dependent induction of pmtA expression, and a WT-like pmtA induction profile was observed for the trans-complemented strain (Fig. 4A). However, the transcription of pmtA was fully derepressed in the absence of iron in the isogenic ΔperR mutant, and no additional induction of pmtA expression was observed upon the addition of iron (Fig. 4A). Together, these data indicate that PerR negatively regulates pmtA expression, and the repression of pmtA by PerR is relieved in response to iron accumulation.
FIG 4.

Upregulation of pmtA is dependent on the metallated state of PerR. (A) Transcript levels of pmtA in the indicated GAS strains grown in THY-C medium supplemented with 1 mM Fe(II) compared to those in untreated WT GAS as measured by qRT-PCR. (B) Transcript level analysis of pmtA in WT GAS grown in medium supplemented with 0.25 mM FeSO4 and challenged with increasing concentrations of Mn(II). (C) Transcript level analysis of pmtA in WT GAS and the ΔperR mutant supplemented with 0.25 mM FeSO4 and challenged with increasing concentrations of Mn(II). Three biological replicates were performed and analyzed in triplicate. Data graphed are means ± standard deviations. Average values for the growth of the WT in unsupplemented medium were used as a reference, and fold changes in the transcript levels in the indicated strains relative to those in the reference sample are shown. *, P < 0.0001.
PerR can exist in different metallated states at its regulatory metal-binding site. Although PerR can mediate transcription repression in either PerR:Zn,Fe or PerR:Zn,Mn states, only the Fe(II)-bound form is responsive to the oxidative stress-dependent derepression of the perR regulon. Thus, to determine whether the induction of pmtA expression by PerR is due to an Fe(II)-catalyzed oxidation of PerR, we performed challenge studies with excess Mn(II). When cells were grown in the presence of excess iron, pmtA expression was induced. However, this induction can be reversed to the levels observed for untreated cells by the addition of excess Mn(II) (Fig. 4B). To ensure that the reversal of induction by Mn(II) requires PerR, we performed similar experiments using the ΔperR strain. Consistent with our hypothesis, no reversal of the Fe(II)-dependent induction of pmtA expression was observed in the ΔperR mutant upon the addition of Mn(II) (Fig. 4C), suggesting that the upregulation of pmtA transcription in the presence of excess iron is likely due to the Fe(II)-dependent oxidation of PerR and the resulting loss of repression.
PmtA is critical for bacterial survival during infection and contributes significantly to GAS virulence.
To test the hypothesis that PmtA-dependent iron efflux is critical for GAS survival in vivo and contributes to GAS pathogenesis, we compared the WT (WT::pDC), the isogenic ΔpmtA mutant (ΔpmtA::pDC), and the isogenic ΔpmtA mutant trans-complemented with pmtA (ΔpmtA::pDC-pmtA) in two mouse models of invasive infection (Fig. 5A to E). Consistent with our hypothesis, the virulence of the ΔpmtA mutant was significantly attenuated compared to that of the WT (WT::pDC) and trans-complemented (ΔpmtA::pDC-pmtA and ΔpmtA::pDC) strains in both intramuscular and subcutaneous models of infection (Fig. 5A and D). To correlate the virulence phenotype at the cellular and tissue levels, we also performed visual macroscopic and microscopic examinations of the tissue lesions. In accordance with the near-mortality data, mice infected with the isogenic ΔpmtA mutant strain displayed confined lesions with minimal tissue damage compared to the WT and the trans-complemented strains (Fig. 5B and E). Since PmtA is involved in iron export and GAS oxidative stress resistance, we hypothesized that the ΔpmtA mutant strain may be defective in survival in vivo due to its efficient clearance from the host by immune effectors compared to the WT. To this end, we compared the GAS CFU of the above-mentioned strains recovered from infected muscular lesions. The results indicated that significantly fewer CFU per gram of tissue were recovered from lesions of mice infected with the ΔpmtA mutant than from lesions of mice infected with the WT and trans-complemented strains (P < 0.05) (Fig. 5C). Collectively, these data demonstrate that the iron efflux activity of PmtA is critical for GAS survival during infection and contributes significantly to GAS virulence.
FIG 5.

PmtA is critical for survival during infection and GAS virulence. (A) Twenty outbred CD-1 mice per strain were injected intramuscularly with 1 × 107 CFU of each strain. Shown is a Kaplan-Meier survival curve with P values derived by a log rank test. (B) Macroscopic (top) and histopathological (bottom) analyses of hind-limb lesions from mice infected with the indicated strains at 48 h postinfection. Areas of host tissue damage are boxed (white boxes). Areas of disseminated lesions in the WT and trans-complemented strains are boxed (black box), whereas confined, less destructive lesions are circled. (C) Twenty mice were infected intramuscularly, and mean CFU recovered from infected muscle tissue at 96 h postinfection are shown, with P values as determined by a t test. (D) Twenty immunocompetent hairless mice were infected with the indicated strains, and the lesion area produced by each strain was determined. The lesion area was measured daily and graphed (means ± standard error of the means). The P value was derived by a log rank test. (E) Histopathological analysis of subcutaneous lesions of mice infected with the indicated strains at 48 h postinfection. Areas of disseminated lesions and ulcerations on the skin surfaces of mice infected with the WT and trans-complemented strains are boxed, whereas confined, less destructive lesions in tissues infected with the ΔpmtA mutant are circled.
DISCUSSION
Here, we report that the inactivation of pmtA results in reduced iron efflux, increased intracellular accumulation of iron, and increased sensitivity to iron toxicity and oxidative stress. These properties likely account for the reduced survival of the ΔpmtA mutant during infection and attenuated GAS virulence. Previous studies have shown that the constitutive expression of pmtA in a ΔperR mutant contributes to GAS survival during oxidative stress, possibly due to the Zn(II) efflux activity of PmtA (28, 32). However, the results presented here support a model in which PmtA is a P1B4-type ATPase that exports Fe(II), as shown recently for the PmtA orthologs PfeT and FrvA (13, 37). In addition, we demonstrated that pmtA expression is not induced by Zn(II) and that the ΔpmtA mutant is not sensitive to Zn(II) intoxication. Although these observations do not support a physiologically relevant role for PmtA in Zn(II) efflux, this does not exclude some export activity for PmtA with other metals. We suggest that the previously observed contribution of PmtA to GAS survival during Zn(II) excess is likely a result of low-level Zn(II) efflux by PmtA that become significant only when pmtA is constitutively expressed in the ΔperR mutant. In WT GAS, any role for PmtA in Zn(II) detoxification is likely masked by the presence of Zn(II)-inducible efflux systems. Consistent with this, the contribution of FrvA to Zn(II) resistance became apparent only in strains lacking the endogenous Zn(II) efflux transporters (13).
Bacterial pathogens counter oxidative stress by replacing ROS-sensitive iron at the catalytic centers of metalloenzymes with unreactive, noncognate metals such as Zn(II) and Mn(II) (17). Although replacement of Fe(II) with Mn(II) at metal centers can functionally complement metalloproteins, replacement with zinc can be deleterious, as mismetallation with Zn(II) leads to dysfunctional proteins (17, 40, 41). Thus, removal of excess Zn(II) during oxidative stress as a microbial antioxidant defense remains a formal possibility. However, GAS encodes a dedicated zinc exporter, CzcD, which is upregulated specifically in response to Zn(II) toxicity and aids GAS proliferation during zinc surplus (8). Furthermore, our findings that PmtA is required for growth during iron toxicity but dispensable for GAS survival under conditions of excess Zn suggest that a role for PmtA-mediated Zn export in GAS oxidative stress resistance is unlikely (Fig. 1 and 2; see also Fig. S3 in the supplemental material). Thus, we conclude that PmtA is an iron efflux pump that promotes in vivo bacterial fitness by reducing cytosolic iron accumulation and aiding GAS antioxidant defense.
The concept of iron withholding as a host nutritional immune mechanism to control microbial growth has been extensively studied. The host recruits numerous extracellular antimicrobial factors, such as lipocalin, lactoferrin, and transferrin, to impose iron limitation on pathogens (4, 42, 43). In addition, the host recruits NRAMP-1 transporters to the phagosomal membrane to shuttle iron away from phagocytosed pathogens to limit their growth within phagosomes (44–46). However, recent evidence indicates that hosts also deploy defense strategies to increase intracellular Cu(II) and Zn(II) levels and impose metal toxicity on microbial pathogens (8, 9, 11, 12, 47). However, given the significance of iron to bacterial physiology and the presence of redundant host nutritional defense mechanisms to impose iron limitation both in the extracellular compartment and within phagosomes, our observations that GAS possesses machinery to counter iron toxicity were surprising. Although it is possible that the host also employs iron toxicity as a nutritional immune defense, experimental evidence in this regard is limited (48, 49). However, our finding that increased intracellular iron accumulation in the ΔpmtA mutant increases its sensitivity to oxidative stress raises another possibility. During host-induced oxidative stress, GAS encounters acute increases in the concentrations of superoxide anions and hydrogen peroxide. Superoxide anions attack the iron-sulfur clusters of bacterial proteins, which results in the rapid release of iron and elevated cytosolic free-iron levels. Given the high reactivity of the free-iron pool with hydrogen peroxide to generate toxic hydroxyl radicals, it can act as a biocatalyst to promote oxidative damage and cause cell death. Thus, transient expulsion of cytosolic free iron by PmtA may provide immediate relief by removing the key reactant of the Fenton reaction, thus reducing the production of hydroxyl radicals and minimizing oxidative damage. Given that GAS lacks classical bacterial oxidative stress defense mechanisms such as catalases and heme-biosynthetic pathways, it is likely that PmtA constitutes the first line of GAS antioxidant defense.
The orthologs of PmtA in B. subtilis and L. monocytogenes are controlled by both PerR and the regulator of iron homeostasis, Fur (ferric uptake regulator) (13, 37). Importantly, there is evidence to suggest that Fur may activate the expression of pfeT and frvA in response to excess iron, which is consistent with a primary role for PfeT and FrvA in protection against Fe toxicity (13, 37). However, PmtA is primarily regulated by PerR, and GAS lacks a homolog of Fur. Consistent with this, the inactivation of perR was sufficient to decouple the iron-dependent induction of pmtA expression, and no additional induction of pmtA was observed in the ΔperR mutant during iron toxicity (Fig. 4A to C). Furthermore, PerR-dependent pmtA induction occurs only during iron surplus, and this induction can be reversed to the repressed state by the addition of excess Mn(II) to the growth medium (Fig. 4B and C), suggesting that the specific metallated state of PerR, PerR:Zn,Mn or PerR:Zn,Fe, determines the regulatory outcome. Such coupling of pmtA transcription regulation to the oxidative stress sensor PerR lends further support to our working model that PmtA-dependent iron efflux is a critical component of GAS antioxidant defense. Based on these observations, we propose a model that links PmtA-mediated iron efflux to GAS oxidative stress resistance. Under physiological growth conditions, PerR exists in either the PerR:Zn,Mn or PerR:Zn,Fe state and mediates the repression of pmtA. However, during oxidative stress, the disruption of iron-sulfur clusters by ROS causes the cytosolic accumulation of iron and generates an environment conducive for the Fenton reaction (Fig. 6). In addition, the increase in the intracellular free-iron pool likely favors the PerR:Zn,Fe metallated state over PerR:Zn,Mn. As a result, ROS-sensitive PerR:Zn,Fe undergoes metal-catalyzed oxidation, and the ensuing conformational changes in PerR lead to the derepression of pmtA (Fig. 6). Such an upregulation of PmtA, in addition to the regulation of other components of the perR regulon, aids GAS survival during oxidative stress by detoxifying the cytosol of iron and reducing the oxidative stress susceptibility of GAS.
FIG 6.

Proposed model for regulation and contribution of PmtA to GAS pathogenesis. In wild-type GAS (left), iron released from Fe-S cluster-containing proteins by ROS stress causes an increase in the GAS intracellular iron concentration. As a result, the iron-metallated form of PerR senses ROS, and oxidized PerR (Oxi-PerR) causes the derepression of pmtA by its dissociation from the pmtA promoter. PmtA aids GAS antioxidant defense by exporting iron out of the cytosol using the energy derived from ATP hydrolysis. In the ΔpmtA mutant, a failure to prevent the cytosolic accumulation of iron results in increased sensitivity to oxidative stress, increased oxidative damage, reduced bacterial survival, and attenuated GAS virulence.
Conclusions.
During infection, bacterial pathogens undergo repeated cycles of metal deficiency and metal toxicity in different host compartments and during different stages of infection. Thus, it is imperative that pathogens possess fine-tuned regulatory programs to sense the changes in metal availability and alternate between the expressions of metal uptake and metal efflux systems. Although iron limitation is a well-studied host nutritional immune strategy, emerging evidence indicates that pathogens may encounter iron toxicity during infection. Bacteria may experience iron intoxication due to (i) host imposition, by mechanisms not yet elucidated; (ii) transient iron overload during the transition from iron-sparse to iron-rich environments; or (iii) iron release from metalloenzymes in response to host-imposed oxidative stress. Regardless, the results presented here demonstrate that iron efflux by PmtA is important for bacterial survival in the host and GAS virulence, providing a direct link between iron export and bacterial pathogenesis. Furthermore, PmtA is highly conserved among pathogenic streptococci, suggesting a similar role for iron efflux in streptococcal pathogenesis. Thus, continued investigation into the role of PmtA orthologs in other streptococci will not only yield novel insights into the role of iron efflux in bacterial pathogenesis but also provide a foundation for the development of novel therapeutics targeting bacterial iron efflux pumps.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table S3 in the supplemental material. Strain MGAS10870 is a previously described invasive serotype M3 isolate whose genome has been fully sequenced (50). MGAS10870 is representative of serotype M3 strains that cause invasive infections and has wild-type sequences for all major regulatory genes (50). The Escherichia coli DH5α strain was used as the host for plasmid constructions. GAS was grown routinely on Trypticase soy agar containing 5% sheep blood (bovine serum albumin [BSA]; Becton Dickinson) or in Todd-Hewitt broth containing 0.2% (wt/vol) yeast extract (THY broth; Difco). THY-C broth (THY medium supplemented with 1% citrate trisodium dehydrate) was used in iron toxicity experiments to minimize iron precipitation. When required, spectinomycin or chloramphenicol was added to a final concentration of 150 μg/ml or 5 μg/ml, respectively. All GAS growth experiments were done in triplicate on three separate occasions for a total of nine replicates. Cultures grown overnight were inoculated into fresh medium to achieve an initial optical density at 600 nm (OD600) of 0.03. Bacterial growth was monitored by measuring the A600.
Construction of the pmtA-inactivated mutant strain.
The insertional inactivation of the pmtA gene in wild-type strain MGAS10870 was performed by using methods described previously (51, 52). The pmtA gene is not part of a multigene operon; thus, it is unlikely that the insertional inactivation of pmtA causes a polar effect on the expression of flanking genes (see Fig. S1 in the supplemental material). Briefly, a PCR fragment containing a spectinomycin resistance (spc) cassette with the fragment of the gene to be deleted on either side was generated in a three-step PCR process. Subsequently, the plasmid with the spc gene disruption cassette was introduced into the parent strain by electroporation, and the gene was disrupted by homologous recombination. The isogenic mutant strains were selected by growth on spectinomycin-containing medium. Inactivation of the gene was confirmed by DNA sequencing. Primers used for the construction of strain 10870 ΔpmtA are listed in Table S4 in the supplemental material.
Construction of the pmtA trans-complementation plasmid.
To complement the isogenic ΔpmtA mutant strain, the coding sequence of the full-length pmtA gene together with the pmtA promoter region were cloned into the E. coli-GAS shuttle vector pDC123 (53). By using the primers listed in Table S4 in the supplemental material, the respective fragments were amplified from GAS genomic DNA by PCR, digested with BglII and NdeI, and ligated into digested vector pDC123. The inserts were verified by DNA sequencing and electroporated into the appropriate mutant derivatives of strain MGAS10870.
Growth studies during metal toxicity.
Strains were grown overnight in THY broth, and the culture diluted in THY medium was grown overnight to an A600 of 0.4. A 1:100 dilution of the cells was made in THY-C medium supplemented with various concentrations of each metal. Cells were aliquoted into a 96-well plate, and growth was monitored by measuring the A600 using a microplate reader at 37°C.
Transcript analysis by qRT-PCR.
GAS strains were grown to mid-exponential phase (OD600 of ∼0.5) and incubated with 2 volumes of RNAprotect (Qiagen) for 10 min at room temperature. Bacteria were harvested by centrifugation, and the cell pellets were snap-frozen with liquid nitrogen. RNA isolation and purification were performed by using an RNeasy kit (Qiagen). Purified RNA was analyzed for quality and concentration with an Agilent 2100 bioanalyzer. cDNA was synthesized from the purified RNA by using Superscript III (Invitrogen), and TaqMan qRT-PCR was performed with an ABI 7500 Fast system (Applied Biosystems). Comparison of transcript levels was done by using ΔCT method of analysis, using tufA as the endogenous control gene (54, 55). The TaqMan primers and probes used are listed in Table S4 in the supplemental material.
Metal content analysis by ICP-MS.
To measure the intracellular metal concentration, GAS strains were grown to the mid-exponential phase of growth (A600 of ∼0.5) in THY medium. Cells were diluted in THY-C medium and grown to an A600 of 0.5. Untreated cells of each strain were collected, and the remaining culture was exposed to 4 mM FeSO4 for 15 min. Cells were harvested by centrifugation, washed once with 2 volumes of phosphate-buffered saline (PBS) containing 1 mM nitrilotriacetic acid, and washed twice with PBS supplemented with 0.1 M EDTA. Subsequently, cells were washed with Chelex-treated PBS, and cells were stored at 80°C. Cell pellets resuspended in 0.4 ml of PBS were lysed by ballistic disintegration (lysing matrix B and Fastprep96 automated homogenizer; MP Biomedicals), and the cytosolic content was separated by centrifugation at 13,000 rpm for 30 min. The total protein concentration was measured by using the Bradford assay (56). Samples were mixed with a buffer containing 5% HNO3 and 0.1% Triton X-100 and heated in a sand bath at 95°C for 30 min. Samples were centrifuged, and supernatants were diluted in HNO3 (2% [vol/vol] final concentration). Metal levels were analyzed by ICP-MS (Elan DRC II; Perkin-Elmer) using Ga as an internal standard. The total concentration of metal ions is expressed as micrograms of ion per gram of protein.
RNA sequencing.
GAS was grown in THY-C medium to the mid-exponential growth phase and supplemented with or without 1 mM FeSO4 for 15 min. RNA isolation and purification were performed by using an RNeasy minikit (Qiagen) according to the manufacturer's protocol. RNA was analyzed for quality and concentration with an Agilent 2100 bioanalyzer. The rRNA was then removed by using a Ribo-zero treatment kit (Epicenter) according to the manufacturer's protocol and further purified by using the Min-Elute RNA purification kit (Qiagen). The ribosomally depleted RNA was then used to synthesize adaptor-tagged cDNA libraries using the ScriptSeq V2 RNA-seq library preparation kit (Epicenter). cDNA libraries were then run on a NextSeq instrument using the Illumina v2 reagent kit (Illumina). Approximately 2 million reads were obtained per sample, and the reads were mapped to the MGAS315 genome by using CLC-Genomics WorkBench, version 5 (CLC Bio). For RNA-seq analysis, the total number of reads per gene between the replicates was normalized by TPKM [(reads/kilobase of gene)/(million reads aligning to the genome)]. Using the TPKM values, pairwise comparisons were carried out between the two samples to identify the differentially expressed genes. Genes with a 2-fold difference and a P value of <0.05 after applying Bonferroni's correction were considered to be statistically significant.
Animal infection studies.
Mouse experiments were performed according to protocols approved by the Houston Methodist Hospital Research Institute Institutional Animal Care and Use Committee. These studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals (57). The virulence of the isogenic mutant GAS strains was assessed by using two mouse models. For intramuscular infection, 20 3- to 4-week-old female CD1 mice (Harlan Laboratories) were inoculated in the right hind limb with 1 × 107 CFU of each strain and monitored for near mortality. Results were graphically displayed as a Kaplan-Meier survival curve and analyzed by using the log rank test. For subcutaneous infection, 20 4- to 5-week-old immunocompetent female SKH1-hrBR hairless mice (Charles River BRF, Houston, TX) were inoculated subcutaneously with 1 × 107 CFU of each strain, and lesion areas were measured daily. Data were graphically displayed as mean lesion area over time ± the standard error of the mean (SEM) (Prism4) and analyzed by using analysis of variance (ANOVA) (XLStat). For histopathology, infected hind limbs were examined at 24 and 48 h postinoculation. Tissues from excised lesions were fixed in 10% phosphate-buffered formalin, decalcified, serially sectioned, and embedded in paraffin by using standard automated instruments. Hematoxylin-and-eosin- and Gram-stained sections were examined in a blind fashion with a BX5 microscope and photographed by using a DP70 camera (Olympus). Micrographs of tissues taken from the inoculation sites that showed pathology characteristic of each strain were selected for publication. The GAS burden (n = 20 mice/strain) in infected limbs was determined as previously described (55). Results were graphically displayed as the mean number of CFU recovered per gram of tissue at 96 h postinfection, with a P value of <0.05, as determined by using the Mann-Whitney log rank test, considered to be statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Health (grants 1R01AI109096-01A1 to M.K. and GM059323 to J.D.H.).
We declare no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00091-17.
REFERENCES
- 1.Nairz M, Schroll A, Sonnweber T, Weiss G. 2010. The struggle for iron—a metal at the host-pathogen interface. Cell Microbiol 12:1691–1702. doi: 10.1111/j.1462-5822.2010.01529.x. [DOI] [PubMed] [Google Scholar]
- 2.Weinberg ED. 2000. Modulation of intramacrophage iron metabolism during microbial cell invasion. Microbes Infect 2:85–89. doi: 10.1016/S1286-4579(00)00281-1. [DOI] [PubMed] [Google Scholar]
- 3.Nakashige TG, Zhang B, Krebs C, Nolan EM. 2015. Human calprotectin is an iron-sequestering host-defense protein. Nat Chem Biol 11:765–771. doi: 10.1038/nchembio.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. 2004. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 5.Cassat JE, Skaar EP. 2013. Iron in infection and immunity. Cell Host Microbe 13:509–519. doi: 10.1016/j.chom.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaible UE, Kaufmann SHE. 2004. Iron and microbial infection. Nat Rev Microbiol 2:946–953. doi: 10.1038/nrmicro1046. [DOI] [PubMed] [Google Scholar]
- 7.Crouch M-LV, Castor M, Karlinsey JE, Kalhorn T, Fang FC. 2008. Biosynthesis and IroC-dependent export of the siderophore salmochelin are essential for virulence of Salmonella enterica serovar Typhimurium. Mol Microbiol 67:971–983. doi: 10.1111/j.1365-2958.2007.06089.x. [DOI] [PubMed] [Google Scholar]
- 8.Ong C-LY, Gillen CM, Barnett TC, Walker MJ, McEwan AG. 20 January 2014. An antimicrobial role for zinc in innate immune defense against group A Streptococcus. J Infect Dis doi: 10.1093/infdis/jiu053. [DOI] [PubMed] [Google Scholar]
- 9.White C, Lee J, Kambe T, Fritsche K, Petris MJ. 2009. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J Biol Chem 284:33949–33956. doi: 10.1074/jbc.M109.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Achard MES, Tree JJ, Holden JA, Simpfendorfer KR, Wijburg OLC, Strugnell RA, Schembri MA, Sweet MJ, Jennings MP, McEwan AG. 2010. The multi-copper-ion oxidase CueO of Salmonella enterica serovar Typhimurium is required for systemic virulence. Infect Immun 78:2312–2319. doi: 10.1128/IAI.01208-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams CL, Neu HM, Gilbreath JJ, Michel SLJ, Zurawski DV, Merrell DS. 2016. Copper resistance of the emerging pathogen Acinetobacter baumannii. Appl Environ Microbiol 82:6174–6188. doi: 10.1128/AEM.01813-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, Niederweis M. 2011. Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 108:1621–1626. doi: 10.1073/pnas.1009261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pi H, Patel SJ, Argüello JM, Helmann JD. 2016. The Listeria monocytogenes Fur-regulated virulence protein FrvA is an Fe(II) efflux P1B4-type ATPase. Mol Microbiol 100:1066–1079. doi: 10.1111/mmi.13368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olsen RJ, Shelburne SA, Musser JM. 2009. Molecular mechanisms underlying group A streptococcal pathogenesis. Cell Microbiol 11:1–12. doi: 10.1111/j.1462-5822.2008.01225.x. [DOI] [PubMed] [Google Scholar]
- 15.Olsen RJ, Musser JM. 2010. Molecular pathogenesis of necrotizing fasciitis. Annu Rev Pathol 5:1–31. doi: 10.1146/annurev-pathol-121808-102135. [DOI] [PubMed] [Google Scholar]
- 16.Storz G, Imlay JA. 1999. Oxidative stress. Curr Opin Microbiol 2:188–194. doi: 10.1016/S1369-5274(99)80033-2. [DOI] [PubMed] [Google Scholar]
- 17.Imlay JA. 2008. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem 77:755–776. doi: 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nunoshiba T, Obata F, Boss AC, Oikawa S, Mori T, Kawanishi S, Yamamoto K. 1999. Role of iron and superoxide for generation of hydroxyl radical, oxidative DNA lesions, and mutagenesis in Escherichia coli. J Biol Chem 274:34832–34837. doi: 10.1074/jbc.274.49.34832. [DOI] [PubMed] [Google Scholar]
- 19.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. doi: 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makthal N, Rastegari S, Sanson M, Ma Z, Olsen RJ, Helmann JD, Musser JM, Kumaraswami M. 2013. Crystal structure of peroxide stress regulator from Streptococcus pyogenes provides functional insights into the mechanism of oxidative stress sensing. J Biol Chem 288:18311–18324. doi: 10.1074/jbc.M113.456590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J-W, Helmann J. 2007. Functional specialization within the Fur family of metalloregulators. Biometals 20:485–499. doi: 10.1007/s10534-006-9070-7. [DOI] [PubMed] [Google Scholar]
- 22.Dubbs JM, Mongkolsuk S. 2012. Peroxide-sensing transcriptional regulators in bacteria. J Bacteriol 194:5495–5503. doi: 10.1128/JB.00304-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacquamet L, Traoré DAK, Ferrer JL, Proux O, Testemale D, Hazemann JL, Nazarenko E, El Ghazouani A, Caux-Thang C, Duarte V, Latour JM. 2009. Structural characterization of the active form of PerR: insights into the metal-induced activation of PerR and Fur proteins for DNA binding. Mol Microbiol 73:20–31. doi: 10.1111/j.1365-2958.2009.06753.x. [DOI] [PubMed] [Google Scholar]
- 24.Traoré DAK, El Ghazouani A, Ilango S, Dupuy J, Jacquamet L, Ferrer J-L, Caux-Thang C, Duarte V, Latour J-M. 2006. Crystal structure of the apo-PerR-Zn protein from Bacillus subtilis. Mol Microbiol 61:1211–1219. doi: 10.1111/j.1365-2958.2006.05313.x. [DOI] [PubMed] [Google Scholar]
- 25.Traore DAK, Ghazouani AE, Jacquamet L, Borel F, Ferrer J-L, Lascoux D, Ravanat J-L, Jaquinod M, Blondin G, Caux-Thang C, Duarte V, Latour J-M. 2009. Structural and functional characterization of 2-oxo-histidine in oxidized PerR protein. Nat Chem Biol 5:53–59. doi: 10.1038/nchembio.133. [DOI] [PubMed] [Google Scholar]
- 26.Herbig AF, Helmann JD. 2001. Roles of metal ions and hydrogen peroxide in modulating the interaction of the Bacillus subtilis PerR peroxide regulon repressor with operator DNA. Mol Microbiol 41:849–859. doi: 10.1046/j.1365-2958.2001.02543.x. [DOI] [PubMed] [Google Scholar]
- 27.Lee J-W, Helmann JD. 2006. The PerR transcription factor senses H2O2 by metal-catalysed histidine oxidation. Nature 440:363–367. doi: 10.1038/nature04537. [DOI] [PubMed] [Google Scholar]
- 28.Brenot A, Weston BF, Caparon MG. 2007. A PerR-regulated metal transporter (PmtA) is an interface between oxidative stress and metal homeostasis in Streptococcus pyogenes. Mol Microbiol 63:1185–1196. doi: 10.1111/j.1365-2958.2006.05577.x. [DOI] [PubMed] [Google Scholar]
- 29.Le Breton Y, Mistry P, Valdes KM, Quigley J, Kumar N, Tettelin H, McIver KS. 2013. Genome-wide identification of genes required for fitness of group A Streptococcus in human blood. Infect Immun 81:862–875. doi: 10.1128/IAI.00837-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grifantini R, Toukoki C, Colaprico A, Gryllos I. 2011. Peroxide stimulon and role of PerR in group A Streptococcus. J Bacteriol 193:6539–6551. doi: 10.1128/JB.05924-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gryllos I, Grifantini R, Colaprico A, Cary ME, Hakansson A, Carey DW, Suarez-Chavez M, Kalish LA, Mitchell PD, White GL, Wessels MR. 2008. PerR confers phagocytic killing resistance and allows pharyngeal colonization by group A Streptococcus. PLoS Pathog 4:e1000145. doi: 10.1371/journal.ppat.1000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brenot A, King KY, Caparon MG. 2005. The PerR regulon in peroxide resistance and virulence of Streptococcus pyogenes. Mol Microbiol 55:221–234. doi: 10.1111/j.1365-2958.2004.04370.x. [DOI] [PubMed] [Google Scholar]
- 33.King KY, Horenstein JA, Caparon MG. 2000. Aerotolerance and peroxide resistance in peroxidase and PerR mutants of Streptococcus pyogenes. J Bacteriol 182:5290–5299. doi: 10.1128/JB.182.19.5290-5299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ricci S, Janulczyk R, Björck L. 2002. The regulator PerR is involved in oxidative stress response and iron homeostasis and is necessary for full virulence of Streptococcus pyogenes. Infect Immun 70:4968–4976. doi: 10.1128/IAI.70.9.4968-4976.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Argüello JM. 2003. Identification of ion-selectivity determinants in heavy-metal transport P1B-type ATPases. J Membr Biol 195:93–108. doi: 10.1007/s00232-003-2048-2. [DOI] [PubMed] [Google Scholar]
- 36.Arguello JM, González-Guerrero M, Raimunda D. 2011. Bacterial transition metal P1B-ATPases: transport mechanism and roles in virulence. Biochemistry 50:9940–9949. doi: 10.1021/bi201418k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guan G, Pinochet-Barros A, Gaballa A, Patel SJ, Argüello JM, Helmann JD. 2015. PfeT, a P1B4-type ATPase, effluxes ferrous iron and protects Bacillus subtilis against iron intoxication. Mol Microbiol 98:787–803. doi: 10.1111/mmi.13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pesakhov S, Benisty R, Sikron N, Cohen Z, Gomelsky P, Khozin-Goldberg I, Dagan R, Porat N. 2007. Effect of hydrogen peroxide production and the Fenton reaction on membrane composition of Streptococcus pneumoniae. Biochim Biophys Acta 1768:590–597. doi: 10.1016/j.bbamem.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 39.DiRusso CC, Black PN, Weimar JD. 1999. Molecular inroads into the regulation and metabolism of fatty acids, lessons from bacteria. Prog Lipid Res 38:129–197. doi: 10.1016/S0163-7827(98)00022-8. [DOI] [PubMed] [Google Scholar]
- 40.McDevitt CA, Ogunniyi AD, Valkov E, Lawrence MC, Kobe B, McEwan AG, Paton JC. 2011. A molecular mechanism for bacterial susceptibility to zinc. PLoS Pathog 7:e1002357. doi: 10.1371/journal.ppat.1002357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imlay JA. 2014. The mismetallation of enzymes during oxidative stress. J Biol Chem 289:28121–28128. doi: 10.1074/jbc.R114.588814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Theurl I, Fritsche G, Ludwiczek S, Garimorth K, Bellmann-Weiler R, Weiss G. 2005. The macrophage: a cellular factory at the interphase between iron and immunity for the control of infections. Biometals 18:359–367. doi: 10.1007/s10534-005-3710-1. [DOI] [PubMed] [Google Scholar]
- 43.Weiss G, Schett G. 2013. Anaemia in inflammatory rheumatic diseases. Nat Rev Rheumatol 9:205–215. doi: 10.1038/nrrheum.2012.183. [DOI] [PubMed] [Google Scholar]
- 44.Barton CH, Biggs TE, Baker ST, Bowen H, Atkinson PG. 1999. Nramp1: a link between intracellular iron transport and innate resistance to intracellular pathogens. J Leukoc Biol 66:757–762. [DOI] [PubMed] [Google Scholar]
- 45.Cellier MF, Courville P, Campion C. 2007. Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect 9:1662–1670. doi: 10.1016/j.micinf.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 46.Blackwell JM, Searle S. 1999. Genetic regulation of macrophage activation: understanding the function of Nramp1 (= Ity/Lsh/Bcg). Immunol Lett 65:73–80. doi: 10.1016/S0165-2478(98)00127-8. [DOI] [PubMed] [Google Scholar]
- 47.Samanovic MI, Ding C, Thiele DJ, Darwin KH. 2012. Copper in microbial pathogenesis: meddling with the metal. Cell Host Microbe 11:106–115. doi: 10.1016/j.chom.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frawley ER, Crouch M-LV, Bingham-Ramos LK, Robbins HF, Wang W, Wright GD, Fang FC. 2013. Iron and citrate export by a major facilitator superfamily pump regulates metabolism and stress resistance in Salmonella Typhimurium. Proc Natl Acad Sci U S A 110:12054–12059. doi: 10.1073/pnas.1218274110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frawley ER, Fang FC. 2014. The ins and outs of bacterial iron metabolism. Mol Microbiol 93:609–616. doi: 10.1111/mmi.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beres SB, Carroll RK, Shea PR, Sitkiewicz I, Martinez-Gutierrez JC, Low DE, McGeer A, Willey BM, Green K, Tyrrell GJ. 2010. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc Natl Acad Sci U S A 107:4371–4376. doi: 10.1073/pnas.0911295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lukomski S, Hoe NP, Abdi I, Rurangirwa J, Kordari P, Liu M, Dou S-J, Adams GG, Musser JM. 2000. Nonpolar inactivation of the hypervariable streptococcal inhibitor of complement gene (sic) in serotype M1 Streptococcus pyogenes significantly decreases mouse mucosal colonization. Infect Immun 68:535–542. doi: 10.1128/IAI.68.2.535-542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuwayama H, Obara S, Morio T, Katoh M, Urushihara H, Tanaka Y. 2002. PCR-mediated generation of a gene disruption construct without the use of DNA ligase and plasmid vectors. Nucleic Acids Res 30:e2. doi: 10.1093/nar/30.2.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chaffin DO, Rubens CE. 1998. Blue/white screening of recombinant plasmids in Gram-positive bacteria by interruption of alkaline phosphatase gene (phoZ) expression. Gene 219:91–99. doi: 10.1016/S0378-1119(98)00396-5. [DOI] [PubMed] [Google Scholar]
- 54.Virtaneva K, Porcella SF, Graham MR, Ireland RM, Johnson CA, Ricklefs SM, Babar I, Parkins LD, Romero RA, Corn GJ, Gardner DJ, Bailey JR, Parnell MJ, Musser JM. 2005. Longitudinal analysis of the group A Streptococcus transcriptome in experimental pharyngitis in cynomolgus macaques. Proc Natl Acad Sci U S A 102:9014–9019. doi: 10.1073/pnas.0503671102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanson M, Makthal N, Gavagan M, Cantu C, Olsen RJ, Musser JM, Kumaraswami M. 2015. Phosphorylation events in the multiple gene regulator of group A Streptococcus significantly influence global gene expression and virulence. Infect Immun 83:2382–2395. doi: 10.1128/IAI.03023-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 57.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.