

ABSTRACT
Staphylococcus aureus remains a causative agent for morbidity and mortality worldwide. This is in part a result of antimicrobial resistance, highlighting the need to uncover novel antibiotic targets and to discover new therapeutic agents. In the present study, we explored the possibility that iron-sulfur (Fe-S) cluster synthesis is a viable antimicrobial target. RNA interference studies established that Suf (sulfur mobilization)-dependent Fe-S cluster synthesis is essential in S. aureus. We found that sufCDSUB were cotranscribed and that suf transcription was positively influenced by sigma factor B. We characterized an S. aureus strain that contained a transposon inserted in the intergenic space between sufC and sufD (sufD*), resulting in decreased transcription of sufSUB. Consistent with the transcriptional data, the sufD* strain had multiple phenotypes associated with impaired Fe-S protein maturation. They included decreased activities of Fe-S cluster-dependent enzymes, decreased growth in media lacking metabolites that require Fe-S proteins for synthesis, and decreased flux through the tricarboxylic acid (TCA) cycle. Decreased Fe-S cluster synthesis resulted in sensitivity to reactive oxygen and reactive nitrogen species, as well as increased DNA damage and impaired DNA repair. The sufD* strain also exhibited perturbed intracellular nonchelated Fe pools. Importantly, the sufD* strain did not exhibit altered exoprotein production or altered biofilm formation, but it was attenuated for survival upon challenge by human polymorphonuclear leukocytes. The results presented are consistent with the hypothesis that Fe-S cluster synthesis is a viable target for antimicrobial development.
KEYWORDS: iron, sulfur, cluster, Staphylococcus aureus, Suf, neutrophil
INTRODUCTION
Staphylococcus aureus is a human commensal that causes morbidity and mortality worldwide. While it is responsible for low-morbidity maladies, such as folliculitis, it is also capable of causing fatal afflictions, such as endocarditis, bacteremia, and toxic shock syndrome (1, 2). Bacterial antibiotic resistance continues to increase and to be problematic. Infections caused by antibiotic-resistant S. aureus result in increased mortality, increased stress on the health care system, and an increased financial burden (3, 4). Current FDA-approved antibacterials target a limited number of metabolic processes (5). Developing antibacterials that target alternate processes would expand treatment options and aid in multidrug therapy. These facts highlight the need for (i) continued investigations into novel antimicrobial targets and (ii) the discovery of new antimicrobials.
Iron (Fe) is a required nutrient for human bacterial pathogens. Not surprisingly, S. aureus strains defective in acquiring or processing intracellular Fe have decreased virulence (6, 7). Upon acquisition, S. aureus uses Fe to metalate proteins, produce heme, and synthesize inorganic iron-sulfur (Fe-S) cluster prosthetic groups. Three Fe-S cluster synthesis machineries (Suf [sulfur mobilization], Isc, and Nif) that are, for the most part, functionally redundant but biochemically distinct have been described in bacteria (8–10). S. aureus utilizes the SufCDSUB machinery to synthesize Fe-S clusters from monoatomic Fe2+, S0, and electrons (7). SufBCD acts as a molecular scaffold for Fe-S cluster synthesis (11). SufC is an ATPase that has homology with membrane-associated ATPases, SufD participates in Fe acquisition, and SufB is thought to be the site of Fe-S cluster synthesis (12–14). SufS is a cysteine desulfurase that catalyzes the removal of elemental sulfur from cysteine, producing alanine and a SufS-bound persulfide (15). The persulfide is transferred to SufU, which is a sulfur transfer protein that provides the sulfur to SufBCD (16). After synthesis, the Fe-S cluster is transferred directly to either an apoprotein or an Fe-S cluster carrier that traffics the cofactor to the target apoprotein (11, 17). SufA and Nfu function as Fe-S cluster carriers in S. aureus (7, 18). Genetic evidence suggests that SufT and bacillithiol also have roles in the maturation of Fe-S proteins (18–20).
The SufCDSUB Fe-S cluster synthesis machinery is fundamentally different from the synthesis machinery used by mammals. Mammals synthesize Fe-S clusters in two cellular locations (reviewed in reference 21). In mammals, Fe-S clusters are synthesized in mitochondria using machinery that is similar to the bacterial Isc system, as well as in the cytosol using the cytosolic iron-sulfur cluster assembly (CIA) machinery, which does not share homology with described bacterial synthetic systems. Therefore, if a therapeutic agent that inhibits SufCDSUB is developed, it is unlikely that the agent would affect the essential process of Fe-S cluster synthesis in humans.
Proteins containing Fe-S prosthetic groups are widely distributed throughout the proteomes of most organisms and are necessary for diverse cellular processes. Because of the substantial reliance on Fe-S proteins, we hypothesize that disruption of Fe-S cluster synthesis in S. aureus will result in metabolic standstill and eventual cell death. This hypothesis is supported by results from high-density transposon mutant screens showing that the sufCDSUB gene products are important for S. aureus fitness and possibly survival (22–24).
This study was initiated to determine if Fe-S cluster biogenesis is a viable antimicrobial target in S. aureus. RNA interference studies confirmed that the Suf Fe-S cluster biosynthetic system is essential for S. aureus viability. An S. aureus strain with decreased sufSUB transcription had a decreased capability to mature Fe-S proteins. Decreased Suf function resulted in global metabolic defects and reduced survival in human polymorphonuclear neutrophils (PMNs), but it did not alter biofilm formation or exoprotein production.
RESULTS
Expression of antisense RNAs to the sufC or sufU transcripts decreases S. aureus viability.
The conditional expression of an antisense RNA targeted to a corresponding mRNA is an effective means to deplete cells of a specific gene product (25). The essentiality of Suf was examined using mRNA depletion. DNA fragments corresponding to sufC or sufUB were shotgun cloned into a plasmid under the transcriptional control of an anhydrotetracycline (Atet)-inducible promoter. Two clones corresponding to sufC and two clones corresponding to sufU that resulted in decreased growth in tryptic soy broth (TSB) medium upon expression of the plasmid insert were isolated. The plasmids contained fragments that expressed an RNA that was antisense to the 3′ coding region of either the sufC or sufU mRNA. sufC is 762 nucleotides in length, and the psufCKD plasmids contained fragments corresponding to bases 515 to 762 (psufCKD1) and 572 to 750 (psufCKD2). sufU is 465 nucleotides in length, and one clone contained a fragment corresponding to bases 216 to 465 plus 32 bp of intergenic sufUB DNA (psufUKD1) while the second clone corresponded to bases 353 to 465 plus 32 bp of intergenic sufUB DNA (psufUKD2). S. aureus strain RN4220 containing the empty vector or the psufKD plasmids did not exhibit growth abnormalities when cultured on solid medium lacking inducer (Fig. 1). As the concentration of Atet was increased, viability decreased in the cells containing psufKD plasmids, but not in cells containing the empty vector. The efficiency of the knockdown plasmids was decreased in S. aureus strain LAC, and this effect was independent of SigB or Agr, which are known to be defective in RN4220 (data not shown) (26, 27). It is currently unknown why the plasmids behave differently in these two genetic backgrounds.
FIG 1.
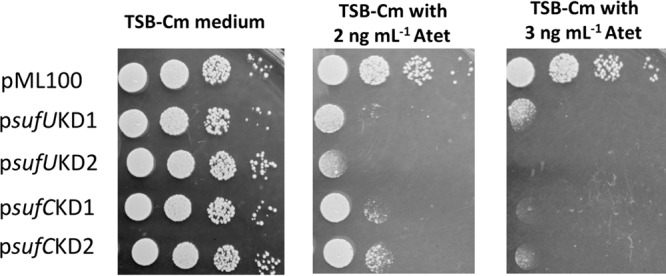
sufC or sufU depletion decreases S. aureus viability. S. aureus RN4220 containing pML100 (empty vector), psufUKD1, psufUKD2, psufCKD1, or psufCKD2 was serially diluted and spot plated on TSA-chloramphenicol medium with and without Atet (inducer). Images from a representative experiment are shown.
A transposon insertion between sufC and sufD results in decreased transcription of downstream suf genes.
Two strains that contain bursa aurealis mariner-based transposons inserted into the sufCDSUB operon between annotated genes were obtained (28). The transposons were located 62 and 63 bp upstream of sufD (sufD*) or sufS (sufS*), respectively (Fig. 2A). We were able to reconstruct the sufD* strain in the S. aureus LAC background, but we were unable to reconstruct the sufS* strain. Therefore, the sufS* strain is not discussed further in this study. We assessed the effects of the sufD* transposon on transcription of sufCDSUB. The transcripts corresponding to the gene upstream of the sufD* transposon were increased (Fig. 2B). In contrast, there was no effect on the sufD transcript, but the transcripts corresponding to sufSUB were decreased.
FIG 2.
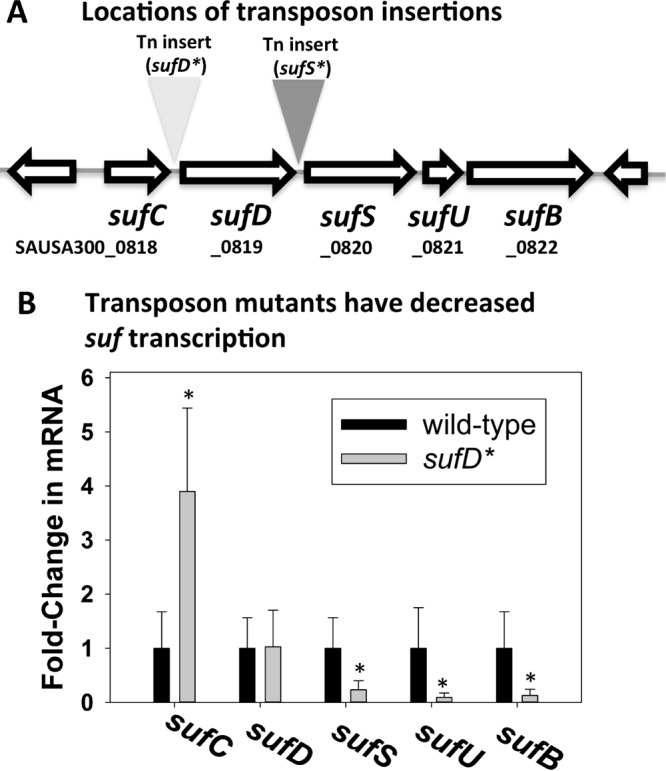
A transposon insertion between sufC and sufD decreases transcription of sufSUB. (A) Locations of the individual sufS* and sufD* transposon insertion sites. The sufS* transposon insertion is located between sufD and sufS, and the sufD* transposon insertion is located between sufC and sufD. (B) The sufD* insertion decreases transcription of sufSUB. Total RNA was isolated from the WT and sufD* strains, and the transcription of the individual sufCDSUB genes was quantified. The data represent average mRNA abundances from cells cultured in biological triplicates; cDNA libraries were analyzed in duplicate. The error bars represent standard deviations; *, P < 0.5 relative to the WT strain using a two-tailed Student t test.
sufCDSUB are cotranscribed, and transcription is modulated by sigma factor B (σB).
A previously published transcriptome sequencing (RNA-seq) data set (29) was analyzed to further understand how the sufD* transposon decreased transcription of sufSUB. The reads that mapped to sufCDSUB were relatively evenly distributed (Fig. 3A), leading to the hypothesis that sufCDSUB are transcribed as an operon using a common promoter. To test this, a cDNA library was generated from DNase-treated wild-type (WT) RNA. We used oligonucleotides that bridged various suf genes to test whether multiple genes existed on the same cDNA (Fig. 3B). The resulting amplicons suggested that sufCDSUB are cotranscribed. As a control, we included a condition under which reverse transcriptase was not added to rule out possible DNA contamination. Reaction mixtures lacking reverse transcriptase did not generate any detectable product, indicating that the amplicons were not the result of contaminating genomic DNA (Fig. 3B).
FIG 3.

The sufCDSUB genes are cotranscribed, and transcription is positively influenced by sigma factor B. (A) Analysis of a previously published RNA-seq data set (29) indicating that sufCDSUB are cotranscribed. (B) The suf genes are cotranscribed. (Top) Schematic of the suf operon; the locations of the amplicons are shown as black bars, and the predicted sizes of the amplicons (generated using the following primer pairs: lanes 2 and 3, sufCRT5 and sufDrevRT; lanes 4 and 5, sufDfwdRT and sufinternal3; lanes 6 and 7, sufinternal5 and sufSrevRT; lanes 8 and 9, sufSfwdRT and sufBrevRT) are shown. (Bottom) Amplicons were generated from cDNA libraries using RNAs isolated from the WT and separated using agarose gel electrophoresis. The samples analyzed in lanes 3, 5, 7, and 9 were generated using a template that was not treated with reverse transcriptase. (C) The promoter of the suf operon contains potential sigma factor A (green) and sigma factor B (red) recognition sites. The predicted transcriptional start site is shown in blue and was determined by analyzing previously published RNA-seq data (29). The annotated sufC translational start site is in purple and underlined. (D) The transcriptional activity of the sufC promoter is modulated by sigma factor B (SigB). The transcriptional activity of sufC was monitored in the WT and ΔsigB strains containing pCM11_suf. The data shown represent the averages of biological triplicates with standard deviations. *, P < 0.5 relative to the WT strain, using a two-tailed Student t test.
The reads from the RNA-seq experiment (29) were further analyzed to ascertain the transcription start site and to determine the extent of the suf 5′ untranslated region (UTR). The distal reads started at an adenine located 82 bp upstream from the predicted translation start site (Fig. 3C). This analysis allowed us to identify putative σA and σB recognition sequences 14 and 28 bp upstream from the proposed transcription start site, respectively (30).
Sigma factor B is a general stress response transcriptional regulator in S. aureus (31). The transcriptional activity of sufC was monitored in the WT and ΔsigB strains during growth. The transcriptional activity of sufC was decreased in the ΔsigB strain (Fig. 3D), confirming that SigB positively influences sufC transcription.
Decreased suf transcription results in lower activities of Fe-S cluster-requiring enzymes.
Aconitase (AcnA) requires an Fe-S cluster for function (32). The AcnA activity in the sufD* strain was ∼20% of that in the WT (Fig. 4A). Returning sufCDSUB to the chromosome of the sufD* strain at a secondary location via episome (sufD* suf+) fully restored AcnA activity.
FIG 4.
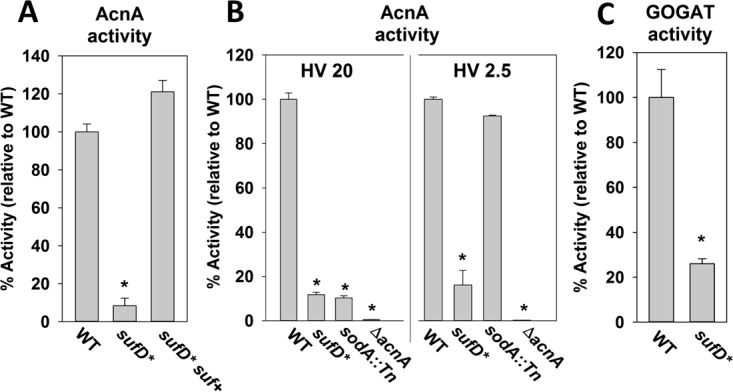
Iron-sulfur cluster-requiring proteins have decreased activity in S. aureus strains with decreased suf transcription. (A) AcnA activity was assessed in the WT, sufD*, and sufD* suf+ strains. (B) AcnA activity is decreased in the sufD* strain irrespective of culture aeration. The AcnA assays were conducted in cell lysates from the WT, sufD*, sodA::Tn, and ΔacnA strains cultured in TSB with altered HV ratios. (C) Glutamate dehydrogenase activities were assessed in the WT and sufD* strains. The data shown represent the averages of biological triplicates with standard deviations. *, P < 0.5 relative to the WT strain using a two-tailed Student t test.
S. aureus increases the transcription of genes necessary to metabolize reactive oxygen species (ROS) when cultured under high aeration, suggesting that endogenous ROS accumulates under these growth conditions (33). Consequently, S. aureus strains deficient in the maturation of Fe-S proteins or scavenging endogenously produced ROS display severe defects in AcnA activity when the dioxygen tension is increased (19). The effect of dioxygen tension on AcnA activity in the WT, ΔacnA, and sufD* strains was assessed. A sodA::Tn mutant that lacks the major superoxide dismutase was included as an experimental control. To modulate the concentration of dioxygen in the culture medium, we varied the ratio of liquid medium volume to culture vessel to gaseous headspace (HV ratio). The higher the HV ratio, the higher the concentration of dissolved dioxygen (34). The sodA::Tn mutant had decreased AcnA activity when cultured at an HV ratio of 20, but the AcnA activity was comparable to that of the WT when cultured at an HV ratio of 2.5 (Fig. 4B). AcnA activity was greatly decreased in the sufD* strain, and AcnA activity was not significantly altered as the culture HV ratio was varied.
Like AcnA, the enzyme glutamate synthase (GOGAT, or GltBD) requires Fe-S clusters for function (35). The sufD* strain displayed ∼25% of the GOGAT activity of the WT (Fig. 4C). Taken together, these findings suggest the sufD* strain has decreased Fe-S enzyme activity and that the Suf system is the dominant Fe-S cluster synthetic system under multiple culture conditions.
Decreased Suf function results in a reduced rate of carbon flux through the TCA cycle.
AcnA catalyzes the first committed step in the tricarboxylic acid (TCA) cycle and therefore acts as a gatekeeper for flux through the TCA cycle. We tested the hypothesis that TCA cycle function would also be decreased in the sufD* strain.
The WT, ΔacnA, and sufD* strains were cultured in TSB, and growth was monitored over time. The growth rates of the WT, sufD*, and ΔacnA strains were similar during the exponential growth phase (<6 h) (Fig. 5A). During the postexponential growth phase (>6 h), the WT and sufD* strains displayed slower growth, but the sufD* strain displayed an extended lag phase before postexponential growth commenced. The ΔacnA strain did not grow after this time, confirming that growth beyond this inflection point requires TCA cycle function. The sufD* suf+ strain did not display growth abnormalities in TSB (data not shown).
FIG 5.

Decreased Suf function results in a reduced rate of carbon flux through the TCA cycle. (A) Growth profiles of the WT, sufD*, and ΔacnA strains. (B) AcnA activity throughout growth. (C) Concentrations of acetate in culture supernatants throughout growth. (D) Spent medium pH throughout growth. The data represent averages of biological triplicates with standard deviations.
The activity of AcnA was also monitored at specific time points throughout growth. AcnA activity was decreased in the sufD* strain throughout growth (Fig. 5B). The largest difference in AcnA activity between the WT and sufD* strains occurred at the start of postexponential outgrowth (∼8 h). Acetate accumulation in culture media from all strains was examined. Consistent with decreased TCA cycle function, acetate uptake was decreased and was nonexistent in the sufD* and ΔacnA strains, respectively (Fig. 5C). All the strains acidified the culture medium at similar rates during the initial growth period. After ∼6 h, the WT and sufD* strains basified the medium, but the rate of basification was lower in the sufD* strain (Fig. 5D). The pH of the medium used to culture the ΔacnA strain did not increase after the initial acidification. Taken together, these findings are consistent with the hypothesis that the decreased AcnA activity of the sufD* strain resulted in decreased flux through the TCA cycle.
Decreased Fe-S cluster synthesis results in decreased growth in media lacking specific amino acids or lipoic acid.
We assayed the growth of the WT, sufD*, and sufD* suf+ strains on chemically defined solid media. The sufD* strain grew poorly on chemically defined media supplemented with the 20 canonical amino acids (20 aa medium), whereas the sufD* suf+ strain grew like the WT. The enzyme lipoyl synthase requires Fe-S clusters (36). Supplementing the 20 aa growth medium with lipoic acid alleviated this growth defect of the sufD* strain. Isoleucine, leucine, and glutamate/glutamine synthesis also requires Fe-S enzymes (37–39). Compared to the WT, the sufD* strain displayed poor to no growth on chemically defined solid medium containing lipoic acid but lacking isoleucine, leucine, or glutamate and glutamine (Fig. 6). These phenotypes could be genetically complemented.
FIG 6.

Decreased Fe-S cluster synthesis causes decreased growth in media lacking specific amino acids or lipoic acid. Auxotrophic analyses were conducted using the WT, sufD*, and sufD* suf+ strains. The strains were grown in TSB before plating on solid chemically defined medium containing the 20 canonical amino acids (A), 20 aa with lipoic acid (B), 19 aa minus isoleucine with lipoic acid (C), 19 aa minus leucine with lipoic acid (D), and 18 aa minus glutamate and glutamine with lipoic acid (E). Images from a representative experiment are shown.
Decreased Suf function results in increased DNA damage and a decreased ability to repair damaged DNA.
The DNA repair enzymes MutY (40), Nth (41), and AddAB (42) require an Fe-S cluster for function. Mutations in rpoB, which encodes RNA polymerase, provide resistance to rifampin (Rif) (43). The rate of spontaneous Rif resistance was determined for the WT and sufD* strains by plating upon tryptic soy agar (TSA) with or without rifampin. The sufD* strain had an ∼20-fold increase in rifampin-resistant cells compared to the WT strain (Fig. 7A).
FIG 7.
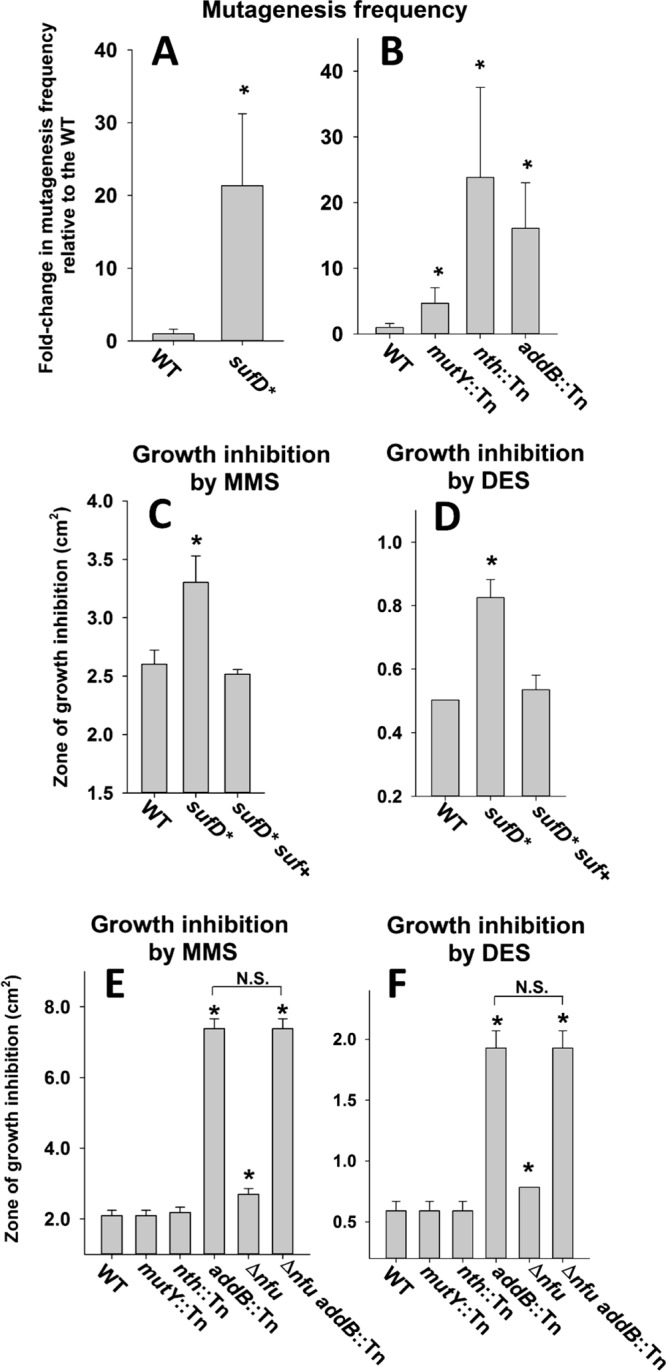
Effect of decreased Suf function on DNA metabolism. (A) The frequency of spontaneous rifampin resistance was measured in the WT and sufD* strains. (B) The frequency of spontaneous rifampin resistance was measured in the WT, mutY::Tn, nth::Tn, and addB::Tn strains. (C) Sensitivity to MMS was assessed in the WT, sufD*, and sufD* suf+ strains. (D) Sensitivity to DES was assessed in the WT, sufD*, and sufD* suf+ strains. (E) Sensitivity to MMS was assessed in the WT, mutY::Tn, nth::Tn, addB::Tn, Δnfu, and Δnfu addB::Tn strains. (F) Sensitivity to DES was assessed in the WT, mutY::Tn, nth::Tn, addB::Tn, Δnfu, and Δnfu addB::Tn strains. The data presented in panels A and B represent the averages of 10 biological replicates with standard deviations. The data presented in panels C, D, E, and F represent the averages of biological triplicates with standard deviations. Student t tests (two tailed) were performed on the data; *, P < 0.05 relative to the WT strain unless otherwise indicated; N.S., not significant (P > 0.05).
We next examined if one or more of the described Fe-S cluster-requiring DNA repair enzymes had a role in preventing rpoB mutations when cultured under standard laboratory conditions. The rate of rifampin resistance was determined in the WT, mutY::Tn, nth::Tn, and addB::Tn mutant strains. The mutY::Tn, nth::Tn, and addB::Tn strains had increased rates of rifampin resistance (Fig. 7B).
We next assayed the susceptibility of the WT, sufD*, and sufD* suf+ strains to chemical mutagens. The sufD* strain had increased sensitivity to methyl methanesulfonate (MMS) (Fig. 7C) and diethyl sulfate (DES) (Fig. 7D) compared to the WT and sufD* suf+ strains. We also examined the necessity for Fe-S cluster-requiring DNA repair proteins for growth in the presence of MMS or DES. The nth::Tn and mutY::Tn strains showed resistance to MMS and DES similar to that of the WT (Fig. 7E and F), but the addB::Tn strain displayed greatly increased sensitivity to both mutagens.
We sought genetic evidence to lend support to the hypothesis that decreased Fe-S cluster assembly resulted in decreased AddAB activity and increased sensitivity to DNA-damaging agents. Despite multiple attempts, we were unsuccessful in constructing the sufD* addB::Tn double-mutant strain, suggesting that the strain may not be viable. However, we were able to create a Δnfu addB::Tn double mutant. Like the sufD* strain, the Δnfu strain displayed increased sensitivity to MMS and DES (Fig. 7E and F). The phenotypic effects of the Δnfu and addB::Tn mutations were not additive. Although not conclusive, these data are consistent with the hypothesis that defects in Fe-S cluster assembly result in diminished ability to repair damaged DNA because of decreased functionality of Fe-S clusters requiring DNA repair enzymes.
Decreased Suf function increases sensitivity to reactive oxygen and reactive nitrogen species.
Oxidation of solvent-accessible Fe-S clusters can result in cluster disintegration and impaired protein function. Proteins requiring Fe-S cluster cofactors are targets for ROS and reactive nitrogen species (RNS) (44, 45).
The growth of the sufD* strain in the presence of methyl viologen was monitored. Methyl viologen is a redox cycling agent that produces superoxide. The sufD* strain had decreased growth when plated upon solid medium containing methyl viologen (Fig. 8A), and the phenotype could be genetically complemented. A strain lacking the major superoxide dismutase (sodA::Tn) displayed decreased growth, verifying superoxide generation. The sufD* strain also displayed decreased survival after challenge with a bolus of hydrogen peroxide (H2O2), and the phenotype could be genetically complemented (Fig. 8B). A katA::Tn strain that is unable to produce functional catalase also displayed decreased survival upon H2O2 challenge.
FIG 8.

Decreased Suf function results in increased sensitivity to RNS and ROS. (A) Methyl viologen sensitivity was monitored in the WT, sufD*, sufD* suf+, and sodA::Tn strains. The cells were cultured in TSB before serial dilution and spot plating on solid TSA supplemented with 40 mM methyl viologen or vehicle control. (B) H2O2 sensitivity was assessed in the WT, sufD*, sufD* suf+, and katA::Tn strains. The cells were challenged with 500 mM H2O2 before the reaction was quenched, and the cells were serially diluted and spot plated on solid TSA medium. (C) Nitroprusside (NP) sensitivity was assessed in the WT, sufD*, and sufD* suf+ strains. Representative growth profiles in the presence and absence of 15 mM nitroprusside in TSB medium are shown. The data are from representative experiments.
Next, we examined the effects of RNS on the sufD* strain. We examined the growth profiles of WT, sufD*, and sufD* suf+ strains in chemically defined medium in the presence and absence of nitroprusside, which interacts with intracellular thiols, resulting in the release of RNS (46). The sufD* mutant had a severe growth defect when exposed to nitrosative stress, and the phenotype could be genetically complemented (Fig. 8C).
The sufD* strain has altered Fe homeostasis.
An S. aureus strain lacking the Fe-S cluster maturation factor Nfu is perturbed in intracellular Fe homeostasis. We examined whether defective Fe-S cluster synthesis also results in perturbed intracellular Fe homeostasis. Growth of the WT, sufD*, and sufD* suf+ strains was monitored in the presence of 2,2-dipyridyl (DIP), which is a cell-permeable divalent metal chelator with specificity for Fe (47). An fhuC::Tn mutant that is defective in Fe scavenging was included as an experimental control (48). The sufD* and fhuC::Tn strains displayed decreased growth compared to the WT when cultured in the presence of DIP, and the phenotype of the sufD* mutation could be genetically complemented (Fig. 9A).
FIG 9.

Decreased Suf function destabilizes intracellular Fe homeostasis. (A) The WT, sufD*, sufD* suf+, and fhuC::Tn strains were spot plated on solid TSA medium with and without 900 mM 2,2-dipyridyl. (B) The WT, sufD*, and sufD* suf+ strains were plated as top agar overlays on solid TSA, and the zones of growth inhibition resulting from streptonigrin intoxication were measured. (C) The WT, nth::Tn, mutY::Tn, addB::Tn, Δnfu, and Δnfu addB::Tn strains were plated as top agar overlays on solid TSA, and the zones of growth inhibition resulting from streptonigrin intoxication were measured. The data presented in panels B and C represent the averages of biological triplicates with standard deviations. Student t tests (two tailed) were performed on the data; *, P < 0.05 compared to the WT unless otherwise indicated.
The antibiotic streptonigrin, in combination with Fe and an intracellular electron donor, causes DNA damage resulting in cell death (49). Higher incidences of cell death are correlated with an increased concentration of nonchelated intracellular Fe (50). The sufD* strain displayed increased sensitivity to growth in the presence of streptonigrin, and the phenotype could be genetically complemented (Fig. 9B).
Streptonigrin, in conjunction with Fe, can catalyze double-stranded DNA breaks (51). Strains defective in Fe-S cluster assembly were defective in repairing damaged DNA (Fig. 7). We examined whether the increased streptonigrin sensitivity of strains defective in maturing Fe-S proteins was the result of defective DNA repair. The streptonigrin sensitivities of the nth::Tn, mutY::Tn, and addB::Tn strains were determined. The nth::Tn and mutY::Tn mutants had streptonigrin sensitivities similar to that of the WT, but the addB::Tn mutant displayed increased sensitivity to streptonigrin (Fig. 9C). The streptonigrin sensitivities of the Δnfu and Δnfu addB::Tn mutants were also assessed. The streptonigrin sensitivity phenotypes attributed to the Δnfu and addB::Tn mutations were additive. These findings suggested that the streptonigrin sensitivity phenotype of strains defective in Fe-S cluster assembly was not exclusively due to defective AddAB function.
Exoprotein production and biofilm formation are not significantly altered in the sufD* mutant.
S. aureus produces and secretes a number of exoproteins, including toxins, adhesins, proteases, and invasins that are crucial for pathogenesis (52). The total abundance of exoproteins was quantified in the spent culture medium obtained from the WT and sufD* strains. S. aureus strains lacking a functional Agr system are deficient in exoprotein production, and therefore, an agrA::Tn strain was included as a control (53). The agrA::Tn strain had decreased exoprotein production, and the phenotype of the sufD* strain was not statistically significant (P = 0.049) (Fig. 10A).
FIG 10.

Decreased Suf function does not significantly affect exoprotein production or biofilm formation. Total exoprotein production (A), hemolysin activity (B), and biofilm formation (C) were assessed in the WT, sufD*, agrA::Tn, and ΔsigB strains. The data presented in panels A and B represent the averages of spent medium supernatants from three biological replicates, and the data in panel C represent averages of eight wells with standard deviations. Student t tests (two tailed) were performed on the data; *, P < 0.05; N.S., P > 0.05 relative to the WT (not significant); ND, not detectable.
The activities of hemolytic toxins present in the spent media from WT and sufD* strains were assessed by examining the ability of spent culture medium to lyse rabbit erythrocytes. An agrA mutant has decreased production of hemolytic toxins and was included as a control (53). The WT and sufD* strains showed similar hemolytic activities (Fig. 10B), whereas exoproteins from the agrA::Tn mutant did not cause detectable lysis.
S. aureus forms surface-associated communities referred to as biofilms. Biofilm-associated cells serve as the etiologic agents of recurrent staphylococcal infections (54). Biofilm formation was monitored aerobically using the WT and sufD* strains. The agrA::Tn and ΔsigB strains were included as experimental controls for increased and decreased biofilm formation, respectively (55, 56). The WT and sufD* strains formed similar amounts of biofilm, whereas the agrA::Tn and ΔsigB strains formed more and less biofilm than the WT, respectively (Fig. 10C).
Effective Fe-S cluster biosynthesis is necessary for survival in human PMNs.
PMNs phagocytize invading bacteria and subject them to toxic chemical species, including ROS (57). The finding that strains defective in Fe-S cluster synthesis have global metabolic defects, including increased sensitivity to ROS and increased nonchelated Fe, led us to hypothesize that decreased Fe-S cluster synthesis would result in decreased survival in human PMNs.
We examined the abilities of the WT, sufD*, and lacB::Tn strains to survive challenge by human PMNs. The lacB::Tn strain was included to evaluate the contribution of the bursa aurealis transposon to bacterial survival. The strains were individually combined with human PMNs, and bacterial survival was monitored at various time points. The sufD* strain had decreased survival compared to that of the WT upon challenge with PMNs (Fig. 11). The survival of the lacB::Tn strain was indistinguishable from that of the WT. Moreover, while the WT and lacB::Tn strains were able to rebound (120 and 180 min), minimal growth rebound was observed with the sufD* strain.
FIG 11.
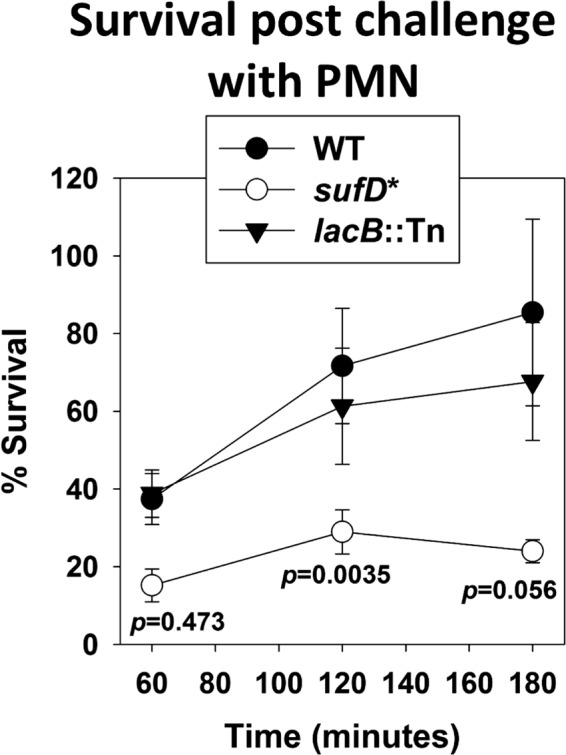
A strain with decreased Suf function has decreased survival in neutrophils. The WT, sufD*, and lacB::Tn strains were opsonized with 20% human serum, washed, and then diluted to 2.5 × 107 CFU/ml and used to infect 250,000 PMNs per well in a 96-well plate. The neutrophils were lysed upon addition of 1% saponin, and CFU were determined at various time points by plating. The data presented represent the averages of biological triplicates, with error bars representing standard errors of the mean. Student t tests (two tailed) were performed on the data, and P values are shown for the sufD* strain relative to the WT.
DISCUSSION
The present study confirmed that the Suf Fe-S cluster synthesis system is essential for S. aureus under standard laboratory growth conditions. These findings imply that Suf is the only Fe-S cluster synthesis system required for growth under these conditions (7). Therefore, if a therapeutic agent is developed that inhibits SufCDSUB, there may not be an alternate synthesis system that can compensate for its loss. Similar to S. aureus, a majority of bacterial species are predicted to utilize only one Fe-S cluster biosynthetic system, and the Suf system is the most widely distributed (58). Data from genetic screens suggest that Fe-S cluster synthesis is also required for fitness or survival of a number of additional human bacterial pathogens during routine laboratory growth (see Table 4), including Mycobacterium tuberculosis and the ESKAPE pathogens Enterococcus faecalis, Pseudomonas aeruginosa, and Acinetobacter baumannii. Not surprisingly, bioinformatics analysis suggests that the individual genomes of these organisms encode only one described Fe-S cluster synthetic system.
TABLE 4.
Fe-S biosynthesis systems in select bacterial strains
| Bacterial species | Fe-S assembly machinery | Fe-S biogenesis system predicted to be essential | Reference |
|---|---|---|---|
| Acinetobacter baumanniia | Isc | Yes | 83 |
| Bacillus subtilis | Suf | Yes | 84 |
| Bacteroides fragilis | Suf | Yes | 85 |
| Burkholderia pseudomallei | Suf and Isc | Yesc | 86 |
| Campylobacter jejuni | Nif | Yes | 87 |
| Clostridium difficile | Suf | Yes | 88 |
| Enterococcus faecalisa | Suf | Yesb | 89 |
| Escherichia coli | Suf and Isc | No | 61 |
| Francisella novicida | Suf | Yes | 90 |
| Haemophilus influenzae | Isc | Yes | 91 |
| Helicobacter pylori | Nif | Yes | 92 |
| Klebsiella pneumoniaea | Suf, Nif and Isc | No | 60 |
| Mycobacterium tuberculosis | Suf | Yes | 93 |
| Porphyromonas gingivalis | Suf | Yes | 94 |
| Pseudomonas aeruginosaa | Isc | Yes | 95 |
| Salmonella enterica | Suf and Isc | No | 96 |
| Staphylococcus aureusa | Suf | Yes | 22 |
| Streptococcus pneumoniae | Suf | Yes | 97 |
| Streptococcus pyogenes | Suf | Yes | 98 |
| Vibrio cholerae | Isc | Yes | 99 |
ESKAPE pathogen, capable of escaping the biocidal effects of antibiotics.
Limited data set.
The IscS cysteine desulfurase is predicted to be essential.
Unlike S. aureus, some bacteria utilize multiple Fe-S cluster assembly machineries (e.g., Suf and Isc) that are biochemically dissimilar but, for the most part, functionally redundant (reviewed in reference 59). Lesions in genes necessary for the function of an individual Fe-S cluster synthesis system are not lethal in these organisms (Escherichia coli and Klebsiella pneumoniae [see Table 4]) (60, 61); therefore, therapeutic agents targeting a single Fe-S cluster synthesis system would be less effective in preventing the growth or survival of these bacteria.
Iron-sulfur cluster synthesis is also essential in mammals, but importantly, mammals use Fe-S cluster synthesis machinery that is fundamentally different from the Suf system (reviewed in reference 21). This decreases the likelihood that a potential therapeutic agent that inhibits Suf function would have adverse effects on Fe-S cluster synthesis in mammals.
We utilized an S. aureus strain (sufD*) with decreased transcription of sufSUB to examine the effects of decreased Suf function on S. aureus physiology. Not surprisingly, the strain had decreased activities of Fe-S cluster-dependent enzymes and global metabolic defects. Decreased Fe-S cluster synthesis reduced growth on media lacking metabolites that require Fe-S proteins for synthesis. Protein-associated and solvent-exposed Fe-S clusters are a primary target of ROS and RNS damage (44, 45, 62), and the sufD* strain displayed increased sensitivity to H2O2, methyl viologen, and nitroprusside. Decreased Suf function also resulted in reduced flux through the TCA cycle and a destabilized nonchelated Fe pool. The sufD* strain had increased mutagenesis and decreased ability to repair DNA, which were likely the result of decreased AddAB and Nth activities.
Two scenarios could explain the essentiality of SufCDSUB for S. aureus survival during standard laboratory growth. There may be a described or unidentified essential Fe-S protein(s). To exmine this, we analyzed the results from high-density transposon screens in hopes of determining why Suf is essential in S. aureus (22–24). With the exception of Fe-S cluster synthesis proteins, the only described Fe-S proteins predicted to be essential are those encoded by fdx (ferredoxin), hemH (ferrochelatase), and addB (DNA helicase/exonuclease). Fdx and AddB were reported to be essential in one of the three studies, whereas HemH was reported to be essential in two of the studies. Here, we report that the LAC addB::Tn mutant is viable. Alternatively, the wide variety of metabolic defects resulting from defective Fe-S protein maturation may result in metabolic standstill and cell death. The numerous metabolic defects of the sufD* strain support this argument. If the inhibition of numerous metabolic functions leads to the death of cells lacking Suf function, it lowers the probability that a mutation will arise, other than mutations that affect SufCDUSB function, which would provide metabolic bypass to these processes.
Decreased Fe-S cluster synthesis did not alter exoprotein accumulation, alpha-toxin production, or biofilm formation. However, the sufD* strain displayed decreased survival in human PMNs. Further emphasizing the importance of Fe-S protein maturation for pathogenesis, an S. aureus strain lacking the Fe-S cluster carrier Nfu also displayed decreased survival in PMNs and decreased tissue colonization in a mouse model of infection (7). A recent study used transposon sequencing (Tn-seq) to identify S. aureus genes that are necessary for fitness in various models of infection (22). A number of described Fe-S proteins were required for fitness, including AddAB, Nth (DNA repair), MiaB (RNA modification), AcnA, Fdx, SdaA (central metabolism), HemN, and HemH (heme synthesis). The Nfu, SufA, and SufT Fe-S protein assembly factors were also required for fitness during infection (22).
In summary, the data presented in the present study confirm that Suf-dependent Fe-S cluster biosynthesis is essential for S. aureus survival under standard laboratory conditions. We show that an S. aureus strain with decreased Suf function has broad metabolic defects and reduced survival upon challenge with human PMNs. The mutant strains and genetic constructs described comprise a valuable toolbox for the identification of potential Suf inhibitors and for further characterization of Fe-S cluster assembly in S. aureus.
MATERIALS AND METHODS
Materials.
Phusion DNA polymerase, deoxynucleoside triphosphates, the quick DNA ligase kit, and restriction enzymes were purchased from New England BioLabs. The plasmid miniprep kit, gel extraction kit, and RNA Protect were purchased from Qiagen. TRIzol and High-Capacity cDNA reverse transcription kits were purchased from Life Technologies. Oligonucleotides, obtained from Integrated DNA Technologies, are listed in Table 1. DNase I was purchased from Ambion. Lysostaphin was purchased from Ambi Products. TSB was purchased from MP Biomedical. Difco BiTek agar was added (15 g liter−1) for solid medium. Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich and were of the highest purity obtainable.
TABLE 1.
Primers used in this study
| Name | Sequence |
|---|---|
| sufDfwdRT | CAAGTTGATGATAATGCATCGAAAG |
| sufDrevRT | ATGGTTCATAAGAGCGTCTGCTAA |
| sufSfwdRT | AACCATTGCAGAAATAGCTCATCA |
| sufSrevRT | GCTTGCGCCCCATCAAC |
| sufUfwdRT | AATGGCAAGTGCATCGATGA |
| sufUrevRT | GCATTGCTTCTCCAAGTGAATG |
| sufBfwdRT | CTGTTGTGGAAATCATTGTGCAT |
| sufBrevRT | GTTCGCCCAGTTTTGAATCG |
| sufCRT5 | GATGAAATCGATTCAGGGTTAGACA |
| sufCRT3 | TTCCCCACGCATTTGGTTA |
| sufCup | TTATTCAGCTGAACCGAACTCTTC |
| sufCdwn | CTCGTTCCCATAGCAAAACCT |
| sufUBup | GTATTTGTGTTGTCGCTTTATCCACC |
| sufUBdwn | CGGGTCTATGACAGTAGATATG |
| pML100rev | GCCTGCAGGTCGACTCTAGAGG |
| pML100for | GGCGTATCACGAGGCCCTTTCG |
| sufinternal5 | GACGTTAATGAAGTAATCAAGGATTTTCCGATATTAGA |
| sufinternal3 | TCTAATATCGGAAAATCCTTGATTACTTCATTAACGTC |
| pLLYCC5 | CTGTAATGGGCCCAATCACTAGTGAATTCCCGAAGCGGTGGCACTTTTCGGGGAAA |
| sufYCC3 | TCTCACGACGTTTTTGGCCGGTACCACGCGTTCCGGACTATATTACCCTGTTATCCCTA |
| YccSuf | TAGGGATAACAGGGTAATATAGTCCGGAACGCGTGGTACCGGCCAAAAACGTCGTGAGA |
| sufpLL39 | GTGCTAAAGAAGTTGTAGGTAATAAAAAAGCTTGCTAGCCGGAAGTCAAGAATGGCTTA |
Bacterial growth and media.
The chemically defined minimal medium was described previously (63) and where noted was supplemented with 0.5 μg ml−1 lipoic acid. S. aureus strains cultured in TSB were grown at 37°C with shaking at 200 rpm in 10-ml culture tubes containing 1 ml of liquid medium unless otherwise stated. Top agar overlays were made by diluting overnight cultures grown in TSB (1:100 in phosphate-buffered saline [PBS]) and then adding 100 μl to 4 ml of 3.5% TSA before pouring it on top of TSA plates. Where noted, 1 μl of 2.5-mg ml−1 streptonigrin dissolved in dimethyl sulfoxide (DMSO), 4 μl of neat diethyl sulfate, or 2 μl of neat methyl methanesulfonate was spotted in the centers of the plates. Antibiotics were added to TSB at the following concentrations: 3 to 5 ng ml−1 Atet, 30 μg ml−1 chloramphenicol (Cm), 1.25 μg ml−1 Rif, and 10 μg ml−1 erythromycin (Erm). To maintain plasmids, the medium was supplemented with 15 μg ml−1 or 5 μg ml−1 Cm or Erm, respectively. Methyl viologen and 2,2-dypyridyl were added to solid media at 40 mM and 900 mM, respectively.
Liquid phenotypic analysis was conducted in 96-well microtiter plates containing 200 μl of medium per well using a BioTek 808E visible absorption spectrophotometer, and culture densities were read at 600 nm. The cells used for inoculation were cultured for 18 h in TSB medium, and the cells were washed with PBS. The optical densities (OD) of the cell suspensions were adjusted to 2.5 (A600) with PBS. Two microliters of the washed cells was added to 198 μl of medium. Where noted, sodium nitroprusside was added to liquid media at 15 mM.
Genetic and recombinant DNA techniques.
The bacterial strains and plasmids used in this study are listed in Tables 2 and 3. Unless otherwise noted, these strains, including the sufD* strain, were constructed in the community-associated methicillin-resistant S. aureus (MRSA) USA300 strain LAC (JMB strains) that had been cured of the plasmid conferring resistance to erythromycin (pUSA03) (64). All transductions were conducted using phage 80α (65). All the S. aureus mutant strains and plasmids were verified using PCR or by sequencing PCR products or plasmids. All DNA sequencing was performed by Genewiz (South Plainfield, NJ).
TABLE 2.
Strains used in this study
| Strain name | Genotype | Source/reference |
|---|---|---|
| WT | USA300_LAC | A. R. Horswill (79) |
| JMB1102 | ΔsigB (SAUSA300_2022) | 55 |
| JMB1163 | ΔacnA::tetM (SAUSA300_1246) | 80 |
| JMB1165 | Δnfu (SAUSA300_0839) | 7 |
| JMB2078 | kat::Tn (ermB) (SAUSA300_1232) | V. Torres |
| JMB2763 | nth::Tn (ermB) (SAUSA300_1343) | BEI (28) |
| JMB2726 | mutY::Tn (ermB) (SAUSA300_1849) | BEI (28) |
| JMB2950 | agrA::Tn (ermB) (SAUSA300_1992) | BEI (28) |
| JMB3298 | addB::Tn (ermB) (SAUSA300_0869) | BEI (28) |
| JMB5853 | sodA::Tn (ermB) (SAUSA300_1513) | BEI (28) |
| None | sufD*::Tn (ermB) (Fig. 2A) | P. Fey |
| None | sufS*::Tn (ermB) (Fig. 2A) | P. Fey |
| JMB8464 | sufD*::Tn (ermB) | This study |
| JMB8472 | sufD*::Tn (ermB), pLL39_sufCDSUB | This study |
| JMB7237 | lacB::Tn (ermB) (SAUSA300_2154) | BEI (28) |
| JMB7525 | fhuC::Tn (ermB) (SAUSA300_0633) | BEI (28) |
| JMB7592 | Δnfu addB::Tn (ermB) | This study |
| RN4220 | Restriction minus | 81 |
| E. coli PX5 | Used for gene cloning | Protein Express |
TABLE 3.
Plasmids used in this study
| Plasmid name | Insert | Function | Reference |
|---|---|---|---|
| pCM11_sufCp | sufC promoter | sufC transcriptional activity | 19 |
| pML100 | None | Gene expression | 67 |
| psufCKD1 | sufC DNA | Suf depletion | |
| psufCKD2 | sufC DNA | Suf depletion | |
| psufUKD1 | sufU DNA | Suf depletion | |
| psufUKD2 | sufU DNA | Suf depletion | |
| pLL39 | None | Genetic complementation | 82 |
| pLL39_sufCDSUB | sufCDSUB | Genetic complementation | |
| pCR2.1_TOPO | None | Cloning |
The Suf depletion plasmids were created as described previously (66). Briefly, the sufC gene and its 5′ untranslated region were amplified using the sufCup and sufCdwn primers. The sufUB amplicon was created using the sufUBup and sufUBdwn primers. The resulting amplicons were gel purified and treated with 0.03 U DNase I (Ambion, Carlsbad, CA) for 5 min. The digested DNAs were separated using agarose gel chromatography, and DNAs of approximately 250 bp were purified. The purified fragments were treated with T4 DNA polymerase (NEB, Ipswich, MA) and subsequently treated with Taq DNA polymerase (NEB). The DNA fragments were cloned into pCR2.1_TOPO (Thermo-Fisher). After transformation and selection, the colonies were pooled and the plasmids were purified. The plasmids were digested with EcoRI, and the insert fragments were gel purified and subsequently subcloned into pML100 (67). After transformation and selection, colonies containing pML100 were pooled, and plasmids were purified and transformed into S. aureus RN4220 and plated on TSA-Cm. Individual chloramphenicol-resistant RN4220 colonies were inoculated into 200 μl of TSB-Cm medium in 96-well microtiter plates and cultured overnight. The cells were subcultured into liquid TSB media with and without Atet, and strains with decreased growth in the presence of Atet were retained. Four positive clones were identified, and the inserts were confirmed by DNA sequencing.
The pLL39_sufCDUSB plasmid was created using yeast recombinational cloning as previously described (68, 69). The amplicons for pLL39_sufCDSUB were created using the following primer pairs: pLLYCC5 and sufYCC3, YccSuf and Sufinternal3, and Sufinternal5 and sufpLL39. pLL39 was linearized using SalI.
RNA-seq analysis of the suf operon.
RNA-seq data were downloaded from the Gene Expression Omnibus (GEO) (accession number GSE48896), corresponding to NCTC8325-4 (29). The downloaded Sequence Read Archive (SRA) files were converted to fastq format using the SRA toolkit and then mapped to the S. aureus genome using Tophat (70, 71). The resulting bam files were sorted and indexed using SAMtools (72) and then converted to tdf format using Integrative Genomics Viewer (IGV) tools (73). The image of the suf operon was acquired using IGV (73).
Protein analysis and GOGAT assays.
GOGAT assays were conducted as previously described with slight modifications (18). Briefly, strains were cultured overnight in TSB, and the cells were pelleted by centrifugation and resuspended in PBS (1:1). The resuspended cells were used to inoculate 5 ml (in a 30-ml tube) of chemically defined medium containing 20 aa and lipoic acid to an OD of 0.1 (A600). Strains were cultured at 37°C with shaking to an OD of 0.8 (A600), and the cells were harvested by centrifugation and resuspended in lysis buffer (50 mM Tris-HCl, pH 7.7). The cells were lysed anaerobically by the addition of 4 μg lysostaphin and 8 μg DNase. The cells were incubated at 37°C until full lysis was observed (∼1 h). The cell debris was removed by centrifugation. GOGAT was assayed by the addition of 60 μl of 50 mM glutamine (pH 7.7), 60 μl of 5 mM α-ketoglutarate (pH 7.7), 60 μl of cell extract, and 60 μl of 0.75 mM NADP (NADPH) to 600 μl of lysis buffer. GOGAT activity was determined by monitoring the rate of NADPH oxidation at 340 nm for 5 min (extinction coefficient at 340 nm [ε340] = 6.22 mM−1 cm−1 [74]).
Aconitase assays.
AcnA assays were conducted as previously described with slight modifications (19). Strains were cultured overnight in TSB before diluting them in fresh TSB to an optical density of 0.1 (A600). The cultures were diluted in 0.5 ml or 4 ml of TSB in 10-ml culture tubes. The cells were cultured for 8 h (Fig. 4), or samples were removed throughout growth (Fig. 5) before they were harvested by centrifugation, and the cell pellets were stored at −80°C. The cells were thawed anaerobically, resuspended with 200 μl of AcnA buffer (50 mM Tris, 150 mM NaCl, pH 7.4), and lysed by the addition of 4 μg lysostaphin and 8 μg DNase. The cells were incubated at 37°C until full lysis was observed (∼1 h). The cell debris was removed by centrifugation, and AcnA activity was assessed as previously described (32).
Protein concentration determination.
The protein concentration was determined using a copper-bicinchoninic acid-based colorimetric assay modified for a 96-well plate (75).
RNA isolation and quantification of mRNA transcripts.
Bacterial strains were cultured overnight in TSB (∼18 h) and diluted in 80 ml of fresh TSB to a final OD of 0.05 (A600) in 300-ml flasks in order to mimic the growth conditions used for the growth and acetate accumulation experiments shown in Fig. 5. The cells were cultured for 8 h before harvesting by centrifugation. The cells were treated with RNAProtect (Qiagen) for 10 min at room temperature and pelleted by centrifugation, and the cell pellets were stored at −80°C. The pellets were thawed and washed twice with 0.5 ml of lysis buffer (50 mM RNase-free Tris, pH 8). The cells were lysed by the addition of 20 μg of lysostaphin and incubated for 30 min at 37°C. RNA was isolated using TRIzol reagent (Ambion-Life Technologies) according to the manufacturer's instructions. DNA was digested with the Turbo DNA-free kit (Ambion-Life Technologies). The cDNA libraries were constructed using isolated RNA as a template and a High Capacity RNA-to-cDNA kit (Applied Biosysytems). An Applied Biosystems StepOnePlus thermocycler and Power SYBR green PCR master mix (Applied Biosystems) were used to quantify DNA abundance. The primers for quantitative real-time PCR of the sufC, sufD, sufS, sufU, and sufB transcripts, designed using Primer Express 3.0 software from Applied Biosystems, are listed in Table 1.
H2O2 killing assays.
Bacterial strains were cultured for 12 h in TSB. The cells were pelleted by centrifugation and resuspended in an equal volume of PBS. The optical densities of the strains were adjusted to an OD of 0.7 (A600) in a total volume of 1 ml of PBS. The cells were subsequently challenged with a bolus of H2O2 (500 mM) and incubated for 1 h at room temperature. Fifty microliters of the reaction mixture was diluted 1:20 in PBS buffer containing catalase (1,300 units ml−1) and incubated for 5 min. Cell viability was visualized by serial dilution of cells and spot plating upon TSA.
Determination of pH profiles and acetic acid concentrations in spent media.
Strains cultured overnight in TSB (∼18 h) were diluted in 80 ml of fresh TSB to a final OD of 0.05 (A600) in 300-ml flasks. At the indicated times, aliquots of the cultures were removed, the culture OD (A600) was determined, and the cells and culture media were partitioned by centrifugation at 14,000 rpm for 1 min. Two milliliters of either the culture supernatant or sterile TSB, which served to provide a pH reading for the point of inoculation, were combined with 8 ml of distilled and deionized water, and the pH was determined using a Fisher Scientific Accumet AB15 pH mV meter. The concentration of acetic acid in the spent medium was determined using a BioVision acetate colorimetric assay kit (K658) according to the manufacturer's instructions.
Static model of biofilm formation.
Biofilm formation was examined as described previously, with minor changes (19, 55). Briefly, overnight cultures were diluted in biofilm medium to a final optical density of 0.05 (A590), added to the wells of a 96-well microtiter plate, and incubated statically at 37°C for 22 h. Prior to harvesting the biofilms, the optical densities (A590) of the cultures were determined. The plate was subsequently washed with water, the biofilms were heat fixed at 60°C, and the plates were allowed to cool to room temperature. The biofilms were stained with 0.1% crystal violet and washed to remove unbound stain. The plates were dried and subsequently destained by the addition of 33% acetic acid, and the absorbance at 570 nm of the resulting solution was recorded. The absorbance (A570) was standardized to an acetic acid blank and subsequently to the optical density of the cells upon harvest. Finally, the data were normalized with respect to the WT strain to obtain relative biofilm formation.
Total exoprotein analyses.
Spent medium supernatants were obtained from overnight cultures, filter sterilized with a 0.22-μm-pore-size syringe filter, and standardized with respect to culture optical densities (A600), as previously described (19). Exoproteins were extracted from the spent medium supernatant using standard trichloroacetic acid precipitation. The resultant protein pellets were resuspended, and protein concentrations were determined using a biuret assay. The data were subsequently normalized with respect to the WT strain.
Hemolysis assays.
The hemolytic activities of staphylococcal exoproteins were determined as previously described (76). The data were subsequently normalized with respect to the WT strain.
Mutagenesis frequency.
Overnight cultures (n = 10) were grown in TSB medium before dilution (1:100) in fresh TSB (A600, ∼0.1). The cells were cultured with shaking for 48 h at 37°C. One hundred microliters of culture was spread plated on TSA supplemented with 1.25 μg ml−1 of rifampin, and CFU were determined after 36 h of incubation. Cultures were also serially diluted and spot plated on TSA to determine total CFU. The mutagenesis frequency was calculated by dividing the number of rifampin-resistant colonies by the total number of CFU.
Transcriptional reporter analyses.
Strains containing the psufCp (19) transcriptional reporter plasmid were grown in TSB-Erm medium overnight. The cultures were then diluted (1:100) in 5 ml of fresh TSB-Erm and allowed to grow for 30 h, during which 200-μl aliquots were removed at various time points and fluorescence and culture OD (A600) were measured with a PerkinElmer HTS 7000 Bio Assay reader. Green fluorescent protein (GFP) was excited at 485 nm, and emission was read at 535 nm. Fluorescence was standardized with respect to the culture OD.
Opsonophagocytic killing assay.
Strains were cultured overnight in TSB and subcultured in TSB (1:100) the following day for 3 h. Human primary PMNs were isolated by dextran gradient as described previously (77). Prior to infection, 96-well plates were coated with 20% human serum in RPMI 1640 (10 mM HEPES plus 0.1% human serum albumin [HSA]) for 30 min at 37°C. Following subculture of the bacteria, the strains were opsonized with 20% human serum for 30 min at 37°C, washed, and diluted to an approximate density of 2.5 × 107 CFU ml−1. Approximately 250,000 PMNs per well in a 96-well plate were infected with approximately 2.5 × 106 CFU to generate a multiplicity of infection (MOI) of 10. With the exception of time zero, the infections were centrifuged at 1,500 RPM for 7 min to synchronize the bacteria with the PMNs. During centrifugation, 1% saponin was added to the time zero infections to lyse the PMNS, and CFU were then determined by serial dilution and plating on TSA. This procedure was followed for the remaining time points up to 180 min.
Blood samples were obtained from anonymous healthy donors as buffy coats (New York City Blood Center). The New York City Blood Center obtained written informed consent from all participants involved in the study. The research was approved by the New York University School of Medicine institutional human subjects board.
Bioinformatic and statistical analyses.
The analyses presented in Table 4 were generated by first using BLAST (78) to identify the homologues of S. aureus SufBCD, E. coli IscU, or Azotobacter vinelandii NifU in the genomes of various bacterial pathogens. The corresponding locus tags were then used to determine whether the genes were predicted to be essential, using published data sets. The data presented were analyzed and plotted using SigmaPlot version 12, and statistical analyses were conducted using Microsoft Excel.
ACKNOWLEDGMENTS
W.E.S. was supported in part by Public Health Service Grant T32 AI007180. V.J.T.'s laboratory was supported by Public Health Service Grants AI099394 and AI105129. V.J.T. is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases. The Belden laboratory is supported by National Institutes of Health grant GM101378. The Boyd laboratory is supported by Rutgers University, the Charles and Johanna Busch Foundation, and USDA MRF project NE-1028 (J.M.B.). Z.R.-C. was supported by a James Macmillan Endowed Fellowship from the Microbial Biology Graduate Program. A.A.M. is supported by a Douglas Eveleigh fellowship from the Microbial Biology Graduate Program and an Excellence Fellowship from Rutgers University. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 2.Daum RS. 2007. Clinical practice. Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. N Engl J Med 357:380–390. [DOI] [PubMed] [Google Scholar]
- 3.Lodise TP Jr, McKinnon PS. 2007. Burden of methicillin-resistant Staphylococcus aureus: focus on clinical and economic outcomes. Pharmacotherapy 27:1001–1012. doi: 10.1592/phco.27.7.1001. [DOI] [PubMed] [Google Scholar]
- 4.Cosgrove SE, Sakoulas G, Perencevich EN, Schwaber MJ, Karchmer AW, Carmeli Y. 2003. Comparison of mortality associated with methicillin-resistant and methicillin-susceptible Staphylococcus aureus bacteremia: a meta-analysis. Clin Infect Dis 36:53–59. doi: 10.1086/345476. [DOI] [PubMed] [Google Scholar]
- 5.Lewis K. 2013. Platforms for antibiotic discovery. Nat Rev Drug Discov 12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 6.Skaar EP, Humayun M, Bae T, DeBord KL, Schneewind O. 2004. Iron-source preference of Staphylococcus aureus infections. Science 305:1626–1628. doi: 10.1126/science.1099930. [DOI] [PubMed] [Google Scholar]
- 7.Mashruwala AA, Pang YY, Rosario-Cruz Z, Chahal HK, Benson MA, Mike LA, Skaar EP, Torres VJ, Nauseef WM, Boyd JM. 2015. Nfu facilitates the maturation of iron-sulfur proteins and participates in virulence in Staphylococcus aureus. Mol Microbiol 95:383–409. doi: 10.1111/mmi.12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng L, Cash VL, Flint DH, Dean DR. 1998. Assembly of iron-sulfur clusters. Identification of an iscSUA-hscBA-fdx gene cluster from Azotobacter vinelandii. J Biol Chem 273:13264–13272. [DOI] [PubMed] [Google Scholar]
- 9.Yuvaniyama P, Agar JN, Cash VL, Johnson MK, Dean DR. 2000. NifS-directed assembly of a transient [2Fe-2S] cluster within the NifU protein. Proc Natl Acad Sci U S A 97:599–604. doi: 10.1073/pnas.97.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi Y, Tokumoto U. 2002. A third bacterial system for the assembly of iron-sulfur clusters with homologs in archaea and plastids. J Biol Chem 277:28380–28393. doi: 10.1074/jbc.C200365200. [DOI] [PubMed] [Google Scholar]
- 11.Wollers S, Layer G, Garcia-Serres R, Signor L, Clemancey M, Latour JM, Fontecave M, Ollagnier de Choudens S. 2010. Iron-sulfur (Fe-S) cluster assembly: the SufBCD complex is a new type of Fe-S scaffold with a flavin redox cofactor. J Biol Chem 285:23331–23341. doi: 10.1074/jbc.M110.127449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nachin L, El Hassouni M, Loiseau L, Expert D, Barras F. 2001. SoxR-dependent response to oxidative stress and virulence of Erwinia chrysanthemi: the key role of SufC, an orphan ABC ATPase. Mol Microbiol 39:960–972. doi: 10.1046/j.1365-2958.2001.02288.x. [DOI] [PubMed] [Google Scholar]
- 13.Saini A, Mapolelo DT, Chahal HK, Johnson MK, Outten FW. 2010. SufD and SufC ATPase activity are required for iron acquisition during in vivo Fe-S cluster formation on SufB. Biochemistry 49:9402–9412. doi: 10.1021/bi1011546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Layer G, Gaddam SA, Ayala-Castro CN, Ollagnier-de Choudens S, Lascoux D, Fontecave M, Outten FW. 2007. SufE transfers sulfur from SufS to SufB for iron-sulfur cluster assembly. J Biol Chem 282:13342–13350. doi: 10.1074/jbc.M608555200. [DOI] [PubMed] [Google Scholar]
- 15.Selbach B, Earles E, Dos Santos PC. 2010. Kinetic analysis of the bisubstrate cysteine desulfurase SufS from Bacillus subtilis. Biochemistry 49:8794–8802. doi: 10.1021/bi101358k. [DOI] [PubMed] [Google Scholar]
- 16.Selbach BP, Chung AH, Scott AD, George SJ, Cramer SP, Dos Santos PC. 2014. Fe-S cluster biogenesis in Gram-positive bacteria: SufU is a zinc-dependent sulfur transfer protein. Biochemistry 53:152–160. doi: 10.1021/bi4011978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chahal HK, Outten FW. 2012. Separate FeS scaffold and carrier functions for SufB(2)C(2) and SufA during in vitro maturation of [2Fe2S] Fdx. J Inorg Biochem 116:126–134. doi: 10.1016/j.jinorgbio.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosario-Cruz Z, Chahal HK, Mike LA, Skaar EP, Boyd JM. 2015. Bacillithiol has a role in Fe-S cluster biogenesis in Staphylococcus aureus. Mol Microbiol 98:218–242. doi: 10.1111/mmi.13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mashruwala AA, Bhatt S, Poudel S, Boyd ES, Boyd JM. 2016. The DUF59 containing protein SufT is involved in the maturation of iron-sulfur (FeS) proteins during conditions of high FeS cofactor demand in Staphylococcus aureus. PLoS Genet 12:e1006233. doi: 10.1371/journal.pgen.1006233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosario-Cruz Z, Boyd JM. 2016. Physiological roles of bacillithiol in intracellular metal processing. Curr Genet 62:59–65. doi: 10.1007/s00294-015-0511-0. [DOI] [PubMed] [Google Scholar]
- 21.Lill R. 2009. Function and biogenesis of iron-sulphur proteins. Nature 460:831–838. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 22.Valentino MD, Foulston L, Sadaka A, Kos VN, Villet RA, Santa Maria J Jr, Lazinski DW, Camilli A, Walker S, Hooper DC, Gilmore MS. 2014. Genes contributing to Staphylococcus aureus fitness in abscess- and infection-related ecologies. mBio 5:e01729–01714. doi: 10.1128/mBio.01729-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santiago M, Matano LM, Moussa SH, Gilmore MS, Walker S, Meredith TC. 2015. A new platform for ultra-high density Staphylococcus aureus transposon libraries. BMC Genomics 16:252. doi: 10.1186/s12864-015-1361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaudhuri RR, Allen AG, Owen PJ, Shalom G, Stone K, Harrison M, Burgis TA, Lockyer M, Garcia-Lara J, Foster SJ, Pleasance SJ, Peters SE, Maskell DJ, Charles IG. 2009. Comprehensive identification of essential Staphylococcus aureus genes using transposon-mediated differential hybridisation (TMDH). BMC Genomics 10:291. doi: 10.1186/1471-2164-10-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji Y, Woodnutt G, Rosenberg M, Burnham MK. 2002. Identification of essential genes in Staphylococcus aureus using inducible antisense RNA. Methods Enzymol 358:123–128. doi: 10.1016/S0076-6879(02)58084-8. [DOI] [PubMed] [Google Scholar]
- 26.Traber K, Novick R. 2006. A slipped-mispairing mutation in AgrA of laboratory strains and clinical isolates results in delayed activation of agr and failure to translate delta- and alpha-haemolysins. Mol Microbiol 59:1519–1530. doi: 10.1111/j.1365-2958.2006.04986.x. [DOI] [PubMed] [Google Scholar]
- 27.Nair D, Memmi G, Hernandez D, Bard J, Beaume M, Gill S, Francois P, Cheung AL. 2011. Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J Bacteriol 193:2332–2335. doi: 10.1128/JB.00027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537–00512. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osmundson J, Dewell S, Darst SA. 2013. RNA-Seq reveals differential gene expression in Staphylococcus aureus with single-nucleotide resolution. PLoS One 8:e76572. doi: 10.1371/journal.pone.0076572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pane-Farre J, Jonas B, Forstner K, Engelmann S, Hecker M. 2006. The sigmaB regulon in Staphylococcus aureus and its regulation. Int J Med Microbiol 296:237–258. doi: 10.1016/j.ijmm.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 31.Gertz S, Engelmann S, Schmid R, Ziebandt AK, Tischer K, Scharf C, Hacker J, Hecker M. 2000. Characterization of the sigma(B) regulon in Staphylococcus aureus. J Bacteriol 182:6983–6991. doi: 10.1128/JB.182.24.6983-6991.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kennedy MC, Emptage MH, Dreyer JL, Beinert H. 1983. The role of iron in the activation-inactivation of aconitase. J Biol Chem 258:11098–11105. [PubMed] [Google Scholar]
- 33.Mashruwala AA, Boyd JM. 2017. The Staphylococcus aureus SrrAB regulatory system modulates hydrogen peroxide resistance factors, which imparts protection to aconitase during aerobic growth. PLoS One 12:e0170283. doi: 10.1371/journal.pone.0170283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ledala N, Zhang B, Seravalli J, Powers R, Somerville GA. 2014. Influence of iron and aeration on Staphylococcus aureus growth, metabolism, and transcription. J Bacteriol 196:2178–2189. doi: 10.1128/JB.01475-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanoni MA, Curti B. 2008. Structure-function studies of glutamate synthases: a class of self-regulated iron-sulfur flavoenzymes essential for nitrogen assimilation. IUBMB Life 60:287–300. doi: 10.1002/iub.52. [DOI] [PubMed] [Google Scholar]
- 36.Ollagnier-de Choudens S, Fontecave M. 1999. The lipoate synthase from Escherichia coli is an iron-sulfur protein. FEBS Lett 453:25–28. doi: 10.1016/S0014-5793(99)00694-8. [DOI] [PubMed] [Google Scholar]
- 37.Flint DH, Emptage MH. 1988. Dihydroxy acid dehydratase from spinach contains a [2Fe-2S] cluster. J Biol Chem 263:3558–3564. [PubMed] [Google Scholar]
- 38.Hentze MW, Argos P. 1991. Homology between IRE-BP, a regulatory RNA-binding protein, aconitase, and isopropylmalate isomerase. Nucleic Acids Res 19:1739–1740. doi: 10.1093/nar/19.8.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller RE. 1974. Glutamate synthase from Escherichia coli–an iron-sulfur flavoprotein. Separation and analysis of non-identical subunits. Biochim Biophys Acta 364:243–249. [DOI] [PubMed] [Google Scholar]
- 40.Porello SL, Cannon MJ, David SS. 1998. A substrate recognition role for the [4Fe-4S]2+ cluster of the DNA repair glycosylase MutY. Biochemistry 37:6465–6475. doi: 10.1021/bi972433t. [DOI] [PubMed] [Google Scholar]
- 41.Fu W, O'Handley S, Cunningham RP, Johnson MK. 1992. The role of the iron-sulfur cluster in Escherichia coli endonuclease III. A resonance Raman study. J Biol Chem 267:16135–16137. [PubMed] [Google Scholar]
- 42.Yeeles JT, Cammack R, Dillingham MS. 2009. An iron-sulfur cluster is essential for the binding of broken DNA by AddAB-type helicase-nucleases. J Biol Chem 284:7746–7755. doi: 10.1074/jbc.M808526200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ezekiel DH, Hutchins JE. 1968. Mutations affecting RNA polymerase associated with rifampicin resistance in Escherichia coli. Nature 220:276–277. doi: 10.1038/220276a0. [DOI] [PubMed] [Google Scholar]
- 44.Jang S, Imlay JA. 2007. Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J Biol Chem 282:929–937. doi: 10.1074/jbc.M607646200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duan X, Yang J, Ren B, Tan G, Ding H. 2009. Reactivity of nitric oxide with the [4Fe-4S] cluster of dihydroxyacid dehydratase from Escherichia coli. Biochem J 417:783–789. doi: 10.1042/BJ20081423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grossi L, D'Angelo S. 2005. Sodium nitroprusside: mechanism of NO release mediated by sulfhydryl-containing molecules. J Med Chem 48:2622–2626. doi: 10.1021/jm049857n. [DOI] [PubMed] [Google Scholar]
- 47.Rauen U, Springer A, Weisheit D, Petrat F, Korth HG, de Groot H, Sustmann R. 2007. Assessment of chelatable mitochondrial iron by using mitochondrion-selective fluorescent iron indicators with different iron-binding affinities. Chembiochem 8:341–352. doi: 10.1002/cbic.200600311. [DOI] [PubMed] [Google Scholar]
- 48.Speziali CD, Dale SE, Henderson JA, Vines ED, Heinrichs DE. 2006. Requirement of Staphylococcus aureus ATP-binding cassette-ATPase FhuC for iron-restricted growth and evidence that it functions with more than one iron transporter. J Bacteriol 188:2048–2055. doi: 10.1128/JB.188.6.2048-2055.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolzan AD, Bianchi MS. 2001. Genotoxicity of streptonigrin: a review. Mutat Res 488:25–37. doi: 10.1016/S1383-5742(00)00062-4. [DOI] [PubMed] [Google Scholar]
- 50.White JR, Yeowell HN. 1982. Iron enhances the bactericidal action of streptonigrin. Biochem Biophys Res Commun 106:407–411. doi: 10.1016/0006-291X(82)91125-1. [DOI] [PubMed] [Google Scholar]
- 51.DeGraff W, Hahn SM, Mitchell JB, Krishna MC. 1994. Free radical modes of cytotoxicity of adriamycin and streptonigrin. Biochem Pharmacol 48:1427–1435. doi: 10.1016/0006-2952(94)90567-3. [DOI] [PubMed] [Google Scholar]
- 52.Bartlett AH, Hulten KG. 2010. Staphylococcus aureus pathogenesis: secretion systems, adhesins, and invasins. Pediatr Infect Dis J 29:860–861. doi: 10.1097/INF.0b013e3181ef2477. [DOI] [PubMed] [Google Scholar]
- 53.Recsei P, Kreiswirth B, O'Reilly M, Schlievert P, Gruss A, Novick RP. 1986. Regulation of exoprotein gene expression in Staphylococcus aureus by agar. Mol Gen Genet 202:58–61. doi: 10.1007/BF00330517. [DOI] [PubMed] [Google Scholar]
- 54.Otto M. 2008. Staphylococcal biofilms. Curr Top Microbiol Immunol 322:207–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lauderdale KJ, Boles BR, Cheung AL, Horswill AR. 2009. Interconnections between sigma B, agr, and proteolytic activity in Staphylococcus aureus biofilm maturation. Infect Immun 77:1623–1635. doi: 10.1128/IAI.01036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boles BR, Horswill AR. 2008. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog 4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nauseef WM. 2007. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev 219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 58.Boyd ES, Thomas KM, Dai Y, Boyd JM, Outten FW. 2014. Interplay between oxygen and Fe-S cluster biogenesis: insights from the Suf pathway. Biochemistry 53:5834–5847. doi: 10.1021/bi500488r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Py B, Barras F. 2010. Building Fe-S proteins: bacterial strategies. Nat Rev Microbiol 8:436–446. doi: 10.1038/nrmicro2356. [DOI] [PubMed] [Google Scholar]
- 60.Bachman MA, Breen P, Deornellas V, Mu Q, Zhao L, Wu W, Cavalcoli JD, Mobley HL. 2015. Genome-wide identification of Klebsiella pneumoniae fitness genes during lung infection. mBio 6:e00775. doi: 10.1128/mBio.00775-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006 0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flint DH, Tuminello JF, Emptage MH. 1993. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J Biol Chem 268:22369–22376. [PubMed] [Google Scholar]
- 63.Mashruwala AA, Roberts CA, Bhatt S, May KL, Carroll RK, Shaw LN, Boyd JM. 2016. Staphylococcus aureus SufT: an essential iron-sulfur cluster assembly factor in cells experiencing a high demand for lipoic acid. Mol Microbiol 102:1099–1119. doi: 10.1111/mmi.13539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pang YY, Schwartz J, Bloomberg S, Boyd JM, Horswill AR, Nauseef WM. 2014. Methionine sulfoxide reductases protect against oxidative stress in Staphylococcus aureus encountering exogenous oxidants and human neutrophils. J Innate Immun 6:353–364. doi: 10.1159/000355915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Novick RP. 1963. Analysis by transduction of mutations affecting penicillinase formation in Staphylococcus aureus. J Gen Microbiol 33:121–136. doi: 10.1099/00221287-33-1-121. [DOI] [PubMed] [Google Scholar]
- 66.Eidem TM, Lounsbury N, Emery JF, Bulger J, Smith A, Abou-Gharbia M, Childers W, Dunman PM. 2015. Small-molecule inhibitors of Staphylococcus aureus RnpA-mediated RNA turnover and tRNA processing. Antimicrob Agents Chemother 59:2016–2028. doi: 10.1128/AAC.04352-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lei MG, Cue D, Roux CM, Dunman PM, Lee CY. 2011. Rsp inhibits attachment and biofilm formation by repressing fnbA in Staphylococcus aureus MW2. J Bacteriol 193:5231–5241. doi: 10.1128/JB.05454-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Joska TM, Mashruwala A, Boyd JM, Belden WJ. 2014. A universal cloning method based on yeast homologous recombination that is simple, efficient, and versatile. J Microbiol Methods 100:46–51. doi: 10.1016/j.mimet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mashruwala AA, Boyd JM. 2016. De novo assembly of plasmids using yeast recombinational cloning. Methods Mol Biol 1373:33–41. doi: 10.1007/7651_2015_275. [DOI] [PubMed] [Google Scholar]
- 70.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup . 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dougall DK. 1974. Evidence for the presence of glutamate synthase in extracts of carrot cell cultures. Biochem Biophys Res Commun 58:639–646. doi: 10.1016/S0006-291X(74)80466-3. [DOI] [PubMed] [Google Scholar]
- 75.Olson BJ, Markwell J. 2007. Assays for determination of protein concentration. Curr Protoc Protein Sci Chapter 3:Unit 3.4. [DOI] [PubMed] [Google Scholar]
- 76.Blevins JS, Beenken KE, Elasri MO, Hurlburt BK, Smeltzer MS. 2002. Strain-dependent differences in the regulatory roles of sarA and agr in Staphylococcus aureus. Infect Immun 70:470–480. doi: 10.1128/IAI.70.2.470-480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.DuMont AL, Yoong P, Surewaard BG, Benson MA, Nijland R, van Strijp JA, Torres VJ. 2013. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun 81:1830–1841. doi: 10.1128/IAI.00095-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 79.Boles BR, Thoendel M, Roth AJ, Horswill AR. 2010. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5:e10146. doi: 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sadykov MR, Mattes TA, Luong TT, Zhu Y, Day SR, Sifri CD, Lee CY, Somerville GA. 2010. Tricarboxylic acid cycle-dependent synthesis of Staphylococcus aureus type 5 and 8 capsular polysaccharides. J Bacteriol 192:1459–1462. doi: 10.1128/JB.01377-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 82.Luong TT, Lee CY. 2007. Improved single-copy integration vectors for Staphylococcus aureus. J Microbiol Methods 70:186–190. doi: 10.1016/j.mimet.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gallagher LA, Ramage E, Weiss EJ, Radey M, Hayden HS, Held KG, Huse HK, Zurawski DV, Brittnacher MJ, Manoil C. 2015. Resources for genetic and genomic analysis of emerging pathogen Acinetobacter baumannii. J Bacteriol 197:2027–2035. doi: 10.1128/JB.00131-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P, Boland F, Brignell SC, Bron S, Bunai K, Chapuis J, Christiansen LC, Danchin A, Debarbouille M, Dervyn E, Deuerling E, Devine K, Devine SK, Dreesen O, Errington J, Fillinger S, Foster SJ, Fujita Y, Galizzi A, Gardan R, Eschevins C, Fukushima T, Haga K, Harwood CR, Hecker M, Hosoya D, Hullo MF, Kakeshita H, Karamata D, Kasahara Y, Kawamura F, Koga K, Koski P, Kuwana R, Imamura D, Ishimaru M, Ishikawa S, Ishio I, Le Coq D, Masson A, Mauel C, Meima R, Mellado RP, Moir A, Moriya S, Nagakawa E, Nanamiya H, Nakai S, Nygaard P, Ogura M, Ohanan T, O'Reilly M, O'Rourke M, Pragai Z, Pooley HM, Rapoport G, Rawlins JP, Rivas LA, Rivolta C, Sadaie A, Sadaie Y, Sarvas M, Sato T, Saxild HH, Scanlan E, Schumann W, Seegers JF, Sekiguchi J, Sekowska A, Séror SJ, Simon M, Stragier P, Studer R, Takamatsu H, Tanaka T, Takeuchi M, Thomaides HB, Vagner V, van Dijl JM, Watabe K, Wipat A, Yamamoto H, Yamamoto M, Yamamoto Y, Yamane K, Yata K, Yoshida K, Yoshikawa H, Zuber U, Ogasawara N. 2003. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A 100:4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Veeranagouda Y, Husain F, Tenorio EL, Wexler HM. 2014. Identification of genes required for the survival of B. fragilis using massive parallel sequencing of a saturated transposon mutant library. BMC Genomics 15:429. doi: 10.1186/1471-2164-15-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moule MG, Hemsley CM, Seet Q, Guerra-Assuncao JA, Lim J, Sarkar-Tyson M, Clark TG, Tan PB, Titball RW, Cuccui J, Wren BW. 2014. Genome-wide saturation mutagenesis of Burkholderia pseudomallei K96243 predicts essential genes and novel targets for antimicrobial development. mBio 5:e00926–00913. doi: 10.1128/mBio.00926-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Metris A, Reuter M, Gaskin DJ, Baranyi J, van Vliet AH. 2011. In vivo and in silico determination of essential genes of Campylobacter jejuni. BMC Genomics 12:535. doi: 10.1186/1471-2164-12-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dembek M, Barquist L, Boinett CJ, Cain AK, Mayho M, Lawley TD, Fairweather NF, Fagan RP. 2015. High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. mBio 6:e02383. doi: 10.1128/mBio.02383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garsin DA, Urbach J, Huguet-Tapia JC, Peters JE, Ausubel FM. 2004. Construction of an Enterococcus faecalis Tn917-mediated-gene-disruption library offers insight into Tn917 insertion patterns. J Bacteriol 186:7280–7289. doi: 10.1128/JB.186.21.7280-7289.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gallagher LA, Ramage E, Jacobs MA, Kaul R, Brittnacher M, Manoil C. 2007. A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc Natl Acad Sci U S A 104:1009–1014. doi: 10.1073/pnas.0606713104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Akerley BJ, Rubin EJ, Novick VL, Amaya K, Judson N, Mekalanos JJ. 2002. A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc Natl Acad Sci U S A 99:966–971. doi: 10.1073/pnas.012602299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Salama NR, Shepherd B, Falkow S. 2004. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J Bacteriol 186:7926–7935. doi: 10.1128/JB.186.23.7926-7935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huet G, Daffe M, Saves I. 2005. Identification of the Mycobacterium tuberculosis SUF machinery as the exclusive mycobacterial system of [Fe-S] cluster assembly: evidence for its implication in the pathogen's survival. J Bacteriol 187:6137–6146. doi: 10.1128/JB.187.17.6137-6146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Klein BA, Tenorio EL, Lazinski DW, Camilli A, Duncan MJ, Hu LT. 2012. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics 13:578. doi: 10.1186/1471-2164-13-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee SA, Gallagher LA, Thongdee M, Staudinger BJ, Lippman S, Singh PK, Manoil C. 2015. General and condition-specific essential functions of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 112:5189–5194. doi: 10.1073/pnas.1422186112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Knuth K, Niesalla H, Hueck CJ, Fuchs TM. 2004. Large-scale identification of essential Salmonella genes by trapping lethal insertions. Mol Microbiol 51:1729–1744. doi: 10.1046/j.1365-2958.2003.03944.x. [DOI] [PubMed] [Google Scholar]
- 97.van Opijnen T, Camilli A. 2012. A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res 22:2541–2551. doi: 10.1101/gr.137430.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Le Breton Y, Belew AT, Valdes KM, Islam E, Curry P, Tettelin H, Shirtliff ME, El-Sayed NM, McIver KS. 2015. Essential genes in the core genome of the human pathogen Streptococcus pyogenes. Sci Rep 5:9838. doi: 10.1038/srep09838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kamp HD, Patimalla-Dipali B, Lazinski DW, Wallace-Gadsden F, Camilli A. 2013. Gene fitness landscapes of Vibrio cholerae at important stages of its life cycle. PLoS Pathog 9:e1003800. doi: 10.1371/journal.ppat.1003800. [DOI] [PMC free article] [PubMed] [Google Scholar]