

Abstract
Numerous studies have been published in the past years investigating the transcriptome of the zebrafish embryo (ZFE) upon being subjected to chemical stress. Aiming at a more mechanistic understanding of the results of such studies, knowledge about commonalities of transcript regulation in response to chemical stress is needed. Thus, our goal in this study was to identify and interpret genes and gene sets constituting a general response to chemical exposure. Therefore, we aggregated and reanalyzed published toxicogenomics data obtained with the ZFE. We found that overlap of differentially transcribed genes in response to chemical stress across independent studies is generally low and the most commonly differentially transcribed genes appear in less than 50% of all treatments across studies. However, effect size analysis revealed several genes showing a common trend of differential expression, among which genes related to calcium homeostasis emerged as key, especially in exposure settings up to 24 h post-fertilization. Additionally, we found that these and other downregulated genes are often linked to anatomical regions developing during the respective exposure period. Genes showing a trend of increased expression were, among others, linked to signaling pathways (e.g., Wnt, Fgf) as well as lysosomal structures and apoptosis. The findings of this study help to increase the understanding of chemical stress responses in the developing zebrafish embryo and provide a starting point to improve experimental designs for this model system. In future, improved time- and concentration-resolved experiments should offer better understanding of stress response patterns and access to mechanistic information.
Keywords: meta-analysis, microarray, stress response, transcriptome, toxicogenomics, zebrafish embryo
Technologies measuring the entirety of gene transcripts, proteins or metabolites in a sample (“omics”) are increasingly used in toxicological research and are discussed to be included into chemical regulatory assessments in the future (Marx-Stoelting et al., 2015). In toxicology, these methods are primarily used to obtain insight into a compound’s mode of action, to group similarly acting compounds or to define new biomarkers (Afshari et al., 2011). Several studies have been published demonstrating responses to chemical exposure on transcriptome, proteome, metabolome, or epigenome level in different organisms. Here, we review the achievements made with the zebrafish embryo (ZFE) in the field of toxicogenomics (measuring gene transcript abundance, often also termed “gene expression”). ZFEs as well as adult zebrafish are increasingly used in biomedical (Lieschke and Currie, 2007; Lin et al., 2016) and environmental research (Scholz et al., 2008) due to the advantageous combination of biological complexity of a whole organism and the potential for high throughput handling (Driessen et al., 2014). The added value of ZFE transcriptome analyses for human toxicology testing has been discussed before (Driessen et al., 2015; Hermsen et al., 2011) and several studies have analyzed the embryo’s transcriptome after it was subjected to chemical exposure (Williams et al., 2014). However, to our knowledge, results of ZFE toxicogenomics studies have not been aggregated or compared so far. In many studies, it could not be resolved, which of the observed effects were specific for the compound or compound class studied, and which would be expected to be of a more general nature, e.g. related to disturbance of embryonic development or global homeostasis. We assume that compounds mostly act via a range of different molecular mechanisms (e.g., different target molecules), but we also expect effect cascades to converge into smaller and more general sets of toxicity pathways (Villeneuve et al., 2014). This should also be reflected on the transcriptome level so that chemical-induced differential transcription of at least some genes or gene sets should be independent of compound or embryonic stage. To be able to derive a meaningful and reproducible grouping of compounds and later on elucidate connections of molecular effects with higher level effects, which are of interest not only for regulatory issues, such knowledge about common responses is of utter importance. Thus, in this meta-analysis we aimed at the identification of genes or gene sets showing a general response towards chemical exposure. In the study presented here, we compared 33 microarray studies including exposures of the ZFE to 60 different compounds (948 arrays in total) and aimed at identifying common transcriptional regulation across all experiments as well as across meaningful subgroups. We followed two complementary approaches (see Figure 1 and Supplementary Fig. 1). First, we re-analyzed each experimental dataset separately, identified differentially expressed genes (DEGs) using moderated t-statistics and then determined the overlap of DEGs across datasets. In a second approach, we aggregated normalized data from all studies and determined a summary effect size using a random effects model. Functional enrichment analysis allowed us to derive conclusions about some general stress responses. Finally, we discuss what is needed for future experiments in order to allow for more powerful interpretations of omics profiles after chemical exposure.
FIG. 1.

Approach of the meta-analysis of transcriptome studies of the zebrafish embryo after chemical exposure. For details also see Supplementary Figure 1.
MATERIALS AND METHODS
Study selection
For our analysis, we followed a strategy similar to that proposed by Ramasamy et al. (2008). Studies were selected for the meta-analysis in which microarray measurements of global gene transcription changes in the ZFE after exposure to chemical compounds were performed (gene knock-down studies were not included). A database query was conducted in Gene Expression Omnibus and ArrayExpress (no search term, Filters: Organism: Danio rerio, Data type: expression profiling by array, one-color array or two-color common reference) and all studies satisfying above restrictions were manually selected. This resulted in 33 studies reporting on 60 compounds tested in 948 arrays (see Table 1).
TABLE 1.
List of Chemicals Included in the Meta-Analysis Together With Assigned Mode of Action (MoA), Study ID, and Corresponding References
A, reactive, teratogenic, carcinogenic; B, neuroactive; C, endocrine; NA, not assigned.
Included in effect size analysis.
Data import, quality control, normalization, and cleaning
Raw data of each study were downloaded from Gene Expression Omnibus or Array Express and imported into R (version 3.2.2, R Core Team, 2015), which was used together with packages from the Bioconductor repository (Huber et al., 2015) for all subsequent analysis. For each dataset, quality control and normalization were conducted as described in Supplementary Material, p.3.
Annotation
Since the microarrays used for the different studies have been designed and annotated using different genome versions (and possibly different annotation strategies), it was necessary to renew the annotation for all arrays. All array probes were mapped against the recent Danio rerio genome (DanRer10, September 2014) and annotated using the Ensembl Database (Ensembl Release 80, May 2015). The annotation strategy was based on Arnold et al. (2014) and is described in Supplementary Material, p.3.
Grouping of contrasts
To be able to derive biologically meaningful information from the large number of different treatments included in the analysis, treatments were grouped according to experimental factors. Those factors were: (1) observation time points, (2) modes of action of compounds, and (3) exposure concentration. The groups were assigned using a rather broad perspective. This way groups included enough different treatments and studies to be able to detect general patterns and not just specific results of one treatment:
Observation time point: the diverse exposure windows (Figure 2a) were grouped into three categories according to observation time point in the ZFE (which was the exposure end in most cases) with early exposures ending at latest at 24 hpf, intermediate exposures ending after 24 hpf and before 50 hpf and late exposures ending later than 50 hpf.
Modes of action: modes of action or effect categories were retrieved from literature for the 60 chemicals used in the different studies. Three groups were analyzed in more detail, namely reactive, teratogenic or carcinogenic substances (“A”), neuroactive substances (“B”) and endocrine disrupting chemicals (“C”). To achieve maximum consistence, chemicals were only assigned to a group if strong evidence for the assignment existed. See Table 1 for the assignments.
Chemical concentration: all considered studies reported the molar concentrations of the applied exposure solution. However, for being able to compare the exposure concentrations of different substances in a quantitative way, it is necessary to relate the exposure concentration to a comprehensive effect scale (such as lethal concentration). Since this was only available for a few studies, experiments were grouped into 3 sets with respect to effect concentrations on the ZFE phenotype: the “no effect” group included all treatments using arbitrarily chosen no effect concentrations and treatments using No Observed Effect Concentration (NOEC), No Observed Adverse Effect level (NOAEL) or fractions of NOEC or NOAEL for exposure; the “LOEC” group contained all treatments using exposure concentrations reported as LOEC, treatments leading to not precisely defined low effects, as well as treatments with exposure concentrations of EC10 and lower as well as BMCGMS1 −BMCGMS10 (as defined by Hermsen et al. (2011)). Finally, the “EC” group contained all treatments with exposure concentrations reported as inducing visible effects, if quantified larger than EC10 as well as BMCGMS ≥ 10 and all reported lethal concentrations (min. = LC5).
FIG. 2.

Metadata of experiments included in the meta-analysis. A, Onset and duration of chemical exposure, each bar represents exposure window of one experiment, bar colors indicate different studies, experiments are grouped as in meta-analysis into early (exposure end before 24 hpf), middle (exposure end before 50 hpf) and late exposures (exposure end after 50 hpf). B, Association plot of experimental subgroups. Width of bar proportional to expected counts, height of bar proportional to Pearson residuals. Black bars indicate significant dependence. This plot shows, that some of the experimental subgroups are not independent, e.g. early experiments were often conducted with LOEC concentrations of neuroactive substances. Mode of Action: A = reactive, teratogenic, B = neuroactive, C = endocrine. Effect concentration: no effect = applied concentration had no reported phenotypic effect in the experiment, LOEC = applied concentration was lowest observed effect concentration or some not precisely defined low effects up to EC10. EC = applied concentration reported to induce visible or lethal effects.
Analysis
The analyzed studies highly varied with regard to the number of measured time points and applied concentrations. Studies with many concentrations or time points measured would therefore have a biased impact on the analysis. This is why in each subgroup only the highest concentration and latest time point of each study and compound was included (suspected to represent the treatment showing the strongest and most general toxicity profile). The meta-analysis was performed with 2 complementary approaches (Figure 1). (1) For separate significance testing each dataset was quantile-normalized and DEGs were identified with a moderated t-test using the R-package “limma” (Ritchie et al., 2015). Cutoff criteria for DEG were |log2 fold change (logFC)|>1 and adjusted P-value (adj P) < 0.05. The proportion of DEGs for each treatment was determined and related to experimental factors. Additionally, those genes were determined which were identified as DEG in most treatments (across all treatments and subgroups). (2) In order to identify genes with a common trend of differential transcription, common significance testing was applied: Each array was normalized by cumulative proportion transformation using the R-package “YuGene”, a normalization method specifically designed for cross-platform normalization considering different dynamic ranges for different platforms (Lê Cao et al., 2014). Then, random effect models were used to determine a summary effect size for each gene based on the fold-change and significance was estimated using a permutation analysis (1000 permutations) based on Significance Analysis of Microarrays (Tusher et al., 2001) applying the R-packages “MAMA “(Ihnatova, 2013) in combination with “GeneMeta” (Choi et al., 2003; Lusa et al., 2015). Cutoff criteria for genes with a significant summary effect size (“meta-genes”) were |effect size|>1 and adj. P < 0.05. This method is described in detail by Choi et al. (2003).
To derive a mechanistic understanding from the lists of genes identified as meta-genes, functional annotation was performed using the Bioconductor package “clusterProfiler” (Yu et al., 2012). A manually combined library of zebrafish gene sets from KEGG (Release 75.0, Kanehisa and Goto 2000), ZFIN (April 2015, Howe et al, 2013), GeneOntology (Ensembl release 80, Gene Ontology Consortium, 2015), WikiPathways (Ensembl release 80, Kelder et al., 2012) and Interpro-domains (Ensembl release 80, Mitchell et al., 2015) was created for enrichment analysis (for details see Supplementary Information on methods).
RESULTS
This meta-analysis aggregated microarray data from 33 studies in which transcriptome responses in ZFE upon exposures to an overall number of 60 different chemicals were investigated. We systematically compared experimental settings with regard to exposure time, concentration and microarray platform. Since the number of DEGs can give a first hint on the extent of molecular disturbance in the embryo (Hermsen et al., 2012), we compared the proportion of DEGs between the studies. We also investigated the influence of different experimental factors on the extent of gene regulation. In the last step we strived to identify commonly regulated genes in the ZFE after chemical exposure.
Heterogeneous exposure settings
A first important finding is that exposure settings across studies are quite heterogeneous with respect to exposure time, concentration and the applied microarray platform.
Exposure times
A summary of exposure onset and durations of all treatments is depicted in Figure 2a. Studies include very early exposures immediately after fertilization (e.g., Hermsen et al., 2012; Schiller et al., 2013b), as well as exposures of stages after hatching of the embryo (e.g., Driessen et al., 2015; Faltermann et al., 2014; Hahn et al., 2014; Kawabata et al., 2015). The exposure durations cover a range between 1 h (Alexeyenko et al., 2010) to several days (e.g., Garcia-Reyero et al., 2014; Lam et al., 2011; Park et al., 2012). RNA extraction for microarray analysis usually took place immediately after the end of exposure, with only one exception: Alexeyenko et al. (2010) analyzed molecular responses to dioxin at several time points during depuration after a short exposure time.
Exposure concentrations
Since studies are based on different motivations and assumptions, the experimental strategies also differed with respect to exposure concentrations. Five of 33 studies used an exposure concentration showing no phenotypic effects in the embryo (e.g., Bonventre et al., 2013; Driessen et al., 2015; Hao et al., 2013), 3 studies used the lowest concentration leading to a sublethal phenotypic or lethal effect (e.g., Saili et al., 2013) and 8 studies used concentrations causing death of 5–20% of all embryos (e.g., Klüver et al., 2011; Lam et al., 2011; Oliveira et al., 2013). Five studies recorded phenotypic effects for the applied exposure concentrations but did not quantify the effects (e.g., Alexeyenko et al., 2010; White et al., 2011; Tal et al., 2012). Three studies used an exposure concentration, which was related to concentrations found in the environment (e.g., Park et al., 2012; Garcia-Reyero et al., 2014) and 8 studies did not explicitly relate their applied concentration to a reference concentration or phenotypic effect (e.g., Hahn et al., 2014; Xu et al., 2014; Kawabata et al., 2015).
Microarray platforms
The studies considered in this meta-analysis made use of different microarray platforms. Commercially available microarrays manufactured by Agilent or Affymetrix were used most frequently. Besides general differences in array design between those companies, the different generations of arrays also appear to differ substantially in terms of transcriptome coverage as depicted in Supplementary Figure 3a. Here, a summary of transcriptome coverage of all used arrays shows that only few platforms achieve to cover most known Danio rerio genes. Roughly only 20% (∼5,000) of known genes are covered across all platforms and can be compared across all conditions. However, if genes are grouped into gene sets using ontology and pathway information from databases, most gene sets are covered with at least 30% of their genes on all arrays (see Supplementary Figure 3b). For our effect size analysis, we used a reduced dataset from fewer studies with the intention to increase the overlap in covered genes across studies and thus the number of genes that could be analyzed. The number of genes could be increased to a coverage of about 45% (∼12 000 genes). We included data from different platforms since a limitation to only one platform would have reduced the analysis to too few compounds to derive any general conclusions.
Association of experimental factors
Figure 2b shows that in this meta-analysis not all experimental groups can be considered to be independent. Shaded bars show a significant correlation between experimental factors. For example, substances in MoA group A (“reactive, teratogenic”) were often investigated in no effect concentrations at late time points.
Extent of gene regulation depends on exposure concentration
As a first analysis step, the proportion of DEGs among all measured genes was determined for all treatments (adjusted P < 0.05, |logFC|>1). The proportion of DEGs ranged from no genes in 77 of 225 contrasts (treatment-control) up to a maximum of roughly 9% DEGs (Figure 3a). Treatments ending at time points between 24 and 50 hpf seem to induce a slightly higher number of genes than those ending at earlier or later time points (see Figure. 3b). Additionally, experiments using no-effect concentrations showed fewer significantly regulated genes than experiments using a concentration reported to cause visible effects on the phenotype (Figure 3d). The fact that some contrasts do not show any significantly regulated genes goes in hand with the published studies about the corresponding datasets, since some of these studies chose not to correct their statistical test for multiple hypothesis testing for deriving a list of regulated genes (Bonventre et al., 2013; Garcia-Reyero et al., 2014; Saili et al., 2013; Xu et al., 2014). Other studies tested a range of concentrations with the lower concentrations not always causing significant transcriptional changes. Additionally, the mode of action seems to have an effect on the extent of gene regulation. In Figure 3c, it is illustrated that endocrine acting substances seem to induce a comparatively larger response. However, this might be due to the fact, that endocrine substances were mostly tested at higher effect concentrations (Figure 2b). We grouped the proportion of regulated genes according to concentration and time (Supplementary Figure 4a) and according to concentration and mode of action (Supplementary Figure 4b) and it appears that both, mode of action as well as concentration play a role for the extent of gene regulation. Therefore and as explained above, it is hardly possible to consider the groups independently. Additionally, it should be noted that some of the treatments in all considered groups show none or only few DEGs. This explains the increased variability within all groups with an increased median proportion of DEGs (leading to “higher” boxes in Figure 3b–d).
FIG. 3.

Proportion of differentially expressed genes (DEGs) among all measured genes in respective treatments: A, Individual experiments, each bar represents one individual treatment. B, Subgroups according to observation time points. C, Mode of action subgroups. D, Subgroups according to the applied effect concentrations.
Low overlap in DEGs between studies
In the next analysis step, we strived to identify genes commonly differentially expressed in the embryo in response to chemical exposure in general. In a first approach, DEGs were identified for each treatment and subsequently overlaps of DEGs were determined across all treatments as well as across subgroups of treatments. In order to reduce study bias, only those treatments with at least one DEG and only the highest concentration and latest time point investigated per substance and study were considered (resulting in a maximum of 60 contrasts/33 studies). Across all treatments and in all subgroups some overlapping DEGs could be identified with an overlap higher than would be expected by chance (P < 0.05). However, even the most commonly differentially transcribed genes appeared in less than 50% of analyzed treatments and regularly showed a study bias, if several compounds of one study were included in the dataset (see Supplementary Figs. 5–14). Each list of DEGs (if there were any detected) for each treatment was also analyzed for functional enrichment using the R-package clusterProfiler (Yu et al., 2012). Again, the overlap of enriched terms was determined, which was found to be in the same range as for genes—with no gene set appearing in more than 50% of the treatments. Since the evidence for general regulation of the genes or gene sets with the highest overlap was weak, no further biological conclusions were derived from the findings of this analysis step. Rather, in a second step genes were searched for that showed a common trend of regulation irrespective of statistical significance in the single studies.
Effect size analysis: common trends of gene regulation
Our expectation to find some genes identified to be differentially expressed in all treatments (and accordingly in experimental subgroups) as evidence for common gene regulation was not met. We reasoned that there might still be genes showing a common trend of differential expression across treatments, without being identified as significantly differentially expressed in each individual treatment. Those genes would not be identified using the overlap method described above. Thus, in a second approach we obtained a summary effect size for each gene across all contrasts before determining its significance by permutation analysis. The summary effect size (bars in Figure 4) can be regarded similarly to a weighted average of the effect sizes of individual treatments, while the single effect sizes of each treatment and gene (circles in Figure 4) were obtained from the fold changes (treatment(s) vs respective control(s)) and normalized by the standard deviation. This method only allowed those genes to be included in the analysis which were tested in all treatments. Therefore, only studies with at least 15 000 investigated genes were included in the analysis (this cutoff was chosen as a trade-off between number of analyzable studies (17 studies containing 176 treatments) and number of analyzable genes (11 679/∼45% coverage)). Again, to reduce study bias the dataset was reduced to those treatments with highest concentration and latest time point for each compound and study. The results of this analysis are summarized in Table 2, where the number of genes with a summary effect size deviating significantly from zero (FDR < 0.05, |effect size| > 1, here called “meta-genes”) is given for each group as well as the number of enriched functional terms within this group of meta-genes. Summary effect sizes across all contrasts were significant for 11 genes, including 4 genes with positive (plekhf1, nars, atf3, phgdh) and 7 genes with negative summary effect size (csf1b, ppp1r27a, lrrn1, pvalb8, atp2a1l, neuro6b, nr4a1). In spite of statistical significant summary effect sizes, single effect sizes of each contrast still show large variation between treatments (Figure 4a). Several treatments even show a reverse effect size in comparison to the summary effect size, meaning for example that the gene is upregulated in some studies whereas the summary effect size shows a significant repression.
FIG. 4.

Estimated effect sizes for selected meta-genes (= genes significantly differentially expressed in a subgroup as indicated by effect size analysis). Values above 0 = increased expression after chemical exposure, values below 0 = decreased expression after chemical exposure. A, Meta-genes for all experiments. B, Top 10 meta-genes in the subgroup of experiments with neuroactive substances. C, Meta-genes associated with the lysosome pathway (KEGG) in the EC subgroup (exposures with concentrations causing apical effects). D, Meta-genes associated with calcium binding (GeneOntology annotation) in the subgroup of early exposures. A, B, Circles represent single effect sizes, arrows single effect sizes smaller −5 or larger 5, colors represent study affiliation, bars summary effect sizes, grey circles and bars single effect sizes and summary effect sizes of experiments not in the group, Study IDs as given in Table 1. C, D, values clipped below −5 and above 5.
TABLE 2.
Number of “Meta-Genes” and Enriched Gene Sets in Experimental Subgroups and Number of Contrasts and Studies Contributing to the Subgroup
| Category | Group | Genes |
Gene sets |
Number of Contrasts | Number of Studies | ||
|---|---|---|---|---|---|---|---|
| Up | Down | Up | Down | ||||
| Time | Early | 164 | 413 | 75 | 62 | 14 | 6 |
| Middle | 177 | 209 | 4 | 7 | 15 | 7 | |
| Late | 12 | 12 | 0 | 0 | 21 | 8 | |
| Mode of action | Reactive | 28 | 15 | 5 | 1 | 6 | 5 |
| Endocrine | 126 | 249 | 0 | 1 | 13 | 6 | |
| Neuroactive | 120 | 168 | 4 | 32 | 7 | 3 | |
| Concentration | No effect | 11 | 14 | 2 | 0 | 16 | 5 |
| LOEC | 411 | 531 | 0 | 21 | 15 | 5 | |
| EC | 151 | 327 | 11 | 35 | 21 | 9 | |
| All | All | 4 | 7 | 17 | 1 | 45 | 17 |
Meta-genes for experimental subgroups
As mentioned above, we also calculated the summary effect sizes for all included genes across different experimental subgroups. For 6 of the 9 subgroups we identified more than 100 genes showing a common trend of increased or decreased expression (see Table 2 and Supplementary Table 1, FDR < 0.05, |effect size|>1). The distribution of effect size and FDR was different between subgroups, as depicted in the volcano plots in Supplementary Figure 15. All genes with a significant summary effect size and respectively enriched gene sets are summarized in Supplementary Tables 1 and 2 for all experimental subgroups. Heatmaps for each subgroup summarizing the single effect sizes of up to 100 of the respective “meta-genes” are provided as supplemental material (Supplementary Figs. 16–24). Figure 4b illustrates exemplary for the top 10 genes responding to neuroactive substances, that meta-genes identified in the subgroups at least partially show a consistent trend (all individual treatments show either increased or decreased expression compared to control). Furthermore, it demonstrates that summary effect sizes for experimental subgroups may substantially differ from the summary effect size for all treatments (light grey bars in Figure 4b indicating no effect across all studies) which may indicate a group specific response.
Observation time point
For early observation time points (up to an embryo age of 24 hpf) 164 genes had an estimated summary effect size larger than 1 (increased expression, FDR < 0.05) and 413 genes had an effect size smaller than −1 (decreased expression, FDR< 0.05). In contrast to other time related subgroups, numerous functional terms were enriched for those genes. Among genes with increased expression, terms related to mesoderm development as well as several T-box transcription factors (tbx6, tbx6l, tbx16, ta) were predominantly affected. Two signaling pathways were overrepresented, namely the Wnt-pathway (e.g., fibronectin 1b, wnt inhibitory factor 1, wnt11r, dact2) and the Fgf-pathway (e.g., fgf receptor 1a, fgf 17, fgf receptor like 1b). Both pathways play an important role in ZFE development (Verkade and Heath, 2008; Ota et al., 2009). Additionally, 10 of the genes with increased expression were coding for proteins with EGF-like domains. The motif enrichment performed with human orthologue gene annotations indicated potential upstream regulation for 10 of the induced genes by the vitamin D receptor (VDR) and the hepatic transcription factor 1 (TCF1). Among the terms enriched for genes with decreased expression one cluster contained genes for beta- and gamma crystallins. Another cluster could be linked to neuron expression, myotome and muscle development. An effect on muscle development is also indicated by the enriched transcription factor motifs of MEF2A (Myocyte Enhancer Factor 2A) and MYOD (myogenic differentiation 1). Besides myogenesis and muscle cell differentiation those potential upstream regulators are involved in neuronal differentiation, cell growth control, apoptosis and cell cycle arrest (Berkes and Tapscott, 2005; McKinsey et al., 2002; Naya and Olson, 1999; Weintraub et al., 1991). Additionally, there were clusters of genes enriched for calcium ion binding proteins (e.g., calpains, parvalbumins, calsequestrins) and for metabolic processes like glycolysis. Furthermore, a cluster of genes coding for homeobox-domain containing proteins was found to show decreased expression in early treatments (e.g., hoxb13a, lhx1b, lhx9, nkx2.1). This protein class is highly conserved and plays an essential role in morphogenesis and early development (Kimmel, 1989; Mallo et al., 2010). A clusterplot showing the downregulated terms of the early exposure group is shown in Supplementary Figure 25.
In observations between 24 and 50 hpf (group: “middle”), 177 genes showed an average increase in expression but only the terms blood, leucine zipper domain, steroid biosynthesis and cellular amino acid metabolic process were enriched for this group of genes. Among the 209 genes with decreased expression only a small group of 6 genes could be annotated to be connected with dentary expression. In late measurements 12 genes showed an average increase and 12 an average decrease in expression, but no functional terms were enriched.
Concentration
Next to the consideration of observation time points we also grouped the experiments according to the applied exposure concentrations. We built 3 groups of experiments, namely concentrations with no reported apical effect (“no effect”), concentrations in the LOEC range (“LOEC”) and concentrations with a reported apical effect (“EC”). Eleven genes showed an average expression increase and 14 an average decrease in the “no effect” concentration group. Enrichment analysis for this group resulted in only 2 terms connected to the fin bud, of which 2 genes were among the genes with increased expression. Experiments of the “LOEC” group showed 410 genes with increased and 531 genes with decreased expression. However, in spite of the large number of genes showing increased expression, only genes with decreased expression could be functionally annotated. Here, diverse anatomical terms were enriched, including musculature system, heart tube, dentary, blood and fin bud. Additionally, genes coding for proteins involved in glycolysis (e.g., fbp2, pfkma, pfkmb, gapdh) were decreased in expression as well as genes coding for globin-like and EF-hand domain containing proteins (e.g., parvalbumins). Experiments with reported apical effects of EC10 and higher, showed 151 genes with increased expression. These were enriched among others for steroid biosynthesis, the protein domain of ABC transporters, and a group of lysosome genes including a range of peptidases (cathepsins, legumain) and glycosidases (hexb, naga). The expression increase of these genes indicates a raise in degradation of damaged proteins and macromolecules as a response to cell damage. In Figure 4c, the single effect sizes of significantly regulated lysosomal genes in “EC” experiments are exemplary illustrated in a heatmap. Here, it can be seen that not all compounds applied in a concentration causing an apical effect induced lysosomal genes in a similar way; however, an overall trend of induction is obvious. The 327 genes with decreased expression in the “EC” group showed enrichment for muscle, dentary and heart expression as well as the troponin domain (similar to the “LOEC” group). Additionally, the terms focal adhesion and ECM-receptor interaction were enriched, possibly indicating destabilized tissue structures. Transcription factor motif enrichment is similar to the “early” group of genes with decreased expression.
Mode of action
The studies included in the effect size analysis investigated 40 chemicals of which 22 were grouped into 3 broad groups with similar modes of action, namely reactive, carcinogenic or teratogenic compounds (group “A”); neuroactive compounds (group “B”); and compounds known to act on the endocrine system (“C”).
We identified 28 genes with increased and 15 genes with decreased expression in the group of reactive substances (“A”) of which 2 genes (nfkb2, tnfrsfa) are coding for proteins containing the death domain and 3 genes are coding for transporters of the major facilitator superfamily (slc37a2, slc17a9b, svolp). Among the genes with increased expression we also found acy3, a gene coding for aminoacylase 3, an enzyme which is known to deacetylate mercapturic acids (Newman et al., 2007), which are common transformation products of reactive/electrophile chemicals (Vermeulen, 1989). Neuroactive compounds (“B”) induced an increased expression of 120 genes, of which 8 were found to be related to the neural rod (wnt4a, gdf6a, cxcl12a, hspa4b, greb1, heyl, hoxd13a, mdkb) and 3 are connected to neuron migration (prickle1b, cxcl12a, hdac1). The 168 genes with decreased expression showed enrichment for muscle and heart expression, glycolysis, the insulin signaling pathway, and fatty acid degradation. The third group of endocrine acting chemicals (“C”) showed common trends of increased expression of 126 genes and decreased expression of 249 genes but no functional term was enriched besides glycolysis for decreased genes.
DISCUSSION
In our meta-analysis, we found that overlap of gene expression profiles is generally low between analyzed studies. However, common trends of differential expression, especially in meaningful subgroups, hint at important stress response mechanisms in the zebrafish embryo (ZFE) which we will discuss below. At the same time, we also identified limitations in experimental design which we will briefly discuss.
The studies compared here all sought to use global gene transcription analysis to analyze the molecular response provoked in the ZFE by exposure to specific compounds. Some studies investigated in substance related effects at a specific embryonic stage like gastrulation, others looked at further developed stages, e.g. with developed hepatocytes. Some studies aimed to identify the mode of action of a specific chemical, others aimed to demonstrate the suitability of the microarray technique for toxicological studies using well-known model chemicals. Those different specific research interests resulted in many different experimental designs. This heterogeneity in experimental factors is a challenge when comparing the data. Since the setup does not give a balanced design regarding the analyzed subgroups (see Figure 2b), it is almost impossible to trace back differences between experiments to a single factor. Another confounding factor was the difference in array design with only a small set of genes being jointly observed on all arrays. Moreover, to obtain comparable data, all data were “forced” into one analysis design here which was restricted to one treatment per chemical and study in order to reduce study bias. This, however, reduced power in our analysis for those studies, which chose to investigate more concentrations or time points and less replicates which is actually a favorable and strong study design.
In the analysis of overlap of regulated genes between treatments none of the most frequently significantly affected genes was found in more than 50% of the treatments. Also in smaller groups of studies no larger overlap was detected. This finding goes in line with earlier meta-analyses of microarray data of tissue samples of ovarian cancer, also showing low overlap between observed expression profiles (Györffy et al., 2008) as well as earlier studies in ecotoxicology indicating, that the number of commonly regulated genes gets smaller as the group of experiments is getting larger (Hermsen et al., 2013). In a meta-analysis of changes within zebrafish proteomes after chemical exposure overlap of regulated proteins was even smaller and found to be below 30% (Groh and Suter, 2015). A recent study by Vidal-Dorsch et al. (2016) showed a lower than expected overlap in an inter-laboratory comparison of ecotoxicological microarray analyses. All these results indicate that analyses of top-lists are always prone to bias. Especially when evaluating single studies, stronger proof of biological regulation e.g., by enrichment analysis (as performed by Driessen et al. (2014)) or molecular interaction network analyses is needed. However, a functional enrichment analysis as performed here also revealed a maximum overlap of only 50% in the subgroups, which might be expected to be larger for more consistent experimental designs.
In spite of a generally low overlap of significantly regulated genes between studies we could detect common trends of gene regulation using an effect-size based approach and found indications for certain molecular responses to chemical stress in the ZFE. Molecular responses were assigned to a range of anatomical regions, biological processes and signaling pathways.
The 11 genes showing a significant trend across all treatments already hint at a few general molecular processes in response to chemical stress. The gene atf3 (activating transcription factor 3) plays a role in cell cycle arrest and apoptosis (Lu et al., 2006) and shows a trend of upregulation across all treatments (Figure 4a). The same is true for plekhf1 (pleckstrin homology and FYVE domain containing 1) which is connected to lysosome dependent apoptosis (Chen et al., 2005). In the “EC” group the importance of lysosomes in stress response is even more pronounced with several lysosomal genes upregulated (Figure 4c).
Among the 7 genes with a trend for downregulation across all treatments 3 genes are connected to calcium homeostasis. The gene pvalb8 (parvalbumin 8) is the zebrafish orthologue of the human OCM2 (oncomodulin 2) coding for a high-affinity calcium ion-binding protein that belongs to the superfamiliy of calmodulin proteins containing an EF-hand protein domain. Also, the expression of the gene for the orphan nuclear receptor nr4a1 showed a trend of downregulation across all studies. It has been shown that the transcriptional regulation of this endocrine receptor is dependent on calcium level (Abdou et al., 2016; Medzikovic et al., 2015). Additionally, the zebrafish gene atp2a1l is an orthologue of the SERCA gene, coding for an intracellular Ca2+ transporting ATPase in the sarcoplasmatic reticulum of muscle cells. This pump has recently been shown to be inhibited by lipophilic compounds in Daphnia magna and it was hypothesized that intracellular calcium level plays an essential role in basal toxicity (Antczak et al., 2015). Finally, the ryanodine receptors (RYR2 and RYR3) functioning to release calcium from the sarcoplasmic reticulum, e.g., in the context of muscle contraction (Pessah et al., 2010), had a significant negative effect size in 4 of the 9 subgroups. Indeed, repression of genes connected to calcium binding or transport was a recurring observation in our analysis and most prominent in early exposures. Downregulation of genes of calcium binding proteins in the early exposure group is displayed in a heatmap in Figure 4d. Here, it can also be seen that negative control compounds like saccharin or mannitol but also nanoparticles did not have an effect on this group of genes, while all other compounds led to differing degrees of downregulation in early exposed ZFE. A downregulation of calcium binding proteins as response to chemical stress was also described as potential key event in mouse embryonic stem cells after progesterone exposure (Kang et al., 2016) and in ZFE exposed to Vitamin D3 (Zhang, 2015). Furthermore, in a proteomics study calcium signaling proteins were found among the most frequently differentially expressed proteins besides actins, myosins, crystallins and metabolic enzymes in a meta-analysis of zebrafish proteome data obtained after chemical stress (Groh and Suter, 2015). Brette et al. (2014) showed that the decrease of intracellular calcium current and calcium cycling leading to disruption of excitation-contraction coupling in fish cardiomyocytes after crude oil exposure is a key mechanism in crude oil caused cardiotoxicity in fish.
Calcium has many different regulatory functions, one of which is its role during cell cycle progression (e.g., Kahl and Means 2003; Roderick and Cook, 2008). In a study with Saccharyomyces cerevisiae, O’Duibhir et al. (2014) detected cell cycle arrest as a general stress response towards environmental perturbations. A cell cycle arrest would also go in line with our finding that, in the subgroups related to observation time, many downregulated genes are connected to those anatomical regions, which develop during the respective developmental stage. This is true for heart, musculature system, parts of the brain, and the eyes all of which start to develop before 24 hpf (Kimmel et al., 1995). Those regions are all connected to downregulated genes in early treatments (ending before 24 hpf). In the second group (exposure ending between 24 and 50 hpf) the dentary system seems to be affected, which is starting to develop just before 2 dpf (Van der Heyden and Huysseune, 2000). In the late exposure group (measurement later than 50 hpf), when most of the zebrafish morphogenesis is completed, no anatomical terms were enriched among the meta-genes. Overall, this could indicate a general delay in development, but also a reduction in growth and metabolism (with the mentioned anatomical regions mostly affected). This is further supported by a significant downregulation of many genes involved in glucose metabolism in 5 of the 9 subgroups. In summary, it might be hypothesized that the repression of calcium binding proteins indicates an early stress response which induces a cell cycle arrest and suppression of differentiation (as shown by Kang et al., 2016). However, it remains to be elucidated whether the downregulation of calcium binding transcripts and proteins is indeed the cause or rather the result of a perturbation of development and differentiation. Most calcium binding genes are cell type specific and might occur as repressed just because the cell type is not as developed as in the respective control.
Additionally, genes can be identified showing the same trend of differential expression for compounds which are expected to act in a similar way. Induced genes in the group of neuroactive compounds could partly be functionally annotated with the terms neural rod and neuron migration. For endocrine disruptors and reactive compounds, the meta-genes could not be functionally annotated, which indicates a knowledge gap in functional annotation of signaling pathways and gene-gene interconnections during zebrafish development. Missing knowledge about relevant sets of genes involved in responses towards toxicants is also indicated by the fact, that more downregulated meta-genes are annotated in gene sets (representing known metabolic and homeostatic functions repressed by chemical stress) than upregulated meta-genes which might represent specific but unknown responses to chemical–biomolecule interactions during development. However, a detailed functional analysis of single affected genes might also give evidence for a specific response as shown for the gene acy3 (aminoacylase 3), which was exclusively affected in the group of reactive compounds and identified as a potential biomarker indicating biotransformation of electrophiles (Newman et al., 2007; Vermeulen, 1989).
Apart from mechanistic evidence, the findings of our study should help to improve the design of toxicogenomic experiments in the future. The analysis of proportions of regulated genes revealed that experiments using concentrations which did not induce visible effects in the embryo showed a low proportion of genes differentially transcribed. This should be considered in toxicogenomics experiments as well as in discussions about the sensitivity of responsive transcriptomes. To foster the interpretation of toxicogenomics studies the distinction between general responses versus specific responses as well as primary versus secondary and early versus late responses induced by individual substances is important. This is especially true, if such data should become useful for risk assessment in the framework of the adverse outcome pathway (AOP) concept (Ankley et al., 2010). This might become much easier in the future if studies are designed to advance from “snapshot” experimental designs covering only single concentrations and time points to a design including a range of concentrations and time points for each study as illustrated in Figure 5. A minimum number of 5 time points and 5 different exposure concentrations would be recommended as this would allow for the application of simple descriptive modeling approaches. We recommend equal spacing of time points and concentrations on a logarithmic scale. The exposure time frame should be chosen according to the scientific question (for instance, if one is specifically interested in disturbance of early embryonic development, exposure should start immediately after fertilization. Otherwise we recommend to start not before 24 hpf, when the embryo has already developed to its phylotypic appearance). Furthermore, exposure concentrations should be anchored to a phenotypic effect (e.g., LCX). This will foster the establishment of causal relationships between gene expression and adverse outcome in the future.
FIG. 5.
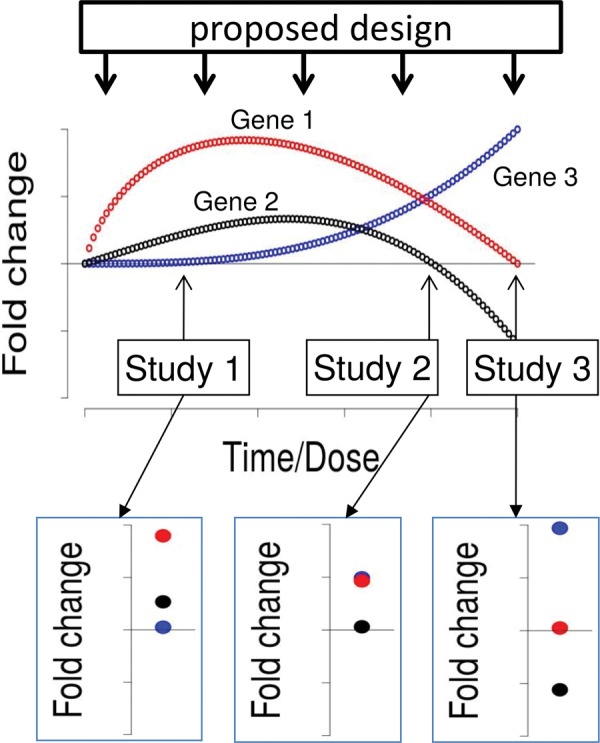
Schematic representation of concentration and time dependent gene expression. Current experimental designs (“Study 1–Study 3”) only give snapshot information and can lead to seemingly contradicting results in toxicological experiments. Concentration and time variation measurements (“proposed design”) would allow establishing systematic relationships.
CONCLUSION
In a meta-analysis of 33 toxicogenomic zebrafish embryo (ZFE) studies, we could not identify a general uniform stress response. We found that the analyzed transcriptome studies show great heterogeneity in experimental settings that we take responsible for heterogeneous results. However, in selected experimental subgroups we could identify common trends of gene regulation. We identified gene sets connected to anatomical development, metabolism and calcium homeostasis as downregulated in different subgroups whereas upregulation of gene sets seemed to be more diverse and thus more specific. Induced genes could, among others, be linked to signaling pathways (e.g. Wnt, Fgf) as well as lysosomal structures and apoptosis.
This analysis shows some methodological constraints of existing ZFE transcriptome studies but at the same time it provides a starting point to substantially increase our understanding of toxic effects of chemicals and stress responses in the developing ZFE.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Till Luckenbach for proofreading and commenting on the manuscript. Furthermore, we would like to thank two anonymous reviewers for their constructive comments on the manuscript.
FUNDING
This work was supported by the German Federal Environmental Foundation Scholarship Program (AZ: 20014/350) and the European Union 7th Framework Programme project SOLUTIONS (grant agreement no. 603437).
REFERENCES
- Abdou H. S., Robert N. M., Tremblay J. J. (2016). Calcium-dependent Nr4a1 expression in mouse Leydig cells requires distinct AP1/CRE and MEF2 elements. J. Mol. Endocrinol. 56, 151–161. [DOI] [PubMed] [Google Scholar]
- Afshari C. A., Hamadeh H. K., Bushel P. R. (2011). The evolution of bioinformatics in toxicology: Advancing toxicogenomics. Toxicol. Sci. 120, S225–S237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexeyenko A., Wassenberg D. M., Lobenhofer E. K., Yen J., Linney E., Sonnhammer E. L. L., Meyer J. N. (2010). Dynamic zebrafish interactome reveals transcriptional mechanisms of dioxin toxicity. PLoS One 5, e10465.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankley G. T., Bennett R. S., Erickson R. J., Hoff D. J., Hornung M. W., Johnson R. D., Mount D. R., Nichols J. W., Russom C. L., Schmieder P. K., et al. (2010). Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 29, 730–741. [DOI] [PubMed] [Google Scholar]
- Antczak P., White T. a., Giri A., Michelangeli F., Viant M. R., Cronin M. T. D., Vulpe C., Falciani F. (2015). Systems biology approach reveals a calcium-dependent mechanism for basal toxicity in Daphnia magna. Environ. Sci. Technol. 49, 11132–11140. [DOI] [PubMed] [Google Scholar]
- Arnold C., Externbrink F., Hackermüller J., Reiche K. (2014). CEM designer : design of custom expression microarrays in the post-ENCODE Era. J. Biotechnol. 189, 154–156. [DOI] [PubMed] [Google Scholar]
- Berkes C. A., Tapscott S. J. (2005). MyoD and the transcriptional control of myogenesis. Semin. Cell Dev. Biol. 16, 585–595. [DOI] [PubMed] [Google Scholar]
- Bonventre J. A., Kung T. S., White L. A., Cooper K. R. (2013). Manipulation of the HIF-Vegf pathway rescues methyl tert-butyl ether (MTBE)-induced vascular lesions. Toxicol. Appl. Pharmacol. 273, 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brette F., Machado B., Cros C., Incardona J. P., Scholz N. L., Block B. A. (2014). Crude Oil Impairs Cardiac Excitation-Contraction Coupling in Fish. Science 343, 772–776. [DOI] [PubMed] [Google Scholar]
- Büttner A., Busch W., Klüver N., Giannis A., Scholz S. (2012). Transcriptional responses of zebrafish embryos exposed to potential sonic hedgehog pathway interfering compounds deviate from expression profiles of cyclopamine. Reprod. Toxicol. 33, 254–263. [DOI] [PubMed] [Google Scholar]
- Chen W., Li N., Chen T., Han Y., Li C., Wang Y., He W., Zhang L., Wan T., Cao X. (2005). The lysosome-associated apoptosis-inducing protein containing the pleckstrin homology (PH) and FYVE domains (LAPF), representative of a novel family of PH and FYVE domain-containing proteins, induces caspase-independent apoptosis via the lysosomal-mitocho. J. Biol. Chem. 280, 40985–40995. [DOI] [PubMed] [Google Scholar]
- Choi J. K., Yu U., Kim S., Yoo O. J. (2003). Combining multiple microarray studies and modeling interstudy variation. Bioinformatics 19, i84–i90. [DOI] [PubMed] [Google Scholar]
- DiBiase A., Li P., Zhou Y. (2012). Zebrafish whole embryos at 36 hpf treated with DMSO or 5 um 11,12-EET. ArrayExpress Database. Available at: https://www.ebi.ac.uk/arrayexpress/experiments/E-GEOD-39707/. Accessed February 27, 2017.
- Driessen M., Kienhuis A. S., Vitins A. P., Pennings J. L a., Pronk T. E., Brandhof E. J., Van Den, Roodbergen M., Water B. V. D., Ven L. T. M. V. D. (2014). Gene expression markers in the zebrafish embryo reflect a hepatotoxic response in animal models and humans. Toxicol. Lett. 230, 48–56. [DOI] [PubMed] [Google Scholar]
- Driessen M., Vitins A. P., Pennings J. L. A., Kienhuis A. S., Water B. V. D., Ven L. T. M. V. D. (2015). A transcriptomics-based hepatotoxicity comparison between the zebrafish embryo and established human and rodent in vitro and in vivo models using cyclosporine A, amiodarone and acetaminophen. Toxicol. Lett. 232, 403–412. [DOI] [PubMed] [Google Scholar]
- Faltermann S., Zucchi S., Kohler E., Blom J. F., Pernthaler J., Fent K. (2014). Molecular effects of the cyanobacterial toxin cyanopeptolin (CP1020) occurring in algal blooms: global transcriptome analysis in zebrafish embryos. Aquat. Toxicol. 149, 33–39. [DOI] [PubMed] [Google Scholar]
- Garcia-Reyero N., Escalon B. L., Prats E., Stanley J. K., Thienpont B., Melby N. L., Barón E., Eljarrat E., Barceló D., Mestres J., et al. (2014). Effects of BDE-209 contaminated sediments on zebrafish development and potential implications to human health. Environ. Int. 63, 216–223. [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium (2015). Gene Ontology Consortium: going forward. Nucleic Acids Res. 43, D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodale B. C., Tilton S. C., Corvi M. M., Wilson G. R., Janszen D. B., Anderson K. A., Waters K. M., Tanguay R. L. (2013). Structurally distinct polycyclic aromatic hydrocarbons induce differential transcriptional responses in developing zebrafish. Toxicol. Appl. Pharmacol. 272, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh K. J., Suter M. J. F. (2015). Stressor-induced proteome alterations in zebrafish: A meta-analysis of response patterns. Aquat. Toxicol. 159, 1–12. [DOI] [PubMed] [Google Scholar]
- Györffy B., Dietel M., Fekete T., Lage H. (2008). A snapshot of microarray-generated gene expression signatures associated with ovarian carcinoma. Int. J. Gynecol. Cancer 18, 1215–1233. [DOI] [PubMed] [Google Scholar]
- Hahn M. E., McArthur A. G., Karchner S. I., Franks D. G., Jenny M. J., Timme-Laragy A. R., Stegeman J. J., Woodin B. R., Cipriano M. J., Linney E. (2014). The transcriptional response to oxidative stress during vertebrate development: effects of tert-butylhydroquinone and 2,3,7,8-tetrachlorodibenzo-p-dioxin. PLoS One 9, e113158.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao R., Bondesson M., Singh A. V., Riu A., McCollum C. W., Knudsen T. B., Gorelick D. a., Gustafsson J. (2013). Identification of estrogen target genes during zebrafish embryonic development through transcriptomic analysis. PLoS One 8, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermsen S. a B., van den Brandhof E.-J., van der Ven L. T. M., Piersma A. H. (2011). Relative embryotoxicity of two classes of chemicals in a modified zebrafish embryotoxicity test and comparison with their in vivo potencies. Toxicol. In Vitro 25, 745–753. [DOI] [PubMed] [Google Scholar]
- Hermsen S. a B., Pronk T. E., van den Brandhof E.-J. J., van der Ven L. T. M., Piersma A. H. (2012). Concentration-response analysis of differential gene expression in the zebrafish embryotoxicity test following flusilazole exposure. Toxicol. Sci. 127, 303–312. [DOI] [PubMed] [Google Scholar]
- Hermsen S. a B., Pronk T. E., van den Brandhof E.-J., van der Ven L. T. M., Piersma A. H. (2013). Transcriptomic analysis in the developing zebrafish embryo after compound exposure: individual gene expression and pathway regulation. Toxicol. Appl. Pharmacol. 272, 161–171. [DOI] [PubMed] [Google Scholar]
- Herrero-Turrión M., Rodríguez-Martín I., López-Bellido R., Rodríguez R. E. (2014). Whole-genome expression profile in zebrafish embryos after chronic exposure to morphine: identification of new genes associated with neuronal function and mu opioid receptor expression. BMC Genomics 15, 874.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe D. G., Bradford Y. M., Conlin T., Eagle A. E., Fashena D., Frazer K., Knight J., Mani P., Martin R., Moxon S. A. T., et al. (2013). ZFIN, the Zebrafish Model Organism Database: increased support for mutants and transgenics. Nucleic Acids Res. 41, D854–D860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W., Carey V. J., Gentleman R., Anders S., Carlson M., Carvalho B. S., Bravo H. C., Davis S., Gatto L., Girke T., et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihnatova I. (2013). MAMA: an R package for Meta-Analysis of MicroArray. Available at: https://cran.r-project.org/web/packages/MAMA/index.html. Accessed February 27, 2017.
- Kahl C. R., Means A. R. (2003). Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr. Rev. 24, 719–736. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H. Y., Choi Y.-K., Jeung E.-B. (2016). Inhibitory effect of progesterone during early embryonic development: Suppression of myocardial differentiation and calcium-related transcriptome by progesterone in mESCs: Progesterone disturb cardiac differentiation of mESCs through lower cytosolic Ca2+. Reprod. Toxicol. 64, 169–179. [DOI] [PubMed] [Google Scholar]
- Kawabata M., Umemoto N., Shimada Y., Nishimura Y., Zhang B., Kuroyanagi J., Miyabe M., Tanaka T. (2015). Downregulation of stanniocalcin 1 is responsible for sorafenib-induced cardiotoxicity. Toxicol. Sci. 143, 374–384. [DOI] [PubMed] [Google Scholar]
- Kelder T., van Iersel M. P., Hanspers K., Kutmon M., Conklin B. R., Evelo C. T., Pico A. R. (2012). WikiPathways: building research communities on biological pathways. Nucleic Acids Res. 40, D1301–D1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C. B. (1989). Genetics and early development of zebrafish. Trends Genet. 5, 283–288. [DOI] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995). Stages of Embryonic Development of the Zebrafish. Dev. Dyn. 203, 253–310. [DOI] [PubMed] [Google Scholar]
- Klüver N., Yang L., Busch W., Scheffler K., Renner P., Strähle U., Scholz S. (2011). Transcriptional response of zebrafish embryos exposed to neurotoxic compounds reveals a muscle activity dependent hspb11 expression. PLoS One 6, e29063.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam S. H., Hlaing M. M., Zhang X., Yan C., Duan Z., Zhu L., Ung C. Y., Mathavan S., Ong C. N., Gong Z. (2011). Toxicogenomic and Phenotypic Analyses of Bisphenol-A Early-Life Exposure Toxicity in Zebrafish. PLoS One 6, e28273.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê Cao K. A., Rohart F., McHugh L., Korn O., Wells C. a. (2014). YuGene: A simple approach to scale gene expression data derived from different platforms for integrated analyses. Genomics 103, 239–251. [DOI] [PubMed] [Google Scholar]
- Lieschke G. J., Currie P. D. (2007). Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353–367. [DOI] [PubMed] [Google Scholar]
- Lin C.-Y., Chiang C.-Y., Tsai H.-J. (2016). Zebrafish and Medaka: new model organisms for modern biomedical research. J. Biomed. Sci. 23, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D., Wolfgang C. D., Hai T. (2006). Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J. Biol. Chem. 281, 10473–10481. [DOI] [PubMed] [Google Scholar]
- Lusa L., Gentleman R., Ruschhaupt M. (2015). GeneMeta: MetaAnalysis for High Throughput Experiments. R package version 1.44.0. Available at: https://www.bioconductor.org/packages/release/bioc/html/GeneMeta.html. Accessed February 27, 2017.
- Mallo M., Wellik D. M., Deschamps J. (2010). Hox genes and regional patterning of the vertebrate body plan. Dev. Biol. 344, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx-Stoelting P., Braeuning A., Buhrke T., Lampen A., Niemann L., Oelgeschlaeger M., Rieke S., Schmidt F., Heise T., Pfeil R., et al. (2015). Application of omics data in regulatory toxicology: report of an international BfR expert workshop. Arch. Toxicol. 89, 2177–2184. [DOI] [PubMed] [Google Scholar]
- McKinsey T. A., Zhang C. L., Olson E. N. (2002). MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 27, 40–47. [DOI] [PubMed] [Google Scholar]
- Medzikovic L., Schumacher C. A., Verkerk A. O., van Deel E. D., Wolswinkel R., van der Made I., Bleeker N., Cakici D., van den Hoogenhof M. M. G., Meggouh F., et al. (2015). Orphan nuclear receptor Nur77 affects cardiomyocyte calcium homeostasis and adverse cardiac remodelling. Sci. Rep. 5, 15404.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell A., Chang H.-Y., Daugherty L., Fraser M., Hunter S., Lopez R., McAnulla C., McMenamin C., Nuka G., Pesseat S., et al. (2015). The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–D221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya F. J., Olson E. (1999). MEF2: a transcriptional target for signaling pathways controlling skeletal muscle growth and differentiation. Curr. Opin. Cell Biol. 11, 683–688. [DOI] [PubMed] [Google Scholar]
- Newman D., Abuladze N., Scholz K., Dekant W., Tsuprun V., Ryazantsev S., Bondar G., Sassani P., Kurtz I., Pushkin A. (2007). Specificity of aminoacylase III-mediated deacetylation of mercapturic acids. Drug Metab. Dispos. 35, 43–50. [DOI] [PubMed] [Google Scholar]
- O’Duibhir E., Lijnzaad P., Benschop J. J., Lenstra T. L., van Leenen D., Groot Koerkamp M. J. A., Margaritis T., Brok M. O., Kemmeren P., Holstege F. C. P. (2014). Cell cycle population effects in perturbation studies. Mol. Syst. Biol. 10, 732.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oggier D. M., Lenard A., Küry M., Hoeger B., Affolter M., Fent K. (2011). Effects of the protein kinase inhibitor PKC412 on gene expression and link to physiological effects in zebrafish Danio rerio eleuthero embryos. Toxicol. Sci. 119, 104–115. [DOI] [PubMed] [Google Scholar]
- Oliveira E., Casado M., Faria M., Soares A. M. V. M., Navas J. M., Barata C., Piña B. (2013). Transcriptomic response of zebrafish embryos to polyaminoamine (PAMAM) dendrimers. Nanotoxicology 5390, 92–99. [DOI] [PubMed] [Google Scholar]
- Ota S., Tonou-Fujimori N., Yamasu K. (2009). The roles of the FGF signal in zebrafish embryos analyzed using constitutive activation and dominant-negative suppression of different FGF receptors. Mech. Dev. 126, 1–17. [DOI] [PubMed] [Google Scholar]
- Park J.-W., Heah T. P., Gouffon J. S., Henry T. B., Sayler G. S. (2012). Global gene expression in larval zebrafish (Danio rerio) exposed to selective serotonin reuptake inhibitors (fluoxetine and sertraline) reveals unique expression profiles and potential biomarkers of exposure. Environ. Pollut. 167, 163–170. [DOI] [PubMed] [Google Scholar]
- Pelayo S., Oliveira E., Thienpont B., Babin P. J., Raldúa D., André M., Piña B. (2012). Triiodothyronine-induced changes in the zebrafish transcriptome during the eleutheroembryonic stage: Implications for bisphenol A developmental toxicity. Aquat. Toxicol. 110-111, 114–122. [DOI] [PubMed] [Google Scholar]
- Penn A., Kleinow K., Xin F., Bui A., Xiao R., Perveen Z., Polk P. (2013). Microarray data from zebrafish embryos (ZFE) exposed for 48-hr periods to oil emulsions collected from GoM shoreline sites following the Deepwater Horizon blowout. GSE52873.
- Pessah I. N., Cherednichenko G., Lein P. J. (2010). Minding the calcium store: Ryanodine receptor activation as a convergent mechanism of PCB toxicity. Pharmacol. Ther. 125, 260–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prykhozhij S. V., Marsico A., Meijsing S. H. (2013). Zebrafish Expression Ontology of Gene Sets (ZEOGS): a tool to analyze enrichment of zebrafish anatomical terms in large gene sets. Zebrafish 10, 303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2015). R: A Language and Environment for Statistical Computing. [Google Scholar]
- Ramasamy A., Mondry A., Holmes C. C., Altman D. G. (2008). Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 5, 1320–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderick H. L., Cook S. J. (2008). Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 8, 361–375. [DOI] [PubMed] [Google Scholar]
- Saili K. S., Tilton S. C., Waters K. M., Tanguay R. L. (2013). Global gene expression analysis reveals pathway differences between teratogenic and non-teratogenic exposure concentrations of bisphenol A and 17 β-estradiol in embryonic zebrafish. Reprod. Toxicol. 38, 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmah S., Muralidharan P., Curtis C. L., McClintick J. N., Buente B. B., Holdgrafer D. J., Ogbeifun O., Olorungbounmi O. C., Patino L., Lucas R., et al. (2013). Ethanol exposure disrupts extraembryonic microtubule cytoskeleton and embryonic blastomere cell adhesion, producing epiboly and gastrulation defects. Biol. Open. 2, 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller V., Wichmann A., Kriehuber R., Schäfers C., Fischer R., Fenske M. (2013a). Transcriptome alterations in zebrafish embryos after exposure to environmental estrogens and anti-androgens can reveal endocrine disruption. Reprod. Toxicol. 42, 210–223. [DOI] [PubMed] [Google Scholar]
- Schiller V., Wichmann A., Kriehuber R., Muth-Köhne E., Giesy J. P., Hecker M., Fenske M. (2013b). Studying the effects of genistein on gene expression of fish embryos as an alternative testing approach for endocrine disruption. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 157, 41–53. [DOI] [PubMed] [Google Scholar]
- Scholz S., Fischer S., Gündel U., Küster E., Luckenbach T., Voelker D. (2008). The zebrafish embryo model in environmental risk assessment–applications beyond acute toxicity testing. Environ. Sci. Pollut. Res. Int. 15, 394–404. [DOI] [PubMed] [Google Scholar]
- Smetanová S., Riedl J., Zitzkat D., Altenburger R., Busch W. (2015). High-throughput concentration-response analysis for omics datasets. Environ. Toxicol. Chem. 34, 2167–2180. [DOI] [PubMed] [Google Scholar]
- Tal T. L., Franzosa J. A., Tilton S. C., Philbrick K. A., Iwaniec U. T., Turner R. T., Waters K. M., Tanguay R. L. (2012). MicroRNAs control neurobehavioral development and function in zebrafish. FASEB J. 26, 1452–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanguay R. L., Huang C., Chen J., La Du J., Lin K., Waters K. M., Truong L., Dong Q., Tilton S. C. (2011). Expression Analysis of Trimethyltin Chlorides (TMT) Impact on Embryonic Development. E–GEOD–28131.
- Truong L., Tilton S. C., Zaikova T., Richman E., Waters K. M., Hutchison J. E., Tanguay R. L. (2012). Surface functionalities of gold nanoparticles impact embryonic gene expression responses. Nanotoxicology 29, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher V. G., Tibshirani R., Chu G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 98, 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Heyden C., Huysseune A. (2000). Dynamics of tooth formation and replacement in the zebrafish (Danio rerio) (Teleostei, Cyprinidae). Dev. Dyn. 219, 486–496. [DOI] [PubMed] [Google Scholar]
- Verkade H., Heath J. K. (2008). Wnt signaling mediates diverse developmental processes in zebrafish. Methods Mol. Biol. 469, 225–251. [DOI] [PubMed] [Google Scholar]
- Vermeulen N. P. E. (1989). Analysis of mercapturic acids as a tool in biotransformation, biomonitoring and toxicological studies. Trends Pharmacol. Sci. 10, 177–181. [DOI] [PubMed] [Google Scholar]
- Vidal-Dorsch D. E., Bay S. M., Moore S., Layton B., Mehinto A. C., Vulpe C. D., Brown-Augustine M., Loguinov A., Poynton H., Garcia-Reyero N., et al. (2016). Ecotoxicogenomics: microarray interlaboratory comparability. Chemosphere 144, 193–200. [DOI] [PubMed] [Google Scholar]
- Villeneuve D. L., Crump D., Garcia-Reyero N., Hecker M., Hutchinson T. H., LaLone C. A., Landesmann B., Lettieri T., Munn S., Nepelska M., et al. (2014). Adverse outcome pathway development II: best practices. Toxicol. Sci. 142, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weicksel S. E., Xu J., Sagerström C. G. (2013). Dynamic nucleosome organization at hox promoters during Zebrafish embryogenesis. PLoS One 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H., Davis R., Tapscott S., Thayer M., Krause M., Benezra R., Blackwell T., Turner D., Rupp R., Hollenberg S., et al. (1991). The myoD gene family: nodal point during specification of the muscle cell lineage. Science 251, 761–766. [DOI] [PubMed] [Google Scholar]
- White R. M., Cech J., Ratanasirintrawoot S., Lin C. Y., Rahl P. B., Burke C. J., Langdon E., Tomlinson M. L., Mosher J., Kaufman C., et al. (2011). DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature 471, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T. D., Mirbahai L., Chipman J. K. (2014). The toxicological application of transcriptomics and epigenomics in zebrafish and other teleosts. Brief. Funct. Genomics 13, 157–171. [DOI] [PubMed] [Google Scholar]
- Xu T., Zhao J., Hu P., Dong Z., Li J., Zhang H., Yin D., Zhao Q. (2014). Pentachlorophenol exposure causes Warburg-like effects in zebrafish embryos at gastrulation stage. Toxicol. Appl. Pharmacol. 277, 183–191. [DOI] [PubMed] [Google Scholar]
- Yu G., Wang L.-G., Han Y., He Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. (2015). Network analysis reveals stage-specific changes in zebrafish embryo development using time course whole transcriptome profiling and prior biological knowledge. BioData Min. 8, 26.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.