

A new role for the mutlifunctional protein CTCF in the repair of DNA double-strand breaks is discovered.
Keywords: CTCF, BRCA2, DNA damage, Homologous Recombination, PARP inhibitors, poly ADP ribosylation
Abstract
The repair of DNA double-strand breaks (DSBs) is mediated via two major pathways, nonhomologous end joining (NHEJ) and homologous recombination (HR) repair. DSB repair is vital for cell survival, genome stability, and tumor suppression. In contrast to NHEJ, HR relies on extensive homology and templated DNA synthesis to restore the sequence surrounding the break site. We report a new role for the multifunctional protein CCCTC-binding factor (CTCF) in facilitating HR-mediated DSB repair. CTCF is recruited to DSB through its zinc finger domain independently of poly(ADP-ribose) polymers, known as PARylation, catalyzed by poly(ADP-ribose) polymerase 1 (PARP-1). CTCF ensures proper DSB repair kinetics in response to γ-irradiation, and the loss of CTCF compromises HR-mediated repair. Consistent with its role in HR, loss of CTCF results in hypersensitivity to DNA damage, inducing agents and inhibitors of PARP. Mechanistically, CTCF acts downstream of BRCA1 in the HR pathway and associates with BRCA2 in a PARylation-dependent manner, enhancing BRCA2 recruitment to DSB. In contrast, CTCF does not influence the recruitment of the NHEJ protein 53BP1 or LIGIV to DSB. Together, our findings establish for the first time that CTCF is an important regulator of the HR pathway.
INTRODUCTION
DNA double-strand breaks (DSBs) are among the most deleterious DNA lesions, and there is evidence that even a single DSB is sufficient to promote genomic instability and cell death if left unrepaired (1). DSB may arise during physiological processes such as meiosis and T and B cell receptor rearrangement. These lesions can also result from exogenous stress (for example, cytotoxic agents and ionizing radiation) or endogenous insults (reactive oxygen species and replication errors). To overcome their cytotoxic impact, DSBs are primarily repaired through two mutually exclusive pathways: nonhomologous end joining (NHEJ) and homologous recombination (HR) (2). Understanding how these pathways promote DNA repair, and how they can be disrupted in cancer, has led to new therapeutic approaches to treat multiple types of malignancies. HR frequently uses a sister chromatid as a template to repair the damaged sequence. Therefore, HR is carried out predominantly during the S and G2 phases of the cell cycle (3). The initiation of HR, as opposed to NHEJ, relies heavily upon the formation of extensive single-stranded (ss) 3′ DNA overhangs (4, 5), which require the recruitment of CtIP to the break site, the stimulation of the endonuclease activity of MRE11 in complex with RAD50 and NSB1, and the action of the nucleases EXO1 and BLM/DNA2. Replication protein A (RPA) loads onto this ssDNA, thereby protecting it from breakage (6). Subsequently, the concomitant action of BRCA1, PALB2, and BRCA2, in complex with its partner DSS1, promotes the replacement of RPA and the loading of the recombinase RAD51 onto ssDNA (7, 8). RAD51 is critical for maintaining sequence integrity through homology search, strand invasion, and sister chromatid exchange (7). Previous studies have shown that BRCA2 is central for HR-mediated DSB repair by directly binding ssDNA overhangs and catalyzing RAD51 nucleofilament formation. However, recent evidence suggests that BRCA2 is also recruited at early time periods to DSBs to promote EXO1-dependent DNA end resection through the recognition of poly(ADP-ribose) (PAR) polymers at DSB (9). These modifications are catalyzed by PAR polymerases (PARPs), including the founding member of this family, PARP-1, which promotes the attachment of PAR polymers onto target proteins, a process commonly known as PARylation. PARP-1 activity has been traditionally associated with base excision repair, but clear evidence demonstrates that PARP-1 is recruited to DSB and that PARylation plays an important role in DSB repair, primarily by HR (10–12). Protein PARylation at break sites may play multiple roles in orchestrating the repair of DSB. For example, this activity promotes a more open chromatin conformation to facilitate the recruitment of DNA repair factors (13, 14), and the PAR polymer itself can act as a scaffold for the recruitment of key repair proteins such as BRCA2 (9, 15, 16).
Although extensive research has uncovered much about these key steps in the HR-mediated DSB repair pathway, the process is exquisitely complex, with many additional proteins being implicated as playing roles within this network. However, how DSB repair factors are assembled at DNA damage sites in a PARylation-dependent manner and how this process is controlled are largely underexplored. A recent screen identified 62 DNA binding factors recruited to DNA lesions induced by laser micro-irradiation, many in a PARP-1–dependent manner (17). One of the factors identified is the multifunctional nuclear protein CCCTC-binding factor (CTCF). However, the role of this protein, if any, in the repair of DSB has yet to be investigated. CTCF is an 11-zinc finger transcription protein with well-established roles in genome organization and transcriptional regulation (18–20). In vivo evidence from CTCF heterozygous knockout mice suggests that CTCF acts as a haploinsufficient tumor suppressor (21). These CTCF+/− mice demonstrate increased sensitivity to irradiation-induced oncogenesis, which, coupled with the previous finding that CTCF is recruited to laser micro-irradiation tracks (17), strongly suggests a role for CTCF in the repair of damaged DNA. It is clear that CTCF is multifunctional in nature and may mediate disparate functions through incorporation into protein complexes committed to distinct biological processes (22). Similar to numerous proteins involved in the repair of DNA lesions, CTCF is covalently modified by PARylation (23). CTCF PARylation is commonly lost in breast tumors, correlating with cancer progression (24, 25), but the precise functional impact of PARylation on CTCF function remains ambiguous. In Drosophila, CTCF PARylation stabilizes the interaction with its protein binding partners, such as Cp190 (26), but it is unclear whether CTCF PARylation affects its association with other proteins in mammals.
Here, we sought to examine the role of CTCF in DSB repair. We report that CTCF is recruited to DSB via its DNA binding domain independently of PARylation. Once recruited to DNA lesions, CTCF enhances the recruitment of BRCA2 and promotes DSB repair by HR. In line with this observation, loss of CTCF sensitized cells to PARP inhibitors. Mechanistically, we show that CTCF associates with BRCA2 in a PARylation-dependent manner. These data provide insights into a previously undescribed role of CTCF in DSB repair.
RESULTS
CTCF rapidly localizes to sites of DSB
It was previously shown that CTCF accumulates to DNA lesions following ultraviolet (UV) laser micro-irradiation; however, CTCF dynamics at sites of DNA breaks remain unclear (17). Therefore, we first sought to examine the recruitment of endogenous CTCF to laser micro-irradiation–induced DNA lesions in MCF7 cells at different time points. To this end, we initially used laser micro-irradiation to probe the recruitment of endogenous CTCF to tracks of DNA damage in MCF7 cells. We observe that CTCF is rapidly recruited to laser tracks, and its association persists for up to 120 min, in a pattern similar to PARP-1 (Fig. 1A). Using live-cell imaging, we observed similar results, with CTCF being recruited to breakages within seconds of laser damage, where it remains associated for over 1 hour (fig. S1, A and B). Laser micro-irradiation elicits high levels of both single-strand breaks (SSB) and double-strand breaks (DSB). Therefore, it is possible that CTCF might be recruited to SSBs rather than DSBs. To rule out this possibility, we complemented our initial observation by using the previously described mCherry-LacI-FokI reporter system (27, 28). Here, a single locus within U2OS cells is engineered to carry repeats of the Lac operon. Recruitment of the mCherry-FokI nuclease to this locus results in a single red focus, localized DSB, and the accumulation of DNA repair components (27, 28). In agreement with our laser micro-irradiation experiments, CTCF is readily recruited to FokI cut sites, similar to what we observed with other DNA damage response proteins, including BRCA2, RAD51, and 53BP1, as well as the accumulation of the histone modification γH2A.X (Fig. 1B and fig. S1C). The recruitment of CTCF is dependent on the induction of DSB, because a catalytically dead form of FokI, incapable of generating these lesions, does not lead to a detectable colocalization between FokI and CTCF (Fig. 1B). These data indicate that CTCF is rapidly recruited to and persists at DSBs, suggesting a role for CTCF in the DSB response.
Fig. 1. CTCF localizes to DSBs via its zinc finger domain.
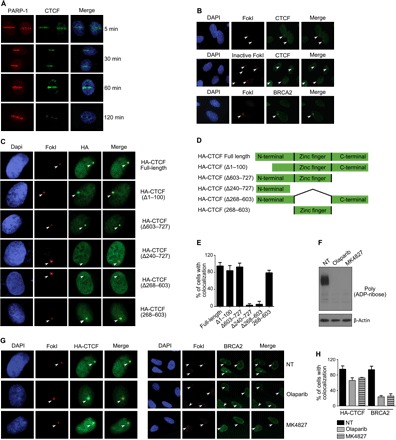
(A) BrdU presensitized MCF7 cells were subjected to laser micro-irradiation using a 405-nm UV laser. Cells were fixed and stained with the indicated antibodies. (B) The U2OS-LacI-FokI-mCherry DSB reporter cell line was transfected with wild-type (WT) mCherry-FokI or an enzymatically inactivate mutant. After 48 hours, cells were fixed and stained with the indicated antibodies. (C) U2OS-LacI cells were cotransfected with HA-tagged full-length CTCF, or various deletions, along with mCherry-FokI. At 48 hours after transfection, cells were fixed and stained with an anti-HA antibody. (D) Schematic of CTCF fragments used to analyze recruitment to FokI cut sites. (E) Bar graph representing the percentage of cells positive for HA at mCherry-FokI foci. Data are means ± SD. (F) The U2OS-LacI-FokI-mCherry DSB reporter cell line was treated with 1 μM of the PARP inhibitor olaparib or MK4827 for 24 hours. Whole-cell extracts were prepared, and Western blotting was carried out using anti-PAR and β-actin antibodies. (G) Staining for BRCA2 and HA-CTCF in the U2OS-LacI-FokI-mCherry DSB reporter cell line before and after exposure to 1 μM PARP-1 inhibitors for 24 hours. (H) Bar graph representing the percentage of cells duo-positive for HA and mCherry-FokI foci or BRCA2 with mCherry-FokI foci. Data are means ± SD.
The CTCF zinc finger domain is required for recruitment to DSB
Numerous proteins involved in the DNA repair process are PARylated (23), and this posttranslational modification can play a significant role in the recruitment of proteins to DNA breakages (9, 29). We observe that the association of CTCF with PARylation is substantially enriched in response to diverse genotoxic insults including γ-irradiation, paclitaxel, and doxorubicin (fig. S2, A to C). Therefore, we speculated that this modification might regulate CTCF recruitment to DSB. To determine the minimal CTCF domain required for recruitment to DSB, we generated a series of hemagglutinin (HA)–tagged CTCF deletion mutants to test for their ability to colocalize with FokI foci (Fig. 1, C to E, and fig. S2D). CTCF deletion mutants composed of only the N-terminal region (CTCF Δ240–727), or of an N-terminal and a C-terminal fusion (CTCF Δ268–603), are unable to colocalize with FokI (Fig. 1, C and E). In contrast, CTCF mutants harboring either the N or C terminus along with the zinc finger domain, or the zinc finger domain alone, readily colocalize to FokI cut sites (Fig. 1, C and E). These experiments reveal that the zinc finger domain of CTCF is necessary and sufficient to promote recruitment to FokI cut sites (Fig. 1, C and E). Previous reports demonstrate that a subset of DNA damage response proteins is dependent on its association with PAR polymers for recruitment to DSB (9, 29). However, our observations suggest that this is not the case for CTCF, whose PARylation sites reside within the N-terminal region, which, based on our data (Fig. 1C), is not sufficient for recruitment to DSB induced by FokI. To further test this idea, we treated cells with two clinically relevant PARP-1 inhibitors, olaparib and MK4827 (Fig. 1, F to H). As expected, PARP-1 inhibition reduces cellular pools of PARylated proteins (Fig. 1F) and diminishes the accumulation of BRCA2 at DSB, as previously described (Fig. 1, G and H) (9). In these experiments, we exposed cells to PARP inhibitors for 24 hours; therefore, both direct and indirect mechanisms of action may account for the significant loss of BRCA2 recruitment to FokI cut sites. In contrast, these inhibitors have little impact on CTCF localization to FokI foci (Fig. 1, G and H). This is consistent with our previous observations showing that the CTCF N terminus, where PARylation sites have been previously identified, is dispensable for its recruitment to DSBs. Overall, our results show that CTCF is recruited to DSB directly via its zinc finger domain, independent of the localized accumulation of PAR chains and the chromatin remodeling activity, catalyzed by PARP-1 (30). However, these data do not exclude that PARylation of CTCF is required for its functions in the DSB response.
Loss of CTCF alters the response to DSB
To further assess the role of CTCF in the DSB response, we targeted the CTCF locus in the normal mammary epithelial cell line MCF10A using the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9) technology. Constitutive deletion of Ctcf in mice leads to embryonic lethality, which likely explained why we were unable to generate a full CTCF knockout cells. However, we obtained single-allele knockouts of CTCF in MCF10A (CTCF+/−), with each clone being derived from a single-cell expansion. We observed a significant reduction of CTCF protein in three distinct clones, ranging from approximately 20 to 50% of controls, dependent on the clone being studied (Fig. 2A). Next, we exposed cells to 2-Gy (gray) γ-irradiation and monitored the repair of DSB over time under control conditions or in three independent CTCF+/− clones. The repair kinetics of DSB, as determined by the disappearance of γH2AX foci (Fig. 2B), is significantly slowed in the three CTCF+/− clones. We observed that γH2AX foci persist significantly in CTCF-depleted cells (Fig. 2, B and C). It is unlikely that this defect is due to altered cell cycle kinetics because CTCF+/− cells showed similar proliferation profiles as control cells (fig. S3A). We validated this experiment in the breast cancer cell line MCF7 using two independent short hairpin RNA (shRNA) constructs to knock down CTCF (Fig. 2, D to F, and fig. S3, B and D). Again, after exposure to 2-Gy (Fig. 2D) or 5-Gy (fig. S3, B and D) irradiation, the kinetics of γH2AX foci resolution was delayed in CTCF knockdown cells. These data suggest that CTCF depletion impairs DSB repair. In support of this possibility, we quantified the resolution of DSBs upon irradiation of MCF7 control or CTCF knockdown cells using the neutral comet assay. Again, we observed the persistence of comet tails at 24 hours after irradiation in CTCF knockdown cells, whereas control cells showed almost complete resolution of comet tails by this time period (Fig. 2, G and H). γ-Irradiation–induced DSBs may be repaired by the NHEJ or HR pathways. Therefore, we monitored the disappearance of the key NHEJ factor 53BP1 at DSBs under the same conditions described above using the MCF7 knockdown cells. In contrast to what we observe for γH2AX, CTCF knockdown has little impact on the timing of 53BP1 foci dissolution (fig. S3, C and E). Overall, these data support the conclusion that CTCF plays a role in the repair of DSBs, likely independent of NHEJ.
Fig. 2. CTCF knockdown leads to altered DNA damage repair kinetics.
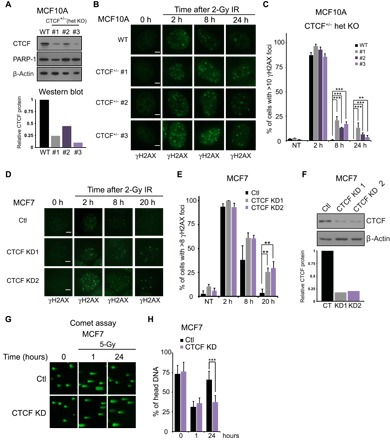
(A) Western blotting analyses for CTCF using lysates from three MCF10A clones with heterozygous deletion of CTCF (CTCF+/−) were subjected to immunoblotting with the indicated antibodies. Bar graph represents the quantification of CTCF signal. (B) MCF10A WT and three CTCF heterozygote (CTCF+/−) clone cell lines were fixed at the indicated times after irradiation (2 Gy) and stained with an anti-γH2AX antibody. (C) Quantification of the percent of cells with more than 10 γH2AX foci. Error bars correspond to means ± SEM (n = 3; ***P ≤ 0.005, ** P ≤ 0.01, χ2 test). (D) MCF7 cells were infected with Ctl shRNA or two constructs directed toward CTCF followed by irradiation (2 Gy). Cells were fixed at the indicated time point and stained with an anti-γH2AX antibody. (E) Quantification of percent of cells with more than eight γH2AX foci. Error bars correspond to means ± SEM (n = 3; **P ≤ 0.01, χ2 test). (F) MCF7 cells infected with scrambled control shRNA, or shRNAs directed toward CTCF, were subjected to immunoblotting with the indicated antibodies. (G) Comet assay was performed on MCF7 Ctl or CTCF-depleted cells following irradiation (5 Gy) at the indicated time points. (H) Quantification of the comet assay in percent of head DNA. Error bars correspond to means ± SEM [n = 3, ***P ≤ 0.005, one-way analysis of variance (ANOVA)]. Scale bars, 5 μm.
Next, we sought to obtain an orthogonal validation of the role of CTCF in the DSB response. Thus, we examined the sensitivity of MCF7 cells to different DNA-damaging agents upon lowering of CTCF levels. We found that the clonogenic survival potential of CTCF knockdown MCF7 cells is significantly impaired following exposure to γ-irradiation or the deoxynucleotide triphosphate–depleting agent hydroxyurea (Fig. 3, A and B). We extended these studies to test the impact of CTCF depletion on the sensitivity to PARP inhibitors. We observed a marked elevation of the cytotoxic response to two PARP inhibitors, olaparib and MK4287, upon CTCF knockdown (Fig. 3, C and D, and fig. S4). It is known that cells having defects in HR are exquisitely sensitive to PARP-1 inhibition. These data, coupled with our finding that CTCF delays the repair kinetics of γH2AX foci but not 53BP1, raise the possibility that CTCF plays a role in DSB repair, specifically through the HR pathway.
Fig. 3. Acute sensitivity to DNA-damaging agents and PARP inhibitors after loss of CTCF.
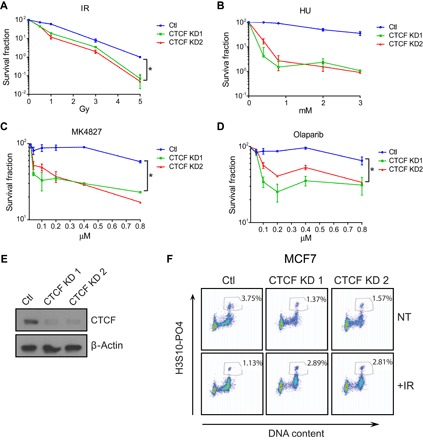
(A to D) Clonogenic survival assay of MCF7 cells comparing CTCF knockdown cells with control cells after treatment with the indicated agents. Cells were grown for 14 days after treatment followed by colony counting and calculation of the survival fraction. Data are means ± SD. (E) Western blot shows CTCF expression in knockdown and control cells. (F) Flow cytometric analysis of phospho-Ser10 histone 3–positive MCF7 cells after irradiation in Ctl and CTCF knockdown cells. Numbers in boxes represent the percentage of cells in mitosis. A representation of three independent experiments is shown.
It is well documented that radiation can lead to the activation of the G2-M cell cycle checkpoint. This checkpoint is often compromised after knockdown of DSB repair proteins, resulting in aberrant progression to mitosis (31). However, little is known about the role of CTCF at the G2-M checkpoint. Therefore, we tested the integrity of this critical checkpoint in MCF7 control and CTCF knockdown cells. As expected, after exposure to γ-irradiation, we observe a characteristic increase in G2-M cells from our control cells (fig. S4D) and a decrease in the proportion of mitotic cells, as assessed by Ser10 phosphorylation of histone 3 (Fig. 3F). In CTCF knockdown cells, this checkpoint is defective, with cells progressing to mitosis irrespective of irradiation exposure (Fig. 3F). We conclude that CTCF is essential for the DSB response, at both the repair of DSBs and the induction of a productive cell cycle arrest at the G2-M checkpoint.
CTCF participates in the HR pathway
On the basis of the hypersensitivity of CTCF-depleted cells to PARP-1 inhibitors, we next investigated the capacity of CTCF to directly regulate HR using two complimentary approaches. First, we used the CRISPR-mClover assay to quantify gene targeting, as previously described (32, 33). Here, insertion of the coding sequence of the mClover fluorescent protein into the 5′ end of the lamin A gene by HR after Cas9-mediated cutting is monitored using flow cytometry (Fig. 4A). We found that the number of mClover-positive cells was reduced by more than twofold in CTCF+/− clones (Fig. 4B) compared to control MCF10A cells. This indicates that gene targeting by HR is suppressed through the loss of CTCF. Next, we reintroduced HA-tagged CTCF in CTCF+/− clones using retroviral transduction and observed a restoration of the mClover signal, confirming that loss of CTCF impairs gene targeting by HR (Fig. 4, B and C). To extend these experiments probing a role of CTCF in HR, we next used the DR-GFP (Direct Repeat-GFP) reporter system in U2OS cells (34). This assay exploits an I–Sce I nuclease site situated in one of two mutated GFP genes, oriented as direct repeats. Expression of I–Sce I generates a DSB, which, when repaired through HR, results in functional GFP expression, quantifiable by flow cytometry (Fig. 4D). As expected, knockdown of RAD51 decreases the frequency of HR to near undetectable levels (Fig. 4, E to G). Down-regulation of CTCF levels with different shRNAs leads to a comparable repression of HR, with the total GFP-positive population decreasing from 3.8% to 1.0 to 1.3% (Fig. 4, E to G). These data indicate that the HR pathway is defective in CTCF knockdown cells and that the involvement of CTCF in the HR process is not tissue-specific.
Fig. 4. CTCF regulates HR at I–Sce I– and CRISPR/Cas9-directed double-strand breaks.
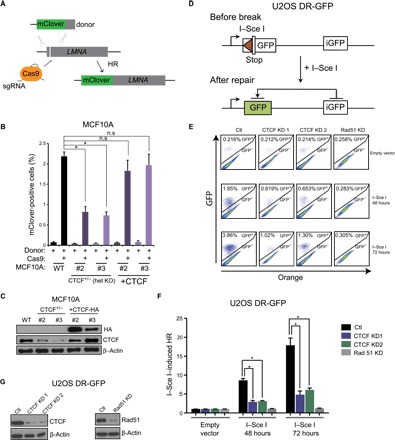
(A) Schematic representing the CRISPR-mClover assay for quantification of gene targeting by HR. (B) Quantification of mClover-positive MCF10A control or CTCF+/− cells by flow cytometry, representing the gene targeting efficiency at the LMNA locus. mClover expression was also quantified in CTCF add-back cells (+CTCF). Data are means ± SEM (n = 3; *P ≤ 0.05, one-way ANOVA). (C) Western blotting of CTCF for cells used in mClover experiments described in (B). (D) Schematic representing the DR-GFP assay for measuring gene targeting at I–Sce I cut sites by HR. (E) U2OS DR-GFP HR reporter cells were infected with control, CTCF, or RAD51 shRNAs and transfected with I–Sce I or an empty vector for 48 or 72 hours. The resultant cells were subjected to flow cytometric analysis for GFP-positive cells. (F) Quantification of data from experiment described in (A) depicted by bar graphs. Error bars correspond to means ± SEM (n = 3; *P ≤ 0.05, two-tailed Student’s t test) (G) Western blot for CTCF and Rad51 expression in cells used for experiments described in (E).
Next, we interrogated whether CTCF regulates the accumulation of repair factors to DSB induced by FokI. As expected, CTCF knockdown has no discernible effect on the colocalization of the NHEJ factors 53BP1 and LIGIV (Fig. 5, A and B), further confirming that CTCF does not play a role in the NHEJ pathway. Little impact is also seen on the initial accumulation of γH2AX at these cut sites, indicating that CTCF acts downstream of this epigenetic mark (Fig. 5, A and B). Strikingly, loss of CTCF leads to a significant reduction in BRCA2 recruitment to FokI-induced DSB (Fig. 5, A and B). This was not due to changes in BRCA2 expression, because BRCA2 protein levels remain constant upon CTCF knockdown (fig. S5A). Previous reports indicate that RAD51 recruitment to DSB is partially dependent on previous recruitment of BRCA2 (35). Therefore, we might expect to see RAD51 accumulation at FokI cut sites comprised in CTCF knockdown cells. Consistent with this concept, we also observe a lack of RAD51 recruitment to FokI-induced DSB upon loss of CTCF (Fig. 5, A and B). Likewise, we also observed a reduction of endogenous RAD51 accumulation at neocarzinostatin (NCS) or irradiation-induced repair foci upon CTCF knockdown in U2OS and MCF7 cells (Fig. 5, C and D, and fig. S5B). As expected, CTCF knockdown had no discernible impact on 53BP1 foci after NCS treatment. The HR factor BRCA1 is still capable of localizing to DSBs following CTCF depletion, suggesting a role for CTCF downstream of BRCA1 in the HR pathway (Fig. 5, C and D).
Fig. 5. CTCF modulates the recruitment of BRCA2 to FokI-induced DSB.
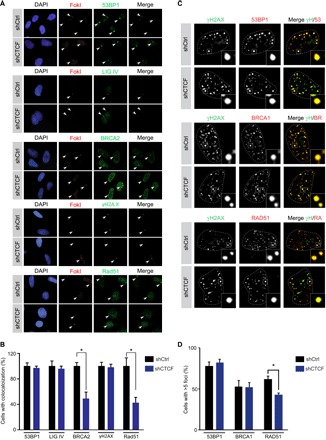
(A) U2OS-LacI-FokI-mCherry DSB reporter cells infected with control or CTCF shRNA were treated with Shield-1 and 4-hydroxytamoxifen for 6 hours to induce FokI expression as per Materials and Methods. Cells were next fixed and immunostained with the indicated antibodies and DAPI. (B) Quantification of colocalization between mCherry-FokI and 53BP1, LIGIV, BRCA2, γH2A.X, and Rad51. Error bars correspond to means ± SEM (n = 3; *P ≤ 0.05, two-tailed Student’s t test). (C) U2OS Ctl or CTCF knockdown cells were treated with NCS (150 ng/ml) for 3 hours followed by fixation and staining with the indicated antibodies. Representative images are shown. (D) Quantification of Ctl or CTCF knockdown cells harboring five or more 53BP1, BRCA1, or RAD51 foci. Error bars correspond to means ± SEM (n = 3; *P ≤ 0.05, paired Student’s t test).
Because CTCF knockdown compromises the recruitment of BRCA2 to FokI-induced DSB, we wanted to identify the domain of CTCF responsible for this activity. The loss of BRCA2 association with FokI foci upon CTCF knockdown is largely rescued through reexpression of full-length CTCF (Fig. 6, A to C) or, likewise, a mutant lacking the C-terminal domain (expression of reconstituted constructs is shown in fig. S6A). This indicates that the loss of BRCA2 upon CTCF knockdown is directly dependent on depletion of CTCF and not off-target effects of the shRNA. In contrast, neither add-back of a CTCF construct with a deletion of the zinc finger domain, which is unable to bind the FokI cut site (Fig. 1, C and D), nor a CTCF PARylation–defective mutant (CTCFPARMUT) (36), which is recruited to the FokI cut site, is capable of rescuing BRCA2 recruitment to FokI-induced DSB (Fig. 6, C and D). From these results, we conclude that CTCF acts downstream of BRCA1 in the HR pathway, likely impairing the recruitment of BRCA2 and the loading of RAD51 to DSBs.
Fig. 6. CTCF association with BRCA2 is PARylation-dependent.
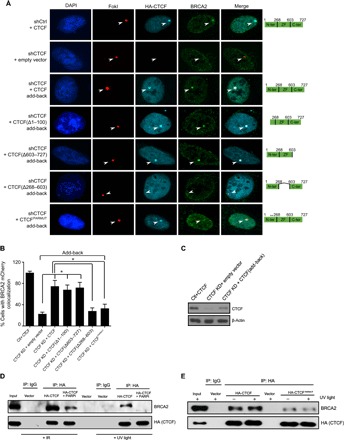
(A) U2OS-LacI-FokI-mCherry DSB reporter cells with CTCF knockdown and reconstituted, full-length CTCF, CTCF deletions, or CTCFPARMUT (defective for PARylation) were probed for BRCA2 colocalization with FokI foci. (B) Histogram representing quantification of colocalization events between mCherry-FokI and BRCA2 for experiments depicted in (A). Error bars correspond to means ± SEM (n = 3; *P ≤ 0.05, two-tailed Student’s t test). (C) Western blot showing reconstitution of full-length CTCF after knockdown with an shRNA targeting the 3′ untranslated region of CTCF. (D) Co-IP showing HA-CTCF interaction with endogenous BRCA2 ± 24 hours of exposure to 5 μM of the PARP-1 inhibitor MK4827 followed by 5-Gy γ-irradiation or light treatment (100 J/m2). Lysates were collected 1 hour after damage. (E) Co-IP showing HA-CTCF or HA-CTCFPARMUT, with endogenous BRCA2 subjected to treatment with UV (100 J/m2).
The capacity of CTCF to coordinate BRCA2 recruitment to DSB, as well as the inability of the CTCFPARMUT construct to rescue BRCA2 binding, suggests an association between BRCA2 and CTCF. Coimmunoprecipitation (Co-IP) experiments reveal an interaction between CTCF and endogenous BRCA2 after exposure to multiple DNA-damaging agents (Fig. 6D and fig. S6, B to D). This association is disrupted by the PARP inhibitor MK4827 (Fig. 6D), suggesting that the interaction between CTCF and BRCA2 might be dependent on CTCF PARylation. Supporting this concept, we find that CTCFPARMUT, unlike the WT counterpart, is unable to strongly associate with BRCA2 (Fig. 6E). Thus, we propose that the principal role of CTCF at sites of DSB is to promote the recruitment of the critical HR factor BRCA2 following CTCF posttranslational modification by PARylation.
DISCUSSION
CTCF is a ubiquitous zinc finger–containing protein involved in several biological processes, including gene activation, insulator activity, and chromatin organization. Previous reports suggest that the multifunctional nuclear protein CTCF might also play a role in the repair of DSB. For example, CTCF heterozygous mice show increased tumorigenesis in response to irradiation (21), and a screen for zinc finger proteins recruited to laser micro-irradiation tracks revealed that CTCF is recruited to these breakages (17). However, it remained unclear whether CTCF plays a functional role at DSB. Here, for the first time, we provide evidence that CTCF acts as an important component of the HR response to DNA damage. In particular, laser micro-irradiation data show that CTCF is recruited within seconds after the appearance of DNA lesions. This association is not transient in nature, as is seen with many chromatin remodelers, such as ALC1 and CHD4 (37, 38), but instead persists for an extended period, with the immobilization of CTCF readily apparent at 60 min after damage. This argues in favor of a functional role for CTCF at sites of DNA damage, rather than recruitment due to the artificial opening and high accessibility of chromatin at laser tracks (30). Our data in the mCherry-LacI-FokI reporter system further confirm that CTCF is mainly recruited to DSBs, although we cannot rule out that SSBs may also favor the mobilization of CTCF to DNA damage. CTCF has a variable consensus sequence of approximately 13 to 15 nucleotides (39), with the core AG G/A G/T GG sequence being highly conserved. It is possible that CTCF is recruited to laser tracks in a sequence-specific manner, with CTCF only being recruited to those DSB appearing proximal to an already existing CTCF binding site. However, the U2OS-LacI system recruits a FokI-LacI repressor fusion protein to 256 integrated Lac operators that does not contain a sequence resembling a CTCF consensus (40). Thus, it is highly unlikely that CTCF is recruited here in a sequence-dependent manner. We know that CTCF is recruited to DSB through its zinc finger domain, but further work is required to decipher whether this targeting is through direct recognition of DSB, through interaction with partner proteins involved in repair, or possibly through the recognition of epigenetic modifications surrounding the breaks. Previous studies indicate that CTCF associates with proteins through its zinc finger domain (41, 42), and we propose that this is the most likely mechanism of CTCF recruitment to DSB.
Although functionally not fully understood, there is much evidence suggesting that dynamic recruitment of PARP-1 to sites of DSB facilitates repair through the HR pathway (12, 43–45). Evidence indicates that following its initial recruitment to DSB, PARP-1 is retained at the break site, allowing the local accumulation of PAR polymers. PARP-1 auto-PARylation and PARylation of target proteins may act as a scaffold to recruit and retain a number of key repair proteins including MRE11 and BRCA2 (9, 29, 46). We see that CTCF modification by PARylation is readily apparent shortly after exposure to DNA-damaging agents. However, the N-terminal domain of CTCF, where the motifs responsible for PARylation are found, is not found to play an important role in its recruitment to DSB. In contrast, the constructs we tested for recruitment to FokI foci harboring the DNA binding zinc finger domain of CTCF aptly colocalized with sites of DSB. Pharmacological inhibition of PARP-1 activity using small-molecule inhibitors also had no noticeable impact on CTCF recruitment. Coupled together, these data strongly suggest that CTCF is directly recruited to DSB through its zinc finger domain, independently of an association with PAR polymers. We previously found that CTCF and PARP-1 biochemically copurify on a DNA resin (22). This complex was distinct from a CTCF complex containing TFII-I and nucleophosmin. We propose that CTCF is organized into multiple complexes and that the complex involved in repairing DSB has a composition unique from the one tethering CTCF to the nucleolus, containing nucleophosmin. Recent reports indicate a growing list of zinc finger proteins to be recruited to DSB (17). However, in contrast to many of the factors within this class of proteins, the recruitment of CTCF to DSB does not depend on PARP-1 enzymatic activity. CTCF is also retained at the break site for an extended period of time, distinct from the transient nature of many other zinc finger proteins (17). Transient recruitment to DSB is commonly observed for chromatin remodelers as well (37, 38, 47). Although CTCF does have the capacity to position nucleosomes, its prolonged accumulation at DSB suggests a more probable role as a scaffold protein, because it has no recognized enzymatic activity.
Functionally, we provide several pieces of evidence supporting a role for CTCF in homology-directed repair. First, CTCF knockdown does not impair the recruitment of 53BP1 or LIGIV to DSB, or the resolution of 53BP1 foci, suggesting a functional NHEJ pathway in CTCF-depleted cells. However, CTCF plays a role in the repair of DSB, as evidenced by the delayed resolution of γH2AX foci in response to irradiation, indicating an NHEJ-independent mechanism of repair. Second, CTCF promotes HR following Cas9 and I–Sce I nuclease–induced DSB, with its loss leading to a greater than 2.5-fold reduction in the fluorescent population for both the CRISPR-mClover and DR-GFP assays, similar to what is observed with RAD51 knockdown cells. The integrity of the G2-M checkpoint, necessary for efficient HR, is dependent on the functionality of proteins in the HR pathway including BRCA2 (48). Thus, a third indication for a role of CTCF in HR is demonstrated by a loss of CTCF closely recapitulating the phenotype of BRCA2 knockdown, where cells aberrantly progress through G2 to mitosis after exposure to irradiation. Fourth, PARP inhibitors demonstrate greatly enhanced cytotoxicity against cells with defects in the HR pathway, first observed against cells harboring BRCA2 mutations (49). We find the same phenotype in CTCF knockdown cells in response to two clinically relevant inhibitors of PARP activity, olaparib and MK4827. The fifth piece of evidence supporting a functional role of CTCF in this pathway is perhaps the most surprising and lends a key insight into the mechanism whereby CTCF modulates HR. We find that CTCF coimmunoprecipitates with BRCA2 in a PARylation-dependent manner, following the induction of DNA damage. Upon CTCF knockdown, BRCA2 recruitment to DSB is greatly compromised, but is rescued through add-back of full-length CTCF, but not CTCF mutants, either devoid of its zinc finger domain, or a PARylation-defective mutant. The capacity of CTCF add-back to restore BRCA2 immobilization at FokI cut sites indicates that the loss is not due to off-target effects of shRNAs or irreversible epigenetic events that would occlude access to DSB. CTCF knockdown had little observable effect on the accumulation of BRCA1 at repair foci, indicating that CTCF acts downstream of BRCA1 in the HR. The association of RAD51 with DSB was also compromised in CTCF knockdown cells, consistent with the loading of RAD51 onto chromatin being primarily dependent on BRCA2 (50). There is evidence that additional proteins, including TOPBP1 and PALB2, also act to enhance the accumulation of genomic RAD51 at DSB (51, 52) and that the nuclear localization of RAD51 may be influenced by the activity of proteins such as IRS1 (53). Differences in the expression level and activity of these proteins among tissue types may explain why more severe defects in RAD51 aggregation at repair foci were observed in U2OS cells than in MCF7 cells. It has been previously reported that BRCA2 recruitment to DSB is dependent on PARP-1 activity (9), a result we repeated here. Although CTCF recruitment to these breaks is not dependent on PARylation, CTCF itself is highly PARylated in response to DNA damage. On the basis of our data, we propose a model where PARylated CTCF acts as a scaffold, participating in the recruitment and stabilization of BRCA2 at sites of repair, and perhaps other proteins additionally, thereby facilitating efficient resolution of the break.
BRCA2 knockdown cells show limited sensitivity to the replication stress–inducing agent hydroxyurea (54). Although DSBs stemming from collapsed replication forks are repaired primarily through HR, the principal role of BRCA2 at replication forks is to stabilize the fork and prevent degradation (55). On the basis of the sensitivity of CTCF knockdown cells to hydroxyurea, we propose that CTCF may play a role beyond recruitment of BRCA2 at stalled replication forks that have degenerated into DSB. PARP-1 activity has been shown to both stabilize replication forks and recruit repair proteins, such as MRE11, XRCC1, and Chk1, to damaged replication forks (56, 57). It is possible that, in this context, PARylated CTCF also acts as an accessory to PARP-1, providing a scaffold to recruit repair proteins. Hence, we propose that CTCF plays a multifaceted role in the response to DNA damage, possibly coordinating both the transcriptional and repair response to damage-inducing agents. For example, it has been previously demonstrated that CTCF plays an essential role in potentiating the transcriptional activation of TP53 in response to the DNA-damaging agent methyl methanesulfonate (58). We also postulate another role for CTCF beyond what we have characterized here. Elegant genome-wide studies reveal a role for CTCF in mediating long-range chromatin contacts (59). It is possible that CTCF establishes new intra-chromosomal links between existing CTCF binding sites and DSB. We envision that these connections may generate chromatin loops, which might not only facilitate the recruitment of repair factors but also additionally define a domain within which epigenetic changes associated with repair processes are contained. Studies to interrogate a potential role for CTCF in mediating these activities are ongoing.
CTCF is found hypo-PARylated in breast tumors, and the loss of this posttranslational modification has been associated with tumor progression (24). Our findings suggest that tumor cells may evolve to lose CTCF PARylation, thereby providing tumors with a means to promote genomic instability through compromising HR. This instability could potentially provide a stochastic mechanism through which cells might gain survival and proliferative advantages.
MATERIALS AND METHODS
Cell culture, viral knockdown, and CRISPR/Cas9
The parental U2OS cell line was maintained in McCoy’s medium supplemented with 10% fetal bovine serum (FBS). The U2OS-LacI-FokI-mCherry DSB reporter cell line (27, 60), U2OS DR-GFP HR reporter cell line (34), human embryonic kidney (HEK)–293 T cells, and MCF7 cells were all maintained routinely in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS. The U2OS-LacI-FokI-mCherry cell line was provided by R. Greenberg (University of Pennsylvania). For induction of FokI expression, cells were treated with 1 μM Shield-1 and 4-hydroxytamoxifen for 6 hours. CTCF and RAD51 knockdown experiments were done using lentiviral shRNA vectors (Sigma) packaged in HEK293T cells, as described previously (22). Briefly, HEK293T cells were transfected with 7 μg of shRNA lentiviral vectors combined with 5 μg of packaging vector MD2G and 2 μg of envelope vector Pax2 using polyethylenimine (1 mg/ml). Viruses were collected at 72 hours after transfection and passed through a 0.45-μm filter. For cell infection, MCF7 and U2OS cells were seeded at 0.3 × 106 and 0.1 × 106 cells/ml, respectively, in six-well dishes and incubated with 300 μl of viral supernatants along with 8 μg of hexadimethrine bromide (Polybrene). Seventy-two hours after infection, cells were subjected to downstream applications. Fresh infections were carried out for each experiment.
For CTCF knockout by CRISPR/Cas9, a U6 promoter and target single-guide RNA (sgRNA) were first cloned into pIDT-SMART (table S1). All constructs were confirmed by sequencing. MCF10A cells were grown in DMEM F12 supplemented with 5% horse serum, epidermal growth factor (10 μg/500 ml), hydrocortisone (250 μg/500 ml), and cholera toxin (50 μg/500 ml). MCF10A cells were cultured to 50 to 60% confluency in six-well plates followed by transfection of 6 μg of total plasmid DNA, including 2 μg of pCas-Guide-ef1a-GFP (OriGene, catalog no. GE100018) and 4 μg of plasmid with sgRNA (2 μg each) using Lipofectamine 3000 (Invitrogen). Two days later, GFP-positive cells were isolated by fluorescence-activated cell sorting into 96-well plates (one cell per well). To screen for heterozygous CTCF cell clones (CTCF+/−), we isolated genomic DNA of each clone and amplified proximal sequences surrounding the Cas9 targets by polymerase chain reaction. Positive clones were first identified using the SURVEYOR Assay Kit (IDT, catalog no. 706020) followed by sequencing. CTCF+/− cells used in this study harbor one WT CTCF allele and one allele with a frameshift mutation, leading to a premature stop codon.
Antibodies and chemicals
A mouse monoclonal antibody recognizing CTCF for Western blotting was from BD Biosciences (612149). Secondary antibodies for Western blot analysis, horseradish peroxidase (HRP)–labeled goat anti-rabbit and HRP-labeled goat anti-mouse, were purchased from KPL (catalog nos. 474-1516 and 474-1806). Mouse monoclonal phospho-Ser139 histone H2AX, PAR, mouse monoclonal antibody recognizing BRCA2, and rabbit polyclonal phospho-Ser10 histone 3 were purchased from Millipore (catalog nos. 05-636, MAB3192, 05-666, and 05-570). Rabbit polyclonal 53BP1 antibodies were purchased from Novus (100-304) and Santa Cruz Biotechnology (sc-22760). Antibodies recognizing Rad51 and nucleophosmin were bought from Santa Cruz Biotechnology (catalog nos. sc-8349 and sc-47725). A mouse monoclonal antibody toward β-actin was purchased from Sigma (A5316). Rabbit polyclonal BRCA1 antibody was purchased from Millipore (07-434). Rabbit polyclonal anti-PARP1 antibody was obtained from Cell Signaling (9542S). Mouse monoclonal antibody recognizing the HA tag was bought from Abcam (AB1424). Secondary antibodies for immunofluorescence, Alexa Fluor 488 goat anti-mouse, Alexa Fluor 594 goat anti-rabbit, Alexa Fluor 680 goat anti-rabbit, and Alexa Fluor 488 goat anti-rabbit were all purchased from Life Technologies (catalog nos. A11029, A11072, A21109, and A11034). Shield-1 was purchased from Clontech (631037). The PARP-1 inhibitor MK4827 was supplied by AdooQ Bioscience (A11026), and olaparib was purchased from Cayman Chemicals (10621-100). Hydroxyurea and 4-hydroxytamoxifen were purchased from Sigma (catalog nos. h8727 and 68047-06-3). Doxorubicin was obtained from Pfizer (DIN 02071002). Paclitaxel was obtained from Biolyse Pharma Corporation (DIN 02244372). GeneJuice transfection reagent was obtained from Millipore (70967). BrdU (5-bromo-2′-deoxyuridine) was purchased from BD Biosciences (347580). DAPI (4′,6-diamidino-2-phenylindole) was purchased from Thermo Fisher (D1306)
Plasmids
HA-tagged CTCF fragments with corresponding deletions of amino acids Δ1–100, Δ603–727, Δ240–727, Δ268–603, and 268–603 were cloned between Eco RI and Xba I restriction sites in PLKO.1 cytomegalovirus multiple cloning site (PLKO.1 CMV MCS, provided by M. Fabian, McGill University). The CTCF PARylation–defective mutant CTCFPARMUT, which has PARylation acceptor sites E233 and E239 mutated to alanine, as previously described (36), was cloned into the Eco RI site of the lentiviral expression vector PMDK208 (61). Plasmids encoding WT and mutant FokI nuclease, HFUW-FokI WT or HFUW-FokI D450A, were provided by R. Greenberg (University of Pennsylvania). Plasmid encoding the nuclease I–Sce I or empty vector, PCAG I–Sce I, and PCAG were provided by S. Richard (McGill University). For knockdown studies, scrambled shRNA (SCH016) or shRNAs targeting CTCF (TRCN0000014548 and TRCN0000014548) or RAD51 (TRCN0000018877) were used (Sigma). For CTCF add-back experiments, an shRNA targeting the 3′ untranslated region of CTCF was used for knockdown (TRCN0000218498; Sigma). All shRNAs are held in the lentiviral vector PLKO.1.
Immunofluorescence, laser micro-irradiation, and live-cell imaging
Immunofluorescence was carried out similarly as described previously (62). All steps were carried out at room temperature. Cells grown on coverslips were fixed in freshly prepared 2% paraformaldehyde for 10 min. Fixed cells were then incubated for 10 min with a combination of permeabilization/blocking buffer [0.3% Triton X-100, 1% bovine serum albumin (BSA), and 1% normal goat serum]. Next, primary antibodies were added for 1 hour in phosphate-buffered saline (PBS) + 1% BSA followed by three washes, each of 5-min duration, with PBS + 1% BSA. Secondary antibody was next added in the same buffer for a period of 1 hour. Nuclei were stained with DAPI (1 μg/ml) for 5 min and subjected to a final wash with 1% BSA in PBS. After this, coverslips were mounted onto glass slides using a ProLong gold antifade reagent (Life Technologies). Images were acquired using a Leica Widefield DM LB2 microscope. Images were analyzed and quantified using ImageJ software [National Institutes of Health (NIH)]. For laser micro-irradiation experiments, cells were cultured in an eight-chamber slides and presensitized with 10 μM BrdU for 24 hours. After washing with PBS once, the culture medium was replaced by phenol red–free DMEM supplemented with 10% FBS, Hoechst 33342 (10 μg/ml), and 10 μM BrdU for 30 min. Finally, this mix was removed and substituted with phenol red–free DMEM (10% FBS). A Zeiss LSM 700 inverted laser scanning confocal microscope equipped with a heated live-cell chamber and a 20× 0.85 numerical aperture objective lens was used for all laser micro-irradiation experiments. The nuclei were irradiated with a 5-mW, 405-nm diode laser set at 50% power, scanning for 20 iterations at a speed of 100 μs/pixel using the zoom bleach function on ZEN software. For staining, cells were subsequently fixed in 2% paraformaldehyde for 15 min and then incubated for 30 min in a permeabilization/blocking buffer (see above). The primary antibodies were added in a PBS + 1% BSA solution overnight at 4°C and then washed three times in PBS + 1% BSA. Secondary antibody was next added in the same buffer for 1 hour at room temperature, followed by three washes with PBS + 1% BSA. Images were acquired as described above. For live-cell imaging, photos were acquired before bleaching, during the bleach, and at 10 s, 30 min, 60 min, and 120 min after the laser micro-irradiation. The fluorescence data were analyzed during the picture acquisition with the ZEN pro software.
Flow cytometry
For flow cytometry–mediated analysis of phospho-Ser10 of histone 3 levels, MCF7 cells were first fixed in ice-cold 70% ethanol. Subsequent steps were carried out at room temperature. Cells were permeabilized using dilution buffer (1% FBS and 0.1% Triton X-100 in PBS), after which primary antibody against phospho-Ser10 of histone 3 was added for 1 hour. Cells were collected at 400 g and washed with dilution buffer. Next, the cells were incubated with an anti-rabbit Alexa Fluor 488 antibody for 1 hour. Again, cells were collected by centrifugation and washed using dilution buffer. Staining was detected with a FACSCalibur platform from BD Biosciences. Data were analyzed using FlowJo 7.6.5. For the mClover assay, MCF10A WT CTCF+/− and HA-CTCF complemented cells were trypsinized, washed with PBS, and electroporated with 1.25 μg of sgRNA plasmid and 1.25 μg of donor template using the 4D-Nucleofector System (Lonza). Cells were subsequently plated in medium and grown for 72 hours before trypsinization and resuspension in PBS. The percentage of mClover-positive cells were determined by flow cytometry, as described above. For the DR-GFP HR assay, U2OS DR-GFP cells were infected with the indicated lentiviral particles in six-well cell culture plates. Forty-eight hours after infection, cells were transfected with empty vector or I–Sce I plasmids for another 48 or 72 hours. Cells were trypsinized, washed in PBS, collected, and resuspended in PBS. The percentage of GFP-positive cells were determined by flow cytometry, as described above.
Western blotting
Western blot analysis was carried out as described previously (63, 64). Cells were washed twice in PBS and lysed in whole-cell lysis buffer [20 mM tris (pH 7.5), 420 mM NaCl, 2 mM MgCl2, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.5% Triton X-100] supplemented with fresh 1 mM dithiothreitol, phenylmethylsulfonyl fluoride, protease inhibitor cocktail (Roche) and phosphatase inhibitors, bis-glycerol phosphate, and NaF. Two volumes of the whole-cell lysis buffer were added to the centrifuged cell pellet and left on ice for 25 min. Lysates were centrifuged at 12,000 rpm at 4°C for 25 min, and supernatants were collected and transferred to a new tube. Protein concentration was measured by the Bradford assay. Protein (20 μg) was loaded onto an 8% acrylamide gel and electrophoresed at 150 V. Transfer of the protein to a nitrocellulose membrane was done overnight at 30 V at 4°C, followed by 30 min at 70 V. Membrane was blocked with 5% nonfat milk in TBST (tris-buffered saline–Tween 20) and incubated with primary antibody overnight at 4°C. Membranes were next washed three times with TBST (20 mM tris base, 137 mM NaCl, and 0.1% Tween 20) for 5, 10, and 15 min. Next, secondary antibodies were added for 1 hour at room temperature in blocking buffer. Western blotting was revealed using a Clarity Western Enhanced Chemiluminescence kit (Bio-Rad) and autoradiography film (Harvard Apparatus).
Coimmunoprecipitation
Co-IP was carried out as described previously (22, 64). Whole-cell lysates were prepared as described above and diluted to 1-ml total volume in IP buffer [20 mM tris (pH 7.5), 100 mM NaCl, 10 mM MgCl2, 2 mM EDTA, 0.5% Triton X-100]. The lysate solutions were then precleared for 2 hours with protein G–agarose beads. After the preclearing stage, beads were pelleted and the supernatant was collected and transferred to a new tube, where capturing antibodies were added and nutated overnight at 4°C. Antibodies with bound proteins were collected by adding fresh protein G–agarose beads and nutated at 4°C for 2 hours. Beads were pelleted, and the supernatant was discarded. The collected beads were washed three times with 1 ml of IP buffer and centrifuged at 1700g to pellet the beads. The final wash was done with IP buffer containing 0.1% Triton X-100 and spun down at 2700g. Proteins were eluted from the beads by adding 25 μl of 2× SDS loading buffer [40 mM tris (pH 6.8), 4% SDS, 20% glycerol, and bromophenol blue] and heated at 100°C for 10 min. Beads were again pelleted, and the resulting supernatant was loaded to acrylamide gel and blotted as described above. Where appropriate, HA-CTCF–transfected HEK293T cells were treated with 5 μM MK4827 for 24 hours before induction of DNA damage.
Cell irradiation
Cells were exposed to γ-irradiation using a clinical linear accelerator (Clinac 21EX, Varian Medical Systems). Cells were placed on a 20-cm stack of solid water (Gammex Inc.), and an additional 5 cm of solid water was placed on top of the holder to provide buildup material. Up to four six-well plates, or three 15-cm petri dishes, could be irradiated at one time. Cells were irradiated using 18–million volt photons. The dose of irradiation to the cells was calculated on the basis of ion chamber measurements and clinical dosimetry data. The dose rate was held constant at approximately 600 cGy/min.
Clonogenic assay
Clonogenic assays were performed similar to those described previously (65, 66). Cells were seeded into six-well plates, in triplicate, and treated with drugs within 16 to 18 hours after plating with the indicated doses. For γ-irradiation, cells were exposed to irradiation before plating. Cells were allowed to grow for 14 days, and the resulting colonies were fixed with 1.5 ml of 6.0% glutaraldehyde and 0.5% crystal violet. Colonies were counted using a GelCount (Optronix) gel quantification system. The surviving fraction (SF) of cells was calculated as follows. First, plating efficiency (PE) was calculated: PE = (number of colonies of untreated or treated cells/number of seeded cells) × 100%. Next, SF was calculated: SF = (number of colonies after treatment)/(number of cells seeded × PE).
Comet assay
Neutral comet assays were carried out as previously described (67). Briefly, MCF7 cells were harvested and embedded in 0.5% low-melting agarose onto Trevigen comet slides (catalog no. 4250-200-03). Samples were then incubated in neutral lysing solution [2% sarkosyl, 0.5 M Na2EDTA, proteinase K (0.5 mg/ml) (pH 8.0)] overnight at 37°C. After overnight lysis, slides were rinsed twice for 30 min each in neutral rinsing buffer [90 mM tris, 90 mM boric acid, 2 mM Na2EDTA (pH 8.5)]. Slides were next subjected to electrophoresis in fresh rinsing buffer for 15 min at 20 V. After electrophoresis, slides were washed in distilled water for 5 min and immersed in 70% ethanol for 5 min to fix. DNA was subsequently stained using the SYBR Gold Nucleic Acid Gel Stain (Fisher, catalog no. S11494). Finally, slides were washed with distilled water for 5 min. Comet tails were quantified using ImageJ software (NIH).
Supplementary Material
Acknowledgments
We thank D. Durocher for insights and stimulating discussions. Funding: This work was supported in part by grants from the Canadian Institute of Health Research, Fonds de recherche Québec—Santé, and the Quebec Breast Cancer Foundation (to M.W. and L.M.). K.H. is supported by a McGill Integrated Cancer Research Training Program. M.M. is supported by a Fonds de recherche Québec—Santé postdoctoral award. A.O., S.R., and M.A.-J. are funded by Canadian Institute of Health Research. A.O. acknowledges support from a Cancer Research Society Operating Grant (2016 to 2018 #21038). Author contributions: K.H., S.R., L.M., A.O., and M.W. conceived and designed the experiments. K.H., M.J., M.M., T.Z., A. Saad, L.M., V.M.L., A. Syme, C.Z., C.R., Z.Y., M.R.F., and A.K. carried out experiments. K.H., M.J., M.M., M.A.-J., S.R., A.O., Z.Y., and M.W. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1601898/DC1
fig. S1. Live-cell imaging of CTCF at laser micro-irradiation tracks.
fig. S2. CTCF association with PARylation increases as a response to DNA-damaging agents.
fig. S3. Impact of CTCF loss on γH2AX and 53BP1 foci resolution.
fig. S4. Loss of CTCF increases sensitivity to PARP inhibitors.
fig. S5. Loss of CTCF impairs Rad51 foci formation following infrared.
fig. S6. DNA damage increases the association between CTCF and BRCA2.
table S1. sgRNA sequences targeting Cas9 to CTCF.
REFERENCES AND NOTES
- 1.Ceccaldi R., Rondinelli B., D’Andrea A. D., Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26, 52–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciccia A., Elledge S. J., The DNA damage response: Making it safe to play with knives. Mol. Cell 40, 179–204 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Escribano-Díaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J. T. F., Tkáč J., Cook M. A., Rosebrock A. P., Munro M., Canny M. D., Xu D., Durocher D., A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Sartori A. A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S. P., Human CtIP promotes DNA end resection. Nature 450, 509–514 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eid W., Steger M., El-Shemerly M., Ferretti L. P., Peña-Diaz J., König C., Valtorta E., Sartori A. A., Ferrari S., DNA end resection by CtIP and exonuclease 1 prevents genomic instability. EMBO Rep. 11, 962–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maréchal A., Zou L., RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 25, 9–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao W., Vaithiyalingam S., San Filippo J., Maranon D. G., Jimenez-Sainz J., Fontenay G. V., Kwon Y., Leung S. G., Lu L., Jensen R. B., Chazin W. J., Wiese C., Sung P., Promotion of BRCA2-dependent homologous recombination by DSS1 via RPA targeting and DNA mimicry. Mol. Cell 59, 176–187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia B., Sheng Q., Nakanishi K., Ohashi A., Wu J., Christ N., Liu X., Jasin M., Couch F. J., Livingston D. M., Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 22, 719–729 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Zhang F., Shi J., Bian C., Yu X., Poly(ADP-ribose) mediates the BRCA2-dependent early DNA damage response. Cell Rep. 13, 678–689 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel A. G., Sarkaria J. N., Kaufmann S. H., Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. U.S.A. 108, 3406–3411 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu Y., Petit S. A., Ficarro S. B., Toomire K. J., Xie A., Lim E., Cao S. A., Park E., Eck M. J., Scully R., Brown M., Marto J. A., Livingston D. M., PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination–mediated DNA repair. Cancer Discov. 4, 1430–1447 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie S., Mortusewicz O., Ma H. T., Herr P., Poon R. Y. C., Helleday T., Qian C., Timeless interacts with PARP-1 to promote homologous recombination repair. Mol. Cell 60, 163–176 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Krishnakumar R., Kraus W. L., The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 39, 8–24 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luijsterburg M. S., de Krijger I., Wiegant W. W., Shah R. G., Smeenk G., de Groot A. J. L., Pines A., Vertegaal A. C. O., Jacobs J. J. L., Shah G. M., van Attikum H., PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol. Cell 61, 547–562 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang H., Jeffrey P. D., Miller J., Kinnucan E., Sun Y., Thomä N. H., Zheng N., Chen P.-L., Lee W.-H., Pavletich N. P., BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 297, 1837–1848 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Li M., Yu X., Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 23, 693–704 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Izhar L., Adamson B., Ciccia A., Lewis J., Pontano-Vaites L., Leng Y., Liang A. C., Westbrook T. F., Harper J. W., Elledge S. J., A systematic analysis of factors localized to damaged chromatin reveals PARP-dependent recruitment of transcription factors. Cell Rep. 11, 1486–1500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filippova G. N., Fagerlie S., Klenova E. M., Myers C., Dehner Y., Goodwin G., Neiman P. E., Collins S. J., Lobanenkov V. V., An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell. Biol. 16, 2802–2813 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valadez-Graham V., Razin S. V., Recillas-Targa F., CTCF-dependent enhancer blockers at the upstream region of the chicken α-globin gene domain. Nucleic Acids Res. 32, 1354–1362 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Recillas-Targa F., de la Rosa-Velázquez I. A., Soto-Reyes E., Insulation of tumor suppressor genes by the nuclear factor CTCF. Biochem. Cell Biol. 89, 479–488 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Kemp C. J., Moore J. M., Moser R., Bernard B., Teater M., Smith L. E., Rabaia N. A., Gurley K. E., Guinney J., Busch S. E., Shaknovich R., Lobanenkov V. V., Liggitt D., Shmulevich I., Melnick A., Filippova G. N., CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell Rep. 7, 1020–1029 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peña-Hernández R., Marques M., Hilmi K., Zhao T., Saad A., Alaoui-Jamali M. A., del Rincon S. V., Ashworth T., Roy A. L., Emerson B. M., Witcher M., Genome-wide targeting of the epigenetic regulatory protein CTCF to gene promoters by the transcription factor TFII-I. Proc. Natl. Acad. Sci. U.S.A. 112, E677–E686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y., Wang J., Ding M., Yu Y., Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat. Methods 10, 981–984 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Docquier F., Kita G.-X., Farrar D., Jat P., O’Hare M., Chernukhin I., Gretton S., Mandal A., Alldridge L., Klenova E., Decreased poly(ADP-ribosyl)ation of CTCF, a transcription factor, is associated with breast cancer phenotype and cell proliferation. Clin. Cancer Res. 15, 5762–5771 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Witcher M., Emerson B. M., Epigenetic silencing of the p16INK4a tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol. Cell 34, 271–284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ong C.-T., Corces V. G., CTCF: An architectural protein bridging genome topology and function. Nat. Rev. Genet. 15, 234–246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shanbhag N. M., Rafalska-Metcalf I. U., Balane-Bolivar C., Janicki S. M., Greenberg R. A., ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang J., Cho N. W., Cui G., Manion E. M., Shanbhag N. M., Botuyan M. V., Mer G., Greenberg R. A., Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 20, 317–325 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haince J. F., McDonald D., Rodrigue A., Déry U., Masson J.-Y., Hendzel M. J., Poirier G. G., PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 283, 1197–1208 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Strickfaden H., McDonald D., Kruhlak M. J., Haince J.-F., Th’ng J. P. H., Rouleau M., Ishibashi T., Corry G. N., Ausio J., Underhill D. A., Poirier G. G., Hendzel M. J., Poly(ADP-ribosyl)ation-dependent transient chromatin decondensation and histone displacement following laser microirradiation. J. Biol. Chem. 291, 1789–1802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu S., Wu Y., Chen Q., Cao J., Hu K., Tang J., Sang Y., Lai F., Wang L., Zhang R., Li S.-P., Zeng Y.-X., Yin Y., Kang T., hSSB1 regulates both the stability and the transcriptional activity of p53. Cell Res. 23, 423–435 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orthwein A., Noordermeer S. M., Wilson M. D., Landry S., Enchev R. I., Sherker A., Munro M., Pinder J., Salsman J., Dellaire G., Xia B., Peter M., Durocher D., A mechanism for the suppression of homologous recombination in G1 cells. Nature 528, 422–426 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinder J., Salsman J., Dellaire G., Nuclear domain ’knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 43, 9379–9392 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakanishi K., Cavallo F., Brunet E., Jasin M., Homologous recombination assay for interstrand cross-link repair. Methods Mol. Biol. 745, 283–291 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jensen R. B., Carreira A., Kowalczykowski S. C., Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467, 678–683 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farrar D., Rai S., Chernukhin I., Jagodic M., Ito Y., Yammine S., Ohlsson R., Murrell A., Klenova E., Mutational analysis of the poly(ADP-ribosyl)ation sites of the transcription factor CTCF provides an insight into the mechanism of its regulation by poly(ADP-ribosyl)ation. Mol. Cell. Biol. 30, 1199–1216 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahel D., Hořejší Z., Wiechens N., Polo S. E., Garcia-Wilson E., Ahel I., Flynn H., Skehel M., West S. C., Jackson S. P., Owen-Hughes T., Boulton S. J., Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325, 1240–1243 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Polo S. E., Kaidi A., Baskcomb L., Galanty Y., Jackson S. P., Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 29, 3130–3139 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plasschaert R. N., Vigneau S., Tempera I., Gupta R., Maksimoska J., Everett L., Davuluri R., Mamorstein R., Lieberman P. M., Schultz D., Hannenhalli S., Bartolomei M. S., CTCF binding site sequence differences are associated with unique regulatory and functional trends during embryonic stem cell differentiation. Nucleic Acids Res. 42, 774–789 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bell C. E., Barry J., Matthews K. S., Lewis M., Structure of a variant of lac repressor with increased thermostability and decreased affinity for operator. J. Mol. Biol. 313, 99–109 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Yao H., Brick K., Evrard Y., Xiao T., Camerini-Otero R. D., Felsenfeld G., Mediation of CTCF transcriptional insulation by DEAD-box RNA-binding protein p68 and steroid receptor RNA activator SRA. Genes Dev. 24, 2543–2555 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chernukhin I. V., Shamsuddin S., Robinson A. F., Carne A. F., Paul A., El-Kady A. I., Lobanenkov V. V., Klenova E. M., Physical and functional interaction between two pluripotent proteins, the Y-box DNA/RNA-binding factor, YB-1, and the multivalent zinc finger factor, CTCF. J. Biol. Chem. 275, 29915–29921 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Beck C., Robert I., Reina-San-Martin B., Schreiber V., Dantzer F., Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 329, 18–25 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Schultz N., Lopez E., Saleh-Gohari N., Helleday T., Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 31, 4959–4964 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krietsch J., Caron M.-C., Gagné J.-P., Ethier C., Vignard J., Vincent M., Rouleau M., Hendzel M. J., Poirier G. G., Masson J.-Y., PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 40, 10287–10301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalisch T., Amé J.-C., Dantzer F., Schreiber V., New readers and interpretations of poly(ADP-ribosyl)ation. Trends Biochem. Sci. 37, 381–390 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ha K., Lee G. E., Palii S. S., Brown K. D., Takeda Y., Liu K., Bhalla K. N., Robertson K. D., Rapid and transient recruitment of DNMT1 to DNA double-strand breaks is mediated by its interaction with multiple components of the DNA damage response machinery. Hum. Mol. Genet. 20, 126–140 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menzel T., Nähse-Kumpf V., Kousholt A. N., Klein D. K., Lund-Andersen C., Lees M., Johansen J. V., Syljuåsen R. G., Sørensen C. S., A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO Rep. 12, 705–712 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bryant H. E., Schultz N., Thomas H. D., Parker K. M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N. J., Helleday T., Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Tarsounas M., Davies D., West S. C., BRCA2-dependent and independent formation of RAD51 nuclear foci. Oncogene 22, 1115–1123 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Moudry P., Watanabe K., Wolanin K. M., Bartkova J., Wassing I. E., Watanabe S., Strauss R., Troelsgaard Pedersen R., Oestergaard V. H., Lisby M., Andújar-Sánchez M., Maya-Mendoza A., Esashi F., Lukas J., Bartek J., TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J. Cell Biol. 212, 281–288 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dray E., Etchin J., Wiese C., Saro D., Williams G. J., Hammel M., Yu X., Galkin V. E., Liu D., Tsai M.-S., Sy S. M.-H., Schild D., Egelman E., Chen J., Sung P., Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat. Struct. Mol. Biol. 17, 1255–1259 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trojanek J., Ho T., Del Valle L., Nowicki M., Wang J. Y., Lassak A., Peruzzi F., Khalili K., Skorski T., Reiss K., Role of the insulin-like growth factor I/insulin receptor substrate 1 axis in Rad51 trafficking and DNA repair by homologous recombination. Mol. Cell. Biol. 23, 7510–7524 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen X., Bosques L., Sung P., Kupfer G. M., A novel role for non-ubiquitinated FANCD2 in response to hydroxyurea-induced DNA damage. Oncogene 35, 22–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M., Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ying S., Chen Z., Medhurst A. L., Neal J. A., Bao Z., Mortusewicz O., McGouran J., Song X., Shen H., Hamdy F. C., Kessler B. M., Meek K., Helleday T., DNA-PKcs and PARP1 bind to unresected stalled DNA replication forks where they recruit XRCC1 to mediate repair. Cancer Res. 76, 1078–1088 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bryant H. E., Petermann E., Schultz N., Jemth A.-S., Loseva O., Issaeva N., Johansson F., Fernandez S., McGlynn P., Helleday T., PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saldaña-Meyer R., González-Buendía E., Guerrero G., Narendra V., Bonasio R., Recillas-Targa F., Reinberg D., CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Genes Dev. 28, 723–734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang Z., Tang Z., Luo O. J., Li X., Zheng M., Zhu J. J., Szalaj P., Trzaskoma P., Magalska A., Wlodarczyk J., Ruszczycki B., Michalski P., Piecuch E., Wang P., Wang D., Tian S. Z., Penrad-Mobayed M., Sachs L. M., Ruan X., Wei C.-L., Liu E. T., Wilczynski G. M., Plewczynski D., Li G., Ruan Y., CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163, 1611–1627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coleman K. A., Greenberg R. A., The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 286, 13669–13680 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaeser M. D., Aslanian A., Dong M.-Q., Yates J. R. III, Emerson B. M., BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J. Biol. Chem. 283, 32254–32263 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Balakrishnan S. K., Witcher M., Berggren T. W., Emerson B. M., Functional and molecular characterization of the role of CTCF in human embryonic stem cell biology. PLOS ONE 7, e42424 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao T., Sun Q., del Rincon S. V., Lovato A., Marques M., Witcher M., Gallotannin imposes S phase arrest in breast cancer cells and suppresses the growth of triple-negative tumors in vivo. PLOS ONE 9, e92853 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marques M., Beauchamp M.-C., Fleury H., Laskov I., Qiang S., Pelmus M., Provencher D., Mes-Masson A.-M., Gotlieb W. H., Witcher M., Chemotherapy reduces PARP1 in cancers of the ovary: Implications for future clinical trials involving PARP inhibitors. BMC Med. 13, 217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu Z., Chen T., Hebert J., Li E., Richard S., A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol. Cell. Biol. 29, 2982–2996 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Franken N. A. P., Rodermond H. M., Stap J., Haveman J., van Bree C., Clonogenic assay of cells in vitro. Nat. Protoc. 1, 2315–2319 (2006). [DOI] [PubMed] [Google Scholar]
- 67.Olive P. L., Banáth J. P., The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 1, 23–29 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/5/e1601898/DC1
fig. S1. Live-cell imaging of CTCF at laser micro-irradiation tracks.
fig. S2. CTCF association with PARylation increases as a response to DNA-damaging agents.
fig. S3. Impact of CTCF loss on γH2AX and 53BP1 foci resolution.
fig. S4. Loss of CTCF increases sensitivity to PARP inhibitors.
fig. S5. Loss of CTCF impairs Rad51 foci formation following infrared.
fig. S6. DNA damage increases the association between CTCF and BRCA2.
table S1. sgRNA sequences targeting Cas9 to CTCF.