

Abstract
After 1 h of lithium-pilocarpine status epilepticus (SE), immunocytochemical labeling of NMDA receptor NR1 subunits reveals relocation of subunits from the interior to the cell surface of dentate gyrus granule cells and CA3 pyramidal cells. Simultaneously, an increase in NMDA-miniature excitatory postsynaptic currents (mEPSC) as well as an increase in NMDA receptor-mediated tonic currents is observed in hippocampal slices after SE. Mean-variance analysis of NMDA-mEPSCs estimates that the number of functional postsynaptic NMDA receptors per synapse increases 38% during SE, and antagonism by ifenprodil suggests that an increase in the surface representation of NR2B-containing NMDA receptors is responsible for the augmentation of both the phasic and tonic excitatory currents with SE. These results provide a potential mechanism for an enhancement of glutamatergic excitation that maintains SE and may contribute to excitotoxic injury during SE. Therapies that directly antagonize NMDA receptors may be a useful therapeutic strategy during refractory SE.
Keywords: NMDA receptor trafficking, Status epilepticus, Epilepsy, Hippocampus, Synaptic excitation
Introduction
Despite the emergence of several new anticonvulsants, status epilepticus (SE) is a major health concern with significant morbidity and a high mortality (DeLorenzo et al., 1996). As seizures persist, SE becomes self-sustaining and increasingly difficult to treat (Alldredge et al., 2001; Mazarati et al., 1998a; Silbergleit et al., 2012; Treiman et al., 1998), even after removal of the initial convulsant trigger (Mazarati et al., 1998b; Morrisett et al., 1987; Suchomelova et al., 2006). Previously we found that trafficking of GABAA receptors, with relocation of subunits from synapses to the cell interior, contributes to a loss of postsynaptic inhibition in hippocampal dentate gyrus granule cells and may explain the initiation of self-sustaining SE and the development of benzodiazepine pharmacoresistance (Naylor et al., 2005). While most GABAergic enhancing agents quickly lose effectiveness during the course of SE (Goodkin and Kapur, 2003; Jones et al., 2002; Kapur and Macdonald, 1997; Mazarati et al., 1998a; Rice and DeLorenzo, 1998, 1999; Treiman et al., 1998), NMDA antagonists retain effectiveness in the late stages of SE (Mazarati and Wasterlain, 1999; McDonald et al., 1990a; Mewasingh et al., 2003; Yen et al., 2004) suggesting that NMDA receptors remain available for pharmacological intervention throughout SE.
Although NMDA receptor trafficking is important in many processes including development/experience (Quinlan et al., 1999a, 1999b; van Zundert et al., 2004), synaptic plasticity/LTP (Carroll and Zukin, 2002; Grosshans et al., 2002; Watt et al., 2004), degenerative disorders (Chen et al., 1999c; Lau and Zukin, 2007; Snyder et al., 2005), and excitotoxicity/cell death (DeRidder et al., 2006; Gee et al., 2006; Guerguerian et al., 2002; Johnston et al., 2002; Lekishvili et al., 2006; Manzerra et al., 2001), the role of NMDA receptor trafficking in SE has not been investigated in detail. To explore the importance of NMDA receptors in maintaining SE, we studied the trafficking of NMDA receptor subunits and found a relocation of NMDA receptor subunits towards the cell surface with an associated increase in excitation involving synaptic as well as tonic currents. This augmented excitation, coupled with a loss of synaptic inhibition (Naylor et al., 2005), provides an important mechanism for seizure maintenance during SE and may contribute to SE-related excitotoxicity (Fujikawa, 1995; Hardingham et al., 2002; Rice and DeLorenzo, 1998). It also may contribute to synaptic plasticity during the epileptogenic process (Delorenzo et al., 2005; Mazarati et al., 2002a) — since progression to a chronic epileptic state frequently follows SE (Lothman et al., 1990; Mazarati et al., 1998b; Rice and DeLorenzo, 1998).
Material and methods
Animal preparation
4–7 week old male Wistar rats (Simonsen Lab, Gilroy, California) received lithium (3 mEq/kg intraperitoneal; night before) and methylscopolamine (1 mg/kg subcutaneous) 30 min before either pilocarpine (40 mg/kg intraperitoneal) or normal saline (Sigma, St. Louis, Missouri). All animal studies were approved by the Institutional Animal Care and Use Committee of the VA Greater Los Angeles Healthcare System. After ~1 h from onset of stage 5 seizures (Racine, 1972), animals were anesthetized with pentobarbital (100 mg/kg) and perfused-fixed with 10% formaldehyde for immunocytochemistry or anesthetized with halothane for decapitation and hippocampal slice preparation.
Induction of SE with neurokinin B (NKB) used a modification of a protocol described previously (Liu et al., 1999). Briefly, rats were implanted under isoflurane anesthesia (Abbott Labs, Chicago, Illinois) with a bipolar stimulating electrode into the perforant path and a bipolar recording electrode, combined with guide cannula, into the granule cell layer of the dorsal dentate gyrus. All experiments were performed in awake animals after at least 7 days recovery. Status epilepticus was induced by injection through a microsyringe (Hamilton Co., Reno, Nevada) inserted into the guide cannula of 1 nmol NKB (Sigma) dissolved in 1 μl DMSO, controls received 1 nmol NKB mixed with 10 nmol of SR142801, a selective NK3 receptor antagonist, or 1 μl DMSO (solvent). EEG was recorded from dentate gyrus during and for 24 h after injection.
Immunocytochemistry
Serial formaldehyde-fixed 40 μm coronal sections were incubated overnight in mouse anti-NR1, NMDA receptor subunit antiserum (5 μg/ml, Chemicon, Temecula, California) and rabbit anti-synaptophysin antiserum (5 μg/ml, Zymed, San Francisco, California). The sections then were incubated for 1 h in FITC-labeled goat anti-rabbit IgG (1:200, Jackson, West Grove, Pennsylvania) and Cy3-labeled donkey anti-mouse IgG (1:500, Jackson) diluted in PBS containing 1% goat serum, horse serum and 0.3% Triton X-100. Confocal images were acquired with a confocal cell-scanning microscope. For this study we used a pinhole of 1.0 Airy units and objectives of 100× oil (NA 1.4), resulting in an estimated optical section thickness (full width at half maximum) of 0.62 μm. Excitation for the green and red signals was provided by 488 nm and 568 nm laser lines. Controls with one label omitted confirmed that the bleed-through between channels with these settings was minimal. Confocal stacks with optical section separation (z-interval) of 0.488 μm were acquired through control and SE hippocampi. Optical sections were averaged three times to reduce noise. Images were acquired at a digital size of 1024×1024 pixels. Images generated in the two channels were merged using green/red pseudo-coloring and were processed further using Adobe Photoshop 5.5 (Adobe Systems Inc., Mountain View, CA). Immunoreactive ‘puncta’ were defined as discrete points along the neuron and dendrite with fluorescence intensity at least twice background. Colocalization was determined by examination by a blinded observer of yellowish ‘overlap’ images of synaptophysin and NR1 subunit immunoreactivity. We counted all NR1 subunit-immunoreactive neuronal cell bodies and 20 CA3 proximal dendrites (25 μm length, more easily distinguished from background in CA3 than DG) per section in 4 sections of dorsal hipporcampus per animal. Data are given as means±SEM (n=3 per group). Statistical significance was determined using a Student’s t-test for unpaired data.
EM post-embedding immunocytochemistry
NR1 localization was evaluated by post-embedding immuno-electron microscopy. One hour after onset of lithium-pilocarpine SE or sham treatment, anesthetized animals (n=3 per group) were perfused with 4% phosphate-buffered paraformaldehyde and 0.1% glutaraldehyde. The brains were then post-fixed in the same fixative for 3 h. Hippocampi were dissected out, subsectioned into approximately 2–4 mm slices and washed in 0.1 M phosphate-buffered saline (PB). Blocks were dehydrated and embedded in London Resin White. Thin sections were picked up on nickel grids, blocked with normal goat serum, cold fish gelatin and nonfat milk in PB buffer, and incubated for 24 h at room temperature with mouse antibody against NR1 (MAB363; Millipore). Grids were then washed in PB, incubated in anti-mouse immunoglobulin conjugated to 20 nm gold particles (1:120 in cold fish gelatin/PB), followed by a wash in PB containing cold fish gelatin and 2.5 M NaCl, then by several washes in water. Grids, stained with uranyl acetate and lead citrate, were viewed using a Philips 201C electron microscope. The outer 25% of asymmetric synapses was defined as ‘lateral’, an area where the label might be perisynaptic rather than located inside the synaptic cleft (Moga et al., 2006).
Electrophysiological recording: whole-cell patch clamp
After decapitation, rat brains were placed in ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): 126 NaCl, 2.5 KCl, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 D-glucose, 1 pyruvate, 0.3 ascorbic acid with pH=7.3 and no added Mg++. 350 μm coronal sections (Leica VT1000S, Bannockburn, Illinois) were held in a chamber at room temperature bubbled with 95% O2 and 5% CO2 until transfer to a chamber perfused at 1.5 ml/min with 33–35 °C ACSF where recordings from somata of visualized neurons were made using an Axoclamp-2B amplifier (Axon Instruments, Foster City, California). Slices from some lithium control animals were incubated in 40 mg/l of pilocarpine for ~1 h to control for a direct pharmacological effect of pilocarpine. Electrode solutions for NMDA-mEPSC recordings contained (mM): 140 Cs gluconate, 4 NaCl, 2 MgCl2, 10 Hepes, 0.05 EGTA, and 2 Mg-ATP (Sigma). The holding potential was −60 mV. Electrode solutions were titrated to pH 7.25 with osmolarity of 280–300 mOsm. Borosilicate electrodes were pulled (Sutter, Novato, California) for a resistance of 4–6 MΩ, and recordings with series resistance changes exceeding 50% were excluded.
Solutions and drugs
ACSF for NMDA-mEPSC recordings contained tetrodotoxin (1 μM; Calbiochem, LaJolla, California), CNQX (10 μM), picrotoxin (30 μM), D-serine (10 μM) (Sigma). ACSF included 3 mM kynurenic acid during cutting and preparation of slices. Ifenprodil (3 μM) selectively antagonized NR2B subunit-containing NMDA receptors, and the non-selective antagonist, APV (50 μM), added at the end of recordings confirmed their NMDA receptor origin (Sigma).
Data analysis
After 3 kHz low-pass filtering with 20 kHz digitizing (Digidata 1200; Axon Instruments), event detection and analysis (e.g. peak amplitude, rise-time, AUC) were with Synaptosoft (Decatur, Georgia), Pclamp 9.2 (Axon Instruments) and software generously provided by I. Mody (UCLA). All data are given as mean±standard deviation. A Student’s t-test was used to assess statistical significance. The slow gating-kinetics of NMDA receptors generates significant variability in NMDA-mEPSC time to peak and can account for the observed differences between the attributes of mean mEPSC traces from a granule cell (Fig. 4A) compared to those of individual mEPSC events (Fig. 4B —cumulative probability distributions). Because events are aligned by onset and not peak amplitude for mean traces, the amplitudes of mean composites will be smaller than the average of the peaks from each individual EPSC event.
Fig. 4.

NMDA-mEPSCs from dentate gyrus granule cells of SE and controls. A, NMDA-mEPSC mean traces from a typical granule cell from a control (black) and a SE animal (gray), demonstrating larger amplitude and area-under-the curve (AUC) in the latter (264 and 229 individual mEPSC events for the mean control and SE cell traces, respectively). B, Cumulative probability distributions for NMDA-mEPSCs from control (black) and SE (gray) granule cells. SE is associated with an increase in absolute peak amplitude and AUC, as indicated by a shift of the curves towards larger absolute values. The 67% decay-time cumulative probability plot also shifts to longer values with SE. A small but significant acceleration of the 10–90% rise-time is noted with SE as well. The cumulative probability distributions include 4328 individual NMDA-mEPSC events (2859 events from 28 lithium-pilocarpine SE cells and 1469 events from 14 lithium-only control cells). Additional controls (14 cells) involving pilocarpine exposure in vitro were not included. The number of events per cell was 102+/−59 for SE and 105+/−59 for controls (mean+/−s.d.).
Computer analysis
Mean and variance traces generated from superimposed NMDA-mEPSCs recorded from a dentate gyrus granule cell were analyzed to estimate the number of postsynaptic NMDA receptors (N), the open single channel current (i), and the channel open probability (Popen). The analysis used personal programs written for Matlab (MathWorks; Natick, Massachusetts). NMDA-mEPSCs with absolute peak amplitudes exceeding 60 pA were excluded. For channels with open probability Popen, the binomial distribution for the population of channels at a synapse generating NMDA-mEPSCs has a mean current (I)
| (1) |
and variance (σ2)
| (2) |
Popen will depend on several factors, including the concentration of glutamate in the synaptic cleft and the kinetics of the individual NMDA receptors. Substituting Eq. (1) into Eq. (2) determines the parabolic equation,
| (3) |
and eliminates Popen as a variable, allowing N and i determination from optimized fits of variance versus mean plots (Fig. 5A). Popen at the peak of the mean trace can be determined subsequently by dividing the peak amplitude of an NMDA-mEPSC by (N·i). Eqs. (1) through (3) were adapted to allow some variability in the number of postsynaptic receptors across the population of synapses that contribute to the mEPSCs of a cell. For example, modifications of the equations that allow two different sizes of synaptic activation (N1, N2) are
Fig. 5.

Mean-variance plots and NMDA-mEPSC peak amplitude distributions with model fits for control (left) and SE (right) granule cells. A, Mean-variance analysis of NMDA-mEPSCs from typical granule cells, with data (grey) and model fit (black). The optimized fit estimates the number of postsynaptic NMDA receptors (N) and individual receptor open current (i) (see Material and methods — Computer analysis). N is predicted as 8±1 per synapse in control cells and 11±2 for SE cell, with no difference in channel conductance noted between SE and controls. B, Histogram for peak amplitudes of individual NMDA-mEPSC events recorded from a control (left) and a SE (right) granule cell with the superimposed predicted distribution (bullet) from simultaneous model fits of A and B.
| (1*) |
| (2*) |
where F1 is the fraction of events with N1 synaptic receptors.
Given the slow kinetics/rise-time of NMDA-mEPSC events with great variability in onset of peak amplitude, the peak of the mean trace is not equal to the mean of the peaks of the individual events. Consequently, additional constraint on optimized fits was provided by simultaneous fit of the distribution of the peak amplitudes of individual NMDA-mEPSC (Fig. 5B) along with the fit of modified Eq. (3) to variance vs. mean plots (Fig. 5A). The assumption of a uniform current (i) for synaptic receptors is not unreasonable as single conductance states primarily characterize the synaptic, NR1-NR2A or NR1-NR2B subunit-containing receptors (Brimecombe et al., 1997; Clark et al., 1997; Erreger et al., 2005; Lieberman and Mody, 1999b).
Numerical methods
The error between model and data was minimized across successive trials using a simplex algorithm (Lagarias et al., 1998).
Results
SE is associated with relocation of NMDA NR1 receptor subunits from the cell interior to synaptic or peri-synaptic areas
NR1 is the essential subunit of NMDA heteromeric receptors (Monyer et al., 1992) and combines primarily with NR2A and/or NR2B subunits in the hippocampus (Wenthold et al., 2003a, 2003b). In granule and pyramidal cells from control hippocampi, NR1 subunit-like immunoreactivity (LI) (red pseudocolor) localizes to the cell interior, which is surrounded by the synaptophysin-LI (Fig. 1A, right upper panel, green pseudocolor). After 1 h of SE, much of this NR1 subunit-LI has relocated to discrete puncta which outline the cell membrane, and frequently colocalizes with synaptophysin-LI, giving these overlaps a yellow color (Fig. 1A, right lower panel). In hippocampal granule cells (Fig. 1A), the number of overlaps between NR1 subunit-LI and synaptophysin-LI increases from 1.85±0.18 per soma in controls to 7.15±0.28 in SE (p<0.001). In CA3 (Fig. 1B), the number of overlaps between synaptophysin-LI and NR1 subunit-LI increases from 3.30±0.33 on the somatic surface of controls to 7.60±0.44 with SE and from 2.50±0.64 for the 25 μm of proximal dendrite of controls to 9.30± 0.20 with SE (p<0.001). In CA1 somas, the number of overlaps increases from 4.00±1.02 in controls to 8.40±0.34 in SE (p=0.002).
Fig. 1.

Subcellular distribution of NMDA NR1 subunits with SE. Hippocampal sections through the dentate gyrus (A) and CA3 (B) of control (upper panels) and SE (lower panels) stained with an antibody against NMDA NR1 subunits (red, left panels) and an antibody against the presynaptic marker synaptophysin (green, middle panels). Overlaps between presynaptic synaptophysin and postsynaptic NR1 subunits appear yellow (right panels). Note increased NR1 subunit-LI colocalization with synaptophysin-LI for SE (right lower panel) compared to controls (right upper panel), suggesting trafficking of NMDA NR1 subunits towards the cell surface during SE. C, The number of colocalizations increase with SE in principal cells of the dentate gyrus (DG), CA3 and CA1 compared to controls (error bars as±SEM).
By electron microscopy immunocytochemistry, the number of asymmetric synapses in the dentate granule cell layer which are immunoreactive for NR1 increases from 8.4±2.8% in controls to 26.9±4.7% in SE (Fig. 2), confirming the light microscopy results. The percentage of asymmetric synapses with 1 gold particle increases from 7.8±2.5% in controls to 22.2±3.8%, and that of synapses with 2–3 gold particles increases from 0.5±0.3% in controls to 4.7±1.2% after SE (p<0.05; 1037 asymmetric control synapse, 724 asymmetric SE synapses). A greater proportion of NR1 particles are located in the periphery of asymmetric synapses after SE, suggesting the possibility of a perisynaptic location and of lateral entry of subunits into synapses (45.3±3.2% for controls versus 58.5±2.7% for SE; p<0.05).
Fig. 2.

Electron microscopy distribution of NR1-LI in asymmetric synapses of the granule cell layer of the dentate gyrus from control (A, upper panels) and SE animals (A, lower panels). Arrows indicate the postsynaptic density (bars=150 nm). Examples of asymmetric synapses showing central/synaptic immunogold labeling (A, left panels) or a mix of central/synaptic and lateral/perisynaptic immunogold labeling of postsynaptic densities (A, right panels) are shown. SE increases the percentage of immunogold-labeled asymmetric synapses compared to controls (B, upper). The lateral distribution of NR1 on the surface of synapses increases with SE (B, lower). (Error bars as±SEM.)
Synaptic relocation of NR1 subunit-LI is also observed in another model of SE induced by intrahippocampal injection of neurokinin B
Double labeling shows, in CA3 pyramidal neurons of control rats, only 3±1 overlaps between synaptophysin- and NMDA NR1-LI on the somatic surface, and 4±1.5 overlaps along the proximal 25-μm of dendrites (Fig. 3A, upper panels). However, at the peak seizure activity 30 min after administration of 1 nmol NKB, the overlaps significantly increase to 11.8±2.4 on the soma and 13.9±2.1 on proximal dendrites respectively (Fig. 3A, lower panels), a 350–390% increase, compared to only a 25±4% increase in number of overlaps in the contralateral hippocampus, suggesting that this phenomenon is not unique to the lithium-pilocarpine model (Fig. 3). By contrast, injection of 1 nmol NKB mixed with 10 nmol SR 142801 (an NKB receptor antagonist; Sanofi-Aventis, Bridgewater, New Jersey) did not significantly increase the number of overlaps between synaptophysin-LI and NMDA NR1-LI on the surfaces of pyramidal neurons neurons (3±1 overlaps on granule cell soma, 3.8±1 overlaps on the proximal 25-μm of dendrites), nor did it cause any seizures (data not shown).
Fig. 3.

Subcellular distribution of NMDA NR1 subunit-LI after NKB-induced SE. A, Hippocampal sections through CA3 of control (upper panels) and SE (lower panels) brains stained with antibodies against the NR1 subunit-LI (red) and against the presynaptic marker synaptophysin-LI (green), with overlaps appearing yellow. Hippocampal sections of CA3 at higher magnification are shown on the right. Note increased NR1 subunit-LI colocalization with synaptophysin-LI in pyramidal cells for NKB SE (bar — 40 μm left panel; 10 μm right panel). B, The number of colocalizations increases with NKB SE at both the soma and proximal dendrites of CA3 pyramidal cells. (error bars as±SEM).
An increase in NMDA-mEPSC amplitude suggests a gain of postsynaptic NMDA receptors during SE
We used hippocampal slices obtained from animals in lithium-pilocarpine SE for 1 h to examine whether the increase in synaptic NR1 puncta reflects an increase in the number of functional postsynaptic NMDA receptors. Mean traces of NMDA-mEPSCs recorded from dentate gyrus granule cells in slices from SE animals (Fig. 4A) increase peak amplitude to 123% of controls (−20.2±2.7 pA for SE vs −16.4±0.64 pA for controls; p<0.001; n=16) and increase the area-under-the-curve (AUC) to 132% of controls (−689±143 pA·ms for SE vs −523±163 pA·ms for controls; p<0.01). Comparison with additional controls with slices obtained from animals injected with lithium the night before and then incubated in aCSF containing 40 mg/L of pilocarpine (n=9) for 1–2 h prior to recording showed significant increases in NMDA-mEPSC amplitude and AUC for SE animals compared to these lithium-pilocarpine controls (pilocarpine control peak amplitude −14.6±2.4 pA and AUC −464±216 pA·ms; p<0.05). This suggests that the NMDA-mEPSC changes we observe with the lithium-pilocarpine model of SE are due to seizures and not a lithium and/or pilocarpine pharmacological effect.
To interpret the basis for the increase in amplitude and AUC with SE, mean-variance analysis of NMDA receptors (see Material and methods — Data analysis) provides estimates of the number of postsynaptic receptors. The basic assumption of this analysis is that NMDA-mEPSCs represent the sum of the individual postsynaptic receptor current responses, and the individual receptors open and close in a probabilistic manner with binomial statistics. Additional constraints during optimized fits of the mean-variance of NMDA-mEPSCs (Fig. 5A) were provided by simultaneous fit of the distribution of NMDA-mEPSC peak amplitudes (Fig. 5B). This analysis suggests that the number of NMDA receptors increases 38% from 8±1 per synapse in controls to 11±2 per synapse with SE (p<0.001; n=16). This relatively small number of NMDA receptors per control synapse is not unexpected (Bekkers and Stevens, 1989; Kohr et al., 2003; Nimchinsky et al., 2004).
SE causes alterations in synaptic NMDA-mEPSC kinetics
In addition to synaptic relocation of NMDA receptors, we find changes affecting NMDA-mEPSC kinetics. For mean traces of NMDA-mEPSCs recorded from granule cells, an acceleration of the 10–90% rise-time occurs (5.32±2.14 ms for SE vs 6.0±2.0 ms for controls; p<0.001; n=14 each group). Also, a prolongation of the 63% decay-time from 21.0±12.8 ms for controls to 24.4±8.3 ms with SE (p<0.01) occurs, and the weighted-tau decay-time (AUC/peak amplitude) also shows a prolongation (40.8±9.3 ms for SE vs 33.0±12.8 ms for controls). For the individual NMDA-mEPSCs events from granule cells, increases in peak amplitude (−27.3±5.25 pA for SE vs −21.8±4.0 pA for controls; p<0.001) and AUC (−766±232 pA·ms for SE vs −575±217 pA·ms for controls; p<0.001) with faster 10–90% rise-times (6.7±2.7 for SE vs 8.4±3.9 ms for controls; p<0.01) are observed. Cumulative probability distributions that pool individual NMDA-mEPSCs from multiple control or SE cells (Fig. 4B) show these results graphically.
These changes may relate to SE-induced alterations of NMDA receptor kinetics and/or effects of SE on the spatiotemporal profile of glutamate release in the synaptic cleft. Results from mean-variance fits provide estimates for the single channel conductance comparable to published values from single channel recordings (Clark et al., 1997; Erreger et al., 2005) and suggest no change in channel conductance with SE (64±2 pS for control vs 65±3 pS with SE; n.s.; n=14 each group), By this analysis, the estimate of channel open probability at the peak of the mean NMDA-EPSC trace is 54±9% for controls vs 45±3% after SE (p<0.001), possibly relating to changes in NMDA receptor kinetics and/or subunit composition.
Other effects of SE include a decrease in mEPSC frequency from 0.96±0.63 Hz for controls to 0.73±0.52 Hz with SE (p<0.001). Some decrease in the resting membrane potential from −68.3±9.1 mV with controls to −65.2±8.6 mV with SE also is noted (p<0.001). Both results may relate to injury and cell/synaptic loss associated with SE.
NR2B subunit content of postsynaptic NMDA receptors increases with SE
In the presence of an antagonist of NR2B containing receptors (ifenprodil; 3 μM; Sigma), mean SE NMDA-mEPSC traces (Fig. 6Aii) decrease in amplitude from −17.38±4.71 pA to −15.16±2.04 (p<0.001; n=29) as the decay-time decreases from 24.4±8.3 to 19.3±9.1 (p<0.001) and AUC decreases from −739.5±243.5 to −636.5±224.5 pA·ms (p<0.001) (Fig. 6Bii). For controls, mean NMDA-mEPSCs (Fig. 6Ai) change from −15.56±1.70 to −15.01±1.87 (n.s.; n=14) with ifenprodil as AUC changes from −553.2±190.5 to −611.21±291.4 (n.s.), and no significant changes are noted with decay-time either (Fig. 6Bi). After ifenprodil, no significant difference between SE and controls exists with respect to NMDA-mEPSC amplitude (−15.2±2.04 vs 15.0±1.9), decay-time (19.3±9.1 ms vs 23.8±21.2), AUC (−636.5±224.5 vs −611.2±291.4), although a faster R-T with SE persists after ifenprodil (5.1±2.3 ms with SE vs 5.99±2.0 ms with controls). This suggests that NR2B-containing receptors account for most of the SE-related increases in NMDA-mEPSC amplitude and AUC and also contribute to NMDA-mEPSC kinetic changes such as a prolongation in decay-time. Mean-variance analysis of NMDA-mEPSCs before and after ifenprodil (Fig. 7) suggests 1.9±1.6 receptors per synapse are ifenprodil-sensitive in slices from SE brains (p<0.001; paired t-test; n=17) vs 0.7±1.6 for controls (n.s.; n=8). These computations further support the conclusion that NR2B-containing receptors mediate the increase in NMDA-mEPSCs with SE.
Fig. 6.

Ifenprodil effect on NMDA-mEPSCs from dentate gyrus granule cells of SE and controls. A, NMDA-mEPSC mean traces from a typical granule cell from a control (i) and a SE (ii) animal. i) NMDA-mEPSC mean traces from a typical control granule cell before (black) and after (grey) addition of ifenprodil (3 μM) to the perfusate show overlap, suggesting a lack of ifenprodil sensitivity for NMDA-mEPSCs in control cells. ii) Unlike control cells, NMDA-mEPSC mean traces from a typical SE granule cell before (black) and after (grey) addition of ifenprodil show significant reduction in the amplitude and the AUC, demonstrating the presence of ifenprodil-sensitive NR2B-containing NMDA receptors at excitatory synapses after SE. B, Changes in cumulative probability distributions for NMDA-mEPSCs from control (i) and SE (ii) granule cells after ifenprodil. i) Control cells prior to perfusion with ifenprodil (black) have cumulative probability distributions for peak amplitude (1), AUC (2), decay-time (3) and rise-time (4) that overlie the corresponding distributions after ifenprodil (grey), suggesting a lack of ifenprodil sensitivity for NMDA-mEPSCs in control cells. ii) Unlike control cells, cumulative distribution plots for SE cells prior to perfusion with ifenprodil (black) are significantly different from distribution plots after ifenprodil (grey). Ifenprodil causes a significant reduction in peak amplitude (1) and AUC, with decreases in the decay-times and rise-times of NMDA-mEPSCs as well. The cumulative probability plots post-ifenprodil include 1044 NMDA-mEPSC events from 16 lithium-pilocarpine SE cells and 571 events from 8 lithium-only control cells. Additional controls (8 cells) involving pilocarpine exposure in vitro were not included.
Fig. 7.

Mean-variance plots and NMDA-mEPSC peak amplitude distributions with model fits for control (A) and SE (B) granule cells before (i) and after (ii) ifenprodil. Mean-variance analysis of NMDA-mEPSCs with data (grey) and optimized model fits (black). The corresponding histogram of NMDA-mEPSC peak amplitudes from that granule is cell shown below each mean-variance plot along with the model-predicted distribution (bullet). Comparison of model results for A) control (n=6) and B) SE (n=10) granule cells before (i) and after (ii) ifenprodil (3 μM) estimates a significant increase of 1.9+/−1.6 ifenprodil sensitive NMDA receptors per synapse for SE, with no significant increase noted in controls.
Increases in NMDA tonic currents during SE are mediated by NR2B-containing receptors
With ifenprodil in the perfusate, a positive tonic current baseline shift from of −76.0±41.1 pA to −66.8±34.5 pA (p<0.05; n=17) for SE granule cells held at −60 mv reveals a depolarizing tonic current of +9.2 pA mediated by NMDA receptors containing NR2B subunits (Fig. 8). No tonic current shift is observed with addition of ifenprodil to controls (−2.6±12.9 pA; n.s.; n=5). After ifenprodil, addition of APV (~50 μM) directly to the chamber produces a similar positive tonic current baseline shift for either SE (+24.9±24.4 pA, p<0.001) or control (+27.8±19.3 pA, p<0.02) granule cell recordings, with no significant difference in the baseline shift between SE and controls. For SE cells, the tonic NMDA current contribution from NR2B-containing receptors is ~37% of the total. This suggests that surface accumulation of NR2B-containing NMDA receptors is primarily responsible for the increase in tonic NMDA currents with SE, similar to our observations for phasic NMDA-mEPSC currents.
Fig. 8.
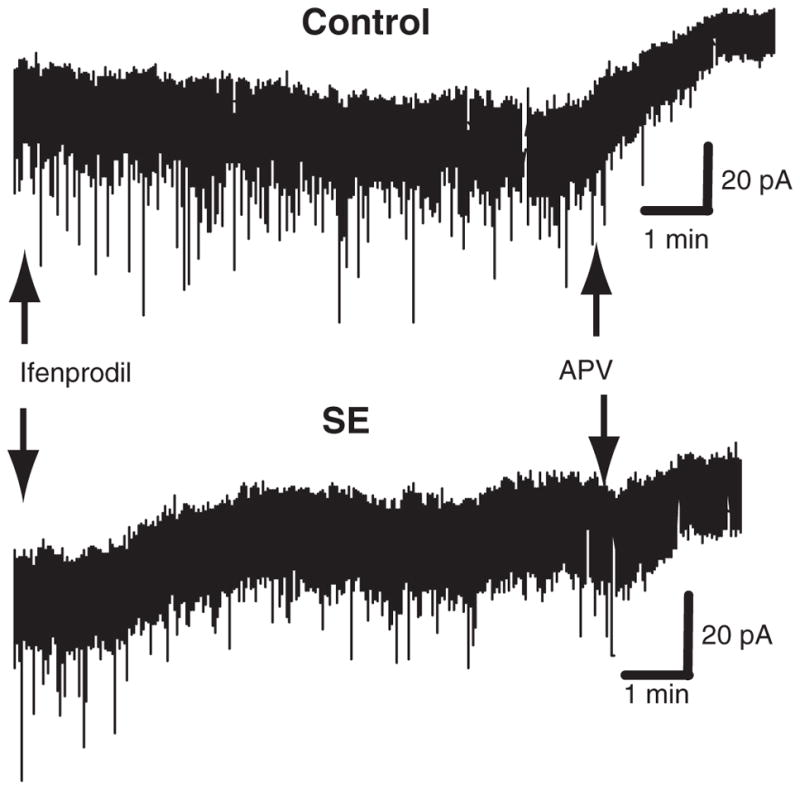
Ifenpodil-sensitive component of NMDA tonic currents of granule cells after SE. Recordings from typical granule cells from control and SE animals. Note an NR2B-dependent component of the NMDA tonic current for SE cells but not controls, as revealed by a significant baseline shift with addition of the NR2B receptor antagonist ifenprodil (3 μM) with no significant baseline shift noted with ifenprodil in controls. Both control and SE cells have significant baseline shifts with the subsequent addition of the non-selective NMDA receptor antagonist, APV.
Discussion
After 1 h of SE, dentate granule cells display a 25% increase in mEPSC amplitude, a 38% increase in the number of physiologically active NMDA receptors per somatic synapse, a two-fold increase in the number of NR1 subunit-LI in the vicinity of the presynaptic marker synaptophysin, and a three-fold increase in NR1-like immunoreactivity associated with postsynaptic densities. The NR1 immunoreactivity is likely a marker for functional heteromeric NMDA receptor complexes because all hippocampal NMDA receptors contain NR1 subunits, and solitary NR1 subunits are retained in the ER (Prybylowski et al., 2002). These changes could in part account for the enhanced glutamatergic excitation that has been described in SE (Wasterlain et al., 2000), and for the therapeutic efficacy of NMDA antagonists in some models of SE (Bertram and Lothman, 1990; Mazarati and Wasterlain, 1999). This movement of NMDA receptor subunits towards the vicinity of synapses coincides with a movement of the GABAA receptor β2/β3 and γ2 subunits from the cell surface to cytoplasmic sites (Goodkin et al., 2005; Naylor et al., 2005). Hence, the direction of trafficking during SE is receptor type-specific. The net movement of NMDA and AMPA (Naylor, unpublished observations) receptors towards the cell surface and towards synapses and the movement of GABAA receptors away from synapses appear maladaptive in a seizing brain, and risk to further exacerbate an imbalance between synaptic inhibition and synaptic excitation.
Normalization of synaptic physiology with slice incubation may partially account for the lower estimate obtained from physiological data compared to immunocyotchemistry. A similar difference between anatomical and physiological measures is observed with GABAA receptor trafficking during SE (Naylor et al., 2005). Alternatively, immunocytochemisty may detect extrasynaptic surface receptors that do not contribute to NMDA-mEPSC responses. It has been shown that delivery of functional NMDA receptors to the cell surface does not necessarily increase the number of functional synaptic receptors (Prybylowski et al., 2002). Many factors, including receptor trafficking from existing cytosolic pools, can regulate surface expression of NMDA receptors at synaptic and extrasynaptic sites. Local de novo synthesis of subunits seems unlikely given the profound inhibition of protein synthesis and the compromised bioenergetic balance during SE (Fujikawa et al., 1988; Wasterlain, 1974), although over-expression of NR1 and NR2B subunits has been reported 24 h after pilocarpine in neuronal cultures (Di Maio et al., 2011).
The increase of synaptic NMDA receptors may characterize an important transition when augmented synaptic excitation, coupled with a major loss of synaptic inhibition (Naylor et al., 2005), provides the framework for progression from single seizures to self-sustaining SE. While loss of inhibition may be important with initiation of SE, augmented glutamatergic excitation is important for the maintenance of SE (Mazarati and Wasterlain, 1999; Rice and DeLorenzo, 1999; Wasterlain et al., 2000). This is consistent with the observation that GABAergic drugs show a significant, time-dependent loss in potency during SE while NMDA antagonists are therapeutically effective even when administered late in SE (Mazarati and Wasterlain, 1999; Yen et al., 2004). Clinically, the NMDA antagonist ketamine was anecdotally effective after 24–48 h of seizure activity in 5 patients (Mewasingh et al., 2003). These findings have therapeutic implications: the standard initial treatment of SE (benzodiazepines) is insufficient because increased glutamatergic excitation is untouched. Furthermore, benzodiazepines are unlikely to fully restore GABAergic inhibition given the dramatic reduction of postsynaptic receptors available for allosteric activation. Combinations of a benzodiazepine with an antagonist at NMDA receptors and another drug which increases inhibition at a non-benzodiazepine site would more fully address the effects of SE-induced receptor trafficking. Indeed, such combinations seem to be more effective than monotherapy at stopping SE (Wasterlain et al., 2011). Certain agents including felbamate (with dual agonist/antagonist actions on GABAergic and glutamatergic receptors, respectively) may have distinct therapeutic potential in SE (Rho et al., 1994).
While we have yet to evaluate the precise temporal and sequential relation for loss of inhibition (Goodkin et al., 2005; Naylor et al., 2005) and gain of excitation during SE, our results suggest that both become significant by 1 h of experimental seizure activity. In addition, significant interplay may exist between the gain of glutamatergic excitation and loss of inhibition. An increase of NMDA receptor activity during SE may contribute to a down-regulation of GABAergic inhibition in some models of epilepsy (Kapur and Lothman, 1990), and NMDA blockade can prevent the loss of effectiveness of GABA enhancing drugs in a model of SE (Rice and DeLorenzo, 1999). We also find that non-NMDA glutamatergic synaptic receptors are upregulated during SE (Naylor, unpublished observations), and the augmentation of NMDA receptors we find here may be crucial in that regard (Lu et al., 2001; Shi et al., 1999).
Kinetic changes at excitatory synapses during SE include an acceleration of the NMDA-mEPSC rise-time along with a prolongation of the tau weighted-decay and a decreased peak open probability. Since subunit subtypes determine NMDA receptor properties, trafficking of postsynaptic NMDA receptors involving shifts in subunit composition can alter receptor kinetics and NMDA-mEPSC attributes. NR2B subunit-containing NMDA receptors have slower deactivation kinetics (Flint et al., 1997; Prybylowski et al., 2002) and greater charge transfer with a lower open probability compared to NR2A subunit-containing receptors (Chen et al., 1999b; Erreger et al., 2005) suggesting the kinetic properties we observe are consistent with a greater proportion of NR2B subunit-containing receptors at synapses. As both NR2A and NR2B subunits can exist at synaptic and extrasynaptic sites (Berberich et al., 2005; Liu et al., 2004; Massey et al., 2004), and a single synapse can contain both NR2A and NR2B subunits as components of NMDA receptor complexes (Kohr et al., 2003), slight shifts in subunit composition could account for the decrease in Popen we observe with SE. Similarly, because receptors containing NR2B subunits have a higher ligand binding affinity than those containing NR2A subunits (Erreger et al., 2005), kinetic changes in NMDA-mEPSC rise-times and decay-times during SE also could be explained by alterations in subunit composition at synapses. Movement of receptors toward the center of synapses could affect NMDA-mEPSC kinetic properties such as rise-time as well (Harney et al., 2008), and although electron microscopy immunocytochemistry reveals a greater proportion of NMDA NR1 subunits at the periphery of synapses, a dramatic increase of subunits is noted at all synaptic locations including synapse centers. Alternatively, the increase in mEPSC-NMDA rise-time associated with a decrease in event frequency partially may relate to selective vulnerability of distal (filtered) excitatory synapses during SE and slice preparation.
Selective pharmacological blockade of NR2B subunit-containing receptors suggests that the increases in both phasic and tonic NMDA receptor-mediated currents during SE is secondary to an increase in NR2B-containing NMDA receptors, probably located at synaptic and extrasynaptic sites, respectively, because similar to tonic GABAergic currents, excitatory tonic currents from NMDA receptors may originate from extrasynaptic receptors (Le Meur et al., 2007). While an increase in synaptic spillover of glutamate to extrasynaptic receptors potentially could contribute to the activation of an increased number of receptors with SE NMDA-EPSCs, our immunocytochemistry findings show clear increases in the synaptic location of NMDA receptor subunits and spill-over should be minimal with miniature EPSC recordings. Furthermore, the NR2B subunit specificity we observe for NMDA-mEPSC current increases with SE would not be expected with spillover, which should indiscriminately activate all available receptors including those containing NR2A as well as NR2B subunits. In fact, no significant phasic or tonic current differences are noted between SE and control cells after antagonism of NR2B-containing receptors. This result also suggests that, unlike the SE-induced increase of a tonic GABAA current attributed to a rise in extracellular [GABA] (Naylor et al., 2005), the increase of a tonic glutamatergic NMDA current in granule cells relates to the de novo appearance of NR2B subunit-containing receptors at extrasynaptic sites. Our pharmacological findings for early SE are compatible with immunohistochemical increases in postsynaptic NR2B subunits that are reported to occur within 2 h for an electrically-induced model of SE (especially for Tyr1472 phosphorylated NR2B that associates with the synaptic scaffolding protein PSD-95), while increases in extrasynaptic NR2B receptors are noted by 96 h after SE (Frasca et al., 2011). Whether NR2B subunit-containing receptors first accumulate extrasynaptically on the neuronal membrane and then migrate to synapses (Chen et al., 1999a; Prybylowski et al., 2002; Tovar and Westbrook, 2002; Triller and Choquet, 2003) is not clear from our analysis.
Other factors can contribute to the changes in receptor kinetics and surface expression that we observe: variations in the dynamics or concentration of transmitter release and uptake during SE, activity-dependent changes in receptor kinetics/desensitization (Krupp et al., 2002; Vissel et al., 2001), and changes in the phosphorylation state of NMDA receptors (Carroll and Zukin, 2002; Friedman et al., 1997; Lan et al., 2001; Li et al., 2001; Lieberman and Mody, 1999a; Lin et al., 2006; Lu et al., 2000; Moussa et al., 2001) that also may affect NMDA receptor expression at synapses (Frasca et al., 2011; Li et al., 2003; Lin et al., 2004, 2006; Rycroft and Gibb, 2004; Wenthold et al., 2003a; Yaka et al., 2002). For example, kinase activation during SE (Beldhuis et al., 1992; Dunah et al., 2004; Huo et al., 2006; Kohira et al., 1992; Niimura et al., 2005; Osonoe et al., 1994; Prybylowski et al., 2005) can affect NMDA receptor channel properties (Ali and Salter, 2001; Moussa et al., 2001) and both receptor interactions with the AP2-clathrin endocytotic complex (Li et al., 2002; Vissel et al., 2001) and delivery to synapses (Grosshans et al., 2002; Lan et al., 2001; Scott et al., 2003). BDNF, also activated during SE (Kornblum et al., 1997), affects NMDA receptor phosphorlyation (Wyneken et al., 2003) and cell-surface stability.
The increased number of NMDA receptors at the cell surface during SE has important implications for cell injury and cell death as well as for the subsequent development of chronic epilepsy. Over-activity of NMDA receptors can increase neuron death during epileptic seizures, stroke, trauma or neurodegenerative disorders (Choi, 1994a, 1994b; During et al., 2000; McDonald et al., 1990a, 1990b), and NMDA receptor blockade can provide neuroprotection with SE, ischemia or excitotoxicity (Brandt et al., 2003; Clifford et al., 1990; Frasca et al., 2011; Fujikawa, 1995; Rice and DeLorenzo, 1998; Seo et al., 2009; Starr and Starr, 1994). In addition, extrasynaptic NMDA receptors that contain NR2B subunits have been implicated with neurotoxicity, degeneration, and cell death (Fong et al., 2002; Hardingham et al., 2002; Milnerwood et al., 2010). Blockade of NMDA receptors reduces CA1 and CA3 cell death after pilocarpine-induced SE (Frasca et al., 2011; Fujikawa, 1995) and mossy fiber sprouting after KA-induced SE (Rice and DeLorenzo, 1998), while also slowing kindling (McNamara et al., 1988) and preventing the development of spontaneous recurrent seizures in some models of epileptogenesis (Prasad et al., 2002; Rice and DeLorenzo, 1998). Of note, felbamate and its close relative fluorofelbamate have a preference for antagonism of NR2B subunit-containing receptors (Harty and Rogawski, 2000) and reduce SE-associated neuronoal injury (Mazarati et al., 2000) and SE-associated epileptogenesis (Mazarati et al., 2002b). NMDA-Ca2+ dependent cell death in the hippocampus may have a role in the development of chronic sequelae after SE (Deshpande et al., 2008).
Conclusion
The increase in excitatory NMDA currents early during SE (primarily from NMDA receptors containing NR2B subunits) and the trafficking of NMDA receptors from the cell interior to synaptic and extrasynaptic sites may contribute to excessive levels of excitation during SE, and may maintain seizure activity initiated by a loss of synaptic GABAA receptors (Goodkin et al., 2008; Naylor et al., 2005). It may aggravate seizure-induced benzodiazepine pharmacoresistance during SE by adding an excitatory component unresponsive to standard treatment with GABAA receptor agonists. The seizure-induced relocation of NMDA receptors to the cell surface helps explain why late treatment with NMDA antagonists is effective in some models of SE (Borris et al., 2000; Mazarati and Wasterlain, 1999; Mewasingh et al., 2003), long after benzodiazepine loss of potency (Kapur and Macdonald, 1997; Mazarati et al., 1998a; Treiman et al., 1998; Walton and Treiman, 1988), and offers a potential site for pharmacological intervention. Agents that directly block glutamatergic receptors or shift the balance of kinase and phosphatase activity to reduce NMDA receptor surface expression deserve investigation in treating SE and in reducing its long-term complications and may be most effective in combination with GABAA agonists and other traditional anticonvulsants (Wasterlain et al., 2011).
Acknowledgments
Support provided by the Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center and a VA Career Development Award to D.N. and by NINDS (N13515) to C.W.
Footnotes
Available online on ScienceDirect (www.sciencedirect.com).
References
- Ali DW, Salter MW. NMDA receptor regulation by Src kinase signalling in excitatory synaptic transmission and plasticity. Curr Opin Neurobiol. 2001;11:336–342. doi: 10.1016/s0959-4388(00)00216-6. [DOI] [PubMed] [Google Scholar]
- Alldredge BK, Gelb AM, Isaacs SM, Corry MD, Allen F, Ulrich S, Gottwald MD, O’Neil N, Neuhaus JM, Segal MR, Lowenstein DH. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med. 2001;345:631–637. doi: 10.1056/NEJMoa002141. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- Beldhuis HJ, Everts HG, Van der Zee EA, Luiten PG, Bohus B. Amygdala kindling-induced seizures selectively impair spatial memory. 2 Effects on hippocampal neuronal and glial muscarinic acetylcholine receptor. Hippocampus. 1992;2:411–419. doi: 10.1002/hipo.450020408. [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby O, Kohr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram EH, Lothman EW. NMDA receptor antagonists and limbic status epilepticus: a comparison with standard anticonvulsants. Epilepsy Res. 1990;5:177–184. doi: 10.1016/0920-1211(90)90036-u. [DOI] [PubMed] [Google Scholar]
- Borris DJ, Bertram EH, Kapur J. Ketamine controls prolonged status epilepticus. Epilepsy Res. 2000;42:117–122. doi: 10.1016/s0920-1211(00)00175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt C, Potschka H, Loscher W, Ebert U. N-methyl-D-aspartate receptor blockade after status epilepticus protects against limbic brain damage but not against epilepsy in the kainate model of temporal lobe epilepsy. Neuroscience. 2003;118:727–740. doi: 10.1016/s0306-4522(03)00027-7. [DOI] [PubMed] [Google Scholar]
- Brimecombe JC, Boeckman FA, Aizenman E. Functional consequences of NR2 subunit composition in single recombinant N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A. 1997;94:11019–11024. doi: 10.1073/pnas.94.20.11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Zukin RS. NMDA-receptor trafficking and targeting: implications for synaptic transmission and plasticity. Trends Neurosci. 2002;25:571–577. doi: 10.1016/s0166-2236(02)02272-5. [DOI] [PubMed] [Google Scholar]
- Chen J, Li H, Luo C, Li Z, Zheng J. Involvement of peripheral NMDA and non-NMDA receptors in development of persistent firing of spinal wide-dynamic-range neurons induced by subcutaneous bee venom injection in the cat. Brain Res. 1999a;844:98–105. doi: 10.1016/s0006-8993(99)01841-7. [DOI] [PubMed] [Google Scholar]
- Chen N, Luo T, Raymond LA. Subtype-dependence of NMDA receptor channel open probability. J Neurosci. 1999b;19:6844–6854. doi: 10.1523/JNEUROSCI.19-16-06844.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Luo T, Wellington C, Metzler M, McCutcheon K, Hayden MR, Raymond LA. Subtype-specific enhancement of NMDA receptor currents by mutant huntingtin. J Neurochem. 1999c;72:1890–1898. doi: 10.1046/j.1471-4159.1999.0721890.x. [DOI] [PubMed] [Google Scholar]
- Choi DW. Calcium and excitotoxic neuronal injury. Ann N Y Acad Sci. 1994a;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994b;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- Clark BA, Farrant M, Cull-Candy SG. A direct comparison of the single-channel properties of synaptic and extrasynaptic NMDA receptors. J Neurosci. 1997;17:107–116. doi: 10.1523/JNEUROSCI.17-01-00107.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford DB, Olney JW, Benz AM, Fuller TA, Zorumski CF. Ketamine, phencyclidine, and MK-801 protect against kainic acid-induced seizure-related brain damage. Epilepsia. 1990;31:382–390. doi: 10.1111/j.1528-1157.1990.tb05492.x. [DOI] [PubMed] [Google Scholar]
- DeLorenzo RJ, Hauser WA, Towne AR, Boggs JG, Pellock JM, Penberthy L, Garnett L, Fortner CA, Ko D. A prospective, population-based epidemiologic study of status epilepticus in Richmond. Virginia Neurology. 1996;46:1029–1035. doi: 10.1212/wnl.46.4.1029. [DOI] [PubMed] [Google Scholar]
- Delorenzo RJ, Sun DA, Deshpande LS. Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintainance of epilepsy. Pharmacol Ther. 2005;105:229–266. doi: 10.1016/j.pharmthera.2004.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRidder MN, Simon MJ, Siman R, Auberson YP, Raghupathi R, Meaney DF. Traumatic mechanical injury to the hippocampus in vitro causes regional caspase-3 and calpain activation that is influenced by NMDA receptor subunit composition. Neurobiol Dis. 2006;22:165–176. doi: 10.1016/j.nbd.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Deshpande LS, Lou JK, Mian A, Blair RE, Sombati S, Attkisson E, DeLorenzo RJ. Time course and mechanism of hippocampal neuronal death in an in vitro model of status epilepticus: role of NMDA receptor activation and NMDA dependent calcium entry. Eur J Pharmacol. 2008;583:73–83. doi: 10.1016/j.ejphar.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio R, Mastroberardino PG, Hu X, Montero L, Greenamyre JT. Pilocarpine alters NMDA receptor expression and function in hippocampal neurons: NADPH oxidase and ERK 1/2 mechanisms. Neurobiol Dis. 2011;42:482–495. doi: 10.1016/j.nbd.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Sirianni AC, Fienberg AA, Bastia E, Schwarzschild MA, Standaert DG. Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol Pharmacol. 2004;65:121–129. doi: 10.1124/mol.65.1.121. [DOI] [PubMed] [Google Scholar]
- During MJ, Symes CW, Lawlor PA, Lin J, Dunning J, Fitzsimons HL, Poulsen D, Leone P, Xu R, Dicker BL, Lipski J, Young D. An oral vaccine against NMDAR1 with efficacy in experimental stroke and epilepsy. Science. 2000;287:1453–1460. doi: 10.1126/science.287.5457.1453. [DOI] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 2005;563:345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neo-cortex. J Neurosci. 1997;17:2469–2476. doi: 10.1523/JNEUROSCI.17-07-02469.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong DK, Rao A, Crump FT, Craig AM. Rapid synaptic remodeling by protein kinase C: reciprocal translocation of NMDA receptors and calcium/calmodulin-dependent kinase II. J Neurosci. 2002;22:2153–2164. doi: 10.1523/JNEUROSCI.22-06-02153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasca A, Aalbers M, Frigerio F, Fiordaliso F, Salio M, Gobbi M, Cagnotto A, Gardoni F, Battaglia GS, Hoogland G, Di Luca M, Vezzani A. Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity. Neurobiol Dis. 2011;43:507–515. doi: 10.1016/j.nbd.2011.04.024. [DOI] [PubMed] [Google Scholar]
- Friedman LM, Matsuda Y, Lazarovici P. The microbial alkaloid toxin staurosporine blocks the phorbol ester-induced increase in beta-amyloid precursor protein in PC12 cells. Nat Toxins. 1997;5:173–179. doi: 10.1002/nt.1. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG. Neuroprotective effect of ketamine administered after status epilepticus onset. Epilepsia. 1995;36:186–195. doi: 10.1111/j.1528-1157.1995.tb00979.x. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Vannucci RC, Dwyer BE, Wasterlain CG. Generalized seizures deplete brain energy reserves in normoxemic newborn monkeys. Brain Res. 1988;454:51–59. doi: 10.1016/0006-8993(88)90802-5. [DOI] [PubMed] [Google Scholar]
- Gee CE, Benquet P, Raineteau O, Rietschin L, Kirbach SW, Gerber U. NMDA receptors and the differential ischemic vulnerability of hippocampal neurons. Eur J Neurosci. 2006;23:2595–2603. doi: 10.1111/j.1460-9568.2006.04786.x. [DOI] [PubMed] [Google Scholar]
- Goodkin HP, Kapur J. Responsiveness of Status Epilepticus to Treatment with Diazepan Decreases Rapidly as Seizure Duration Increases. Epilepsy Curr. 2003;3:11–12. doi: 10.1046/j.1535-7597.2003.03104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Yeh JL, Kapur J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J Neurosci. 2005;25:5511–5520. doi: 10.1523/JNEUROSCI.0900-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J Neurosci. 2008;28:2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans DR, Clayton DA, Coultrap SJ, Browning MD. LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat Neurosci. 2002;5:27–33. doi: 10.1038/nn779. [DOI] [PubMed] [Google Scholar]
- Guerguerian AM, Brambrink AM, Traystman RJ, Huganir RL, Martin LJ. Altered expression and phosphorylation of N-methyl-D-aspartate receptors in piglet striatum after hypoxia-ischemia. Brain Res Mol Brain Res. 2002;104:66–80. doi: 10.1016/s0169-328x(02)00285-1. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Harney SC, Jane DE, Anwyl R. Extrasynaptic NR2D-containing NMDARs are recruited to the synapse during LTP of NMDAR-EPSCs. J Neurosci. 2008;28:11685–11694. doi: 10.1523/JNEUROSCI.3035-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty TP, Rogawski MA. Felbamate block of recombinant N-methyl-D-aspartate receptors: selectivity for the NR2B subunit. Epilepsy Res. 2000;39:47–55. doi: 10.1016/s0920-1211(99)00108-4. [DOI] [PubMed] [Google Scholar]
- Huo JZ, Dykstra CM, Gurd JW. Increase in tyrosine phosphorylation of the NMDA receptor following the induction of status epilepticus. Neurosci Lett. 2006;401:266–270. doi: 10.1016/j.neulet.2006.03.033. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Nakajima W, Hagberg H. Mechanisms of hypoxic neurodegeneration in the developing brain. Neuroscientist. 2002;8:212–220. doi: 10.1177/1073858402008003007. [DOI] [PubMed] [Google Scholar]
- Jones DM, Esmaeil N, Maren S, Macdonald RL. Characterization of pharmacoresistance to benzodiazepines in the rat Li-pilocarpine model of status epilepticus. Epilepsy Res. 2002;50:301–312. doi: 10.1016/s0920-1211(02)00085-2. [DOI] [PubMed] [Google Scholar]
- Kapur J, Lothman EW. NMDA receptor activation mediates the loss of GABAergic inhibition induced by recurrent seizures. Epilepsy Res. 1990;5:103–111. doi: 10.1016/0920-1211(90)90025-q. [DOI] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17:7532–7540. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohira I, Akiyama K, Daigen A, Otsuki S. Enduring increase in membrane-associated protein kinase C activity in the hippocampal-kindled rat. Brain Res. 1992;593:82–88. doi: 10.1016/0006-8993(92)91267-i. [DOI] [PubMed] [Google Scholar]
- Kohr G, Jensen V, Koester HJ, Mihaljevic AL, Utvik JK, Kvello A, Ottersen OP, Seeburg PH, Sprengel R, Hvalby O. Intracellular domains of NMDA receptor subtypes are determinants for long-term potentiation induction. J Neurosci. 2003;23:10791–10799. doi: 10.1523/JNEUROSCI.23-34-10791.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblum HI, Sankar R, Shin DH, Wasterlain CG, Gall CM. Induction of brain derived neurotrophic factor mRNA by seizures in neonatal and juvenile rat brain. Brain Res Mol Brain Res. 1997;44:219–228. doi: 10.1016/s0169-328x(96)00224-0. [DOI] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL. Calcineurin acts via the C-terminus of NR2A to modulate desensitization of NMDA receptors. Neuropharmacology. 2002;42:593–602. doi: 10.1016/s0028-3908(02)00031-x. [DOI] [PubMed] [Google Scholar]
- Lagarias JC, Reeds JA, Wright MH, Wright PE. Convergence properties of the Nelder-Mead simplex method in low dimensions. SIAM J Optim. 1998;9:112–147. [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Le Meur K, Galante M, Angulo MC, Audinat E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol. 2007;580:373–383. doi: 10.1113/jphysiol.2006.123570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekishvili T, Hesketh S, Brazier MW, Brown DR. Mouse galectin-1 inhibits the toxicity of glutamate by modifying NR1 NMDA receptor expression. Eur J Neurosci. 2006;24:3017–3025. doi: 10.1111/j.1460-9568.2006.05207.x. [DOI] [PubMed] [Google Scholar]
- Li BS, Sun MK, Zhang L, Takahashi S, Ma W, Vinade L, Kulkarni AB, Brady RO, Pant HC. Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc Natl Acad Sci U S A. 2001;98:12742–12747. doi: 10.1073/pnas.211428098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Chen N, Luo T, Otsu Y, Murphy TH, Raymond LA. Differential regulation of synaptic and extrasynaptic NMDA receptors. Nat Neurosci. 2002;5:833–834. doi: 10.1038/nn912. [DOI] [PubMed] [Google Scholar]
- Li B, Otsu Y, Murphy TH, Raymond LA. Developmental decrease in NMDA receptor desensitization associated with shift to synapse and interaction with postsynaptic density-95. J Neurosci. 2003;23:11244–11254. doi: 10.1523/JNEUROSCI.23-35-11244.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman DN, Mody I. Casein kinase-II regulates NMDA channel function in hippocampal neurons. Nat Neurosci. 1999a;2:125–132. doi: 10.1038/5680. [DOI] [PubMed] [Google Scholar]
- Lieberman DN, Mody I. Properties of single NMDA receptor channels in human dentate gyrus granule cells. J Physiol. 1999b;518(Pt 1):55–70. doi: 10.1111/j.1469-7793.1999.0055r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Skeberdis VA, Francesconi A, Bennett MV, Zukin RS. Postsynaptic density protein-95 regulates NMDA channel gating and surface expression. J Neurosci. 2004;24:10138–10148. doi: 10.1523/JNEUROSCI.3159-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Jover-Mengual T, Wong J, Bennett MV, Zukin RS. PSD-95 and PKC converge in regulating NMDA receptor trafficking and gating. Proc Natl Acad Sci U S A. 2006;103:19902–19907. doi: 10.1073/pnas.0609924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Mazarati AM, Katsumori H, Sankar R, Wasterlain CG. Substance P is expressed in hippocampal principal neurons during status epilepticus and plays a critical role in the maintenance of status epilepticus. Proc Natl Acad Sci U S A. 1999;96:5286–5291. doi: 10.1073/pnas.96.9.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, Kapur J, Stringer JL. Recurrent spontaneous hippocampal seizures in the rat as a chronic sequela to limbic status epilepticus. Epilepsy Res. 1990;6:110–118. doi: 10.1016/0920-1211(90)90085-a. [DOI] [PubMed] [Google Scholar]
- Lu WY, Jackson MF, Bai D, Orser BA, MacDonald JF. In CA1 pyramidal neurons of the hippocampus protein kinase C regulates calcium-dependent inactivation of NMDA receptors. J Neurosci. 2000;20:4452–4461. doi: 10.1523/JNEUROSCI.20-12-04452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Manzerra P, Behrens MM, Canzoniero LM, Wang XQ, Heidinger V, Ichinose T, Yu SP, Choi DW. Zinc induces a Src family kinase-mediated up-regulation of NMDA receptor activity and excitotoxicity. Proc Natl Acad Sci U S A. 2001;98:11055–11061. doi: 10.1073/pnas.191353598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarati AM, Wasterlain CG. N-methyl-D-asparate receptor antagonists abolish the maintenance phase of self-sustaining status epilepticus in rat. Neurosci Lett. 1999;265:187–190. doi: 10.1016/s0304-3940(99)00238-4. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Baldwin RA, Sankar R, Wasterlain CG. Time-dependent decrease in the effectiveness of antiepileptic drugs during the course of self-sustaining status epilepticus. Brain Res. 1998a;814:179–185. doi: 10.1016/s0006-8993(98)01080-4. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Wasterlain CG, Sankar R, Shin D. Self-sustaining status epilepticus after brief electrical stimulation of the perforant path. Brain Res. 1998b;801:251–253. doi: 10.1016/s0006-8993(98)00606-4. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Baldwin RA, Sofia RD, Wasterain CG. Felbamate in experimental model of status epilepticus. Epilepsia. 2000;41:123–127. doi: 10.1111/j.1528-1157.2000.tb00130.x. [DOI] [PubMed] [Google Scholar]
- Mazarati A, Bragin A, Baldwin R, Shin D, Wilson C, Sankar R, Naylor D, Engel J, Wasterlain CG. Epileptogenesis after self-sustaining status epilepticus. Epilepsia. 2002a;43(Suppl 5):74–80. doi: 10.1046/j.1528-1157.43.s.5.25.x. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Sofia RD, Wasterlain CG. Anticonvulsant and antiepileptogenic effects of fluorofelbamate in experimental status epilepticus. Seizure. 2002b;11:423–430. doi: 10.1053/seiz.2002.0677. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Silverstein FS, Cardona D, Hudson C, Chen R, Johnston MV. Systemic administration of MK-801 protects against N-methyl-D-aspartate- and quisqualate-mediated neurotoxicity in perinatal rats. Neuroscience. 1990a;36:589–599. doi: 10.1016/0306-4522(90)90002-l. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Silverstein FS, Johnston MV. Magnesium reduces N-methyl-D-aspartate (NMDA)-mediated brain injury in perinatal rats. Neurosci Lett. 1990b;109:234–238. doi: 10.1016/0304-3940(90)90569-u. [DOI] [PubMed] [Google Scholar]
- McNamara JO, Russell RD, Rigsbee L, Bonhaus DW. Anticonvulsant and antiepileptogenic actions of MK-801 in the kindling and electroshock models. Neuropharmacology. 1988;27:563–568. doi: 10.1016/0028-3908(88)90176-1. [DOI] [PubMed] [Google Scholar]
- Mewasingh LD, Sekhara T, Aeby A, Christiaens FJ, Dan B. Oral ketamine in paediatric non-convulsive status epilepticus. Seizure. 2003;12:483–489. doi: 10.1016/s1059-1311(03)00028-1. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, Raymond LA. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Moga DE, Shapiro ML, Morrison JH. Bidirectional redistribution of AMPA but not NMDA receptors after perforant path simulation in the adult rat hippocampus in vivo. Hippocampus. 2006;16:990–1003. doi: 10.1002/hipo.20227. [DOI] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- Morrisett RA, Jope RS, Snead OC., III Effects of drugs on the initiation and maintenance of status epilepticus induced by administration of pilocarpine to lithium-pretreated rats. Exp Neurol. 1987;97:193–200. doi: 10.1016/0014-4886(87)90293-7. [DOI] [PubMed] [Google Scholar]
- Moussa RC, Ikeda-Douglas CJ, Thakur V, Milgram NW, Gurd JW. Seizure activity results in increased tyrosine phosphorylation of the N-methyl-D-aspartate receptor in the hippocampus. Brain Res Mol Brain Res. 2001;95:36–47. doi: 10.1016/s0169-328x(01)00231-5. [DOI] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Wasterlain CG. Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci. 2005;25:7724–7733. doi: 10.1523/JNEUROSCI.4944-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura M, Moussa R, Bissoon N, Ikeda-Douglas C, Milgram NW, Gurd JW. Changes in phosphorylation of the NMDA receptor in the rat hippocampus induced by status epilepticus. J Neurochem. 2005;92:1377–1385. doi: 10.1111/j.1471-4159.2005.02977.x. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Yasuda R, Oertner TG, Svoboda K. The number of glutamate receptors opened by synaptic stimulation in single hippocampal spines. J Neurosci. 2004;24:2054–2064. doi: 10.1523/JNEUROSCI.5066-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osonoe K, Ogata S, Iwata Y, Mori N. Kindled amygdaloid seizures in rats cause immediate and transient increase in protein kinase C activity followed by transient suppression of the activity. Epilepsia. 1994;35:850–854. doi: 10.1111/j.1528-1157.1994.tb02522.x. [DOI] [PubMed] [Google Scholar]
- Prasad A, Williamson JM, Bertram EH. Phenobarbital and MK-801, but not phenytoin, improve the long-term outcome of status epilepticus. Ann Neurol. 2002;51:175–181. doi: 10.1002/ana.10085. [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Fu Z, Losi G, Hawkins LM, Luo J, Chang K, Wenthold RJ, Vicini S. Relationship between availability of NMDA receptor subunits and their expression at the synapse. J Neurosci. 2002;22:8902–8910. doi: 10.1523/JNEUROSCI.22-20-08902.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF. Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci U S A. 1999a;96:12876–12880. doi: 10.1073/pnas.96.22.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci. 1999b;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rho JM, Donevan SD, Rogawski MA. Mechanism of action of the anticonvulsant felbamate: opposing effects on N-methyl-D-aspartate and gamma-aminobutyric acidA receptors. Ann Neurol. 1994;35:229–234. doi: 10.1002/ana.410350216. [DOI] [PubMed] [Google Scholar]
- Rice AC, DeLorenzo RJ. NMDA receptor activation during status epilepticus is required for the development of epilepsy. Brain Res. 1998;782:240–247. doi: 10.1016/s0006-8993(97)01285-7. [DOI] [PubMed] [Google Scholar]
- Rice AC, DeLorenzo RJ. N-methyl-D-aspartate receptor activation regulates refractoriness of status epilepticus to diazepam. Neuroscience. 1999;93:117–123. doi: 10.1016/s0306-4522(99)00132-3. [DOI] [PubMed] [Google Scholar]
- Rycroft BK, Gibb AJ. Regulation of single NMDA receptor channel activity by alpha-actinin and calmodulin in rat hippocampal granule cells. J Physiol. 2004;557:795–808. doi: 10.1113/jphysiol.2003.059212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Ehlers MD. Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology. 2003;45:755–767. doi: 10.1016/s0028-3908(03)00250-8. [DOI] [PubMed] [Google Scholar]
- Seo DW, Lopez-Meraz ML, Allen S, Wasterlain CG, Niquet J. Contribution of a mitochondrial pathway to excitotoxic neuronal necrosis. J Neurosci Res. 2009;87:2087–2094. doi: 10.1002/jnr.22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Silbergleit R, Durkalski V, Lowenstein D, Conwit R, Pancioli A, Palesch Y, Barsan W. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med. 2012;366:591–600. doi: 10.1056/NEJMoa1107494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Starr MS, Starr BS. The new competitive NMDA receptor antagonist CGP 40116 inhibits pilocarpine-induced limbic motor seizures and unconditioned motor behaviour in the mouse. Pharmacol Biochem Behav. 1994;47:127–131. doi: 10.1016/0091-3057(94)90121-x. [DOI] [PubMed] [Google Scholar]
- Suchomelova L, Baldwin RA, Kubova H, Thompson KW, Sankar R, Wasterlain CG. Treatment of experimental status epilepticus in immature rats: dissociation between anticonvulsant and antiepileptogenic effects. Pediatr Res. 2006;59:237–243. doi: 10.1203/01.pdr.0000196333.16608.30. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–264. doi: 10.1016/s0896-6273(02)00658-x. [DOI] [PubMed] [Google Scholar]
- Treiman DM, Meyers PD, Walton NY, Collins JF, Colling C, Rowan AJ, Handforth A, Faught E, Calabrese VP, Uthman BM, Ramsay RE, Mamdani MB. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med. 1998;339:792–798. doi: 10.1056/NEJM199809173391202. [DOI] [PubMed] [Google Scholar]
- Triller A, Choquet D. Synaptic structure and diffusion dynamics of synaptic receptors. Biol Cell. 2003;95:465–476. doi: 10.1016/j.biolcel.2003.07.001. [DOI] [PubMed] [Google Scholar]
- van Zundert B, Yoshii A, Constantine-Paton M. Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal. Trends Neurosci. 2004;27:428–437. doi: 10.1016/j.tins.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Vissel B, Krupp JJ, Heinemann SF, Westbrook GL. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat Neurosci. 2001;4:587–596. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]
- Walton NY, Treiman DM. Response of status epilepticus induced by lithium and pilocarpine to treatment with diazepam. Exp Neurol. 1988;101:267–275. doi: 10.1016/0014-4886(88)90010-6. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG. Inhibition of cerebral protein synthesis by epileptic seizures without motor manifestations. Neurology. 1974;24:175–180. doi: 10.1212/wnl.24.2.175. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Liu H, Mazarati AM, Baldwin RA, Shirasaka Y, Katsumori H, Thompson KW, Sankar R, Pereira de Vasconselos A, Nehlig A. Self-sustaining status epilepticus: a condition maintained by potentiation of glutamate receptors and by plastic changes in substance P and other peptide neuromodulators. Epilepsia. 2000;41(Suppl 6):S134–S143. doi: 10.1111/j.1528-1157.2000.tb01572.x. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Baldwin R, Naylor DE, Thompson KW, Suchomelova L, Niquet J. Rational polytherapy in the treatment of acute seizures and status epilepticus. Epilepsia. 2011;52(Suppl 8):70–71. doi: 10.1111/j.1528-1167.2011.03243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AJ, Sjostrom PJ, Hausser M, Nelson SB, Turrigiano GG. A proportional but slower NMDA potentiation follows AMPA potentiation in LTP. Nat Neurosci. 2004;7:518–524. doi: 10.1038/nn1220. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Prybylowski K, Standley S, Sans N, Petralia RS. Trafficking of NMDA receptors. Annu Rev Pharmacol Toxicol. 2003a;43:335–358. doi: 10.1146/annurev.pharmtox.43.100901.135803. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Sans N, Standley S, Prybylowski K, Petralia RS. Early events in the trafficking of N-methyl-D-aspartate (NMDA) receptors. Biochem Soc Trans. 2003b;31:885–888. doi: 10.1042/bst0310885. [DOI] [PubMed] [Google Scholar]
- Wyneken U, Marengo JJ, Villanueva S, Soto D, Sandoval R, Gundelfinger ED, Orrego F. Epilepsy-induced changes in signaling systems of human and rat postsynaptic densities. Epilepsia. 2003;44:243–246. doi: 10.1046/j.1528-1157.2003.17602.x. [DOI] [PubMed] [Google Scholar]
- Yaka R, Thornton C, Vagts AJ, Phamluong K, Bonci A, Ron D. NMDA receptor function is regulated by the inhibitory scaffolding protein, RACK1. Proc Natl Acad Sci U S A. 2002;99:5710–5715. doi: 10.1073/pnas.062046299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen W, Williamson J, Bertram EH, Kapur J. A comparison of three NMDA receptor antagonists in the treatment of prolonged status epilepticus. Epilepsy Res. 2004;59:43–50. doi: 10.1016/j.eplepsyres.2004.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]