

Abstract
Skeletal muscle atrophy is the consequence of protein degradation exceeding protein synthesis. This arises for a multitude of reasons including the unloading of muscle during microgravity, post-surgery bedrest, immobilization of a limb after injury, and overall disuse of the musculature. The development of therapies prior to skeletal muscle atrophy settings to diminish protein degradation is scarce. Mitochondrial dysfunction is associated with skeletal muscle atrophy and contributes to the induction of protein degradation and cell apoptosis through increased levels of ROS observed with the loss of organelle function. ROS binds mtDNA, leading to its degradation and decreasing functionality. Mitochondrial transcription factor A (TFAM) will bind and coat mtDNA, protecting it from ROS and degradation while increasing mitochondrial function. Exercise stimulates cell signaling pathways that converge on and increase PGC-1α, a well-known activator of the transcription of TFAM and mitochondrial biogenesis. Therefore, in the present review we are proposing, separately, exercise and TFAM treatments prior to atrophic settings (muscle unloading or disuse) alleviate skeletal muscle atrophy through enhanced mitochondrial adaptations and function. Additionally, we hypothesize the combination of exercise and TFAM leads to a synergistic effect in targeting mitochondrial function to prevent skeletal muscle atrophy.
Keywords: exercise, mitochondrial transcription factor A, skeletal muscle, atrophy, mitochondrial function
BACKGROUND
Skeletal muscle atrophy is associated with disease, aging, injury, nutritional decrements, and disuse. Many pathways contribute to muscle atrophy but ultimately the imbalance of excessive protein degradation without a corresponding increase in protein synthesis will result in a net muscle tissue loss. Atrophy is also correlated with mitochondrial dysfunction. Many protein degradation and cell apoptosis signaling pathways are stimulated by increased levels of reactive oxygen species, as occurs in mitochondrial dysfunction. Treatments targeting the mitochondria to alleviate high levels of reactive oxygen species and therefore limit protein degradation and apoptosis in skeletal muscle is an attractive area of research. Specifically, treatments that may be prescribed prior to entering known atrophic settings, such as bedrest after elective surgery or microgravity situations, could prevent atrophy and maintain muscle tissue. Maintaining skeletal muscle mass during times of atrophic settings improves health and quality of life.
This review will focus on the role of mitochondrial dysfunction in skeletal muscle atrophy as well as treatments to alleviate mitochondrial dysfunction. Moreover, exercise and mitochondrial transcription factor A both induce mitochondrial biogenesis and increase mitochondrial function. Both of these treatments will be highlighted as potential therapies in the prevention of skeletal muscle atrophy prior to atrophic conditions.
MAIN TEXT
Mitochondria
Mitochondria are cellular organelles responsible for the production of energy in the form of ATP and also play a role in apoptotic signaling. These organelles are believed to be directly descended from pro-bacteria forming an endosymbiotic relationship with an original eukaryotic host cell, leading to the self-replicating nature of mitochondria we observe today (Jornayvaz and Shulman, 2010). The maternal genetics create the mitochondrial lineage of most animals (Cloonan and Choi, 2016). Upon fertilization, paternal mitochondrial DNA (mtDNA) is selectively degraded by a mitochondrial endonuclease. Along with protease and autophagy mechanisms, this degradation leads to mitochondria from the paternal side being effectively abolished, allowing for maternal mtDNA to replicate and normal development of the organism (Zhou et al., 2016).
MtDNA
The mtDNA is a double-stranded, circular genome located in the inner mitochondrial matrix of the organelle, compartmentally separate from the nuclear DNA of the cell. MtDNA is approximately 16.5 kilobases coding for 37 genes. Thirteen of these genes encode functional subunits of the electron transport chain which are essential for oxidative phosphorylation (OXPHOS) (Bolisetty and Jaimes, 2013; Jornayvaz and Shulman, 2010). Twenty-two genes encode transfer RNAs while 2 genes encode ribosomal RNAs, all of which are involved in the synthesis of mitochondrial proteins. The remaining protein of the mitochondria requires nuclear DNA, cytosolic translation, and effective import into the organelle.
MtDNA is essential to maintain energy homeostasis in the organism. Defects in mtDNA are associated with a multitude of mitochondrial related diseases and phenotypes such as diabetes, cancer, aging, and cardiovascular disease (Campbell et al., 2012; Jornayvaz and Shulman, 2010; van Osch et al., 2015) While many diseases may not originate in the mitochondria, there may still be an element of mitochondrial dysfunction involved in the disease genesis or progression. Alterations in the mitochondrial genome change the morphology and physiology of specific tissues. In skeletal muscle, Gehrig et al. observed a fiber-type switching from type I and more oxidative muscle fibers towards type II and more glycolytic muscle fibers in humans with mitochondrial myopathy (Gehrig et al., 2016). This type of mitochondrial defect changes the form and function of skeletal muscle leading to variations in the predominant energy sources used, increased muscle weakness, and decreased muscle health by a loss of oxidative capacity. These abnormalities of mtDNA occur in a variety of ways including DNA methylation, DNA variants, reactive oxygen species (ROS) interactions, and transcriptional machinery defects. The consequence of these issues affects mtDNA copy number availability and interrupts typical synthesis of proteins in this organelle (St John, 2016). In the skeletal muscle of aging humans, increases in mtDNA mutations and decreases in mtDNA has been observed (Short et al., 2005) and is typically associated with decreases in muscle mass (atrophy) and function. If the synthesis of mitochondrial and OXPHOS proteins is disrupted by an abnormality or a decrease in mtDNA copy number, a decrement in the efficient production of ATP (Valero, 2014) and the overproduction of ROS occurs. This increase in ROS further associates with mtDNA and leads to greater mutation and degradation creating a disturbance in energy homeostasis and an increased potential for association with disease. Excess ROS is also implicated in the activation of protein degradation and apoptosis pathways in muscle through ubiquitin-proteosome and caspase pathways, respectively (Siu, 2009; Tisdale, 2005). An unbalanced increase in these pathways is highly correlated to skeletal muscle atrophy (Chopard et al., 2009). Therefore, we conclude maintaining the mtDNA structure and mtDNA copy number in order to transcribe mitochondrial proteins are vital components of proper health and function in skeletal muscle.
Interventions to improve mitochondrial dysfunction and promote normal function have been widely studied. Treatments include pharmacologic, nutritional supplementation, and exercise prescription. These treatments generally attempt to modulate one or more of the various signaling pathways involved in the creation of mitochondrial proteins, also termed mitochondrial biogenesis, to counteract dysfunctional effects (Kang et al., 2013; Liang et al., 2014; Saleem and Hood, 2013). The effects of exercise specifically correlate with an increase in transcription factors of the mitochondria in skeletal muscle. A 15-minute bout of endurance exercise in mice increased nuclear encoded and mitochondrial encoded genes involved in mitochondrial biogenesis including key transcription factors and signaling proteins (Saleem and Hood, 2013). Furthermore, a study inducing contractile activity of the rat tibialis anterior muscle by electrical stimulation revealed a significant increase in mitochondrial transcription factor A (TFAM), a mtDNA regulatory protein. Results revealed increased TFAM mRNA levels after four days of stimulation while also increasing mitochondrial TFAM protein levels and mitochondrial specific enzyme after 5 days (Gordon et al., 2001). This indicated TFAM expressions connection to the creation of new mitochondria with exercise. Thus, the present review will emphasize the role of exercise in inducing mitochondrial biogenesis pathways associated with TFAM as a mechanism to preserve mtDNA and protect skeletal muscle tissue from atrophy.
TFAM
As mentioned previously and indicated in the literature, a key component of properly functioning mtDNA is mitochondrial transcription factor A (TFAM) (Kunkel et al., 2016). TFAM is a diversely functioning protein playing a role in mtDNA transcription, organization, and maintenance. Lack of this transcription factor in systemic TFAM knockout mice resulted in severe mtDNA depletion and embryonic lethality (Stiles et al., 2016). Overexpression of TFAM in cardiac myocytes in a transgenic mouse model increased mtDNA copy number and diminished pathological hypertrophy after myocardial infarction. This subsequently led to an increase in survival rate (Fujino et al., 2012).
TFAM is a low molecular weight molecule (~25 kDa) with non-specific DNA binding properties encoded in the nucleus and transcribed in the cytosol as a pre-protein. From here, it is shuttled to the mitochondrion by a complex of cytosolic heat shock proteins (HSPs), HSP60 and HSP70, that act as protective chaperones and as a guide to locate the mitochondria. HSP protection prevents phosphorylation, promotes proper folding, and deters degradation during transport (Kunkel et al., 2016). The HSP60/HSP70 complex and attached pre-protein then bind to specific cytosolic receptors located on the translocase of the outer membrane complex (TOM) of the mitochondria, which serves as a main entry point for many mitochondrial proteins (Paschen and Neupert, 2001). The TFAM pre-protein will, next, pass from the TOM, through the intermembrane space, to a channel of the translocase of the inner membrane (TIM), specifically TIM23, using the selectivity of this complex for positively charged molecules (Santos and Kowluru, 2013). Entry into the matrix is achieved first by the binding of a mitochondrial heat shock-protein chaperone, mitochondrial HSP70, to TIM23 at a specific docking site (TIM44). TIM44-associated HSP70 causes conformational changes in HSP70 that either passively traps or actively pulls TFAM to the chaperone, as the precise mechanism is not completely understood (Schmidt et al., 2010). A mitochondrial processing peptidase (MPP) will cleave off the targeting sequence of the pre-protein on the matrix side of TIM, while a chaperone, mtHSP60, along with mtHSP70, will re-fold the protein into its mature form inside of the matrix (Hood et al., 2003). Again, in the inner mitochondrial matrix, the mitochondrial heat shock protein complex mtHSP60/mtHSP70 serves as a protector of TFAM from degradation. Specifically, the mitochondrial protease, LON, selectively degrades oxidatively modified and improperly folded TFAM unbound to mtDNA (Matsushima et al., 2010). After effective transport into the matrix, properly folded TFAM is able to come into close proximity with mtDNA.
TFAM Binding: Transcription Initiation and Scaffolding
Free mtDNA and free TFAM are both rapidly degraded in the matrix. As mentioned previously, mtDNA interacting with ROS promotes a decrease in mtDNA copy number and unbound TFAM is cleaved by Lon protease. TFAM interacting with mtDNA will form a protein-DNA complex known as a mitochondrial nucleoid which prevents the degradation of each component (Kang et al., 2007). This complex forms these nucleoid structures as TFAM binds and coats, potentially, the entire mitochondrial genome. This provides the structural stability of the mtDNA. Ekstrand et al. revealed that the levels of total TFAM were directly proportional to the number of mtDNA copies in a mouse embryo model (Ekstrand et al., 2004). Further, work done by Larsson, et al., revealed that heterozygous TFAM depletion resulted in 35–40% decreases in mtDNA copy numbers while homozygous TFAM depletion resulted in embryonic lethality due to severe depletions of mtDNA copies and OXPHOS (Larsson et al., 1998). These results provide strong evidence of the correlation of TFAM and mtDNA and supports the role of TFAM preventing degradation of mtDNA.
TFAM will abundantly bind mtDNA both specifically and non-specifically. Two distinct promoter regions of mtDNA, light strand promoter (LSP) and heavy strand promoter 1 (HSP1), are bound by TFAM while other non-distinct regions of the mitochondrial genome are also able to bind. The specific binding of TFAM to these promoter regions activates transcription of the genome.
Specific binding of TFAM upstream of promoter sites induces bending of the genome and recruitment of transcriptional machinery. Two high mobility group-box domains (HMG-box A and HMG-box B) insert into grooves on the LSP, HSP1, or non-specific regions of mtDNA. These insertions cause distortion of the mtDNA and result in bending (Ngo et al., 2014). Bending allows TFAM to interact with the transcriptional machinery, specifically mitochondrial RNA polymerase (POLRMT). POLRMT has sequence specific binding to TFAM and mtDNA promoter regions as well as upstream regions. This complex encourages binding of mitochondrial transcription factor B2 (TFB2M), resulting in a single RNA precursor in the mitochondrion and is the initiation of transcription (Kühl et al., 2016). This process further advances to transcription elongation, transcription termination, initiation of translation, and translation elongation ultimately resulting in the synthesis of mature mitochondrial proteins (Taanman, 1999).
While the vast majority of mitochondrial proteins are nuclear encoded and require organelle import, intramitochondrial protein synthesis is still a highly necessary process to ensure proper function and maintenance of the mitochondrion and the OXPHOS system. TFAM, as previously described, is a key regulator of this process.
Exercise and TFAM: Upstream Factors
Exercise enhances physical performance and is well known to be associated with many positive health benefits (Warburton et al., 2006). A single bout of exercise causes a change in molecular expression in muscle that promotes specific adaptations. If the appropriate duration, frequency, and intensity of contractile activity in muscle tissue occurs from physical exercise, specific signaling mechanisms will ensue, leading to the tissue adapting to the demand of the physical stress. Levels of circulating inflammatory markers, growth factors, and adrenergic compounds also increase during exercise. Exercise induces phenotypic changes of the muscle that include increased cross sectional area, increased capillary density, fiber type transitioning, and mitochondrial biogenesis resulting in increased mitochondrial density of the muscle cell (Garatachea et al., 2015; Yan et al., 2012). This increased mitochondrial density is a part of an adaptive process allowing the organism experiencing the stress of exercise to increase functional proteins involved in the creation of energy (mitochondria and the OXPHOS system) in order to handle that specific stress in the future. A key component of exercise-induced mitochondrial biogenesis occurs via the effects of skeletal muscle contraction and the upregulation of TFAM (Gordon et al., 2001).
For a skeletal muscle to contract, an action potential(s) must propagate from the motor cortex, through nerve fibers of the central nervous system, synapse at motor neurons located on the spinal cord down to the neuromuscular junction (NMJ). The signal must cross the NMJ and further propagate across the sarcolemma of the muscle and down into the t-tubules where voltage-gated dihydropyridine receptors (DHPRs) are able to interact with ryanodine receptors. Ryanodine receptor activation releases calcium from the sarcoplasmic reticulum (SR) into the cytoplasm of the muscle cell (Rebbeck et al., 2014). From there, calcium is able to bind to troponin and remove tropomyosin from active binding sites of actin contractile proteins. Finally, in the presence of adequate ATP, muscle contractions are able to occur by the recycling of cross-bridging between myosin and actin proteins. During exercise, potentially high rates of this “excitation-contraction coupling” process occurs. The release of calcium from the SR as well as the increased demand for ATP in this process leads to significant molecular changes within the cell.
The amount of cytosolic calcium released from the SR in skeletal myocytes during contraction not only depends on duration, frequency, and intensity of exercise of the muscle but also on the muscle fiber type. Isolated single muscle fiber calcium has been measured previously to be concentrated in the range of 30–50 nM. Upon stimulation, type I (slow-twitch) muscle fibers produced concentrations between 100–300 nM of calcium while type II fibers (fast-twitch) are capable of a drastically higher (~10x) 1–2 μM range (Godin et al., 2010). Baylor and colleagues report fast-twitch fibers capable of 3–4 times the amount of slow twitch (Baylor and Hollingworth, 2012). Nonetheless, cytoplasmic calcium concentrations in contracting fast-twitch muscle appear to be much greater than slow-twitch. This is important because specific types of exercise requiring higher relative intensities and perhaps longer relative durations recruit type II fibers at a greater rate than lower intensity exercise and disuse of the muscle (Krustrup et al., 2004). This leads to high levels of cytosolic calcium and increased intracellular signaling. Specifically, increased calcium activates calcium/calmodulin-dependent protein kinases (CAMKs) that are linked to increases in mitochondrial biogenesis (Chin, 2005; Combes et al., 2015).
Increasing the intensity of physical activity also increases the AMP/ATP ratio in skeletal muscle (King-Himmelreich et al., 2016; Wang et al., 2003). As the previously described contraction process takes places and the cellular energy demand increases, the use of phosphate groups from ATP increases the amount of ADP and AMP, thus increasing the AMP/ATP ratio. AMP interacts with an AMP-activated protein kinase (AMPK) that is interrupted by high levels of ATP (Hardie, 2004). The β-subunit of AMPK has a glycogen binding domain that, when bound, inhibits the activation of AMPK (McBride and Hardie, 2009). Exercise not only increases the AMP/ATP ratio but also depletes muscle glycogen, potentially releasing and activating AMPK. For these reasons AMPK is a cellular energy sensor and has been characterized as a “master switch” of metabolism (Jørgensen et al., 2006). In response to exercise, activation of AMPK leads to downstream activators of mitochondrial biogenesis (Bergeron et al., 2001; Seo et al., 2015).
Exercise will also increase the levels of ROS in muscle (Dimauro et al., 2016). ROS has been implicated in many health disorders including muscle myopathies (Moulin and Ferreiro, 2016). The major contributors to ROS in skeletal muscle include xanthine oxidase (XO), NADPH-oxidases (NOXs), and mitochondria. In mitochondria, ROS production occurs due to electron leakage in the electron transport chain (ETC) (Gutterman, 2005). The ETC passes electrons through a series of protein complexes along the inner mitochondrial membrane with oxygen being the last acceptor of the electron. As oxygen accepts electrons in the ETC, it typically results in the formation of water. Oxygen that undergoes incomplete reductive processes results in radicals and the potential formation of ROS. Complex I and Complex III of the ETC appear to be the greatest contributors of ROS in the mitochondria. Although the precise contributions of each is poorly understood, these locations in muscle cells produce oxidants that are capable of negatively affecting cellular function while also activating potentially beneficial signaling pathways. An example of this was demonstrated in which male Wistar rat groups performed exercise and ate high antioxidant diets. The groups eating high antioxidant diets revealed decreased levels of mitochondrial biogenesis markers, regardless of exercise, indicating a potentially negative outcome of decreasing ROS signaling (Strobel et al., 2011). The maintenance and balance of ROS through antioxidant defense systems and adaptations to the mitochondria are important factors in skeletal muscle health and control potential mitochondrial biogenesis signaling.
Other major signaling mechanisms of exercise and mitochondrial biogenesis include the increased levels of inflammatory markers, epinephrine, and growth factors. Markers such as interleukins (IL-1,6) and tumor necrosis factor-alpha (TNF-α) bind cellular receptors and lead to increases in ROS production (Kim et al., 2010). Adrenal glands release epinephrine in response to exercise. Speculatively, this could be because of both the physical stress that is perceived as well as an emotional response that potentially occur during more intense forms of exercise, causing the release of epinephrine. Binding of epinephrine to β-adrenergic receptors of the cell lead to increases in cyclic AMP (cAMP), involved in biogenesis pathways. Growth factors such as insulin-like growth factor-1 (IGF-1) and insulin enter the cell and activate protein synthesis pathways, also implicated in the creation of mitochondrial proteins (Morita et al.).
Exercise and TFAM: Downstream Factors
The various effects of skeletal muscle contraction and the stress of exercise connect to mitochondrial biogenesis in several ways. These upstream signals converge downstream to ultimately increase TFAM transcription. This convergence point that is a major link in mitochondrial biogenesis, and has received a great deal of attention in the literature, is the peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC-1α) and is activated in a multitude of ways (Fernandez-Marcos and Auwerx, 2011; Olesen et al., 2010; Suwa et al., 2008).
PGC-1α is a transcriptional coactivator with formidable evidence of its correlation exercise. Endurance exercise increases levels of PGC-1α in rat soleus muscle 18 hours post-exercise (Suwa et al., 2008). Kang et al. subjected female Sprague-Dawley rats to anaerobic sprinting exercise and noticed a 5.6-fold increase in PGC-1α in the exercise group compared to the controls (Kang et al., 2009). Another group subjected rats to 20 minutes of aerobic treadmill running and increased PGC-1α mRNA levels 1.5–5 fold in soleus and gastrocnemius muscles while also noting 6 weeks of chronic exercise increased PGC-1α mRNA levels by ~25% in rat soleus muscle (Bocco et al., 2016).
The aforementioned increase in cytosolic calcium with exercise activates CAMK which upregulates PGC-1α (Chin, 2005). CAMK is also a potent activator of myocyte-enhancer factor-2 (MEF2) (McKinsey et al.). MEF2 is capable of binding to the nuclear promoter regions of the PGC-1α gene, enhancing its transcription (Liang and Ward, 2006). As AMP levels increase due to the demand of physical activity it interacts with AMPK, which also directly activates PGC-1α (Seo et al., 2015). ROS increases during exercise activate p38 mitogen-activated protein kinase (p38MAPK) that upregulates PGC-1α (Bartlett et al., 2014). ROS signaling also includes the activation of AMPK indirectly, although it is possible this is due to the decrease in ATP associated with stressful events (Irrcher et al., 2009). While ROS signaling leads to biogenesis of mitochondrial proteins, excessive ROS is detrimental to the cell. Furthermore, silent mating type information regulator 2 homolog (SIRT1) upregulation due to increased levels of nicotinamide adenine dinucleotide (NAD+) caused by exercise will deacetylate PGC-1α leading to its activation (Srivastava, 2016). Epinephrine released in response to exercise increases adenylyl cyclase leading to increased cAMP. A cAMP dependent protein kinase (PKA) activates with increased cAMP, and further interacts and upregulates cAMP response element binding protein (CREB) that plays a role in PGC-1α upregulation. Growth factors (GFs) such as IGF-1 and insulin, as well as mechanical loading of muscle, activate mammalian target of rapamycin (mTOR), a major protein synthesis pathway that also promotes PGC-1α activation (Cunningham et al., 2007; Morita et al., 2015).
PGC-1α is able to co activate nuclear respiratory factors (NRFs), specifically NRF-1 and NRF-2, nuclear transcription factors responsible for activating many mitochondrial proteins (Tokusumi et al., 2004). When this occurs, TFAM mRNA is transcribed from the nuclear genome by the NRFs where it is able to be exported to the cytosol for translation, imported into the mitochondria, and maintain mtDNA and activate transcription of mitochondrial proteins (refer to Figure 1).
Figure 1.

Mitochondrial Import of TFAM
An exercise stimulus is a powerful metabolic signal, as we can see, that activates a multitude of signaling pathways in skeletal muscle leading to the transcription of TFAM, a key modulator of mitochondrial biogenesis. For these reasons, exercise and TFAM are attractive variables to study in skeletal muscle pathology.
Skeletal Muscle Atrophy
Skeletal muscle atrophy is caused by a host of factors including disease, aging, injury, nutritional decrements, and disuse. It is characterized by a decrease in the cross sectional area of the muscle, a decline in force generative capabilities, decrease in functional proteins of the muscle mass, and a loss of oxidative ability making the tissue less resistant to fatigue. There is a clear link in the literature with the dysfunction of the mitochondria and skeletal muscle atrophy (Kang et al., 2016; Mukai et al., 2016). The cause of both depends on the originating factor (disease, starvation, etc.).
The disuse of muscle occurs for many reasons including bed rest from disease, cast immobilization due to injury, spinal cord injury, weightlessness environments, sedentary lifestyle. Ultimately, the lack of muscle contraction and lack of stimuli loading the muscle encompasses disuse. When this happens, increased activation of protein signaling pathways leading to protein degradation and apoptosis arise with corresponding downregulation of protein synthesis pathways. When protein degradation exceeds protein synthesis over time, atrophy occurs along with a decrease in mitochondrial function.
Protein Degradation
Mechanical stress of contracting skeletal muscle under a load, as occurs in exercise, causes many changes throughout skeletal myocytes. Contraction induced by myosin and actin cross-bridging is sensed by the cell through proteins such as dystrophin and integrin complexes (Barton and Morris, 2003). Dystrophin is a cytoplasmic protein that interacts with actin fibers under the cell surface and further associates with other related proteins to form a dystrophin complex that spans the entire sarcolemmal membrane (Gao and McNally, 2015). This acts as a “shock absorber” and provides stiffness to the contracting myocyte in order to prevent the force from being transferred into the sarcolemma inappropriately, causing deformation of the membrane.
Pasternak’s group demonstrated this by using dystrophin knockout (mdx) mice lacking this protein (similar to humans with Duchenne muscular dystrophy). Mdx mice displayed fragile myotubes with roughly 4 times less stiffness than normal myotubes (Pasternak et al., 1995). This important protein and its respective complex, along with other sensory complexes (integrin), activates focal adhesion kinases (FAKs) and mediates the prevention of apoptosis and activation of growth pathways (Barton and Morris, 2003). With disuse, mechanical stress does not occur and the corresponding growth signaling and apoptosis prevention is prohibited. Further, lack of contraction leads to the activation of catabolic pathways in skeletal muscle.
The ubiquitin-proteasome pathway is a main regulator of protein degradation. Muscle ring finger-1 (Murf-1) and Atrogin-1/MAFbx are E3 ligases in skeletal muscle that play a key role in ubiquitin marked degradation (Gumucio and Mendias, 2013). These ligases are activated by multiple stimuli, in particular ROS, inflammatory markers, and the unloading of muscle. Activation of IκB kinase (IKK) occurs via these stimuli, which is associated with the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (Bodine and Baehr, 2014). NF-κB is a transcription factor responsible for transcription of many degradative genes. In this case, NF-κB is able to translocate into the nucleus and play a role in the transcription of Murf-1 and Atrogin-1/MAFbx. Mature Murf-1 and Atrogin-1/MAFbx proteins polyubiquinate polypeptides in the muscle as a method of targeting the protein for degradation. Skeletal muscles consist mostly of the myofibrillar proteins myosin and actin involved in the functional aspect of the cells. Therefore, for significant atrophy to occur, these proteins must be degraded.
Murf-1 is involved in targeting myosin light chains for degradation while atrogin-1/MAFbx targets actin molecules (Bilodeau et al., 2016). After these myofibrillar proteins are polyubiquinated, proteasomes attach and degrade the protein resulting in atrophy. Tawa et al. demonstrated the importance of the ubiquitin pathway by inhibiting proteasome activity in the rat soleus muscle of three atrophy inducing pathological states (denervation, hyperthyroidism, sepsis). Without altering protein synthesis expression, protein balance increased with high percentages of reductions in proteolysis and an increase in ubiquinated proteins of the muscle was observed (Tawa et al., 1997). This study, among others, delineated the ubiquitin-proteasome pathway as the key modulator of protein degradation and thus, should be considered when working with skeletal muscle atrophy models. It is possible that different pathways contribute to degradation in different settings based upon the conditions of the model and should also be considered when reviewing data.
Other protease contributors in skeletal muscle protein degradation include calpains and lysosomes (Chopard et al., 2009). Calpains are intracellular proteases that are regulated by Ca2+. In skeletal muscle, calpain-1, calpain-2, and calpain-3 are mainly expressed. Calpain-3 is downregulated during atrophy as well as in exercise suggesting that its absence is necessary for atrophy (Bartoli and Richard, 2005). Recently, Shenkman and colleagues used an inhibiting agent to diminish calpain-1 in a rat model while inducing skeletal muscle atrophy by hindlimb suspension for 3 days. The soleus muscle size was significantly reduced in rats that were only suspended while those suspended and treated with the calpain-1 inhibitor retained muscle size (Shenkman et al., 2015). Calpain-1 levels were increased in the suspension only model. Interestingly, levels of calpain-2 were not affected by hindlimb suspension induced atrophy, indicating a key role of calpain-1 in short-term (3 days) disuse. Inhibiting calpain-1 decreases MAFbx expression and inhibits protein ubiquitination. Studies increasing the atrophy duration should be done to further elucidate these changes in protein expression.
Lysosomal activity is also a contributing factor to protein degradation in muscle. Autophagy using lysosomal machinery (autophagy-lysosomal system) is upregulated during catabolic states of skeletal muscle (Sandri, 2013). Normal contraction of muscle tissue over time causes damage to the contracting proteins as well as organelles of the cell. In order for these damaged proteins and organelles to be degraded and recycled to continue normal or enhanced function, the cells must have a self-selecting degradative system. This system is autophagy.
Again, NF-κB pathway is an important transcription factor inducing autophagy. Autophagosomes consume defective proteins of the cell and transport the defective material to lysosomes. Lysosomes are membrane-bound vesicles containing acid hydrolases that, when released, degrade the cellular components of the autophagosome (Bechet et al., 2005). The process is also induced by multiple types of exercise(Ju et al., 2016; Schwalm et al., 2015). This could be to enhance the removal of damaged proteins due to the stress of exercise contractions or may also be to provide energy for sustained contractions in the cell. While this process positively impacts the function of skeletal muscle tissue, excessive autophagy-lysosomal activity, as seen in many myopathies, leads to atrophy.
Cell Death (Apoptosis)
Cells undergo a programmed cell death (apoptosis) due to specific biochemical interactions that cause blebbing of the cell membrane, condensation of chromatin, and cell fragment lysis resulting in the death of the cell. When skeletal muscle is not contracting over time, as occurs in disuse, an increase in cell apoptosis has been observed (Siu et al., 2009). As load and contractions of the muscle diminish, the removal of myonuclei occurs and is associated with apoptosis (Barton and Morris, 2003) along with the destruction of cellular proteins. The myonuclear domain is reduced, resulting in diminished function in the myofiber.
Caspases are proteases within the cell that play a role in the disassembly of the nucleus and cytoskeleton while cleaving many other cellular proteins. Caspases are categorized as “initiators” and “executioners” with caspase-2, -6, -9, -10 being characterized as initiators while caspase-3, -7, and -8 are executioners (Connolly et al., 2014). Specifically, caspase-3 is a major executioner involved in skeletal muscle atrophy. Signalling for caspase-3 includes both extrinsic and intrinsic factors. Extrinsic factors include binding of the tumor necrosis factor (TNF) ligand to TNF death receptors on transmembrane proteins. This TNF binding causes a signal cascade that results in procaspase-8 and the activation of the caspase-3 executioner. Capase-3 activates an endonuclease (CAD) that degrades DNA (Elmore, 2007) and thus, the execution of apoptosis.
A major intrinsic pathway leading to caspase-3 activation includes the release of cytochrome c (Cyt c) from the mitochondria. Signals such as elevated levels of ROS and reactive-nitrogen species (RNS) increase mitochondrial membrane permeability that release Cyt c from the mitochondria (Dupont-Versteegden, 2006). B-cell leukemia/lymphoma 2 (Bcl-2) and Bcl-XL block the release of Cyt c and serve an anti-apoptotic role. Members of the Bcl-2 associated death promoter (BAD) family, such as BAX and BAK, serve as pro-apoptotic factors and increase pore permeability of the mitochondria allowing the release of Cyt c (Yang et al., 1997). Cyt c is then involved in the activation of the initiator caspase-9, forming an apoptosome complex. This leads to the activation of the executioner caspase-3 and degradation of cellular components (Wang and Youle, 2009). Leeuwenburgh et al. performed hindlimb suspension on young and old rats over a 14-day period. Their results indicated an 84% increase in caspase-3 activity in the soleus of young rats after inducing atrophy over 14 days (Leeuwenburgh et al., 2005). Furthermore, a study by Nagano and colleagues observed active caspase-3 activity in male Wistar rats after 3-weeks of hindlimb unloading (Nagano et al., 2008). These studies indicate an apoptotic link between disuse-induced muscular atrophy and caspase-3.
There are caspase-dependent and caspase-independent pathways that induce apoptosis. Apoptosis-inducing factor (AIF) is released from the mitochondria upon stimulation from pro-apoptotic signals. This AIF activates apoptosis through chromosomal condensation and DNA fragmentation in a caspase-independent manner (Joza et al., 2009). EndonucleaseG (EndoG) is a protein released from the mitochondria after pro-apoptotic signaling. EndoG is caspase-independent and is associated with DNA fragmentation that coincides with apoptosis during muscle atrophy induced by hindlimb suspension in aged mice (Dupont-Versteegden, 2006). With a multitude of cell signaling pathways involved in cell apoptosis, cross-talk is likely to occur between the various cascades.
Protein Synthesis
The counterbalance to protein degradation in muscle cells is protein synthesis. As stated previously, when degradation exceeds synthesis over time, atrophy occurs. Signals for protein synthesis include many stimulatory factors such as mechanical loading of the muscle, hormone circulation, and nutrition. Conversely, protein synthesis signaling is inhibited by fasting, augmentation of growth factor release or binding, and muscular disuse. Many proteins play a role in the synthesis pathway but the previously mentioned mammalian target of rapamycin (mTOR) is consistently viewed as the major regulator of this process.
Two functional complexes are formed from mTOR termed mTOR complex-1 and mTOR complex-2 (mTORC1 and mTORC2). Although both mTORC1 and mTORC2 have a unique role, both complexes will be encompassed and referenced as ‘mTOR’ for the scope of the current review. Signaling from extracellular growth factors, such IGF-1 and insulin, and amino acids (specifically branched-chain amino acids) lead to the activation of the PI3K/AKT pathway that is associated with the activation of mTOR (Laplante and Sabatini, 2009). Mechanical loading via resistance exercise of the skeletal muscle also activates this pathway to activate mTOR (Drummond et al., 2009; Ogasawara et al., 2016).
Activated mTOR interacts with many downstream effectors including two well-established proteins in particular, S6 kinase (S6K) ribosomal protein and eukaryotic translation initiation factor-4 (eIF-4E) binding protein (4E-BP) (Morita et al., 2015). The phosphorylation of 4E-BP by mTOR allows the unbinding and activation of eIF-4E. Similarly, other eIF family proteins are activated with the phosphorylation and activation of S6K by mTOR. These signaling cascades increase translation initiation rates and thus, overall protein synthesis.
Speculatively, one could assume during skeletal muscle atrophy the rates of protein synthesis would decrease due to the lack of loading stimuli. In this case you could also assume you would observe diminished mTOR signaling. However, results from You et al. revealed an increase in mTOR activity after 7 days of hind limb immobilization in mice (You et al., 2015). Similarly, in a mouse hindlimb unloading model, Liu and colleagues observed increased 4E-BP binding and decreased eIF-4E activation (decreased protein synthesis) after 3 days and diminished binding and enhanced eIF-4E activation (increased protein synthesis) after 14 days. These results may be due to a compensatory or safety mechanism in which protein synthesis signaling is induced in order to protect or save the muscle tissue from wasting after prolonged periods of disuse. Furthermore, as noted previously, during muscle atrophy we observe a decline in normal muscle tissue morphology and function. With variation in protein synthesis during atrophy, it appears the degradation process accompanying muscle disuse and the associated atrophy outweighs any countering protein synthesis that is occurring.
Mitochondrial Dysfunction Associated with Atrophy
Mitochondria play an increasingly important role in the atrophy process. A definitive answer to why atrophy is associated with mitochondrial dysfunction is somewhat unclear. ROS and its damaging effects are associated with the dysfunction of mitochondria and the activation of degradation signaling in muscle. In skeletal muscle, mitochondria are classified as SubSarcolemmal (SS) and InterMyoFibril (IMF), based on their respective locations in the fiber. It appears IMF mitochondria display greater changes in response to exercise. This could be due to their locations, as SS mitochondria are thought to be associated with supplying the cell membrane with energy rather than contractile proteins supplied by IMF mitochondria. During contractile activity, mitochondria of the skeletal muscle cells produce high amounts of ROS. The mitochondria’s ability to withstand the demand of energy from the working cells and its ability to neutralize ROS via local antioxidant defense will determine ROS accumulation in the cell in an acute setting. During chronic stress periods, as occurs in disuse of muscle, the cell’s ability to handle energy demand and neutralize ROS decreases leading to an accumulation of ROS. A group recently investigated the redox balance that occurs during skeletal muscle atrophy using a hindlimb suspension mouse model over 2 weeks. Soleus muscle mass was significantly reduced after 2 weeks, indicating atrophy. ROS generating subunits (Nox1) were significantly elevated indicating ROS is increased during atrophy. Also, a decrease in the expression of antioxidants that scavenge ROS and a decrease in subunits of the mitochondrial complex 1 were observed, further indicating mitochondrial dysfunction associated with atrophy (Nuoc et al., 2016).
A wealth of evidence reveals the negative effects of ROS in the mitochondria including induction of cell apoptosis, protein oxidation, and mtDNA mutations (Lenaz, 1998). As previously explained, the mitochondria serve an important role in cell apoptosis signaling with the release of Cyt c and the change in permeability of mitochondrial membranes. Numerous studies also show changes in mitochondrial density, size, activity, and shape during periods of disuse are associated with muscular atrophy. Mitochondrial autophagy (mitophagy) and mitochondrial dynamics are both observed to change during skeletal muscle atrophy.
The function of mitophagy is to degrade defective mitochondrial proteins. In skeletal muscle atrophy, excessive mitophagy results as functional proteins are damaged due to the effects of increased ROS in the mitochondria. This increase arises with deficiencies in the electron transport chain. Mitophagy occurs under normal, healthy conditions but can also become excessive and lead to the destruction of a disproportionate amount of mitochondrial protein. Keeping mitophagy in balance by limiting excessive ROS production through the maintenance of mitochondrial proteins may be key in limiting dysfunction during periods of muscle disuse.
Mitochondrial homeostatic maintenance through mitochondrial dynamics is also affected during atrophy. This includes the processes of mitochondrial fusion and fission. Mitochondria are constantly merging and dividing, which is important in maintaining normal mitochondrial function. Fusion is particularly important in this process. The outer membrane mitofusin proteins (Mfn1 and Mfn2) and the inner membrane protein optic atrophy 1 (Opa1) play a key role here. During periods of stress, fusion processes are able to mix mitochondrial contents of separate, possibly damaged, mitochondria as a way of complementing each other to increase function (Youle and van der Bliek, 2012). In Mfn2 heart knockout mice, disruption of mitochondrial respiration and a decrement in ATP production was observed (Mourier et al., 2015). Other studies reveal that down regulating these genes hinders embryonic development leading to embryonic death.
Fission processes are thought to be linked to mitophagy and serve as a “quality control” mechanism (Youle and van der Bliek, 2012). With an accumulation of damaged protein in the organelle, a dynamin-related protein 1 (Drp1) is recruited from the cytosol. Drp1 acts as a constrictor of mitochondria, effectively severing portions of the organelle with damaged proteins. This allows for autophagy activity to occur and the normal function of the organelle to continue. Excessive fission or unbalanced fusion and fission processes is detrimental to mitochondrial function.
We elucidated the expression of Drp1 and Mfn-2 in the hyperhomocysteinemia (HHcy) mouse model. Mice with HHcy revealed decreased levels of Mfn-2 and increased Drp1 expression with an associated increase in oxidative stress (Familtseva et al., 2014). Additionally, in a second study we observed a decrease in the Mfn-2 to Drp1 ratio during a heart failure mouse model which suggested an increase in mitophagy (Givvimani et al., 2014). These two studies reveal imbalances in fusion and fission processes associated with disease states.
Cannavino et al. showed during hindlimb suspension the Drp1 protein was upregulated in mouse soleus muscle while levels of Mfn-2 protein were downregulated in gastrocnemius muscle. Furthermore, the same group used a mouse model over-expressing PGC-1α during hindlimb suspension and revealed a prevention of skeletal muscle atrophy and a decrease in Mfn-2 of the gastrocnemius (Leduc-Gaudet et al., 2015). These results indicate a potential tie between atrophy and mitochondrial dynamics. As the evidence elucidating roles of mitochondrial dynamics associated with skeletal muscle atrophy is scarce, this potential link should be studied further in the future.
The diverse role of mitochondria and wealth of evidence connecting the mitochondria to skeletal muscle atrophy has led to many studies targeting this organelle to alleviate negative effects of atrophy from conditions of disuse. Recently, atrophy induced by dexamethasone treatment was shown to induce mitochondrial dysfunction prior to the onset of protein degradation processes (Liu et al., 2016). This treatment caused a decrement of intracellular ATP and loss of mitochondrial components involved in cellular respiration. Treatment with the mitochondrial nutrient resveratrol enhanced mitochondrial function while simultaneously ameliorating muscle atrophy. As discussed previously, resveratrol activates SIRT1. This activation is associated with the deacetylation and activation of PGC-1α, leading to mitochondrial biogenesis. Resveratrol treatment also increases running times and muscle oxygen consumption in mice while inducing mitochondrial biogenesis related mRNA expression, particularly PGC-1α and TFAM (Lagouge et al., 2006). As shown earlier, both of these molecules are also activated by exercise.
Exercise: Skeletal Muscle Protection
Exercise induces a hormetic response in animals. That is, an appropriate dose of exercise over time leads to beneficial cellular adaptations to withstand the stress of exercise. This process of adaptation is at the essence of evolutionary biology. At extreme or high dosages, exercise can be toxic. When appropriate dosages of exercise are applied to an organism, the hormetic response plays a role in the prevention of pathological conditions.
Exercise has been suggested to increase mitochondrial volume by up to 40% (Lundby and Jacobs, 2016). This is due to factors of mitochondrial biogenesis being increased with exercise, signaling the increase in mitochondrial proteins to be synthesized (refer to figure 2). In aging skeletal muscle, a rapid decline in muscle mass and muscle performance parameters are observed as are decreases in mitochondrial volume and biogenesis. Moderate exercise reverses or attenuates the decline in mitochondrial biogenesis markers and reduce the age-associated reduction in skeletal muscle mass (Koltai et al., 2012). Although aging is its own respective form of skeletal muscle atrophy, this elucidates the idea that exercise is able to reverse the effects of atrophy through the induction of mitochondrial biogenesis.
Figure 2.

Exercise Signaling Pathways-TFAM and Mitochondrial Biogenesis
The type of exercise, volume, intensity, and frequency as well as genetic capabilities of the organism will ultimately determine the extent to which muscle cells will adapt. Athletes that undergo intense physical exercise, especially endurance training, drastically increase the amount of mitochondrial proteins, antioxidant defense, and oxidative capabilities as an adaptation that more efficiently produces ATP to meet the demands of training. Those participating in lower levels of exercise and physical training will also increase mitochondrial proteins and efficiency (Koltai et al., 2012), but generally to a lesser degree as stress is reduced and does not require higher levels of adaptation to meet the energy demand of the cell. With increased mitochondrial capabilities from exercise training, muscle cells will have a greater capacity to produce ATP and greater antioxidant scavenging enzymes (Steinbacher and Eckl, 2015). This adaptation allows the muscle cells to withstand larger levels of stress that would be incurred through physical training by a specific mechanism of reducing and neutralizing ROS. As stated previously, increased ROS activates many pathways that cause skeletal muscle cellular proteins to be degraded, resulting in a net loss of protein over time and in the appearance of the negative effects of atrophy.
If the previously discussed processes that contribute to protein degradation, cell apoptosis, and variation in protein synthesis signaling in skeletal muscle atrophy are diminished during an atrophic setting, a reduction in atrophy is observed. Electronic stimulation of muscle tissue causing contraction throughout a 14 day hindlimb suspension protocol resulted in significant increases in soleus muscle mass compared to controls and to an increase in Bcl-2, preventing mitochondrial release of CytC and apoptosis (Guo et al., 2012). As we know, contractile activity, as occurs in exercise, also stimulates mitochondrial biogenesis and protein synthesis potentially leading to these results. The administration of branched chained amino acids (BCAAs) throughout a 14 day hindlimb suspension model of male Wistar rats diminished atrogin-1 and murf-1 protein expression and prevented a decrease in soleus mass (Jang et al., 2015). BCAAs are known to activate mTOR pathway signaling and protein synthesis while reducing degradation, resulting in the protection of muscle mass, while mTOR activation is implicated in mitochondrial biogenesis (Figure 2). In aging mice overexpressing catalase, a ROS reducing enzyme in the mitochondria, an improvement in muscle performance and lowered protein oxidation was observed compared to aged control mice indicating a crucial role for mitochondrial function protecting skeletal muscle (Umanskaya et al., 2014).
The results above clearly indicate that different methods associated with improving mitochondrial function are capable of improving or preventing skeletal muscle loss during atrophy. A question that remains is whether or not the skeletal muscle can be protected from atrophy by interventions that occur prior to the atrophic setting? The abundance of evidence suggesting a correlation between mitochondrial dysfunction and atrophy indicate it may be beneficial to target mitochondrial biogenesis to reduce excess ROS and alleviate the dysfunction of mitochondria and the efficiency of ATP production as a mechanism to diminish the processes of atrophy. If the muscle cells’ adaptations are able to last for any significant time during a period of unloading, atrophy may be at least diminished. Based on the current literature supporting increased mitochondrial biogenesis and function in skeletal muscle cells, we hypothesize exercising training prior to acute periods of unloading and disuse will provide protection to skeletal muscle from atrophy.
One particular group exercised male Wistar rats with treadmill running for 25 minutes prior to a 14 day hindlimb suspension protocol. In rats with no exercise intervention, muscle weight and fiber diameter, capillary to fiber ratio, and type I fiber ratio were all reduced after hindlimb suspension compared to control rats under normal activity and no suspension. In the group that exercised prior to suspension, all of the previous parameters were significantly improved, comparably (Tasaki et al., 2007). The results indicate exercise is as a counter measure to skeletal muscle atrophy prior to times of unloading. Although these findings are interesting, a single session of exercise may not be an appropriate amount of training time to induce adaptations that lead to long-term protection. A typical beginner exercising animal generally will not exercise completely during its first session, as it has not become familiarized to the procedure. Multiple training sessions over time allow the animal to understand the demands of the procedure while also allowing an exercise protocol to be applied that emphasizes a progression over time, leading to increased levels of adaptation. These heightened adaptations could be key in protecting the muscle from atrophy during unloading and disuse.
Another group used swimming exercise, 3 sessions a week for 4 months, on female Wistar rats before immobilizing a hindlimb for 14 days to assess the effect of exercise on rehabilitation of atrophied skeletal muscle. The exercise prevented loss of soleus muscle mass compared to rats that did not exercise. A feature of the present study was the use of swimming exercise, common in rehabilitation programs, as it does not load particular areas of the body that were injured previously while still allowing the muscle to contract. This also disallows increased mechanical loading that would be experienced in land based exercise, potentially providing different results. Nonetheless, this study further indicates a powerful role of exercise in preventing atrophy during acute disuse.
TFAM and Exercise: Skeletal Muscle Atrophy Protection
Mitochondrial biogenesis and function require TFAM to protect mtDNA from ROS and degradation while activating mtDNA transcription of specific proteins. TFAM proteins precede activity of mitochondrial biogenesis. Electrical stimulation of rat muscle at 10 Hz for 3 hours/day resulted in an initial 55% increase in TFAM by day 4 of stimulation. This was followed by a significant import of TFAM into IMF mitochondria by day 5 and 7 and increased import protein machinery content. Further, by day 7 TFAM binding to the mitochondrial promoter increased 49%, the mitochondrial transcript cytochrome c oxidase (COX) III subunit protein content increased 65%, and COX enzyme activity increased by 72% (Gordon et al., 2001). This study reveals a time schedule of TFAM preceding mitochondrial biogenesis processes and the intricate role TFAM plays in mitochondrial biogenesis that occurs during skeletal muscle contractile activity. Contractile activity occurs in exercise, and thus, the data of this study promotes further connection of TFAM and exercise.
Exercise creates an internal environment producing many signals inducing mitochondrial biogenesis. The activation of PGC-1α is of particular importance, as it is a convergence point of many of the signaling cascades activated by exercise. Kang et al. overexpressed PGC-1α in the tibialis anterior muscle of mice through in vivo transfection before immobilizing a randomly selected hindlimb for 14 days followed by 5 days of remobilization creating an acute period of unloading and disuse of the musculature. After 7 and 14 days of immobilization, control mice revealed a steady decline in PGC-1α and TFAM mRNA expression levels. PGC-1α overexpression resulted in a decrease in ROS, increase in mitochondrial functional markers, and reduction in inflammatory markers after immobilization and remobilization of the muscle. There was also an increase in mtDNA and mitochondrial density. This resulted in the preservation of soleus muscle mass after remobilizing previously immobilized muscle (Kang et al., 2016).
With the increase in TFAM being a downstream product of upregulating PGC-1α, the results of increased mtDNA copy number, mitochondrial density, and mitochondrial function are expected. Here we see creating a condition, i.e. overexpressing PGC-1α, correlating with TFAM and mitochondrial biogenesis prior to an atrophic protocol in skeletal muscle resulted in the protection of the muscle. Since PGC-1α is upstream of TFAM, we also hypothesize that the upregulation or overexpression of TFAM prior to skeletal muscle unloading or disuse will protect the muscle from atrophy.
Nishiyama and colleagues mated transgenic mice overexpressing TFAM with mice of a mitochondrial disease model with mutant mtDNA. This resulted in the amelioration of mitochondrial disease and prolonged lifespan by increasing mtDNA copy number due to the effects of overexpressing TFAM (Nishiyama et al., 2010). This reveals a correlation between mtDNA copy number and treating a pathological condition. Moreover, Fujino et al. overexpressed a recombinant form of human TFAM (rhTFAM) in cultured cardiac myocytes. This rhTFAM rapidly entered the mitochondria and increased mtDNA copy number. Electron transport chain proteins (COX I and COX III) were elevated, as they are mtDNA encoded while nuclear DNA encoded proteins were not significantly elevated. Overexpression of rhTFAM also attenuated pathological hypertrophy by blocking the activation of nuclear factor of activated T-cells protein, known to induce pathological hypertrophy in cardiac myocytes (Fujino et al., 2012). Again, the data shows a correlation between mtDNA copy number and attenuating pathology with this example being in cardiac muscle cells, implicating the possibility of TFAM overexpression in the treatment of mitochondrial related pathologies in skeletal muscle cells.
Similarly, Ikeda et al. used transgenic mice overexpressing TFAM in a volume-overload induced heart failure model to observe the effects of upregulated TFAM on cardiac muscle after heart failure. TFAM mice exhibited improved cardiac function, diminished pathological hypertrophy, and a reduction in cardiac muscle ROS compared to control, heart failure mice. Mitochondrial enzymatic activity did not decline in the TFAM model, as is seen with the control heart failure mice. This group also used rat isolated cardiomyocytes to show an increase in mitochondrial ROS resulted in an increase in harmful, degrading proteinases (metallomatrix proteinases, specifically MMP-2 and MMP-9). Myocytes overexpressing human TFAM resulted in increased mtDNA copy number and a suppression of mitochondrial ROS and proteinase activity (Ikeda et al., 2015). The results of this study indicate TFAM increases mtDNA copy number in cardiac muscle while also limiting oxidative stress. This also implicates a potential use in skeletal muscle to treat ROS induced pathological conditions, such as atrophy.
CONCLUSIONS
Exercise induces mitochondrial biogenesis and efficient mitophagy, increases the ability to create ATP and neutralize ROS, reduces cell apoptosis, increases growth and protein synthesis signaling, and decreases protein degradation pathways. These effects reveal the role exercise plays in skeletal muscle atrophy prevention and its potential use as a treatment prior to atrophic situations, such as prescribing exercise pre-surgery to a patient that will be on bedrest or immobilized post-surgery. TFAM protects mtDNA from degradation via ROS and initiates mitochondrial protein transcription while improving mitochondrial function. This reveals the role TFAM plays in preventing mitochondrial dysfunction and highlights the connection this dysfunction has with skeletal muscle atrophy. This knowledge can lead to future targeting of the mitochondria as a method of treating the negative effects associated with skeletal muscle disuse. Further, we believe the combination of exercise training with the overexpression of TFAM will synergistically enhance mitochondrial function and prevent skeletal muscle atrophy.
Figure 3.

The Imbalance of Atrophy
Figure 4.
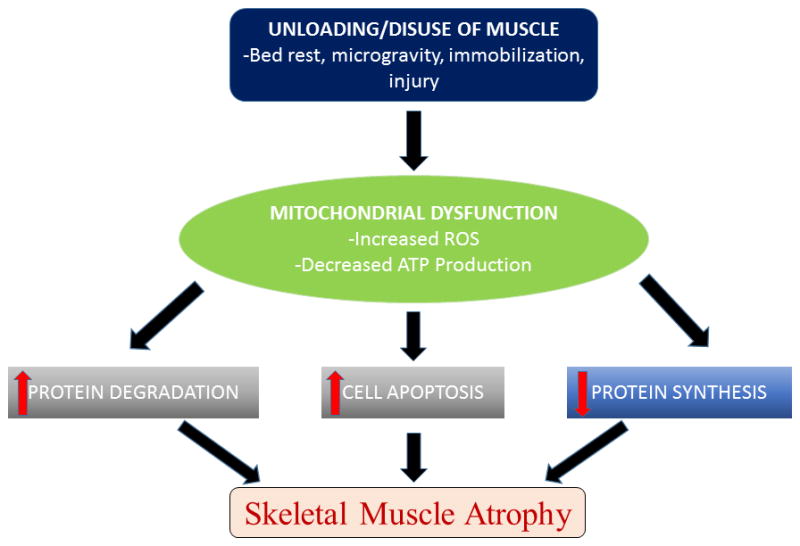
Pathways to Atrophy
Figure 5.

Atrophy Treatment Plan
Acknowledgments
Funding
The study was supported by NIH Grant HL74185 to SCT and NIH F31 Grant 1F31HL132527-01 to GHK.
Contract grant sponsor: NIH, Contract grant number: HL74185
Contract grant sponsor: NIH, Contract grant number: 1F31HL132527-01
Footnotes
Competing Interests
The authors declare that they have no competing interests.
Authors’ Contributions
Author Nicholas T. Theilen prepared the manuscript and it was reviewed by Suresh C. Tyagi and George H. Kunkel. All authors read and reviewed the manuscript.
Acknowledgements
Not applicable.
References
- Bartlett JD, Close GL, Drust B, Morton JP. The emerging role of p53 in exercise metabolism. Sports medicine (Auckland, NZ) 2014;44(3):303–309. doi: 10.1007/s40279-013-0127-9. [DOI] [PubMed] [Google Scholar]
- Bartoli M, Richard I. Calpains in muscle wasting. The international journal of biochemistry & cell biology. 2005;37(10):2115–2133. doi: 10.1016/j.biocel.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Barton E, Morris C. Mechanisms and strategies to counter muscle atrophy. The journals of gerontology Series A, Biological sciences and medical sciences. 2003;58(10):M923–926. doi: 10.1093/gerona/58.10.m923. [DOI] [PubMed] [Google Scholar]
- Baylor SM, Hollingworth S. Intracellular calcium movements during excitation-contraction coupling in mammalian slow-twitch and fast-twitch muscle fibers. J Gen Physiol. 2012;139(4):261–272. doi: 10.1085/jgp.201210773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechet D, Tassa A, Taillandier D, Combaret L, Attaix D. Lysosomal proteolysis in skeletal muscle. The international journal of biochemistry & cell biology. 2005;37(10):2098–2114. doi: 10.1016/j.biocel.2005.02.029. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. American journal of physiology Endocrinology and metabolism. 2001;281(6):E1340–1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- Bilodeau PA, Coyne ES, Wing SS. The Ubiquitin Proteasome System in Atrophying Skeletal Muscle - Roles & Regulation. American journal of physiology Cell physiology. 2016 doi: 10.1152/ajpcell.00125.2016. ajpcell.00125.02016. [DOI] [PubMed] [Google Scholar]
- Bocco BM, Louzada RA, Silvestre DH, Santos MC, Anne-Palmer E, Rangel IF, Abdalla S, Ferreira AC, Ribeiro MO, Gereben B, Carvalho DP, Bianco AC, Werneck-de-Castro JP. Thyroid hormone activation by type 2 deiodinase mediates exercise-induced peroxisome proliferator-activated receptor-gamma coactivator-1alpha expression in skeletal muscle. The Journal of physiology. 2016;594(18):5255–5269. doi: 10.1113/JP272440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. American Journal of Physiology - Endocrinology and Metabolism. 2014;307(6):E469–E484. doi: 10.1152/ajpendo.00204.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolisetty S, Jaimes EA. Mitochondria and reactive oxygen species: physiology and pathophysiology. International journal of molecular sciences. 2013;14(3):6306–6344. doi: 10.3390/ijms14036306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CT, Kolesar JE, Kaufman BA. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochimica et biophysica acta. 2012;1819(9–10):921–929. doi: 10.1016/j.bbagrm.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Chin ER. Role of Ca2+/calmodulin-dependent kinases in skeletal muscle plasticity. Journal of applied physiology (Bethesda, Md : 1985) 2005;99(2):414–423. doi: 10.1152/japplphysiol.00015.2005. [DOI] [PubMed] [Google Scholar]
- Chopard A, Hillock S, Jasmin BJ. Molecular events and signalling pathways involved in skeletal muscle disuse-induced atrophy and the impact of countermeasures. Journal of cellular and molecular medicine. 2009;13(9b):3032–3050. doi: 10.1111/j.1582-4934.2009.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan SM, Choi AM. Mitochondria in lung disease. The Journal of clinical investigation. 2016;126(3):809–820. doi: 10.1172/JCI81113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes A, Dekerle J, Webborn N, Watt P, Bougault V, Daussin FN. Exercise-induced metabolic fluctuations influence AMPK, p38-MAPK and CaMKII phosphorylation in human skeletal muscle. Physiological reports. 2015;3(9) doi: 10.14814/phy2.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly PF, Jager R, Fearnhead HO. New roles for old enzymes: killer caspases as the engine of cell behavior changes. Frontiers in physiology. 2014;5:149. doi: 10.3389/fphys.2014.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450(7170):736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- Dimauro I, Mercatelli N, Caporossi D. Exercise-induced ROS in heat shock proteins response. Free radical biology & medicine. 2016;98:46–55. doi: 10.1016/j.freeradbiomed.2016.03.028. [DOI] [PubMed] [Google Scholar]
- Drummond MJ, Fry CS, Glynn EL, Dreyer HC, Dhanani S, Timmerman KL, Volpi E, Rasmussen BB. Rapamycin administration in humans blocks the contraction-induced increase in skeletal muscle protein synthesis. The Journal of physiology. 2009;587(Pt 7):1535–1546. doi: 10.1113/jphysiol.2008.163816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont-Versteegden EE. Apoptosis in skeletal muscle and its relevance to atrophy. World Journal of Gastroenterology : WJG. 2006;12(46):7463–7466. doi: 10.3748/wjg.v12.i46.7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Human molecular genetics. 2004;13(9):935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- Elmore S. Apoptosis: A Review of Programmed Cell Death. Toxicologic pathology. 2007;35(4):495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Familtseva A, Kalani A, Chaturvedi P, Tyagi N, Metreveli N, Tyagi SC. Mitochondrial mitophagy in mesenteric artery remodeling in hyperhomocysteinemia. Physiological reports. 2014;2(4):e00283. doi: 10.14814/phy2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. The American Journal of Clinical Nutrition. 2011;93(4):884S–890S. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino T, Ide T, Yoshida M, Onitsuka K, Tanaka A, Hata Y, Nishida M, Takehara T, Kanemaru T, Kitajima N, Takazaki S, Kurose H, Kang D, Sunagawa K. Recombinant mitochondrial transcription factor A protein inhibits nuclear factor of activated T cells signaling and attenuates pathological hypertrophy of cardiac myocytes. Mitochondrion. 2012;12(4):449–458. doi: 10.1016/j.mito.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Gao QQ, McNally EM. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Comprehensive Physiology. 2015;5(3):1223–1239. doi: 10.1002/cphy.c140048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garatachea N, Pareja-Galeano H, Sanchis-Gomar F, Santos-Lozano A, Fiuza-Luces C, Morán M, Emanuele E, Joyner MJ, Lucia A. Exercise Attenuates the Major Hallmarks of Aging. Rejuvenation Research. 2015;18(1):57–89. doi: 10.1089/rej.2014.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrig SM, Mihaylova V, Frese S, Mueller SM, Ligon-Auer M, Spengler CM, Petersen JA, Lundby C, Jung HH. Altered skeletal muscle (mitochondrial) properties in patients with mitochondrial DNA single deletion myopathy. Orphanet journal of rare diseases. 2016;11(1):105. doi: 10.1186/s13023-016-0488-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givvimani S, Pushpakumar S, Veeranki S, Tyagi SC. Dysregulation of Mfn2 and Drp-1 proteins in heart failure. Canadian journal of physiology and pharmacology. 2014;92(7):583–591. doi: 10.1139/cjpp-2014-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin R, Ascah A, Daussin FN. Intensity-dependent activation of intracellular signalling pathways in skeletal muscle: role of fibre type recruitment during exercise. The Journal of physiology. 2010;588(Pt 21):4073–4074. doi: 10.1113/jphysiol.2010.195925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JW, Rungi AA, Inagaki H, Hood DA. Effects of contractile activity on mitochondrial transcription factor A expression in skeletal muscle. Journal of applied physiology (Bethesda, Md : 1985) 2001;90(1):389–396. doi: 10.1152/jappl.2001.90.1.389. [DOI] [PubMed] [Google Scholar]
- Gumucio JP, Mendias CL. Atrogin-1, MuRF-1, and sarcopenia. Endocrine. 2013;43(1):12–21. doi: 10.1007/s12020-012-9751-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B-S, Cheung K-K, Yeung SS, Zhang B-T, Yeung EW. Electrical Stimulation Influences Satellite Cell Proliferation and Apoptosis in Unloading-Induced Muscle Atrophy in Mice. PLoS ONE. 2012;7(1):e30348. doi: 10.1371/journal.pone.0030348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutterman DD. Mitochondria and Reactive Oxygen Species: An Evolution in Function. Circulation Research. 2005;97(4):302–304. doi: 10.1161/01.RES.0000179773.18195.12. [DOI] [PubMed] [Google Scholar]
- Hardie DG. The AMP-activated protein kinase pathway – new players upstream and downstream. Journal of Cell Science. 2004;117(23):5479–5487. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- Hood DA, Adhihetty PJ, Colavecchia M, Gordon JW, Irrcher I, Joseph AM, Lowe ST, Rungi AA. Mitochondrial biogenesis and the role of the protein import pathway. Medicine and science in sports and exercise. 2003;35(1):86–94. doi: 10.1097/00005768-200301000-00015. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Ide T, Fujino T, Arai S, Saku K, Kakino T, Tyynismaa H, Yamasaki T, Yamada K, Kang D, Suomalainen A, Sunagawa K. Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS One. 2015;10(3):e0119687. doi: 10.1371/journal.pone.0119687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irrcher I, Ljubicic V, Hood DA. Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. American journal of physiology Cell physiology. 2009;296(1):C116–123. doi: 10.1152/ajpcell.00267.2007. [DOI] [PubMed] [Google Scholar]
- Jang J, Yun HY, Park J, Lim K. Protective effect of branched chain amino acids on hindlimb suspension-induced muscle atrophy in growing rats. Journal of exercise nutrition & biochemistry. 2015;19(3):183–189. doi: 10.5717/jenb.2015.15062704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen SB, Richter EA, Wojtaszewski JFP. Role of AMPK in skeletal muscle metabolic regulation and adaptation in relation to exercise. The Journal of physiology. 2006;574(Pt 1):17–31. doi: 10.1113/jphysiol.2006.109942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays in biochemistry. 2010;47 doi: 10.1042/bse0470069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joza N, Pospisilik JA, Hangen E, Hanada T, Modjtahedi N, Penninger JM, Kroemer G. AIF: not just an apoptosis-inducing factor. Annals of the New York Academy of Sciences. 2009;1171:2–11. doi: 10.1111/j.1749-6632.2009.04681.x. [DOI] [PubMed] [Google Scholar]
- Ju JS, Jeon SI, Park JY, Lee JY, Lee SC, Cho KJ, Jeong JM. Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise-trained condition. The journal of physiological sciences : JPS. 2016;66(5):417–430. doi: 10.1007/s12576-016-0440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, Chung E, Diffee G, Ji LL. Exercise training attenuates aging-associated mitochondrial dysfunction in rat skeletal muscle: role of PGC-1alpha. Experimental gerontology. 2013;48(11):1343–1350. doi: 10.1016/j.exger.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Kang C, O’Moore KM, Dickman JR, Ji LL. Exercise activation of muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha signaling is redox sensitive. Free radical biology & medicine. 2009;47(10):1394–1400. doi: 10.1016/j.freeradbiomed.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Kang C, Yeo D, Ji LL. Muscle Immobilization Activates Mitophagy and Disrupts Mitochondrial Dynamics in Mice. Acta physiologica (Oxford, England) 2016 doi: 10.1111/apha.12690. [DOI] [PubMed] [Google Scholar]
- Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7(1–2):39–44. doi: 10.1016/j.mito.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Lee SB, Park JK, Yoo YD. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L) Cell death and differentiation. 2010;17(9):1420–1434. doi: 10.1038/cdd.2010.19. [DOI] [PubMed] [Google Scholar]
- King-Himmelreich TS, Schramm S, Wolters MC, Schmetzer J, Moser CV, Knothe C, Resch E, Peil J, Geisslinger G, Niederberger E. The impact of endurance exercise on global and AMPK gene-specific DNA methylation. Biochemical and biophysical research communications. 2016;474(2):284–290. doi: 10.1016/j.bbrc.2016.04.078. [DOI] [PubMed] [Google Scholar]
- Koltai E, Hart N, Taylor AW, Goto S, Ngo JK, Davies KJ, Radak Z. Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. American journal of physiology Regulatory, integrative and comparative physiology. 2012;303(2):R127–134. doi: 10.1152/ajpregu.00337.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krustrup P, Soderlund K, Mohr M, Gonzalez-Alonso J, Bangsbo J. Recruitment of fibre types and quadriceps muscle portions during repeated, intense knee-extensor exercise in humans. Pflugers Archiv : European journal of physiology. 2004;449(1):56–65. doi: 10.1007/s00424-004-1304-3. [DOI] [PubMed] [Google Scholar]
- Kühl I, Miranda M, Posse V, Milenkovic D, Mourier A, Siira SJ, Bonekamp NA, Neumann U, Filipovska A, Polosa PL, Gustafsson CM, Larsson N-G. POLRMT regulates the switch between replication primer formation and gene expression of mammalian mtDNA. Science Advances. 2016;2(8):e1600963. doi: 10.1126/sciadv.1600963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel GH, Chaturvedi P, Tyagi SC. Mitochondrial pathways to cardiac recovery: TFAM. Heart failure reviews. 2016;21(5):499–517. doi: 10.1007/s10741-016-9561-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127(6):1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling at a glance. Journal of Cell Science. 2009;122(20):3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nature genetics. 1998;18(3):231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- Leduc-Gaudet JP, Auger MJ, St Jean Pelletier F, Gouspillou G. Towards a better understanding of the role played by mitochondrial dynamics and morphology in skeletal muscle atrophy. The Journal of physiology. 2015;593(14):2993–2994. doi: 10.1113/JP270736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeuwenburgh C, Gurley CM, Strotman BA, Dupont-Versteegden EE. Age-related differences in apoptosis with disuse atrophy in soleus muscle. American journal of physiology Regulatory, integrative and comparative physiology. 2005;288(5):R1288–1296. doi: 10.1152/ajpregu.00576.2004. [DOI] [PubMed] [Google Scholar]
- Lenaz G. Role of mitochondria in oxidative stress and ageing. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1998;1366(1–2):53–67. doi: 10.1016/s0005-2728(98)00120-0. [DOI] [PubMed] [Google Scholar]
- Liang C, Curry BJ, Brown PL, Zemel MB. Leucine Modulates Mitochondrial Biogenesis and SIRT1-AMPK Signaling in C2C12 Myotubes. Journal of nutrition and metabolism. 2014;2014:239750. doi: 10.1155/2014/239750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Ward WF. PGC-1α: a key regulator of energy metabolism. Advances in Physiology Education. 2006;30(4):145–151. doi: 10.1152/advan.00052.2006. [DOI] [PubMed] [Google Scholar]
- Liu J, Peng Y, Wang X, Fan Y, Qin C, Shi L, Tang Y, Cao K, Li H, Long J, Liu J. Mitochondrial Dysfunction Launches Dexamethasone-Induced Skeletal Muscle Atrophy via AMPK/FOXO3 Signaling. Molecular Pharmaceutics. 2016;13(1):73–84. doi: 10.1021/acs.molpharmaceut.5b00516. [DOI] [PubMed] [Google Scholar]
- Lundby C, Jacobs RA. Adaptations of skeletal muscle mitochondria to exercise training. Experimental physiology. 2016;101(1):17–22. doi: 10.1113/EP085319. [DOI] [PubMed] [Google Scholar]
- Matsushima Y, Goto Y, Kaguni LS. Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM) Proceedings of the National Academy of Sciences of the United States of America. 2010;107(43):18410–18415. doi: 10.1073/pnas.1008924107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride A, Hardie DG. AMP-activated protein kinase--a sensor of glycogen as well as AMP and ATP? Acta physiologica (Oxford, England) 2009;196(1):99–113. doi: 10.1111/j.1748-1716.2009.01975.x. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends in Biochemical Sciences. 27(1):40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- Morita M, Gravel S-P, Chénard V, Sikström K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, McLaughlan S, Nouet Y, Pause A, Pollak M, Gottlieb E, Larsson O, St-Pierre J, Topisirovic I, Sonenberg N. mTORC1 Controls Mitochondrial Activity and Biogenesis through 4E-BP-Dependent Translational Regulation. Cell Metabolism. 18(5):698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Morita M, Gravel SP, Hulea L, Larsson O, Pollak M, St-Pierre J, Topisirovic I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell cycle (Georgetown, Tex) 2015;14(4):473–480. doi: 10.4161/15384101.2014.991572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulin M, Ferreiro A. Muscle redox disturbances and oxidative stress as pathomechanisms and therapeutic targets in early-onset myopathies. Seminars in cell & developmental biology. 2016 doi: 10.1016/j.semcdb.2016.08.003. [DOI] [PubMed] [Google Scholar]
- Mourier A, Motori E, Brandt T, Lagouge M, Atanassov I, Galinier A, Rappl G, Brodesser S, Hultenby K, Dieterich C, Larsson NG. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. The Journal of cell biology. 2015;208(4):429–442. doi: 10.1083/jcb.201411100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai R, Matsui N, Fujikura Y, Matsumoto N, Hou DX, Kanzaki N, Shibata H, Horikawa M, Iwasa K, Hirasaka K, Nikawa T, Terao J. Preventive effect of dietary quercetin on disuse muscle atrophy by targeting mitochondria in denervated mice. The Journal of nutritional biochemistry. 2016;31:67–76. doi: 10.1016/j.jnutbio.2016.02.001. [DOI] [PubMed] [Google Scholar]
- Nagano K, Suzaki E, Nagano Y, Kataoka K, Ozawa K. The activation of apoptosis factor in hindlimb unloading-induced muscle atrophy under normal and low-temperature environmental conditions. Acta histochemica. 2008;110(6):505–518. doi: 10.1016/j.acthis.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Ngo HB, Lovely GA, Phillips R, Chan DC. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nature communications. 2014;5:3077. doi: 10.1038/ncomms4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama S, Shitara H, Nakada K, Ono T, Sato A, Suzuki H, Ogawa T, Masaki H, Hayashi J, Yonekawa H. Over-expression of Tfam improves the mitochondrial disease phenotypes in a mouse model system. Biochemical and biophysical research communications. 2010;401(1):26–31. doi: 10.1016/j.bbrc.2010.08.143. [DOI] [PubMed] [Google Scholar]
- Nuoc TN, Kim S, Ahn SH, Lee JS, Park BJ, Lee TH. The analysis of antioxidant expression during muscle atrophy induced by hindlimb suspension in mice. The journal of physiological sciences : JPS. 2016 doi: 10.1007/s12576-016-0444-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara R, Fujita S, Hornberger TA, Kitaoka Y, Makanae Y, Nakazato K, Naokata I. The role of mTOR signalling in the regulation of skeletal muscle mass in a rodent model of resistance exercise. Sci Rep. 2016;6:31142. doi: 10.1038/srep31142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J, Kiilerich K, Pilegaard H. PGC-1alpha-mediated adaptations in skeletal muscle. Pflugers Archiv : European journal of physiology. 2010;460(1):153–162. doi: 10.1007/s00424-010-0834-0. [DOI] [PubMed] [Google Scholar]
- Paschen SA, Neupert W. Protein import into mitochondria. IUBMB life. 2001;52(3–5):101–112. doi: 10.1080/15216540152845894. [DOI] [PubMed] [Google Scholar]
- Pasternak C, Wong S, Elson EL. Mechanical function of dystrophin in muscle cells. The Journal of cell biology. 1995;128(3):355–361. doi: 10.1083/jcb.128.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeck RT, Karunasekara Y, Board PG, Beard NA, Casarotto MG, Dulhunty AF. Skeletal muscle excitation-contraction coupling: who are the dancing partners? The international journal of biochemistry & cell biology. 2014;48:28–38. doi: 10.1016/j.biocel.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Saleem A, Hood DA. Acute exercise induces tumour suppressor protein p53 translocation to the mitochondria and promotes a p53-Tfam-mitochondrial DNA complex in skeletal muscle. The Journal of physiology. 2013;591(14):3625–3636. doi: 10.1113/jphysiol.2013.252791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome()() The international journal of biochemistry & cell biology. 2013;45(10):2121–2129. doi: 10.1016/j.biocel.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JM, Kowluru RA. Impaired Transport of Mitochondrial Transcription Factor and the Metabolic Memory Phenomenon Associated with the Progression of Diabetic Retinopathy. Diabetes/metabolism research and reviews. 2013;29(3):204–213. doi: 10.1002/dmrr.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nature reviews Molecular cell biology. 2010;11(9):655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- Schwalm C, Jamart C, Benoit N, Naslain D, Premont C, Prevet J, Van Thienen R, Deldicque L, Francaux M. Activation of autophagy in human skeletal muscle is dependent on exercise intensity and AMPK activation. Faseb j. 2015;29(8):3515–3526. doi: 10.1096/fj.14-267187. [DOI] [PubMed] [Google Scholar]
- Seo S, Lee M-S, Chang E, Shin Y, Oh S, Kim I-H, Kim Y. Rutin Increases Muscle Mitochondrial Biogenesis with AMPK Activation in High-Fat Diet-Induced Obese Rats. Nutrients. 2015;7(9):8152–8169. doi: 10.3390/nu7095385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkman BS, Belova SP, Lomonosova YN, Kostrominova TY, Nemirovskaya TL. Calpain-dependent regulation of the skeletal muscle atrophy following unloading. Arch Biochem Biophys. 2015;584:36–41. doi: 10.1016/j.abb.2015.07.011. [DOI] [PubMed] [Google Scholar]
- Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(15):5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu PM. Muscle apoptotic response to denervation, disuse, and aging. Medicine and science in sports and exercise. 2009;41(10):1876–1886. doi: 10.1249/MSS.0b013e3181a6470b. [DOI] [PubMed] [Google Scholar]
- Siu PM, Wang Y, Alway SE. Apoptotic signaling induced by H(2)O(2)-mediated oxidative stress in differentiated C2C12 myotubes. Life sciences. 2009;84(13–14):468–481. doi: 10.1016/j.lfs.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S. Emerging therapeutic roles for NAD+ metabolism in mitochondrial and age-related disorders. Clinical and Translational Medicine. 2016;5(1):1–11. doi: 10.1186/s40169-016-0104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John JC. Mitochondrial DNA copy number and replication in reprogramming and differentiation. Seminars in cell & developmental biology. 2016;52:93–101. doi: 10.1016/j.semcdb.2016.01.028. [DOI] [PubMed] [Google Scholar]
- Steinbacher P, Eckl P. Impact of oxidative stress on exercising skeletal muscle. Biomolecules. 2015;5(2):356–377. doi: 10.3390/biom5020356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles AR, Simon MT, Stover A, Eftekharian S, Khanlou N, Wang HL, Magaki S, Lee H, Partynski K, Dorrani N, Chang R, Martinez-Agosto JA, Abdenur JE. Mutations in TFAM, encoding mitochondrial transcription factor A, cause neonatal liver failure associated with mtDNA depletion. Molecular genetics and metabolism. 2016;119(1–2):91–99. doi: 10.1016/j.ymgme.2016.07.001. [DOI] [PubMed] [Google Scholar]
- Strobel NA, Peake JM, Matsumoto A, Marsh SA, Coombes JS, Wadley GD. Antioxidant supplementation reduces skeletal muscle mitochondrial biogenesis. Medicine and science in sports and exercise. 2011;43(6):1017–1024. doi: 10.1249/MSS.0b013e318203afa3. [DOI] [PubMed] [Google Scholar]
- Suwa M, Nakano H, Radak Z, Kumagai S. Endurance exercise increases the SIRT1 and peroxisome proliferator-activated receptor gamma coactivator-1alpha protein expressions in rat skeletal muscle. Metabolism: clinical and experimental. 2008;57(7):986–998. doi: 10.1016/j.metabol.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochimica et biophysica acta. 1999;1410(2):103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- Tasaki H, Matsubara T, Miki A. Effect of exercise and heat stress before hindlimb suspension on prevention of the skeletal muscle atrophy. Bulletin of health sciences Kobe. 2007;23:1–8. [Google Scholar]
- Tawa NE, Jr, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. The Journal of clinical investigation. 1997;100(1):197–203. doi: 10.1172/JCI119513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale MJ. The ubiquitin-proteasome pathway as a therapeutic target for muscle wasting. The journal of supportive oncology. 2005;3(3):209–217. [PubMed] [Google Scholar]
- Tokusumi Y, Zhou S, Takada S. Nuclear respiratory factor 1 plays an essential role in transcriptional initiation from the hepatitis B virus x gene promoter. Journal of virology. 2004;78(20):10856–10864. doi: 10.1128/JVI.78.20.10856-10864.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umanskaya A, Santulli G, Xie W, Andersson DC, Reiken SR, Marks AR. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(42):15250–15255. doi: 10.1073/pnas.1412754111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero T. Mitochondrial biogenesis: pharmacological approaches. Current pharmaceutical design. 2014;20(35):5507–5509. doi: 10.2174/138161282035140911142118. [DOI] [PubMed] [Google Scholar]
- van Osch FHM, Voets AM, Schouten LJ, Gottschalk RWH, Simons CCJM, van Engeland M, Lentjes MHFM, van den Brandt PA, Smeets HJM, Weijenberg MP. Mitochondrial DNA copy number in colorectal cancer: between tissue comparisons, clinicopathological characteristics and survival. Carcinogenesis. 2015;36(12):1502–1510. doi: 10.1093/carcin/bgv151. [DOI] [PubMed] [Google Scholar]
- Wang C, Youle RJ. The Role of Mitochondria in Apoptosis() Annual review of genetics. 2009;43:95–118. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Yang X, López de Silanes I, Carling D, Gorospe M. Increased AMP:ATP Ratio and AMP-activated Protein Kinase Activity during Cellular Senescence Linked to Reduced HuR Function. Journal of Biological Chemistry. 2003;278(29):27016–27023. doi: 10.1074/jbc.M300318200. [DOI] [PubMed] [Google Scholar]
- Warburton DER, Nicol CW, Bredin SSD. Health benefits of physical activity: the evidence. CMAJ : Canadian Medical Association Journal. 2006;174(6):801–809. doi: 10.1503/cmaj.051351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Lira VA, Greene NP. Exercise training-induced Regulation of Mitochondrial Quality. Exercise and Sport Sciences Reviews. 2012;40(3):159–164. doi: 10.1097/JES.0b013e3182575599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275(5303):1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- You JS, Anderson GB, Dooley MS, Hornberger TA. The role of mTOR signaling in the regulation of protein synthesis and muscle mass during immobilization in mice. Disease models & mechanisms. 2015;8(9):1059–1069. doi: 10.1242/dmm.019414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li H, Li H, Nakagawa A, Lin JLJ, Lee E-S, Harry BL, Skeen-Gaar RR, Suehiro Y, William D, Mitani S, Yuan HS, Kang B-H, Xue D. Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science. 2016;353(6297):394–399. doi: 10.1126/science.aaf4777. [DOI] [PMC free article] [PubMed] [Google Scholar]