

Abstract
A number of recent advances in the field of magic-angle-spinning (MAS) solid-state NMR have enabled its application to a range of biological systems of ever increasing complexity. To retain biological relevance, these samples are increasingly studied in a hydrated state. At the same time, experimental feasibility requires the sample preparation process to attain a high sample concentration within the final MAS rotor. We discuss these considerations, and how they have led to a number of different approaches to MAS NMR sample preparation. We describe our experience of how custom-made (or commercially available) ultracentrifugal devices can facilitate a simple, fast and reliable sample preparation process. A number of groups have since adopted such tools, in some cases to prepare samples for sedimentation-style MAS NMR experiments. Here we argue for a more widespread adoption of their use for routine MAS NMR sample preparation.
Keywords: MAS NMR, ultracentrifugation, sample packing, hydration, sedimentation
Introduction
Biological solid-state NMR (ssNMR) has undergone tremendous development in recent years, providing new insights into the structure and dynamics of membrane proteins, protein aggregates, and a variety of other biologically interesting samples (Goldbourt 2013; Knight et al. 2013; Loquet et al. 2013; Porcelli et al. 2013; Weingarth and Baldus 2013; Wang and Ladizhansky 2014; Andreas et al. 2015; Quinn et al. 2015; Ravera et al. 2015). Most of these results stem from the use of magic angle spinning (MAS) ssNMR. The burgeoning application of MAS ssNMR for biological samples has been enabled by technological improvements in the equipment available for ssNMR. This includes higher magnetic fields, faster MAS, and improved probe designs (Dillmann et al. 2007; McNeill et al. 2009; Demers et al. 2011; Lamley et al. 2014; Wickramasinghe et al. 2015; Andreas et al. 2016; Pöppler et al. 2016). An equally important contribution comes from improvements in sample preparation, including an increasing use of selective and extensive isotopic labeling strategies (Straus 2004; Higman et al. 2009; Loquet et al. 2010; Demers et al. 2014; Mehler et al. 2015; Schubeis et al. 2017; Zhang et al. 2016). Together, these factors have improved the sensitivity and resolution that can be obtained by MAS ssNMR.
The improved performance of ssNMR enables the study of a variety of biological systems, whether aggregated proteins, membrane-bound proteins or membranes themselves. A key factor in the application of ssNMR over other structural techniques continues to be the ability to probe proteins (or other biomolecules) in an appropriate biological context. Proper sample hydration is often critical in this respect (Igumenova et al. 2004; Ball 2008; Linden et al. 2011; Siemer et al. 2012; Fragai et al. 2013). For example, enzymatic protein activity is reliant on hydration (Khodadadi et al. 2010; Schirò et al. 2015). Additionally, the proper folding and function of membrane proteins requires not only a lipid bilayer environment, but also a certain degree of hydration (Zhang et al. 2014). Dehydration can have adverse impacts on membranes, as it can cause lateral separation of membrane proteins from lipid-rich domains, formation of non-bilayer lipid phases and an increased propensity for gel phase formation (Wolfe and Bryant 1999; Bryant et al. 2001; Mandal and Van der Wel 2016). Some of these dehydration-induced changes can be directly observed via ssNMR (Ulrich and Watts 1994; Gawrisch et al. 2007; Zhang et al. 2014). In many cases, a lack of proper hydration results in reduced spectral quality, for instance due to the entrapment of multiple structural conformations that manifests itself as increased NMR line widths (Tang et al. 1999; Martin and Zilm 2003; Comellas et al. 2011; Bertini et al. 2013). Thus proper sample hydration is critical in the design and execution of modern biomolecular ssNMR, and needs to be considered during the process of packing or preparing the actual samples.
The rotor packing process should achieve multiple goals: maximize signal/noise, ensure biological relevance, and be robust, reproducible and easy. In order to maximize the NMR signal, one aims to pack the maximum amount of the studied biomacromolecules into the μL-sized volumes of typical MAS rotors. This usually occurs at the expense of excess buffer. Traditional MAS NMR packing methods have taken different approaches. One approach has been to pack and study samples that were completely dry, which maximizes the amount of (labeled) protein or peptide, but also sacrifices biological relevance in the pursuit of maximum signal (Antzutkin et al. 2000; Petkova et al. 2002; Verel et al. 2008). As noted above, this approach often suffers from line broadening and reduced spectral quality. To circumvent this concern, some studies have employed a protocol in which the rotor is packed with dry material, followed by a controlled rehydration of the latter inside the rotor (Kloepper et al. 2007). However, it is currently most common that samples are packed into the rotor in an already hydrated state. In some cases this involves the controlled de- and re-hydration of the sample, which is however completed before, instead of after, packing the sample into the rotor (Sharpe et al. 2006; Vilar et al. 2008). This kind of approach for studying re-hydrated samples has been used in biomolecular solid-state NMR studies of a variety of samples (Kennedy and Bryant 1990; Gregory et al. 1993; Jakeman et al. 1998; Pauli et al. 2000; Seidel et al. 2005; Krushelnitsky et al. 2006; Luchinat et al. 2013; Ravera 2015; Ravera et al. 2016). In other cases, dehydration of the hydrated samples is avoided once the protein (complex) of interest is successfully reconstituted or assembled. In such cases, centrifugation is typically employed to concentrate the biological solids to fit them into the MAS rotor volume. The hydrated sample is then centrifuged into a dense pellet, and then the latter is transferred into the rotor. The transfer can be done by carefully scooping to sample with micro-spatulas (Zhang et al. 2014), but this is time-consuming, and risks both sample losses and partial or local dehydration. An improved approach centrifuges these pre-condensed pellets into the rotor using funnel-shaped devices that fit in a tabletop (micro)centrifuge (Das et al. 2013; Hisao et al. 2016). This sometimes involves a fixed-angle tabletop centrifuge that could lead to an uneven packing of the sample material, which can compromise stable spinning during MAS. It is worth including a warning that use of fixed-angle ultracentrifuges can be even more detrimental, as it may result in physical damage to the MAS rotor that could manifest itself in rotors shattering during packing or during subsequent MAS.
Here we describe the approach that our lab has been using to pack all types of hydrated biological samples for MAS NMR. This approach combines the pelleting of the sample and the packing into the rotor into a single process, enabled by the use of swinging-bucket ultracentrifugal packing tools. It is designed to balance the need to maximize the signal/noise, maintain biological relevance and achieve easy reproducibility. We note that such devices are successfully used by ourselves (Van der Wel et al. 2007; Li et al. 2011; Hoop et al. 2012; Hoop et al. 2014; Mandal et al. 2015; Mandal and Van der Wel 2016) and various other groups (Böckmann et al. 2009; Gardiennet et al. 2012; Bertini et al. 2013; Kunert et al. 2014; Wiegand et al. 2015; Wiegand et al. 2016b), but that there are many others that have not adopted this approach.
To the best of our knowledge, the earliest such application was described in the literature in 2009, when it was used for the packing of crystalline protein samples (Böckmann et al. 2009). In subsequent years this methodology was also applied to the packing of amyloid fibrils (Li et al. 2011; Van Melckebeke et al. 2011). Around the same time the high centrifugal forces during MAS were used to sediment large soluble proteins, in methods known as sedimentation NMR and FROSTY MAS NMR (Mainz et al. 2009; Bertini et al. 2011). The latter abbreviation (FROSTY) stands for freezing rotational diffusion of protein solutions at low temperature and high viscosity, since the detection of CP-based signals was initially attributed to protein immobilization due to these parameters rather than the MAS-induced sedimentation that was later identified (Sarkar et al. 2016). This led to the use of ultracentrifugal packing devices to directly sediment soluble proteins into MAS rotors, during the packing process (Bertini et al. 2012; Gardiennet et al. 2012; Ferella et al. 2013; Fragai et al. 2013). In this work, we argue that these devices serve as ideal packing tools for the routine preparation of all types of (hydrated) biological samples. Design considerations as well as applications are discussed, showing our everyday use of this approach as a standard practice when packing MAS NMR samples.
Methods
Construction of sample packing tool
The ultracentrifugal packing devices were constructed in the Department of Cell Biology machine shop at the University of Pittsburgh, based on designs made by the authors. The packing device was converted from concept to 3D solid model using SolidWorks software package (Waltham, MA). Prior to fabrication, the 3D solid model was programmed for machining using Mastercam (Tolland, CT). Finally, individual parts were machined out of unfilled polyether ether ketone (PEEK) obtained from McMaster Carr (Elmhurst, IL) using a Hardinge GS-150 CNC (computer numerical controlled) lathe (Elmira, NY). As described, the tools are explicitly designed for use with Bruker-style MAS rotors, and for use in a swinging-bucket SW 32 Ti ultracentrifuge rotor from Beckman Coulter (Indianapolis, IN).
Sample packing protocol
An empty MAS NMR rotor, with the rotor drive cap removed, is placed into the open packing tool base. After manual assembly of the packing tool, the aqueous sample suspension is pipetted into the device’s funnel. Depending on preference, the device may be inserted into an empty centrifuge tube, in order to catch inadvertent sample leakage. The tube pictured in Fig. 1a below is a Beckman Coulter (Indianapolis, IN) Ultra-Clear™ centrifuge tube, part number 344058. The filled device is carefully inserted into one of the buckets of the swinging bucket rotor (Beckman Coulter SW 32 Ti). A second, counter-balance bucket is balanced carefully, by filling it with a second packing tool (see Results) and water. The packing process is then performed by ultracentrifugation for the specified times in a Beckman Coulter Optima L-100 XP ultracentrifuge at up to 32,000 RPM (~175,000 × g).
Fig. 1.

Schematics of the ultracentrifugal device for packing 3.2mm MAS rotors. (a) (L – R) Photo showing a 3.2mm Bruker MAS rotor with a cap (red rectangle), packing tool, an empty centrifuge tube and bucket holder for the SW 32 Ti UCR. (b) Exploded view of the packing tool with outer dimensions of the three components. This design drawing does not show the threading of part I (see panel a). (c) X-ray view showing the internal design. The bottom piece (I) holds the rotor, the middle piece (II) is the funnel in which the sample suspension is applied and the top piece (III) stabilizes the ultracentrifuge tube and packing device in the UCR bucket. Pieces I and II thread together for a tight seal (unsealed state shown in panel a). (d) Sliced-through view of parts I–II showing the relative dimensions and locations of the funnel, 3.2 mm Bruker rotor (grey rectangle), and O-ring (red). The parts are shown partly unscrewed for clarity.
Preparation of the reference samples
The cytochrome-c containing sample was prepared as previously described (Mandal et al. 2015). Briefly, tetra-oleoyl-cardiolipin (TOCL) and di-oleoylphosphatidylcholine (DOPC), obtained in chloroform solution from Avanti Polar Lipids (Alabaster, AL), were mixed at a molar ratio of 1:4, dried under N2 gas, further dried under vacuum and resuspended in 20mM HEPES (pH 7.4). The resuspended liposomes were subjected to freeze thawing and extrusion through 200 nm polycarbonate membranes to generate 200-nm diameter large unilamellar vesicles (LUVs), confirmed by dynamic light scattering (DLS) (Mandal et al. 2015). Pre-dissolved uniformly 13C and 15N (U-13C,15N) labeled horse heart cytochrome c, obtained by expression in Escherichia coli, was added to the LUVs. The vesicle-bound proteins were then packed into a MAS rotor using the described ultracentrifugal packing device, either by centrifugation at 143,000 × g for 3 hours and 175,000 × g for 4.5 hours, or by centrifuging at 143,000 × g for 5 hours. For more details see ref. (Mandal et al. 2015).
Mutant huntingtin (htt) exon 1 samples were prepared as described (Hoop et al. 2016). U-15N-labeled and U-13C-labeled fusion proteins were expressed in Escherichia coli and purified. The purified proteins were mixed together after which aggregation was initiated upon cleavage of the fusion protein by factor Xa protease (Promega Corporation, Madison, WI). The resulting amyloid-like fibrils were washed with buffer, and then packed into MAS rotors using the described ultracentrifugal packing device. Packing was done by first centrifuging the sample at 154,000 × g for one hour. Subsequently, 1 mL of buffer was used to rinse the original sample tube in order to recover residual sample that may have been left behind. The sample was then centrifuged a second time under identical conditions as the first round. A second rinse and a third centrifugation cycle was performed before the rotor was sealed for study by MAS NMR.
Nanofibers assembled by the acylated peptide amphiphiles were prepared as described (Merg et al. 2016), starting with site-specifically labeled peptides obtained by solid-phase peptide synthesis. The divalent peptide conjugate molecule featured a C18 organic tail attached to two identical gold-binding peptide sequences. The peptide sequence in the molecule was AYSSGAPPMPPF. In the construct studied, the methionine was oxidized prior to assembly of the entire molecule. The construct, named C18-(PEPAuM-ox)2, featured U-13C,15N-labeling in residues A1 and P10. The mature assembled fibrils were packed into MAS rotors using the ultracentrifugal packing device, in a single step process involving ultracentrifugation for 1 h at 175,000 × g.
MAS ssNMR experiments
The huntingtin exon 1 fibril measurements were performed on an 800 MHz (18.8 T) spectrometer (Bruker Biospin, Billerica, MA), with a 3.2 mm HCN Bruker MAS ssNMR probe (Hoop et al. 2016). Experiments were performed at a spinning rate of 13 kHz, with a set temperature of 275 K. Based on systematic temperature calibration experiments with external samples (Mandal and Van der Wel 2016), we estimate this to reflect a similar sample temperature (within several degrees). The 2D spectrum was acquired with 50 ms of proton-driven spin diffusion recoupling, a 1.5 ms CP contact time, a recycle delay (RD) of 3 s and an applied TPPM decoupling on 1H at 83 KHz. All other experiments were performed on a wide-bore 600 MHz (14.1T) spectrometer (Bruker Biospin, Billerica, MA) using a 3.2mm HCN Bruker MAS ssNMR probe outfitted with an “EFree” coil. MAS NMR on the cytochrome c sample was performed at a sample temperature of 233 K while spinning at 8.33 KHz (Mandal et al. 2015). Using a CP contact time of 1 ms, 256 scans were collected with a RD of 3.2 s and an applied TPPM decoupling on 1H at 83 KHz. The NMR experiments on the C18-dipeptide nanoparticles were performed at a set temperature of 277 K while spinning at 10 KHz (Merg et al. 2016). We estimate this to imply al sample temperature of ~272±3 K. Using a CP contact time of 1 ms, 1000 scans were obtained with a RD of 3 s and an applied TPPM decoupling on 1H at 83 KHz.
Electron Microscopy and Atomic Force Microscopy
Transmission electron microscopy (TEM) and atomic force microscopy (AFM) measurements on htt exon 1 fibrils and C18-dipeptide nanoparticles were performed as previously reported (Hoop et al. 2016; Merg et al. 2016). The previously unpublished TEM data in Fig. 4a were obtained as follows. Unlabeled cyt-c obtained from Sigma Aldrich (St. Louis, MO) was added to liposomes at a lipid to protein molar ratio of 40:1. The liposomes were prepared as described above using a mixture of TOCL, DOPC and cholesterol at a molar ratio of (0.15:0.75:0.10) obtained from Avanti Polar Lipids. The sample was diluted in half with ddH2O and absorbed on to a freshly glow discharged carbon coated copper EM grid for more than one minute. Excess sample was wicked away with a filter paper. The grid was stained with 1% (wt/vol) uranyl acetate. The grid was allowed to fully air dry before images were acquired. The TEM measurements were performed on a Tecnai T12 Spirit transmission electron microscope (FEI, Hillsboro, OR) operating at 120 kV and equipped with an UltraScan 1000 CCD camera (Gatan, Pleasanton, CA).
Fig. 4.
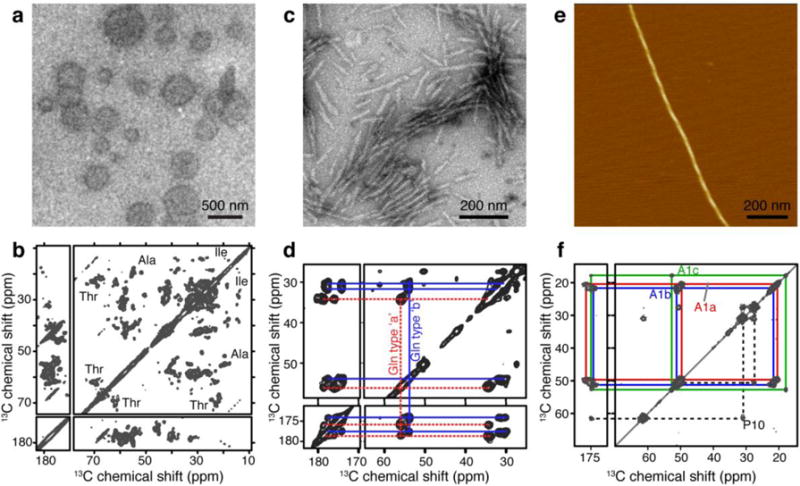
Examples of samples packed with ultracentrifugal devices. (a) TEM of TOCL/DOPC/cholesterol (0.15:0.75:0.10 molar ratio) vesicles with bound cytochrome c. (b) 13C-13C CP DARR spectrum of 1.7 mg of U-13C,15N cytochrome c bound to TOCL/DOPC (1:4 molar ratio) vesicles measured at a sample temperature of 233 K. (Adapted from (Mandal et al. 2015), Copyright 2015, with permission from Elsevier.) (c) TEM on [U-13C-15N]-labeled huntingtin exon 1 fibrils prepared for ssNMR. (d) 13C-13C CP DARR spectrum of 4.4 mg of htt exon1 fibrils measured at 275 K and 13 kHz MAS. Panels (c–d) are adapted from (Hoop et al. 2016). (e) AFM on C18-(PEPAuM-ox)2 fibers. (f) 13C-13C CP DARR spectrum of 1.9 mg of C18 -(PEPAuM-ox)2 peptide nanofibrils measured at 277 K. Panels (e–f) are adapted with permission from (Merg et al. 2016). Copyright 2016 American Chemical Society. The NMR measurements were performed on a 600 and 800 MHz (1H) spectrometer using 3.2 mm MAS rotors.
Results
MAS NMR sample packing design considerations
The process of MAS NMR sample packing essentially constitutes the concentrating of hydrated ‘solid’ samples of interest, ideally while avoiding dehydration of the sample. The most common and convenient way of achieving this is by pelleting the solid (or soluble) macromolecules from their initial aqueous suspension or solution by centrifugation. To avoid dehydration and ensure time efficiency, this ideally is done in a one-step fashion directly into the rotor. Finally, the resulting pellet should not only be compact, but also evenly distributed in the MAS rotor to avoid compromising rotor stability during the MAS. The latter argues against use of fixed-angle centrifuge rotor systems.
All these requirements are conveniently and perfectly met with the use of funnel-shaped packing devices designed for use in swinging bucket rotors. For the most general applicability, the device should permit pelleting of all kinds of (hydrated) ssNMR samples. The time and speed of centrifugation required for pelleting the sample depends on the nature of the sample. Protein crystals and aggregates often have densities that are significantly larger than that of water (1.24 to 1.54 g/mL (Bell et al. 1982; White et al. 2007)), which facilitates pelleting at relatively moderate g-forces, even in small table top centrifuges (Schmidt et al. 2010). Samples featuring lipid LUVs require the use of ultracentrifugation, due to the small density differences (~1.03 g/mL) with the aqueous buffer (see also below). Ultracentrifugation can also be used to sediment soluble protein complexes out of solution, for study by ssNMR (Bertini et al. 2011; Gardiennet et al. 2012).
Thus, in 2009 we set out to design our own ultracentrifugal packing device, with similar features as those used (and described) by others in the ssNMR community (Böckmann et al. 2009; Bertini et al. 2012; Gardiennet et al. 2012; Gelis et al. 2013). Primary goals were to obtain a robust and long-lived tool (i.e. a simple design), limit sample losses during tool usage, assure ease of use, and speed up the sample preparation process. The guiding principle was to start with a typical Eppendorf-style microcentrifuge-tube-sized sample (~1 mL) of a suspension or solution and pellet the “solid” fraction into the μL-sized MAS rotor in a single step.
Packing device design
To allow for the pelleting of a broad range of samples, we designed a tool that can withstand and achieve high g-forces: i.e. it is suitable for use in an ultracentrifuge. As noted above, symmetric and even rotor packing requires the use of swinging bucket ultracentrifuge rotors (UCR), such as the ones listed in Table 1. We reiterate here the comment that use of fixed-angle ultracentrifuges also could cause physical damage to the MAS rotor with associated risks in terms of rotors shattering before or during MAS. The design discussed here fits the 38.5 mL tubes of an SW 32 Ti UCR capable of achieving g-forces up to 175,000 × g on the tool itself. Greater g-forces can be achieved by the use of other UCR types, requiring the design of more compact tools that fit the smaller tube sizes (Table 1). Note that the placement of the MAS rotor within the device determines the g-forces that the sample itself will experience. For example, our packing tool can be spun at a maximum rate of 32,000 RPM corresponding to 175,000 × g forces felt by the device itself. However, the actual MAS rotor within our packing device experiences a maximum g-force of up to 162,000 × g. As discussed in more detail below, it is important to note that these g-forces are significantly less than the typical g-forces experienced by the sample while under MAS (Table 2).
Table 1.
Properties of swinging-bucket rotors available for Beckman Coulter ultracentrifuges.
| Ultracentrifuge rotor (UCR) | Max spinning rate (RPM) | G-force on sample | Maximum swinging bucket volume (ml) |
|---|---|---|---|
| SW 32 Ti | 32,000 | 175,000 | 38.5 |
| SW 40 Ti | 40,000 | 285,000 | 14 |
| SW 60 Ti | 60,000 | 485,000 | 4 |
Table 2.
Characteristics of Bruker MAS rotors.
| Bruker MAS rotor | Max. MAS rate b) | G-force on sample | Sample volume a) (μl) | |
|---|---|---|---|---|
| (kHz) | (RPM) | |||
| 0.7 mm | 111 | 6,660,000 | 11,180,000 | 0.5 |
| 1.3 mm | 67 | 4,020,000 | 8,140,000 | 2.5 – 3 |
| 1.9 mm | 42 | 2,520,000 | 5,330,000 | 10 – 14 |
| 2.5 mm (regular) | 35 | 2,100,000 | 3,700,000 | 12 – 16 |
| 2.5 mm (thinwall) | 35 | 2,100,000 | 4,200,000 | 12 – 20 |
| 3.2 mm (regular) | 24 | 1,440,000 | 2,550,000 | 30 – 40 |
| 3.2 mm (thinwall) b) | 5 | 300,000 | 130,000 | 42 – 50 |
| 8.3 | 500,000 | 360,000 | ||
| 12 | 720,000 | 750,000 | ||
| 24 | 1,440,000 | 3,020,000 | ||
| 4 mm | 15 | 900,000 | 1,360,000 | 89 – 92 |
Sample volume estimates for all rotors (except 0.7mm) were obtained, and calculated from dimensions published by Cortecnet (Brooklyn, NY). The sample volume estimate for 0.7mm rotor was obtained from Bruker Biospin.
Additional lower MAS rates are reported for the 3.2mm MAS rotors.
We designed two devices that fit the SW 32 Ti UCR buckets: one for 3.2 mm and one for 4 mm OD Bruker MAS rotors. The former device is shown in Fig. 1 and Fig. S1 (in the Supporting Information), while the latter is shown only in Fig. S1. Figures S2 and S3 show technical drawings of both devices. The design features three discrete pieces, each of which is machined from unfilled PEEK, in order to withstand the repeated and extensive exposure to high g forces. The total weight of these PEEK-based packing tool components is approximately 36 gram, with the 3.2-mm- and 4-mm-rotor devices having very similar masses within ~0.5 gram. The specifications of the Beckman Coulter SW 32 Ti swinging bucket rotor indicate that it can be operated at maximum speed up to a maximum sample density of 1.2 mg/mL. This reflects a maximum recommended sample weight of 46.2 gram for each 38.5 mL tube. Thus, even with the MAS rotor and up to 1–1.5 mL of sample volume, we stay well below this weight limit. The bottom piece (part I in Fig. 1) holds the 3.2 mm (or 4 mm) flat-bottomed Bruker rotors without the drive cap in place. Piece I is machined for a tight fit around the MAS rotor, in order to avoid excessive local stresses and forces during the packing process. The middle piece screws on to the bottom piece to seal the rotor in place, while also featuring a funnel designed to hold the initial sample suspension (Fig. 1c–d). The funnel is designed to hold a sample up to ~1 mL. Importantly, an O-ring is placed around the rotor to seal the parts and avoid sample losses (Fig. S1). A proper seal is assured by the usage of an O-ring slightly thicker (1.5 mm) than the vertical gap separating the parts I and II (Fig. 1d). The outer dimensions of the assembled device are such that it fits inside a clear centrifuge tube (Fig. 1a; middle), in case there is a concern of accidental spillage or leakage. In the current design, the top piece (part III in Fig. 1b–c) is not threaded or otherwise attached in any specific way to funnel part II. In this implementation, the only function was to prevent possible collapse of the surrounding centrifuge tube if one uses the device within such a tube (the thin-walled tubes are prone to collapse when used partly empty). In principle, one can outfit parts II and III with threading to extend the volume of the funnel, such that even larger initial sample volumes can be conveniently packed in a single run (Bertini et al. 2012; Ferella et al. 2013; Fragai et al. 2013). In this initial design we aimed for maximum simplicity and robustness and decided against a potentially more leak-prone threaded design. We chose to design part II as a somewhat shallow and short funnel (at the expense of maximizing volume) to ensure the most convenient inspection (and handling) of the pelleted sample after the sample packing process.
The external centrifuge tube acts as a backup in case of spills, which might occur if there is a failure of the O-ring or the bottom parts are not assembled properly. This fail-safe has rarely been necessary over the lifetime (~7 years) of our devices.
Usage of the ultracentrifuge packing device
The protocol for packing MAS samples with these ultracentrifugal devices is illustrated in Fig. 2. Since the sample will be centrifuged into the rotor, one end of the rotor is fully pre-assembled with any bottom spacers or bottom caps needed to seal it. For many Bruker-made rotors one end is closed anyway, so this may require no assembly. The empty rotor (lacking the drive cap) is inserting into the device’s bottom part (Fig. 1d). The dimensions are such that the rotor sticks out slightly, such that it will insert into the O-ring (shown red in Fig. 1d). After verifying the positioning and presence of said O-ring, the funnel (part II in Fig. 1b–d) is screwed on tightly by hand. This compresses the O-ring to seal the device against leakage. The device is now ready for administering the sample.
Fig. 2.
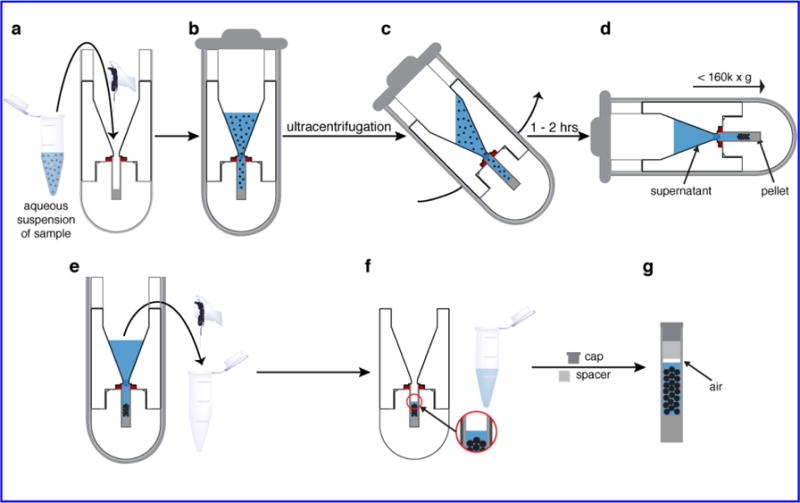
Packing process. (a) The sample suspension is placed in the funnel, up to a 1 mL volume. (b) The packed tool is inserted in its swinging bucket and sealed. (c, d) During UC the bucket achieves a horizontal orientation that ensures even pelleting of the sample within the rotor. (e) After completion of the UC, excess supernatant is removed. (f) A small amount of supernatant is left to maintain excess hydration. (g) After disassembly of the packing tool the rotor is capped. Note that it is important to leave a small gap between the liquid and the spacer or cap (see text). In this figure part III is not shown to scale to best display the sample handling.
The sample of interest is prepared as a hydrated suspension or solution. Up to 1 mL of the aqueous sample is transferred into the funnel (Fig. 2a–b), after which the filled device is encapsulated in the appropriate swinging-bucket tube holder (e.g. Fig. 1a). To avoid drying out of the sample in the evacuated ultracentrifuge, it is critical that the bucket holder itself is carefully sealed. A secondary tube containing either dense solution or a second packing tool is then carefully weight-balanced, and used as a counter balance during centrifugation. During centrifugation (Fig. 2c–d) the sample will be pelleted into an evenly distributed pellet at the bottom of the rotor. While the funnel can hold more than 1 mL, we typically limit the capacity to accommodate potential motion of the liquid sample during the centrifugation process (Fig. 2c). When feasible, samples to be used for packing are designed to have a volume of no more than ~1 mL (i.e. roughly the size of a typical microcentrifuge tube), to facilitate a single-step packing process. Alternatively, one can pack a larger volume by repeating the ultracentrifuge step multiple times (see also below).
The desired end result of a the packing process is to have the “biosolid” forming an even pellet or sediment in the rotor, with a clear supernatant filling the funnel and top of the rotor (Fig. 2e). The packing efficiency is evaluated by visual inspection and through the use of UV-Vis absorbance measurements on small samples taken from the supernatant. Next, the clear supernatant is carefully extracted from the funnel, such that an amount of supernatant remains atop the sample within the rotor (Fig. 2f). Note that, if used, the external clear centrifuge tube is typically deformed to the point where it has to be irreversibly removed by carefully cutting. The packing tool is then carefully disassembled, to avoid accidental spillage of the supernatant remaining in the rotor and to ensure that the pellet is not disturbed. Despite extensive use under high g-forces, our devices show no sign of significant deformation and are quite easily assembled and disassembled by hand. Once the MAS rotor has been extricated, additional supernatant can be carefully removed using a pipette such that there is only a small layer of supernatant on top of the pellet. This supernatant ensures excess hydration, but should be small enough that a gap remains between it and the spacer (or drive cap) used to seal the rotor (Fig. 2g). Performing this step after the disassembly of the tool allows for easier and careful judgment of the depths at which the pellet and supernatant lie in the rotor so as not to disturb the pellet. The supernatant removed in this final step can be added to the pool of supernatant that was removed in prior steps, which can then be analyzed by UV absorbance (or otherwise) to estimate how much protein was packed into the rotor. The gap above the sample is crucial for allowing thermal expansion, which can occur due to frictional heating during the MAS spinning and/or due to heating from radio frequency (RF) pulses (in particular 1H decoupling). A failure to leave room for sample expansion risks the drive cap being dislodged during NMR experiments, which would in turn cause rotor crashes.
Troubleshooting and optimization
Although the described procedures are suitable for a wide variety of samples (see below), the employed sample volume, centrifugation rate and centrifugation times may require some trial-and-error optimization. When packing a new kind of sample one may start at lower g-forces (~100,000 × g) for shorter periods of time (30 minutes to 1 hour), and evaluate the pelleting efficiency. The latter can be done by taking a small aliquot of the supernatant at the end of a run and submitting it to UV-Vis analysis. If there is sample still present in the supernatant, another round of UC either at higher speeds or for longer duration is performed as needed. If subsequent rounds of UC still fail to completely pack the entire sample, some troubleshooting is required. First, for certain samples the braking at the end of the runs may result in a disruption of the pelleted material. Using low or no braking during UC can rectify this problem, at the expense of some of the time efficiency of the packing protocol. Second, there may simply be more sample than can physically fit in the rotor. This may be fairly predictable for nanocrystalline proteins or peptides, but can be harder to predict for samples such as hydrated proteoliposomes. It is also possible that the sample needs to be subjected to higher g-force ultracentrifugation for extended durations. Conversely, if evidence of sample degradation due to very high g-forces is observed, then efficient packing can often be achieved at lower centrifugation speeds. A somewhat cautious approach may also be warranted when using newly designed or commercially obtained packing devices, at least during initial usage, to check for centrifugal-force-induced damage to the device or rotors.
Applications
Centrifugal packing tools have already been used by ourselves and others for preparing a wide variety of MAS NMR samples like protein micro- or nanocrystals (Böckmann et al. 2009), sedimented soluble proteins (Bertini et al. 2012; Gardiennet et al. 2012), protein complexes (Wiegand et al. 2016a; Wiegand et al. 2016b), fibrillar aggregates (Hoop et al. 2014; Hoop et al. 2016), and membrane-based samples (Das et al. 2013; Kunert et al. 2014; Mandal et al. 2015; Hisao et al. 2016). As we discuss below, this approach is particularly beneficial for those samples were ultracentrifugation is critical to achieve efficient pelleting or sedimentation.
Case Study 1 – Membrane-associated proteins
We first examine the application of this approach to the ssNMR study of lipid vesicles, or vesicle-bound proteins, using our recent work as an example (Mandal et al. 2015; Mandal and Van der Wel 2016). Unilamellar lipid vesicles of varying sizes are frequently used to mimic biological membranes in studies of protein-lipid or peptide-lipid interactions (Scalise et al. 2013; Mandal et al. 2015; Stepanyants et al. 2015; LeBarron and London 2016; Veshaguri et al. 2016). Unilamellar vesicles expose a maximum amount of membrane surface for binding by peripherally binding membrane proteins (Mandal et al. 2015) or antimicrobial peptides (Porcelli et al. 2013). Given their common use in vesicle leakage or vesicle fusion assays, ssNMR studies of LUV-based samples also allow a correlation to the results of such functional assays. As illustrated in Fig. 3a, LUVs feature a single spherical lipid bilayer filled with aqueous solution (Fig. 3a), with diameters ranging from 100 nm to ~500 nm (New 1994; Sharma and Sharma 1997). For most common applicable sizes, the water-filled LUVs have densities in the range of 1.02 – 1.08 g/mL (Fig. 3b) compared to protein aggregates or protein crystals with densities ~1.3 g/mL or higher (Bell et al. 1982; White et al. 2007). Note that, as indicated in Fig. 3a, the lipid bilayer itself has a notably higher density, but only makes up a very small fraction of the entire LUV (Mandal and Van der Wel 2016). The low LUV density means that they are not easy to pellet unless one employs ultracentrifugation (Tortorella et al. 1993). This is different for multilamellar vesicles (MLVs), which are more readily pelleted, as they can be 10–100× heavier than typical LUVs (Fig. 3). LUVs, and in particular, LUVs sized less than 200 nm, increasingly require high g-forces that can only be achieved by ultracentrifugation. This has led prior studies to increase their effective density by the inclusion of halogenated (Tortorella and London 1994) or biotinylated lipids (Tortorella et al. 1993). Others achieve a similar effect by loading them with sucrose (Zschörnig et al. 2005; Abe et al. 2011). However, this is not always practical, for instance if the vesicles are made porous by the addition of pre-amyloid oligomers or membrane-active peptides or proteins.
Fig. 3.

Dimensions and densities of lipid vesicles. (a) Schematic LUV shown approximately to scale. The (local) density of the lipid bilayer is higher than that of the water phase, but the overall density of the whole LUV is quite similar to that of water. (b) Typical size, density and molecular weight of differently sized LUVs, MLVs and protein microcrystals.
The use of ultracentrifugal sample packing was especially advantageous in our recent studies of the peripheral membrane protein cytochrome c (Fig. 4a–b) (Mandal et al. 2015). For both functional assays and for MAS ssNMR, the protein was bound to the membrane surface of 200-nm-sized LUVs. To achieve sample pelleting for ssNMR, the packing device containing the protein/LUV mixture was initially spun at 143,000 × g for 3 hours. Since UV measurements on the supernatant indicated that there was still LUV-bound protein left in the supernatant, a second UC step was performed at 175,000 × g for 4.5 hours. The final UV measurements confirmed that the 1.7 mg of (labeled) membrane-bound cytochrome c had completely pelleted in the rotor. A second, smaller sample featuring 0.75mg of protein was fully packed after 143,000 × g for 5 hours. The resultant samples were studies by 1D, 2D and 3D MAS ssNMR, with an example spectrum shown in Fig. 4b (Mandal et al. 2015). As described elsewhere, these NMR studies provided new insights into the membrane-induced structural changes in cytochrome c, as well as the dynamics and phase behavior of the lipids themselves (Mandal et al. 2015; Mandal and Van der Wel 2016).
Case Study 2 – Fibrillar samples
Another application of the packing tool is in the study of fibrillar protein and peptide aggregates, whether in a biological context or for materials-science applications. These can present a challenge for packing due to their elongated and sometimes quite rigid fibrillar structure, which turns the samples into hydrogels. Such hydrogels can resist compaction unless they are subjected to high g-forces. Depending on the sample, pre-treatments involving sonication or ultracentrifugation may be beneficial prior to the actual packing process.
We have successfully used the ultracentrifuge-packing tool to prepare a variety of peptide and protein fibrils (Li et al. 2011; Sivanandam et al. 2011; Hoop et al. 2014; Hoop et al. 2016). Our most recent studies on huntingtin exon1 (Fig. 4c–d) (Hoop et al. 2014; Hoop et al. 2016) involved the packing of htt exon1 fibrils into 3.2 mm MAS rotors. The packing protocol used here was slightly modified, based on the fact that here the sample size was limited by the amount of available protein fibrils rather than the size of the MAS rotor. After the first round of centrifugation at 154,000 × g for one hour, the supernatant was removed. At that time, 1 mL of buffer was used to rinse the original sample tube in order to recover residual sample that may have been left behind. This second batch was then added to the funnel and submitted to another round of centrifugation. A second rinse and centrifugation cycle was performed before the rotor was sealed for study by MAS NMR. At each stage, the supernatant collected was checked by UV-Vis absorbance spectrometry to determine the final amount of sample packed in the rotor. Fig. 4d shows an example spectrum obtained on such a sample, with more data shown in our published work on huntingtin exon 1 fibrils and other polyQ aggregates (Sivanandam et al. 2011; Hoop et al. 2014; Hoop et al. 2016).
Case Study 3 – Nanocrystals and other nanoassemblies
Some samples, especially protein nano-crystals and non-fibrillar aggregates do not necessarily require ultracentrifugation for efficient pelleting. Nonetheless, we find the described approach equally useful for these samples as it renders the process convenient, fast and reproducible. In addition, packing by ultracentrifugation results in a higher packing density that translates into an improved signal-to-noise ratio that is beneficial even for crystalline samples. Thus, we have similarly used this approach for peptide- or protein-based nanocrystals (Van der Wel et al. 2010; Li and van der Wel 2013). Fig. 4e–f illustrates a more recent application to acylated divalent peptide-derived nanoparticles (C18-(PEPAuM-ox)2 where PEP denotes the sequence AYSSGAPPMPPF) that are designed to induce formation of chiroptical gold particle assemblies (Fig. 4e–f) (Merg et al. 2016). The packing protocol used here was very simple. After applying the nanoparticle suspension to the packing tool, it was subjected to ultracentrifugation for 1 hour at 175,000 × g at 4 °C. UV measurements on the supernatant collected at the end of the centrifugation run confirmed that 1.92 mg of particles were packed into the rotor. Various MAS NMR spectra (e.g. Fig. 4f) were then used to probe the structural and motional features of the nanoparticle assemblies.
Discussion
Potential concerns and caveats
We have delineated how ultracentrifugal packing tools facilitate a generally applicable, convenient and effective way to pack hydrated samples for MAS ssNMR. The resulting samples retain their fully hydrated state, which is valuable to assure biological relevance, but also can be beneficial for spectral quality (Tang et al. 1999; Martin and Zilm 2003; Igumenova et al. 2004; Linden et al. 2011; Siemer et al. 2012; Bertini et al. 2013; Fragai et al. 2013; Böckmann and Meier 2014; Hisao et al. 2016). At the same time, one can minimize excess buffer, which not only maximizes the ssNMR signal but also may improve the RF distribution in the sample (Böckmann et al. 2009). In this section, we discuss and address a number of potential concerns with this approach.
Perhaps most importantly, one may express concern about the consequences of submitting potentially sensitive (biological) samples to ultracentrifugation during the packing process. The g-forces associated with the advocated sample packing approach can exceed 100,000 × g (Table 1), which may indeed lead to irrevocable changes in certain samples (Han et al. 2010; Renault et al. 2013; Cai et al. 2014). Whilst this is not an unreasonable concern, it is important to realize that this may be a moot point when samples are used for MAS ssNMR. Table 2 lists maximum (and typical) spin rates for different types of MAS rotors along with the corresponding g-forces that will be exerted on the sample. Comparing Tables 1 and 2, it is clear that that most MAS experiments are done at conditions that result in larger g-forces on the sample than exerted by an ultracentrifugal packing device. In addition, UC packing can often be quite effectively performed at reduced UC spinning rates that stay below the g-forces that cause damage or are achieved during MAS. Thus, it seems unnecessary to avoid ultracentrifugal packing due to concerns of sample damage.
Another concern affecting the design and use of these devices is whether the g-forces involved would (over time) cause damage and deformations in either the packing device or the MAS rotors. The two ultracentrifugal packing devices described in this paper were purposely designed to be simple, solid and robust. Since their construction in January 2010, they have been subjected to hundreds of hours of centrifugation. A few cosmetic scratches notwithstanding the tools continue to function as designed without leakage. Given that the g-forces associated with MAS exceed those of the packing process, one may think that there is no need to worry about rotor damage. However, the direction of the forces during packing will be along the length of the rotor. Depending on the design and execution of the packing device, one may also end up with particularly high forces being applied to specific points on the rotor. However, after almost seven years of usage, we have not observed any evidence or indication of negative consequences for the MAS rotors.
The described designs are based on Bruker design MAS rotors, and in particular their 3.2 and 4 mm rotors. Devices suitable for packing other rotor sizes or rotor designs will require design changes. For instance, some rotors have two open ends. Although one could insert an end-cap before packing the sample, it is likely that these caps may not withstand the forces involved in the packing process. Thus, one may need to replace the end cap after sample packing, or adapt the design of the packing device. For other rotor sizes, and in particular the advent of increasingly small rotors, it is likely that design changes will be needed beyond a mere “scaling down” of the described devices. That said, it may also be that this approach is particularly beneficial for the tinier rotor sizes (more on this below).
Finally, although we focused on the study of hydrated samples, there are certainly scenarios where packing tools are not needed for the ssNMR study of entirely dry samples (Bajaj et al. 2009). When investigating transient or unstable intermediates (Chimon and Ishii 2005; Chimon et al. 2007), one may need to trap and stabilize such states by freezing and lyophilizing the samples. In addition, it is also true that complete dehydration, followed by controlled re-hydration, is likely to remain the best way of retaining complete control of the sample hydration. This is important when low levels of sample hydration are necessary or desired, or for systematic studies of the impact of hydration on protein structure, dynamics and ssNMR spectral quality. Indeed, there is a long-standing body of work describing ssNMR on samples submitted to controlled re-hydration following lyophilization (Kennedy and Bryant 1990; Gregory et al. 1993; Jakeman et al. 1998; Pauli et al. 2000; Seidel et al. 2005; Krushelnitsky et al. 2006; Comellas et al. 2011; Luchinat et al. 2013; Ravera et al. 2016; Tuttle et al. 2016). As such, there are clearly scenarios where ultracentrifugal packing methods may not be necessary or practical.
Alternative designs and commercial availability
As noted above, similar devices have been used in a variety of ssNMR labs and have been described in the literature. Typically these have been custom-built devices that each feature different design choices and priorities. One of the differences in the various designs in the literature so far is the materials used for construction of the packing tool components. It is important to use a material that can be milled to high precision, is strong, durable and protective of the rotor during centrifugation. In our case these considerations and the expertise and tools available to us dictated our choice to use unfilled PEEK to construct all components of our packing tool. Other packing tools have been constructed usually using different materials like polyoxymethylene (POM), PEEK with 30% glass, delrin®, vespel®, aluminum and epoxide glass for the various components (Böckmann et al. 2009; Bertini et al. 2012; Gelis et al. 2013). It is worth noting that the choice of material (and design) should consider the UCR weight limits, as discussed above. There are examples of centrifugal tools designed for packing samples for ultrafast MAS NMR in the literature, for instance those used for packing amyloid fibril samples into 1.3 mm sized rotors (Hoop et al. 2016). Some of these devices are also commercially available. For instance, Giotto Biotech (Sesto Fiorentino, Italy) sells such devices for various types of MAS rotors, including 1.3mm diameter ones. Finally, there are scenarios where one may need to reach higher g-forces than possible in the devices described here. This includes samples containing small LUVs and SUVs, or sedimentation NMR applied to smaller proteins. Such cases require packing tools designed for used in a swinging-bucket UCR that achieves even higher g-forces (Table 1). For instance, a prior report described an ultracentrifugal packing device for use with a SW 60 Ti UCR allowing a maximum g-force up to 485,000 × g (Gardiennet et al. 2012). Given that this still stays well below the g-forces associated with commonly used MAS NMR rates (Table 2), it seems likely that this may be the optimal approach going forward, assuming that these centrifugal forces are found to not affect the structural integrity of the MAS rotors themselves.
Conclusions
In this report we discuss and advocate the more general use of ultracentrifugal packing devices for routine preparation of samples for modern MAS NMR. This approach maintains hydration throughout the packing process while at the same time making the entire process simpler and faster than more manual sample transfer methods. As discussed, these devices have a proven track record in applications to a wide array of sample types, but still are underused in the ssNMR community. We expect that a wider adoption of this methodology will help make ssNMR sample preparation faster, more routine, and more reproducible.
Supplementary Material
Acknowledgments
We thank Mike Delk for his help with the NMR experiments. Funding support was from the University of Pittsburgh and the National Institutes of Health grants R01GM112678 and R01GM113908 (P.v.d.W.), and T32 GM088119 (J.C.B.).
Footnotes
Author Contributions
T.W. and P.v.d.W. designed the packing tool. T.W. fabricated the packing tool. A.M. prepared samples. A.M. packed samples. A.M. and J.C.B. performed MAS NMR experiments. J.C.B. performed TEM measurements. A.M. and P.v.d.W. wrote the manuscript. All authors have read and edited the manuscript.
References
- Abe M, Niibayashi R, Koubori S, Moriyama I, Miyoshi H. Molecular mechanisms for the induction of peroxidase activity of the cytochrome c-cardiolipin complex. Biochemistry. 2011;50(39):8383–8391. doi: 10.1021/bi2010202. [DOI] [PubMed] [Google Scholar]
- Andreas LB, Jaudzems K, Stanek J, Lalli D, Bertarello A, Le Marchand T, Cala-De Paepe D, Kotelovica S, Akopjana I, Knott B, Wegner S, Engelke F, Lesage A, Emsley L, Tars K, Herrmann T, Pintacuda G. Structure of fully protonated proteins by proton-detected magic-angle spinning NMR. Proc Natl Acad Sci USA. 2016;113(33):9187–9192. doi: 10.1073/pnas.1602248113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreas LB, Le Marchand T, Jaudzems K, Pintacuda G. High-resolution proton-detected NMR of proteins at very fast MAS. J Magn Reson. 2015;253:36–49. doi: 10.1016/j.jmr.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Antzutkin ON, Balbach JJ, Leapman RD, Rizzo NW, Reed J, Tycko R. Multiple quantum solid-state NMR indicates a parallel, not antiparallel, organization of β-sheets in Alzheimer’s β-amyloid fibrils. Proc Natl Acad Sci USA. 2000;97(24):13045–13050. doi: 10.1073/pnas.230315097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj VS, Van der Wel PCA, Griffin RG. Observation of a low-temperature, dynamically driven structural transition in a polypeptide by solid-state NMR spectroscopy. J Am Chem Soc. 2009;131(1):118–128. doi: 10.1021/ja8045926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball P. Water as an active constituent in cell biology. Chem Rev. 2008;108(1):74–108. doi: 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- Bell DJ, Heywood-Waddington D, Hoare M, Dunnill P. The density of protein precipitates and its effect on centrifugal sedimentation. Biotechnol Bioeng. 1982;24(1):127–141. doi: 10.1002/bit.260240111. [DOI] [PubMed] [Google Scholar]
- Bertini I, Engelke F, Gonnelli L, Knott B, Luchinat C, Osen D, Ravera E. On the use of ultracentrifugal devices for sedimented solute NMR. J Biomol NMR. 2012;54(2):123–127. doi: 10.1007/s10858-012-9657-y. [DOI] [PubMed] [Google Scholar]
- Bertini I, Luchinat C, Parigi G, Ravera E. SedNMR: on the edge between solution and solid-state NMR. Acc Chem Res. 2013;46(9):2059–2069. doi: 10.1021/ar300342f. [DOI] [PubMed] [Google Scholar]
- Bertini I, Luchinat C, Parigi G, Ravera E, Reif B, Turano P. Solid-state NMR of proteins sedimented by ultracentrifugation. Proc Natl Acad Sci USA. 2011;108(26):10396–10399. doi: 10.1073/pnas.1103854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böckmann A, Gardiennet C, Verel R, Hunkeler A, Loquet A, Pintacuda G, Emsley L, Meier BH, Lesage A. Characterization of different water pools in solid-state NMR protein samples. J Biomol NMR. 2009;45(3):319–327. doi: 10.1007/s10858-009-9374-3. [DOI] [PubMed] [Google Scholar]
- Böckmann A, Meier B. Prions. Prion. 2014;4(2):72–79. doi: 10.4161/pri.4.2.11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant G, Koster KL, Wolfe J. Membrane behaviour in seeds and other systems at low water content: the various effects of solutes. Seed Sci Res. 2001;11:17–25. [Google Scholar]
- Cai H, Chen Y, Cui X, Cai S, Chen Z. High-resolution 1H NMR spectroscopy of fish muscle, eggs and small whole fish via Hadamard-encoded intermolecular multiple-quantum coherence. PLoS ONE. 2014;9(1):e86422. doi: 10.1371/journal.pone.0086422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimon S, Ishii Y. Capturing intermediate structures of Alzheimer’s beta-amyloid, Abeta(1–40), by solid-state NMR spectroscopy. J Am Chem Soc. 2005;127(39):13472–13473. doi: 10.1021/ja054039l. [DOI] [PubMed] [Google Scholar]
- Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, Ishii Y. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s β-amyloid. Nat Struct Mol Biol. 2007;14(12):1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- Comellas G, Lemkau LR, Nieuwkoop AJ, Kloepper KD, Ladror DT, Ebisu R, Woods WS, Lipton AS, George JM, Rienstra CM. Structured Regions of alpha-Synuclein Fibrils Include the Early-Onset Parkinson’s Disease Mutation Sites. J Mol Biol. 2011;411(4):881–895. doi: 10.1016/j.jmb.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das N, Murray DT, Cross TA. Lipid bilayer preparations of membrane proteins for oriented and magic-angle spinning solid-state NMR samples. Nat Protoc. 2013;8(11):2256–2270. doi: 10.1038/nprot.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers J-P, Chevelkov V, Lange A. Progress in correlation spectroscopy at ultra-fast magic-angle spinning: basic building blocks and complex experiments for the study of protein structure and dynamics. Solid State Nucl Magn Reson. 2011;40(3):101–113. doi: 10.1016/j.ssnmr.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Demers J-P, Habenstein B, Loquet A, Vasa SK, Giller K, Becker S, Baker D, Lange A, Sgourakis NG. High-resolution structure of the Shigella type-III secretion needle by solid-state NMR and cryo-electron microscopy. Nat Commun. 2014;5:1–12. doi: 10.1038/ncomms5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillmann B, Elbayed K, Zeiger H, Weingertner M-C, Piotto M, Engelke F. A novel low-E field coil to minimize heating of biological samples in solid-state multinuclear NMR experiments. J Magn Reson. 2007;187(1):10–18. doi: 10.1016/j.jmr.2007.02.018. [DOI] [PubMed] [Google Scholar]
- Ferella L, Luchinat C, Ravera E, Rosato A. SedNMR: a web tool for optimizing sedimentation of macromolecular solutes for SSNMR. J Biomol NMR. 2013;57(4):319–326. doi: 10.1007/s10858-013-9795-x. [DOI] [PubMed] [Google Scholar]
- Fragai M, Luchinat C, Parigi G, Ravera E. Practical considerations over spectral quality in solid state NMR spectroscopy of soluble proteins. J Biomol NMR. 2013;57(2):155–166. doi: 10.1007/s10858-013-9776-0. [DOI] [PubMed] [Google Scholar]
- Gardiennet C, Schütz AK, Hunkeler A, Kunert B, Terradot L, Böckmann A, Meier BH, Meier BH. A Sedimented Sample of a 59 kDa Dodecameric Helicase Yields High-Resolution Solid-State NMR Spectra. Angew Chem Int Ed. 2012;51(31):7855–7858. doi: 10.1002/anie.201200779. [DOI] [PubMed] [Google Scholar]
- Gawrisch K, Gaede HC, Mihailescu M, White SH. Hydration of POPC bilayers studied by 1H-PFG-MAS-NOESY and neutron diffraction. Eur Biophys J. 2007;36(4–5):281–291. doi: 10.1007/s00249-007-0142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelis I, Vitzthum V, Dhimole N, Caporini MA, Schedlbauer A, Carnevale D, Connell SR, Fucini P, Bodenhausen G. Solid-state NMR enhanced by dynamic nuclear polarization as a novel tool for ribosome structural biology. J Biomol NMR. 2013;56(2):85–93. doi: 10.1007/s10858-013-9721-2. [DOI] [PubMed] [Google Scholar]
- Goldbourt A. Biomolecular magic-angle spinning solid-state NMR: recent methods and applications. Curr Opin Biotechnol. 2013;24(4):705–715. doi: 10.1016/j.copbio.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Gregory RB, Gangoda M, Gilpin RK, Su W. The influence of hydration on the conformation of bovine serum albumin studied by solid-state 13C-NMR spectroscopy. Biopolymers. 1993;33(12):1871–1876. doi: 10.1002/bip.360331212. [DOI] [PubMed] [Google Scholar]
- Han Y, Ahn J, Concel J, Byeon I-JL, Gronenborn AM, Yang J, Polenova T. Solid-state NMR studies of HIV-1 capsid protein assemblies. J Am Chem Soc. 2010;132(6):1976–1987. doi: 10.1021/ja908687k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higman VA, Flinders J, Hiller M, Jehle S, Markovic S, Fiedler S, van Rossum B-J, Oschkinat H. Assigning large proteins in the solid state: a MAS NMR resonance assignment strategy using selectively and extensively 13C-labelled proteins. J Biomol NMR. 2009;44(4):245–260. doi: 10.1007/s10858-009-9338-7. [DOI] [PubMed] [Google Scholar]
- Hisao GS, Harland MA, Brown RA, Berthold DA, Wilson TE, Rienstra CM. An efficient method and device for transfer of semisolid materials into solid-state NMR spectroscopy rotors. J Magn Reson. 2016;265:172–176. doi: 10.1016/j.jmr.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoop CL, Lin H-K, Kar K, Hou Z, Poirier MA, Wetzel R, van der Wel PCA. Polyglutamine amyloid core boundaries and flanking domain dynamics in huntingtin fragment fibrils determined by solid-state nuclear magnetic resonance. Biochemistry. 2014;53(42):6653–6666. doi: 10.1021/bi501010q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoop CL, Lin H-K, Kar K, Magyarfalvi G, Lamley JM, Boatz JC, Mandal A, Lewandowski JR, Wetzel R, van der Wel PCA. Huntingtin exon 1 fibrils feature an interdigitated β-hairpin–based polyglutamine core. Proc Natl Acad Sci USA. 2016;113(6):1546–1551. doi: 10.1073/pnas.1521933113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoop CL, Sivanandam VN, Kodali R, Srnec MN, van der Wel PCA. Structural characterization of the caveolin scaffolding domain in association with cholesterol-rich membranes. Biochemistry. 2012;51(1):90–99. doi: 10.1021/bi201356v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igumenova TI, McDermott AE, Zilm KW, Martin RW, Paulson EK, Wand AJ. Assignments of Carbon NMR Resonances for Microcrystalline Ubiquitin. J Am Chem Soc. 2004;126(21):6720–6727. doi: 10.1021/ja030547o. [DOI] [PubMed] [Google Scholar]
- Jakeman DL, Mitchell DJ, Shuttleworth WA, Evans JN. Effects of sample preparation conditions on biomolecular solid-state NMR lineshapes. J Biomol NMR. 1998;12(3):417–421. doi: 10.1023/a:1008305118426. [DOI] [PubMed] [Google Scholar]
- Kennedy SD, Bryant RG. Structural effects of hydration: studies of lysozyme by 13C solids NMR. Biopolymers. 1990;29(14):1801–1806. doi: 10.1002/bip.360291411. [DOI] [PubMed] [Google Scholar]
- Khodadadi S, Roh JH, Kisliuk A, Mamontov E, Tyagi M, Woodson SA, Briber RM, Sokolov AP. Dynamics of biological macromolecules: not a simple slaving by hydration water. Biophys J. 2010;98(7):1321–1326. doi: 10.1016/j.bpj.2009.12.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloepper KD, Hartman KL, Ladror DT, Rienstra CM. Solid-state NMR spectroscopy reveals that water is nonessential to the core structure of alpha-synuclein fibrils. J Phys Chem B. 2007;111(47):13353–13356. doi: 10.1021/jp077036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight MJ, Felli IC, Pierattelli R, Emsley L, Pintacuda G. Magic angle spinning NMR of paramagnetic proteins. Acc Chem Res. 2013;46(9):2108–2116. doi: 10.1021/ar300349y. [DOI] [PubMed] [Google Scholar]
- Krushelnitsky A, Gogolev Y, Golbik R, Dahlquist F, Reichert D. Comparison of the internal dynamics of globular proteins in the microcrystalline and rehydrated lyophilized states. Biochim Biophys Acta Proteins Proteomics. 2006;1764(10):1639–1645. doi: 10.1016/j.bbapap.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Kunert B, Gardiennet C, Lacabanne D, Calles-Garcia D, Falson P, Jault J-M, Meier BH, Penin F, Böckmann A. Efficient and stable reconstitution of the ABC transporter BmrA for solid-state NMR studies. Front Mol Biosci. 2014;1:5. doi: 10.3389/fmolb.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamley JM, Iuga D, Öster C, Sass H-J, Rogowski M, Oss A, Past J, Reinhold A, Grzesiek S, Samoson A, Lewandowski JR. Solid-state NMR of a protein in a precipitated complex with a full-length antibody. J Am Chem Soc. 2014;136(48):16800–16806. doi: 10.1021/ja5069992. [DOI] [PubMed] [Google Scholar]
- LeBarron J, London E. Effect of lipid composition and amino acid sequence upon transmembrane peptide-accelerated lipid transleaflet diffusion (flip-flop) Biochim Biophys Acta. 2016;1858(8):1812–1820. doi: 10.1016/j.bbamem.2016.04.011. [DOI] [PubMed] [Google Scholar]
- Li J, Hoop CL, Kodali R, Sivanandam VN, van der Wel PCA. Amyloid-like fibrils from a domain-swapping protein feature a parallel, in-register conformation without native-like interactions. J Biol Chem. 2011;286(33):28988–28995. doi: 10.1074/jbc.M111.261750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, van der Wel PCA. Spinning-rate encoded chemical shift correlations from rotational resonance solid-state NMR experiments. J Magn Reson. 2013;230:117–124. doi: 10.1016/j.jmr.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden AH, Franks WT, Akbey Ü, Lange S, van Rossum B-J, Oschkinat H. Cryogenic temperature effects and resolution upon slow cooling of protein preparations in solid state NMR. J Biomol NMR. 2011;51(3):283–292. doi: 10.1007/s10858-011-9535-z. [DOI] [PubMed] [Google Scholar]
- Loquet A, Giller K, Becker S, Lange A. Supramolecular Interactions Probed by 13C− 13C Solid-State NMR Spectroscopy. J Am Chem Soc. 2010;132(43):15164–15166. doi: 10.1021/ja107460j. [DOI] [PubMed] [Google Scholar]
- Loquet A, Habenstein B, Lange A. Structural investigations of molecular machines by solid-state NMR. Acc Chem Res. 2013;46(9):2070–2079. doi: 10.1021/ar300320p. [DOI] [PubMed] [Google Scholar]
- Luchinat C, Parigi G, Ravera E. Water and Protein Dynamics in Sedimented Systems: A Relaxometric Investigation. ChemPhysChem. 2013;14(13):3156–3161. doi: 10.1002/cphc.201300167. [DOI] [PubMed] [Google Scholar]
- Mainz A, Jehle S, van Rossum BJ, Oschkinat H, Reif B. Large Protein Complexes with Extreme Rotational Correlation Times Investigated in Solution by Magic-Angle-Spinning NMR Spectroscopy. J Am Chem Soc. 2009;131(44):15968–15969. doi: 10.1021/ja904733v. [DOI] [PubMed] [Google Scholar]
- Mandal A, Hoop CL, DeLucia M, Kodali R, Kagan VE, Ahn J, van der Wel PCA. Structural Changes and Proapoptotic Peroxidase Activity of Cardiolipin- Bound Mitochondrial Cytochrome c. Biophys J. 2015;109(9):1873–1884. doi: 10.1016/j.bpj.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A, Van der Wel PCA. MAS 1H NMR Probes Freezing Point Depression of Water and Liquid-Gel Phase Transitions in Liposomes. Biophys J. 2016;111(9):1965–1973. doi: 10.1016/j.bpj.2016.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RW, Zilm KW. Preparation of protein nanocrystals and their characterization by solid state NMR. J Magn Reson. 2003;165(1):162–174. doi: 10.1016/s1090-7807(03)00253-2. [DOI] [PubMed] [Google Scholar]
- McNeill SA, Gor’kov PL, Shetty K, Brey WW, Long JR. A low-E magic angle spinning probe for biological solid state NMR at 750 MHz. J Magn Reson. 2009;197(2):135–144. doi: 10.1016/j.jmr.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehler M, Eckert CE, Busche A, Kulhei J, Michaelis J, Becker-Baldus J, Wachtveitl J, Dötsch V, Glaubitz C. Assembling a correctly folded and functional heptahelical membrane protein by protein trans-splicing. J Biol Chem. 2015;2015;290(46):27712–27722. doi: 10.1074/jbc.M115.681205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merg AD, Boatz JC, Mandal A, Zhao G, Mokashi-Punekar S, Liu C, Wang X, Zhang P, van der Wel PCA, Rosi NL. Peptide-directed assembly of single-helical gold nanoparticle superstructures exhibiting intense chiroptical activity. J Am Chem Soc. 2016;138(41):13655–13663. doi: 10.1021/jacs.6b07322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New RRC. Influence of liposome characteristics on their properties and fate. In: Philippot JR, Schuber F, editors. Liposomes as Tools in Basic Research and Industry. CRC Press; Boca Raton, FL: 1994. pp. 3–20. [Google Scholar]
- Pauli J, van Rossum B, Förster H, de Groot HJM, Oschkinat H. Sample Optimization and Identification of Signal Patterns of Amino Acid Side Chains in 2D RFDR Spectra of the α-Spectrin SH3 Domain. J Magn Reson Ser A. 2000;143(2):411–416. doi: 10.1006/jmre.2000.2029. [DOI] [PubMed] [Google Scholar]
- Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA. 2002;99(26):16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pöppler A-C, Demers J-P, Malon M, Singh AP, Roesky HW, Nishiyama Y, Lange A. Ultrafast Magic-Angle Spinning: Benefits for the Acquisition of Ultrawide-Line NMR Spectra of Heavy Spin-1/2 Nuclei. ChemPhysChem. 2016;17(6):812–816. doi: 10.1002/cphc.201501136. [DOI] [PubMed] [Google Scholar]
- Porcelli F, Ramamoorthy A, Barany G, Veglia G. On the role of NMR spectroscopy for characterization of antimicrobial peptides. Methods in Molecular Biology. 2013;1063:159–180. doi: 10.1007/978-1-62703-583-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn CM, Lu M, Suiter CL, Hou G, Zhang H, Polenova T. Magic angle spinning NMR of viruses. Prog Nucl Magn Reson Spectrosc. 2015;86–87:21–40. doi: 10.1016/j.pnmrs.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravera E. The bigger they are, the harder they fall: A topical review on sedimented solutes for solid-state NMR. Concepts Magn Reson. 2015;43(6):209–227. [Google Scholar]
- Ravera E, Ciambellotti S, Cerofolini L, Martelli T, Kozyreva T, Bernacchioni C, Giuntini S, Fragai M, Turano P, Luchinat C. Solid-State NMR of PEGylated Proteins. Angew Chem. 2016;128(7):2492–2495. doi: 10.1002/anie.201510148. [DOI] [PubMed] [Google Scholar]
- Ravera E, Schubeis T, Martelli T, Fragai M, Parigi G, Luchinat C. NMR of sedimented, fibrillized, silica-entrapped and microcrystalline (metallo)proteins. J Magn Reson. 2015;253:60–70. doi: 10.1016/j.jmr.2014.12.019. [DOI] [PubMed] [Google Scholar]
- Renault M, Shintu L, Piotto M, Caldarelli S. Slow-spinning low-sideband HR-MAS NMR spectroscopy: delicate analysis of biological samples. Sci Rep. 2013;3:3349. doi: 10.1038/srep03349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar R, Mainz A, Busi B, Barbet-Massin E, Kranz M, Hofmann T, Reif B. Immobilization of soluble protein complexes in MAS solid-state NMR: Sedimentation versus viscosity. Solid State Nucl Magn Reson. 2016;76–77:7–14. doi: 10.1016/j.ssnmr.2016.03.005. [DOI] [PubMed] [Google Scholar]
- Scalise M, Pochini L, Giangregorio N, Tonazzi A, Indiveri C. Proteoliposomes as tool for assaying membrane transporter functions and interactions with xenobiotics. Pharmaceutics. 2013;5(3):472–497. doi: 10.3390/pharmaceutics5030472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirò G, Fichou Y, Gallat F-X, Wood K, Gabel F, Moulin M, Härtlein M, Heyden M, Colletier J-P, Orecchini A, Paciaroni A, Wuttke J, Tobias DJ, Weik M. Translational diffusion of hydration water correlates with functional motions in folded and intrinsically disordered proteins. Nat Commun. 2015;6:6490. doi: 10.1038/ncomms7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HLF, Shah GJ, Sperling LJ, Rienstra CM. NMR Determination of Protein pK(a) Values in the Solid State. J Phys Chem Lett. 2010;1(10):1623–1628. doi: 10.1021/jz1004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubeis T, Nagaraj M, Ritter C. Segmental Isotope Labeling of Insoluble Proteins for Solid-State NMR by Protein Trans-Splicing. Methods Mol Biol. 2016;2017;1495:147–160. doi: 10.1007/978-1-4939-6451-2_10. [DOI] [PubMed] [Google Scholar]
- Seidel K, Etzkorn M, Heise H, Becker S, Baldus M. High-Resolution Solid-State NMR Studies on Uniformly [13C,15N]-Labeled Ubiquitin. ChemBioChem. 2005;6(9):1638–1647. doi: 10.1002/cbic.200500085. [DOI] [PubMed] [Google Scholar]
- Sharma A, Sharma US. Liposomes in drug delivery: progress and limitations. Int J Pharm. 1997;154(2):123–140. [Google Scholar]
- Sharpe S, Yau W-M, Tycko R. Structure and dynamics of the HIV-1 Vpu transmembrane domain revealed by solid-state NMR with magic-angle spinning. Biochemistry. 2006;45(3):918–933. doi: 10.1021/bi051766k. [DOI] [PubMed] [Google Scholar]
- Siemer AB, Huang K-Y, McDermott AE. Protein Linewidth and Solvent Dynamics in Frozen Solution NMR. PLoS ONE. 2012;7(10):e47242. doi: 10.1371/journal.pone.0047242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanandam VN, Jayaraman M, Hoop CL, Kodali R, Wetzel R, van der Wel PCA. The Aggregation-Enhancing Huntingtin N-Terminus Is Helical in Amyloid Fibrils. J Am Chem Soc. 2011;133(12):4558–4566. doi: 10.1021/ja110715f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanyants N, Macdonald PJ, Francy CA, Mears JA, Qi X, Ramachandran R. Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol Biol Cell. 2015;26(17):3104–3116. doi: 10.1091/mbc.E15-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus SK. Recent developments in solid-state magic-angle spinning, nuclear magnetic resonance of fully and significantly isotopically labelled peptides and proteins. Philos Trans R Soc Lond, B, Biol Sci. 2004;359(1446):997–1008. doi: 10.1098/rstb.2003.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Belton PS, Ng A, Ryden P. 13C MAS NMR Studies of the Effects of Hydration on the Cell Walls of Potatoes and Chinese Water Chestnuts. J Agric Food Chem. 1999;47(2):510–517. doi: 10.1021/jf980553h. [DOI] [PubMed] [Google Scholar]
- Tortorella D, London E. Method for efficient pelleting of small unilamellar model membrane vesicles. Anal Biochem. 1994;217(2):176–180. doi: 10.1006/abio.1994.1106. [DOI] [PubMed] [Google Scholar]
- Tortorella D, Ulbrandt ND, London E. Simple centrifugation method for efficient pelleting of both small and large unilamellar vesicles that allows convenient measurement of protein binding. Biochemistry. 1993;32(35):9181–9188. doi: 10.1021/bi00086a025. [DOI] [PubMed] [Google Scholar]
- Tuttle MD, Courtney JM, Barclay AM, Rienstra CM. Preparation of Amyloid Fibrils for Magic-Angle Spinning Solid-State NMR Spectroscopy. Methods in Molecular Biology. 2016;1345:173–183. doi: 10.1007/978-1-4939-2978-8_11. [DOI] [PubMed] [Google Scholar]
- Ulrich AS, Watts A. Molecular response of the lipid headgroup to bilayer hydration monitored by 2H-NMR. Biophys J. 1994;66(5):1441–1449. doi: 10.1016/S0006-3495(94)80934-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Wel PCA, Lewandowski JR, Griffin RG. Solid-State NMR Study of Amyloid Nanocrystals and Fibrils Formed by the Peptide GNNQQNY from Yeast Prion Protein Sup35p. J Am Chem Soc. 2007;129(16):5117–5130. doi: 10.1021/ja068633m. [DOI] [PubMed] [Google Scholar]
- Van der Wel PCA, Lewandowski JR, Griffin RG. Structural characterization of GNNQQNY amyloid fibrils by magic angle spinning NMR. Biochemistry. 2010;49(44):9457–9469. doi: 10.1021/bi100077x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Melckebeke H, Schanda P, Gath J, Wasmer C, Verel R, Lange A, Meier BH, Böckmann A. Probing water accessibility in HET-s(218–289) amyloid fibrils by solid-state NMR. J Mol Biol. 2011;405(3):765–772. doi: 10.1016/j.jmb.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Verel R, Tomka IT, Bertozzi C, Cadalbert R, Kammerer RA, Steinmetz MO, Meier BH. Polymorphism in an amyloid-like fibril-forming model peptide. Angew Chem Int Ed. 2008;47(31):5842–5845. doi: 10.1002/anie.200800021. [DOI] [PubMed] [Google Scholar]
- Veshaguri S, Christensen SM, Kemmer GC, Ghale G, Møller MP, Lohr C, Christensen AL, Justesen BH, Jørgensen IL, Schiller J, Hatzakis NS, Grabe M, Pomorski TG, Stamou D. Direct observation of proton pumping by a eukaryotic P-type ATPase. Science. 2016;351(6280):1469–1473. doi: 10.1126/science.aad6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar M, Chou H-T, Lührs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek R. The fold of alpha-synuclein fibrils. Proc Natl Acad Sci USA. 2008;105(25):8637–8642. doi: 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Ladizhansky V. Recent advances in magic angle spinning solid state NMR of membrane proteins. Prog Nucl Magn Reson Spectrosc. 2014;82:1–26. doi: 10.1016/j.pnmrs.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Weingarth M, Baldus M. Solid-state NMR-based approaches for supramolecular structure elucidation. Acc Chem Res. 2013;46(9):2037–2046. doi: 10.1021/ar300316e. [DOI] [PubMed] [Google Scholar]
- White ET, Tan WH, Ang JM, Tait S, Litster JD. The density of a protein crystal. Powder Technol. 2007;179(1–2):55–58. [Google Scholar]
- Wickramasinghe A, Wang S, Matsuda I, Nishiyama Y, Nemoto T, Endo Y, Ishii Y. Evolution of CPMAS under fast magic-angle-spinning at 100 kHz and beyond. Solid State Nucl Magn Reson. 2015;72:9–16. doi: 10.1016/j.ssnmr.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand T, Cadalbert R, Gardiennet C, Timmins J, Terradot L, Böckmann A, Meier BH. Monitoring ssDNA Binding to the DnaB Helicase from Helicobacter pylori by Solid-State NMR Spectroscopy. Angew Chem Int Ed Engl. 2016a;55(45):14164–14168. doi: 10.1002/anie.201607295. [DOI] [PubMed] [Google Scholar]
- Wiegand T, Gardiennet C, Cadalbert R, Lacabanne D, Kunert B, Terradot L, Böckmann A, Meier BH. Variability and conservation of structural domains in divide-and- conquer approaches. J Biomol NMR. 2016b;65(2):79–86. doi: 10.1007/s10858-016-0039-8. [DOI] [PubMed] [Google Scholar]
- Wiegand T, Gardiennet C, Ravotti F, Bazin A, Kunert B, Lacabanne D, Cadalbert R, Güntert P, Terradot L, Böckmann A, Meier BH. Solid-state NMR sequential assignments of the N-terminal domain of HpDnaB helicase. Biomol NMR Assign. 2015;10(1):13–23. doi: 10.1007/s12104-015-9629-8. [DOI] [PubMed] [Google Scholar]
- Wolfe J, Bryant G. Freezing, drying, and/or vitrification of membrane- solute-water systems. Cryobiology. 1999;39(2):103–129. doi: 10.1006/cryo.1999.2195. [DOI] [PubMed] [Google Scholar]
- Zhang H, Hou G, Lu M, Ahn J, Byeon I-JL, Langmead CJ, Perilla JR, Hung I, Gor’kov PL, Gan Z, Brey WW, Case DA, Schulten K, Gronenborn AM, Polenova T. HIV-1 Capsid Function Is Regulated by Dynamics: Quantitative Atomic-Resolution Insights by Integrating Magic-Angle-Spinning NMR, QM/MM, and MD. J Am Chem Soc. 2016;138(42):14066–14075. doi: 10.1021/jacs.6b08744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Chen Y, Tang X, Li J, Wang L, Yang J. Solid-state NMR shows that dynamically different domains of membrane proteins have different hydration dependence. J Phys Chem B. 2014;118(32):9553–9564. doi: 10.1021/jp503032h. [DOI] [PubMed] [Google Scholar]
- Zschörnig O, Paasche G, Thieme C, Korb N, Arnold K. Modulation of lysozyme charge influences interaction with phospholipid vesicles. Colloids Surf B. 2005;42(1):69–78. doi: 10.1016/j.colsurfb.2005.01.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.