

ABSTRACT
Central nervous system (CNS) inflammation and autophagy dysfunction are known to be involved in the pathology of neurodegenerative diseases. Manganese (Mn), a neurotoxic metal, has the potential to induce microglia-mediated neuroinflammation as well as autophagy dysfunction. NLRP3 (NLR family, pyrin domain containing 3)- CASP1 (caspase 1) inflammasome-mediated neuroinflammation in microglia has specific relevance to neurological diseases. However, the mechanism driving these phenomena remains poorly understood. We demonstrate that Mn activates the NLRP3-CASP1 inflammasome pathway in the hippocampus of mice and BV2 cells by triggering autophagy-lysosomal dysfunction. The autophagy-lysosomal dysfunction is induced by lysosomal damage caused by excessive Mn accumulation, damaging the structure and normal function of these organelles. Additionally, we show that the release of lysosomal CTSB (cathepsin B) plays an important role in Mn-induced NLRP3-CASP1 inflammasome activation, and that the increased autophagosomes in the cytoplasm are not the main cause of NLRP3-CASP1 inflammasome activation. The accumulation of proinflammatory cytokines, such as IL1B (interleukin 1 β) and IL18 (interleukin 18), as well as the dysfunctional autophagy pathway may damage hippocampal neuronal cells, thus leading to hippocampal-dependent impairment in learning and memory, which is associated with the pathogenesis of Alzheimer disease (AD).
KEYWORDS: autophagy, manganese, microglia, neuroinflammation, NLRP3 inflammasome
Introduction
Alzheimer disease (AD) is the most common dementing illness in the world.1 Neuroinflammation is recognized as a fundamental response of the central nerve system (CNS) not only to acute injury, but also to chronic neurodegenerative disease.2 Neuroinflammatory changes in AD include the robust activation of microglia and astrocytes, and the release of proinflammatory cytokines, cell adhesion molecules and chemokines.3 Proinflammatory cytokines, including IL1B (interleukin 1 β), IL18 (interleukin 18) and TNF (tumor necrosis factor), are key initiators of neuroinflammation. IL1B levels are elevated in AD, contributing to its pathology by increasing amyloid precursor gene expression, MAPT/Tau hyperphosphorylation and memory impairment.4
Manganese (Mn) is an essential trace element that is widely distributed in the earth crust. It not only plays a key role in numerous biochemical reactions, including immune response, ATP generation, bone growth, digestion and reproduction, but also serves as a cofactor for various enzymes, such as arginase and glutamine synthetase.5,6 However, excessive Mn accumulation from either environmental or occupational sources may lead to a neurodegenerative disorder referred to as manganism.7,8 Patients with manganism exhibit cognitive and motor dysfunction.9,10 Additionally, Mn overload in brain may be involved in the etiology of AD, since patients with elevated Mn levels exhibit dementia and typical pathological signs of AD characterized by neuritic plaques and neurofibrillary tangles.10,11 In animal models, chronic Mn treatment induces upregulation of amyloid-like protein 1 and diffuse amyloid β plaques in the frontal cortex of macaques,12,13 thus supporting a link between advanced-stage manganism and dementia.11,14
The innate immune system components in the CNS act as the first-line of broader immune response to pathogen-associated molecular patterns from extracellular milieu and endosomal compartments, as well as danger-associated molecular patterns from host-derived danger signals.15 Microglia are the innate immune cells within the CNS and they express a range of pattern recognition receptors, including Toll-like receptors and Nod-like receptors.15 Microglia constantly monitor the proximal environment through pattern recognition receptors. Once microglia sense tissue injury or a foreign (infectious) agent, a complex network of activation pathways activates the innate immune response, releasing immune molecules, including cytokines, chemokines and reactive oxygen species.15 Mn has been shown to induce microglial activation, releasing proinflammatory cytokines, such as IL1B.16 However, the mechanism of Mn-induced microglial activation and its immune response remains unclear. IL1B is synthesized as an inactive cytoplasmic precursor (36 kDa, called pro-IL1B), which is cleaved into the biological active form (17 kDa, called mature IL1B) in response to proinflammatory stimuli. The maturation and release of IL1B requires cysteine proteases.17 The inflammasome involving NLRP3 (NLR family, pyrin domain containing 3)-CASP1 (caspase 1) represents a primary pathway, which can regulate the maturation of IL1B. Belonging to the Nod-like receptor family, NLRP3 has emerged as the most versatile innate immune receptor because of its broad specificity.18 Several studies have reported that a broad array of stimuli can activate the NLRP3-CASP1 inflammasome pathway and release of the proinflammatory factor IL1B in immune cells, which is involved in innate immune responses. However, whether Mn, a naturally occurring heavy metal, is involved in the innate immune response through the NLRP3-CASP1 inflammasome pathway in the microglia has yet to be determined.
Autophagy is a highly conserved homeostatic process by which cytoplasmic macromolecules, excess or damaged organelles, and several pathogens are delivered to lysosomes for degradation. The autophagy machinery is composed of ATG (autophagy-related) genes, of which ATG1 to ATG10, ATG12 to ATG14 and ATG16L1/2, and ATG18 are all required for autophagosome formation.19,20 Autophagosome formation is controlled by the ATG12–ATG5-ATG16L1/2 protein complex, which acts in part as an E3 ligase for MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), an acknowledged autophagosomal marker.21 In most cases, autophagy activation ensures cell survival during starvation or stressful conditions. Once autophagy dysfunction is triggered, it induces cell damage. Lysosome function is critical for the process of autophagy. The release and cytoplasmic accumulation of lysosomal protease CTSB (cathepsin B) result in abnormal lysosomal function. Recent studies have shown that the release of CTSB and the inhibition of autophagy are involved in inflammasome activation.22,23 In the macrophages, the lysosomal damage results in cytosolic release of lysosomal contents-CTSB, which are sensed by the NLRP3 inflammasome, thus activating the NLRP3-CASP1 pathway.22,24,25 In our previous studies, rats injected with MnCl2 into the striatum show dysregulation of autophagy, which was characterized by increased number of mitochondrial vacuoles, swollen and broken endoplasmic reticulum and dysfunctional lysosomes.26 However, whether the autophagy-lysosomal pathway participates in inflammasome activation and Mn-induced cell death has yet to be understood. Therefore, elucidation of the mechanisms involved in Mn-induced autophagy dysfunction and inflammasome activation is a requisite for the better understanding of mechanisms associated with Mn neurotoxicity.
In the present study, we show NLRP3-CASP1 inflammasome and autophagy activation in the hippocampal region in a mouse model of subacute manganism along with cognitive dysfunction. Disrupting the autophagy-lysosomal pathway with ammonium chloride (NH4Cl) attenuated the Mn-induced NLRP3 activation and inflammatory factor (IL1B) release, suggesting that the autophagy-lysosomal pathway and CTSB play important roles in Mn-induced NLRP3-CASP1 inflammasome activation. We demonstrate that Mn-induced the NLRP3-CASP1 inflammasome pathway activation and autophagy play important roles in the subsequent neuroinflammation. The results suggest that there are close relationships between neuroinflammation, autophagy and cognitive dysfunction upon subacute Mn exposure. Taken together, these results suggest that Mn can cause cell injury by destabilizing the autophagy-lysosomal system, leading to NLRP3 inflammasome dependent CASP1 activation and inflammatory factor (IL1B) release. Furthermore, a novel molecular mechanism of Mn-induced cognitive dysfunction was elucidated, whereby sublethal concentrations of Mn activated proinflammatory NLRP3-dependent signaling by disrupting the autophagy-lysosomal pathway.
Results
Mn concentration in blood and hippocampus and Mn exposure affected the ability to learn and memory in mice
To confirm the validity of the Mn model, we tested Mn levels in blood and hippocampal tissues by atomic fluorescence spectrophotometry in various groups of mice injected with the metal for 7 d. The concentration of Mn in blood and in the hippocampal tissue was significantly increased after 7 d of Mn injection (Fig. 1A, B). No significant differences in Mn concentrations were observed in the NaCl-injected and lipopolysaccharide (LPS)-injected groups after 7 d (Fig. 1A, B). Next, 2 sets of protocols (illustrated in Fig. 1C) were used for fear memory test in Mn, LPS, and NaCl groups. LPS, the active component of gram-negative bacteria cell walls, is a potent stimulator of the immune system. It has been demonstrated that LPS selectively impairs contextual fear. Therefore, we used it as a positive control. No significant difference in the percent of frozen time was observed between the 3 groups in each of the training session (Fig. 1D). In the delayed fear conditioning, the mice were tested for contextual and cued fear memory 24 h after training. The percent of frozen time in the NaCl-injected group was markedly increased in the contextual as well as in the cued test compared with the training session (Fig. 1D). However, no significant difference in the percent of frozen time was found in Mn-injected group and LPS-injected group in the contextual test as well as in the cued test compared with the training session (Fig. 1D). In the session of contextual test and cued test, the percentage of frozen time in the NaCl-injected group was markedly increased compared with the Mn-and LPS-injected groups (Fig. 1D). These findings suggest that the injection of Mn and LPS alters fear memory in mice.
Figure 1.
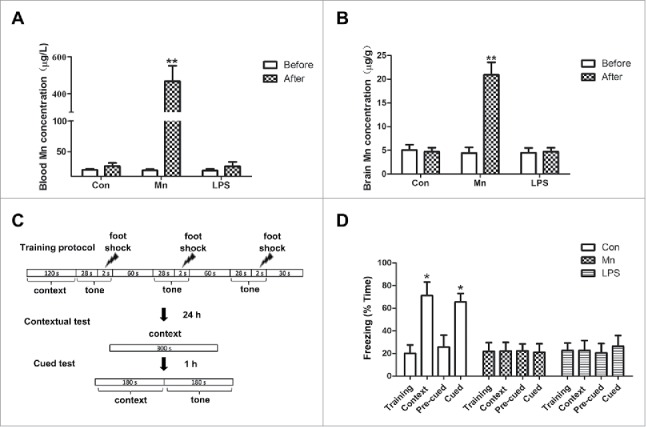
Mn concentration in blood and hippocampus, and Mn effects on memory in mice. (A, B) Mn concentration in blood (A) and in hippocampus (B). (C) Illustration of the protocols used for cue and contextual fear conditioning. (D) Freezing behavior was expressed as the percentage of time in the training and testing session for MnCl2- (n = 10), LPS- (n = 10) and NaCl-injected groups (n = 10). Statistical analysis was performed by using one-way ANOVA. Data are shown as means ± SE. *P < 0.05, **P < 0.01 when compared with the control group.
Mn activates the NLRP3-CASP1 inflammasome both in microglia within the hippocampus of mice and in BV2 cells
To investigate whether Mn induced the activation of the NLRP3-CASP1 inflammasome in hippocampal microglia, immunofluorescence staining was performed. ITGAM was used as a specific marker for microglia. Resting microglia were highly branched with small cell bodies. In the activation state, the length of the branches is reduced and the cell body is enlarged. As shown in Fig. 2A, after 7 d of Mn- or LPS-injection, the hippocampal region displayed strong and extensive staining for NLRP3 compared with the NaCl group. The majority of the microglia were in the resting state with negative staining for NLRP3 in the NaCl group. However, in the Mn and LPS injection group, microglia in the hippocampus were activated displaying a dramatic change in morphology and staining positively for NLRP3. Protein expression of NLRP3, CASP1 and IL1B was analyzed in response to Mn, LPS and NaCl by western blotting (Fig. 2B). The NaCl and LPS groups were set as the negative and positive controls, respectively. CASP1 activation was assessed by the appearance of CASP1 p20. Compared with the NaCl group, the expression of NLRP3, cleaved CASP1 and mature IL1B (Fig. 2B) was significantly increased after Mn injection for 7 d. The LPS induced-group, in which NLRP3, cleaved CASP1 and mature IL1B significantly increased was set as the positive control. In addition, Mn- and LPS-induced hippocampal microglia result in increased IL1B and IL18 secretion in hippocampus compared with NaCl (Fig. 2C).
Figure 2.
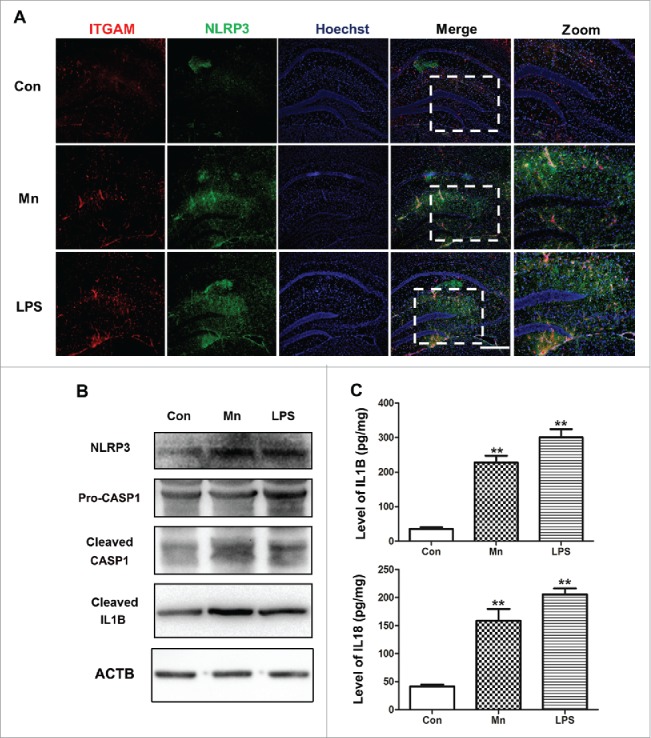
Mn activates the NLRP3-CASP1 inflammasome in murine hippocampal microglia. Mice were treated with MnCl2 for 7 d. (A) The hippocampal sections of mice were stained for NLRP3 (green staining), ITGAM as the microglia marker (red staining). The LPS-induced group and NaCl-induced group were the positive control and negative control, respectively. Nuclei were stained with Hoechst. Bar: 50 µm. (B) The protein level of NLRP3, CASP1 and cleaved IL1B in the hippocampus tissue was analyzed by western blot. (C) Levels of IL1B and IL18 in the hippocampal tissue were measured by ELISA. Statistical analysis was performed with one-way ANOVA. Data are shown as means ± SE. **P < 0.01 when compared with the negative control group.
To further determine the role of Mn in activating the NLRP3-CASP1 inflammasome in microglia, we selected BV2 cells, as a microglia cell line for in vitro studies. The immunocytochemical staining was used to evaluate the expression and localization of NLRP3 in response to Mn and LPS. The optimal Mn concentration necessary to elicit a response was determined. We found that after 6 h Mn treatment, the cells were positively stained for NLRP3 (Fig. 3A) in the cytoplasm compared with the untreated cells. The LPS-induced group, which showed staining for NLRP3 in the cytoplasm, was set as the positive control. Next the protein levels of NLRP3, cleaved CASP1 and mature IL1B in BV2 cells were evaluated in response to Mn and LPS by western blotting. LPS (10 μg/ml) was set as a positive control. A range of Mn concentration (25, 50, 100 μM) was used. After 6 h Mn-treatment, protein expression of NLRP3 was significantly increased compared with the untreated cells (Fig. 3B). CASP1 activation was assessed by the appearance of CASP1 p20. The expression of pro-CASP1 protein was detected in all groups, but the expression of active CASP1 (p20) was detected only in the BV2 cell line, treated with Mn or LPS (Fig. 3B). Furthermore, we evaluated the release of proinflammatory cytokines, including IL1B and IL18 in BV2 cells induced by Mn treatment. A significant increase of IL1B and IL18 was observed by ELISA in BV2 cells after 6 h of Mn- and LPS-induction (Fig. 3C).
Figure 3.
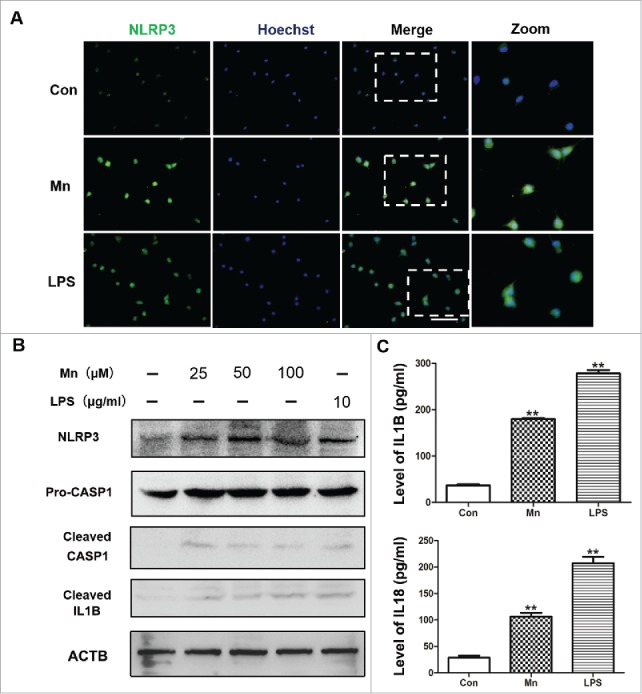
Mn activates the NLRP3-CASP1 inflammasome in BV2 cells. BV2 cells were treated with MnCl2 for 6 h. (A) The BV2 cells were stained for NLRP3 (green staining). The LPS-induced group and untreated group were the positive control and negative control, respectively. Nuclei were stained with Hoechst. Bar: 50 µm. (B) The protein level of NLRP3, CASP1 and cleaved IL1B in the BV2 cells was analyzed by western blot. The relative band intensities were determined by densitometry. (C) Levels of IL1B and IL18 in the BV2 cell culture supernatants were measured by ELISA. Statistical analysis was performed with one-way ANOVA. Data are shown as means ± SE. **P < 0.01 when compared with the negative control group.
The NLRP3 inflammasome contributes to CASP1 activation and IL1B secretion after Mn induction in BV2 cells
To define the role of NLRP3 inflammasome components in activation of CASP1 in response to Mn, Nlrp3 in BV2 cells was knocked down by siRNA. Compared with nontarget siRNA control and untreated cells, the protein level of NLRP3 was significantly reduced in Mn-induced group and LPS-induced group (Fig. 4A). Concomitantly, significant reduction in CASP1 p20 levels was also observed (Fig. 4A). Furthermore, the expression of IL1B and IL18 was downregulated (Fig. 4B, C). Taken together, the results established that NLRP3 was required for Mn-induced CASP1 activation and proinflammatory cytokine (IL1B and IL18) secretion.
Figure 4.

The NLRP3 inflammasome contributes to CASP1 activation and IL1B secretion after Mn induction in BV2 cells. NLRP3 knockdown cells and nontarget control cells were treated with Mn or LPS for 6 h. (A) The protein level of NLRP3, CASP1 and cleaved IL1B were analyzed by western blot. The relative band intensities were determined by densitometry. (B) The mRNA expression of Il1b was analyzed by qPCR. (C) Levels of IL1B and IL18 in the BV2 cell culture supernatants were measured by ELISA. Statistical analysis was performed with one-way ANOVA. Data are shown as means ± SE. * P < 0.05, ** P < 0.01 when compared with the untreated group. @ P < 0.05 when compared with the NC siRNA group. # P < 0.05 when compared with the NC siRNA MnCl2 -induced group. & P < 0.05 when compared with the NC siRNA LPS-induced group. ¥ P < 0.05 when compared with the Nlrp3 siRNA Group.
Mn blocks autophagy flux
Autophagy has been implicated in a variety of neural degeneration diseases. We have already reported that Mn exposure induces autophagy dysfunction in the substantia nigra.27 We therefore studied the effect of Mn on autophagy in BV2 cells by determining LC3-II, ATG5, BECN1 (Beclin 1, autophagy related), SQSTM1 and CTSB protein expression 6 h after Mn exposure. LC3-II is an autophagosomal surface protein marker that is ultimately degraded inside the autolysosomes.28 Results of western blotting showed that the LC3-II level was dramatically increased by Mn treatment. Concomitantly, the accumulation of SQSTM1, a protein degraded by autophagy,29 was observed in Mn-exposed BV2 cells. The massive increase in protein levels of LC3-II and SQSTM1 indicated that autophagic lysosomal degradation activity was impaired by Mn treatment (Fig. 5B). Immunofluorescence staining showed that Mn exposure for 6 h substantially induced the accumulation of LC3-positive autophagic vacuoles (Fig. 5A). Cysteine proteases CTSB is the most abundant lysosomal proteases.24 To investigate the effect of Mn on lysosomal function, the active form of CTSB was determined. Exposure to Mn for 6 h substantially increased the levels of active CTSB in BV2 cells (Fig. 5B). In addition, we found that the autophagy pathway was blocked by Mn in the BV2 cell line, as evidenced by increased typical autophagosomes with double membranes and cellular contents, vacuole-containing autophagosomes observed under TEM (Fig. 5C). Moreover, we found increased number of dysfunctional swollen lysosomes containing high dense particulate materials and mitochondrial vacuoles, indicating lysosomal damage (Fig. 5C). Our results suggest that Mn accumulated in the lysosome and caused compensatory increase in CTSB and autophagy-lysosomal dysfunction 6 h after the exposure, ultimately, leading to interruption of the degradation of autophagy body.
Figure 5.

Alterations of autophagosomes and lysosomes as well as changes of autophagy-related proteins after Mn induction in BV2 cells. (A) Representative immunocytochemistry of BV2 cells after treatment with Mn for 6 h to determine LC3B protein levels. The LPS and sham groups were used as the positive and negative control groups, respectively. Nuclei were stained with Hoechst. Scale bars: 20 μm. (B) The protein level of ATG5, LC3B, BECN1 and CTSB were analyzed by western blot. The relative band intensities were determined by densitometry. (C) TEM shows the ultrastructure of cells treated with Mn and LPS for 6 h. Arrows indicate autophagosomes, including residual digested material and empty vacuoles; n = 3. Scale bars: 1 μm.
The activation of the NLRP3-CASP1 inflammasome induced by Mn is not restrained by controlling the autophagy elongation stage
To further examine whether dysfunction of lysosomes was involved in the activation of the NLRP3-CASP1 inflammasome pathway, BV2 cells were used in an in vitro study. Autophagy consists of several steps: sequestration, transport to lysosomes, degradation, and utilization of degradation products, and each step may exert diverse function. To determine whether stepped-up effect of Mn on NLRP3 inflammosome was mediated by autophagy elongation stage, we evaluated the influence of Atg5 siRNA on the inhibitory effect of Mn exposure on NLRP3 inflammasome formation using western blotting analysis and immunofluorescent staining for LC3. Downregulation of Atg5 prevented the Mn-induced LC3-II accumulation in BV2 cells (Fig. 6). However, knockdown of Atg5 could not inhibit Mn- and LPS-induced increase in NLRP3, cleaved CASP1 and mature IL1B, suggesting that autophagy elongation stage was not involved in the activation of NLRP3-CASP1 inflammasomes (Figs. 6B, 7). Collectively, these data suggested that the increase of autophagosome formation was not responsible for the activation of NLRP3-CASP1 inflammasomes.
Figure 6.

The activation of the NLRP3-CASP1 inflammasome induced by Mn is not restrained by controlling the autophagy elongation stage. BV2 cells were pretreated with Atg5 siRNA for 24 h followed by treatment with 100 μM Mn for 6 h. (A) Representative immunocytochemistry of BV2 cells after treatment with Mn for 6 h to determine LC3 protein levels. Nuclei were stained with Hoechst. Scale bars: 20 μm. (B) The protein level of ATG5, LC3, BECN1 and CTSB were analyzed by western blot. The relative band intensities were determined by densitometry.
Figure 7.

The release of inflammatory cytokines, such as IL1B induced by Mn is not restrained by controlling the autophagy elongation stage. BV2 cells were pretreated with Atg5 siRNA for 24 h followed by treatment with 100 μM Mn for 6 h. (A) The protein level of mature cleaved IL1B was analyzed by western blot. The relative band intensities were determined by densitometry. (B) The mRNA expression of Il1b was analyzed by qPCR. (C) Levels of IL1B and IL18 in the BV2 cell culture supernatants were measured by ELISA. Statistical analysis was performed with one-way ANOVA. Data are shown as means ± SE. *P < 0.05 when compared with the untreated group. # P < 0.05 when compared with the Atg5 siRNA group.
The activation of the NLRP3-CASP1 inflammasome induced by Mn is not restrained by controlling the autophagy maturation stage
Autophagosome-lysosome fusion and autolysosome degradation are required in the late autophagic process to maintain functional autophagic flux and cellular homeostasis. Bafilomycin A1 (BAF) is a specific inhibitor of the subunit of vacuolar-type H+-ATPase. Electron microscopy showed that BAF inhibited the binding of autophagy body to lysosomes and prevented the maturation of autophagy body. Thus, autophagy maturation stage was disrupted by the V-ATPase inhibitor BAF. Herein, we examined whether autophagy maturation stage (autophagosomes fusion with lysosomes) was involved in the activation of the NLRP3-CASP1 inflammasome induced by Mn exposure for 6 h. The BV2 cells were treated with 100 μM Mn or 100 nM BAF alone or in combination for 6 h. The inhibitory effect of BAF on LC3-II degradation was confirmed by western blotting and immunofluorescent staining for LC3 (Fig. 8). Importantly, the addition of BAF enhanced the Mn-induced LC3-II accumulation in BV2 cells (Fig. 8A). Whereas BAF did not influence Mn- and LPS -induced significant increase of NLRP3, cleaved CASP1 and mature IL1B (Figs. 8B, 9). These results suggested that BAF did not affect the ability of Mn to induce the activation of NLRP3-CASP1 inflammasomes.
Figure 8.

The activation of the NLRP3-CASP1 inflammasome induced by Mn is not restrained by controlling the autophagy maturation stage. BV2 cells were pretreated with 100 nM BAF for 24 h followed by treatment with 100 µM Mn for 6 h. (A) Representative immunocytochemistry of BV2 cells after treatment with Mn for 6 h to determine LC3B protein levels. Nuclei were stained with Hoechst. Scale bars: 20 μm. (B) The protein level of ATG5, LC3B, BECN1 and CTSB were analyzed by western blot. The relative band intensities were determined by densitometry.
Figure 9.

The release of inflammatory cytokines, such as IL1B induced by Mn is not restrained by controlling the autophagy maturation stage. BV2 cells were pretreated with 100 nM BAF for 24 h followed by treatment with 100 μM Mn for 6 h. (A) The protein level of mature cleaved IL1B was analyzed by western blot. The relative band intensities were determined by densitometry. (B) The mRNA expression of Il1b was analyzed by qPCR. (C) Levels of IL1B and IL18 in the BV2 cell culture supernatants were measured by ELISA. Statistical analysis was performed with one-way ANOVA. Data are shown as means ± SE. *P < 0.05 when compared with the untreated group. # P < 0.05 when compared with the BAF group.
Mn-induced release of CTSB triggers NLRP3-CASP1 inflammasome activation
Previous studies have reported that crystal-induced (such as monosodium urate and silica) lysosomal damage plays an important role in activating the NLRP3-CASP1 inflammasome.17 Lysosomes contain several proteolytic enzymes, including those of the CTSB family. The release of CTSB has been linked with the activation of the NLRP3 inflammasome. In our in vitro study, we found that CTSB expression in BV2 cells was significantly increased 6 h after Mn exposure compared with the sham group (Fig. 5B). To further examine whether release of CTSB from the lysosomes was involved in the activation of the NLRP3-CASP1 inflammasome pathway, BV2 cells were treated with 20 mM NH4Cl to suppress the activities of CTSB via shifting the lysosomal pH above the optimum of their activities. Treatment with the NH4Cl caused a significant reduction in protein CTSB levels, attenuating the activation of NLRP3-CASP1 inflammasomes (Fig. 10A, B) as well as the release of IL1B and IL18 (Fig. 10C). These results indicated that CTSB was critical for Mn-induced activation of the NLRP3-CASP1 inflammasome and subsequent release of proinflammatory cytokines.
Figure 10.

Mn enhances NLRP3-CASP1 inflammasome activation and release of inflammatory cytokines, such as IL1B in BV2 cells is blocked by pretreatment with NH4Cl. BV2 cells were treated with 20 mM NH4Cl at the indicated concentrations for 30 min, and subsequently treated with Mn or LPS for 6 h. (A, B) The protein level of ATG5, LC3B, CTSB, cleaved CASP1 and cleaved IL1B were analyzed by western blot. The relative band intensities were determined by densitometry. (C) Levels of IL1B and IL18 in the BV2 cell culture supernatants were measured by ELISA. Statistical analysis was performed with one-way ANOVA. Data are shown as means ± SE. *P < 0.05 ** P < 0.01 when compared with the untreated group. #P < 0.05 when compared with the Mn-induced group. & P < 0.05 when compared with the LPS-induced group. @ P < 0.05 when compared with the NH4Cl group.
Discussion
In the past decade, a growing interest has focused on understanding the role of heavy metals in the etiology of neurodegenerative disease, including manganism, Wilson disease, Parkinson disease (PD), Huntington disease and Alzheimer disease (AD).30 Mn, an essential, yet potentially neurotoxic metal, readily crosses the blood-brain barrier and blood-cerebrospinal fluid barrier in both the developing fetus and adult.31 Excessive accumulation of Mn is associated with neurodegeneration, characterized by dysfunctional basal ganglia, leading to a severe neurological disorder analogous to PD.30 AD is clinically characterized by a progressive and irreversible loss of learning and memory ability, and represents the most common dementing illness.32 A potential link between Mn and AD has been advanced.12,13,33 Herein, we demonstrate that Mn accumulates in the hippocampus upon subacute exposure in a murine model. The accumulation of Mn in the hippocampal region significantly reduced contextual fear conditioning, indicating that Mn selectively influences hippocampal-dependent memory consolidation.
The innate immune response protects against injury in the CNS. However, excessive or off-target action can also promote immunopathology. Excessive neuroinflammation causes neuron and oligodendrocyte dysfunction and cell death, thus leading not only to acute injury, but also to chronic neurodegenerative diseases. Innate immune products referred to proinflammatory cytokines that are released in response to the detection of foreign substances, or responses to infectious agents, such as fever and reduced activity (all of these produce a “sickness syndrome”). IL1B, known to play a role in the “sickness syndrome,” exerts influence on the hippocampal-dependent memory consolidation. IL1B is selectively involved in hippocampal-dependent memory as demonstrated with various memory tasks, such as cue and contextual fear conditioning, as well as the Morris water escape task. Additionally, IL1B elevation has been detected in brains of aged AD mouse models,34,35 as well as in AD patients,36 providing a link between IL1B and AD pathogenesis. Herein, we demonstrated that Mn could activate the innate immune response, leading to the release of IL1B. Accordingly, we speculate that the accumulation of IL1B in the hippocampus resulted in impairment of learning and memory ability in mice. Pro-IL1B is produced as the inactive precursor of IL1B in the cytosol. Maturation and release of IL1B requires the activation of the NLRP3-CASP1 inflammasome. The NLRP3 inflammasome, which comprises the NLRP3 scaffold, the PYCARD/ASC adaptor and CASP1, is involved in a range of neurological diseases including infection, acute sterile brain and chronic neurodegenerative disease.37 In acute infections, most of pathogen-associated molecular patterns-like virus (such as Japanese encephalitis virus and West Nile virus) and bacteria (such as S. pneumoniae and S.aureus) can activate the NLRP3 inflammasome in microglia and macrophages within the CNS.38-43 While in the case of chronic sterile inflammation, the NLRP3 inflammasome activation in microglia and macrophages within CNS can be induced by misfolded protein, such as amyloid β, SNCA/α-synuclein and PRNP (prion protein).24,44-47 Here, we found that Mn activated the NLRP3-CASP1 inflammasome pathway both in vivo and in vitro. We provided evidence that NLRP3 and CASP1 were expressed in microglia within the hippocampal region and BV2 cells, a microglial cell line. We analyzed the presence, localization and quantity of NLRP3 in the hippocampus and found that the NLRP3 inflammasome was widely expressed in this region upon Mn treatment, particularly in activated microglia as characterized by morphological changes and intense expression of NLRP3. Additionally, the studies performed in BV2 cells showed that the NLRP3-CASP1 inflammasome can be activated by a range of Mn concentrations in vitro. Downregulation of Nlrp3 significantly reduced the expression of CASP1 p20 as well as the amount of IL1B. Taken together, microglia may be activated by Mn both in vivo and in vitro, thus leading to the activation of the NLRP3-CASP1 inflammasome pathway, release of IL1B and the ensuing innate immune response. Excessive accumulation of IL1B in the hippocampus may cause neuron and oligodendrocyte dysfunction or death thus contributing to the impairment of learning and memory. The process of excessive neuroinflammation has a potential relationship to AD pathogenesis. Therefore, further in-depth investigation should be performed to fully evaluate the neurotoxic effect of IL1B on neurons and oligodendrocytes by using the conditional medium from Mn-exposed microglia, or by coculturing neurons and oligodendrocytes with Mn-exposed microglia.
Given the critical role of the NLRP3 inflammasome in the maturation and release of proinflammatory cytokines, we evaluated whether the NLRP3 inflammasome was involved in Mn-induced release of IL1B, IL18 and TNF from microglia. Using immunofluorescent staining, we showed that Mn exposure induced a significant increase in hippocampal expression of NLRP3. In addition, we confirmed that Mn exposure resulted in a significant increase of the NLRP3 inflammasome, as evidenced by increased expression of NLRP3 and cleaved CASP1 in hippocampal tissues and the BV2 cell line. Moreover, knockdown of Nlrp3 significantly inhibited Mn-induced cleavage of CASP1 and the release of IL1B and IL18. The results demonstrated that the NLRP3 inflammasome was involved in Mn-induced release of proinflammatory cytokines from microglia. We have to point that knockdown of Nlrp3 downregulates the expression of TNF, the maturation and release of which is regulated by Toll-like receptors members.
Autophagy is an important physiological process that influences cellular homeostasis. Autophagy dysfunction is associated with various neurological diseases, including PD, AD and Huntington disease.10,48 In our previous studies, we have found that Mn can result in impairment of motion ability through affecting the function of dopaminergic neurons via decreasing autophagy in substantia nigra and striatum.27 In the present study, we found that activation of the NLRP3 inflammasome was involved in Mn-induced release of proinflammatory cytokines from microglia.16 Next, we explored the possible relationships between autophagy and the formation of the NLRP3 inflammasome in response to Mn. We showed that Mn resulted in a significant increase of the expression of BECN1, LC3-II, and ATG5. Moreover, we found that the expression of SQSTM1, a chaperone protein of autophagy degradation, was increased by Mn, indicating the interruption of autophagy degradation in lysosomes. These results support the notion that Mn induced the formation of autophagosomes, but inhibited autophagy, as illustrated by cytoplasmic accumulation of autophagosomes along with lysosomes containing degradation products. The results of the in vitro experiments also supported the above notion.
Furthermore, we also observed significant increase in the expression of CTSB in microglia, induced by Mn. We speculated that the increase of CTSB may be attributed to 2 reasons: first, the increase of CTSB may be the result of compensatory enhancement of eliminating activity in response to direct Mn stimulation; second, Mn accumulation may result in structural injury to lysosomes, leading to release of CTSB. We further confirmed that Mn could induce autophagy dysfunction, which mediated the activation of the NLRP3 inflammasome.
Autophagy consists of several steps, including initiation, elongation, fusion of autophagosome and lysosome, and degradation.20 We next examined which step was involved in Mn-induced activation of the NLRP3 inflammasome. ATG5 is essential for the recruitment of LC3 and the formation and elongation of autophagosome. We showed that knockdown of Atg5 significantly inhibited Mn-induced transformation from LC3-I to LC3-II, indicating the interruption of the formation of the autophagy precursor. However, the activation of the NLRP3 inflammasome and the subsequent release of proinflammatory cytokines (IL1B and IL18) were not altered by the knockdown of Atg5. These results indicated that the formation of autophagosome was not the essential step toward the activation of the NLRP3 inflammasome.
Next, we examined the possible role of fusion of autophagosome and lysosome and degradation in Mn-induced activation of the NLRP3 inflammasome. BAF, an inhibitor of the proton pump, inhibited the combination of autophagosome and lysosome.49 In the study, we found that inhibition of the late stage of autophagy by BAF had no evident effect on Mn-induced activation of the NLRP3 inflammasome and release of proinflammatory cytokines, indicating that disorder of the fusion of autophagosome and lysosome and degradation was not involved in Mn-induced activation of the NLRP3 inflammasome.
Previous studies have shown that NLRP3 can be activated by CTSB.17 Therefore, we explored whether the increase of CTSB was involved in Mn-induced activation of the NLRP3 inflammasome. NH4Cl inhibits the acidification of the lysosome and decreases the expression of CTSB.50 We found that NH4Cl significantly inhibited the Mn-induced increase of CTSB, NLRP3 and cleaved CASP1, and the release of IL1B and IL18, without a significant effect on autophagy-related proteins. The results indicated that CTSB was critical for Mn-induced activation of the NLRP3 inflammasome and subsequent release of proinflammatory cytokines.
We demonstrated that the NLRP3-CASP1 inflammasome pathway in the hippocampus of mice and BV2 cells can be activated by Mn via autophagy dysfunction and lysosomal CTSB release. Additionally, we found that Mn-induced autophagy dysfunction was caused by the dysfunction of lysosomes which release vast amounts of CTSB. The accumulation of proinflammatory cytokines, such as IL1B and IL18, as well as the dysfunctional autophagy pathway may damage neuronal cells, thus lead to hippocampal-dependent impairment of learning and memory. More work is required to investigate the neurotoxic effect of IL1B on neuronal cells as well as the relationship between the autophagy dysfunction and impairment in learning and memory in the hippocampal region.
Materials and methods
Animals, treatment, hippocampus tissue collection
All procedures involving animals were performed strictly in accordance with the international standards of animal care guidelines and were approved by the institutional care and use committee of Fourth Military Medical University (Permit Number: SCXK-2012–0007). Mice were injected subcutaneously with manganese (II) chloride tetrahydrate (MnCl2 4H2O) in a subacute treatment based on a published protocol.51,52 Briefly, animals were randomly assigned to 3 treatment groups and injected with either 0.1 mL of Mn (100 mg/kg, in saline), saline (0.9% NaCl) and LPS (200 µl, 1mg/ml). This treatment was repeated every third day for a wk, for a total of 3 treatments on 1, 4, and 7 d.
For hippocampal tissue collection following behavioral testing, the mice were killed by rapid cervical dislocation and brains were immediately removed and placed onto a chilled glass petri dish atop a bed of crushed ice. The brain was kept moist with phosphate-buffered saline (PBS; Sigma, P5493). The hippocampus was dissected using microforceps and stored at −80°C in 0.1 M PBS (pH 7.4).
Mn concentration in blood and hippocampus samples
At the end of the behavioral study, blood samples (0.1 ml) and hippocampal samples (0.2 g) were collected from mice after decapitation in each group. The level of Mn was determined by hydride generation atomic fluorescence spectrometry (PerkinElmer PinAAcle 900T Part No. N3160082 Serial No. PTCS12070201, Waltham, MA, USA) using the national standard method.
Cue and contextual fear conditioning
Associative memory was tested using NIR Video Fear Conditioning System for Mouse (Med Associates, Inc., St. Albans, VT, USA). The experimental protocol was performed as previously reported and schematically illustrated in Fig. 1A.53 Briefly, the training session consisted of a 2-min exploration period followed by 2 conditioned stimulus-unconditioned stimulus (CS-US) pairings (tone 90 db white noise 30 sec duration, foot-shock intensity 0.75 mA). Context tests were performed in the same training chamber 24 h after training. Cue tests were performed in a distinct chamber 1 h after training for 6 min. Scoring the animal every 10 sec as freezing or not freezing for different duration in the contextual test, precued test and cued test. The measures were as follows:
Cell, chemical reagents, and siRNA transfection
BV2, one type of microglia cell line, was chosen as experimental model and siRNA oligos against Nlrp3 (GenePharma, Nlrp3-Mus-727 siRNA oligo) were transfected using Lipofectamine 2000 (Invitrogen, 11668027) according to the manufacturer's protocol. LPS (Sigma, L3880) and NH4Cl (Sigma, A9434) were from Sigma-Aldrich.
Transmission electron microscopy (TEM)
TEM was conducted as previously reported.27 Briefly, hippocampal tissue and BV2 cells were fixed in 2% glutaraldehyde in 0.1 M PBS (pH 7.4) for 2 h at 4°C. Subsequently, they were postfixed in 1% osmium tetroxide in 0.1 M PBS (pH 7.4) for 2 h at 4°C. Next, the hippocampal tissue was dissected out (1 mm3) and processed in 1% osmium tetroxide. The tissues and cells then were dehydrated in ascending ethanol solutions ranging from 20 to 100%, and embedded in Epon812 (Sigma, 45347–1L-F). Ultrathin sections (80 nm) were impregnated in 2% uranyl acetate and Reynolds lead citrate. Sections were viewed with a transmission electron microscope (Zeiss Serien-No.3527001716, Baden-Württemberg, Oberkochen Germany).
Real-time quantitative PCR (qPCR)
Total RNA was extracted from BV2 cells using RNA plus (Takara, 9108) and treated with RNase-free DNase I (Promega, M6101). One microgram of total RNA was used as a template to make first-strand cDNA by oligo-dT priming with the Omniscript RT kit (Qiagen, 205113). qPCR analyses were performed in an ABI Prism 7500 fast RealTime PCR System (Applied Biosystems, Foster City, CA, USA) with the SYBR Green PCR master mix reagent (Takara, RR430A) in a 40-cycle PCR. The denaturing, annealing and extension conditions of each PCR cycle were 95°C for 30 s, 95°C for 5 s, 60°C for 34 s and 95°C for 15 s. The relative amount or fold change of the target gene expression was normalized relative to the level of D-glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and relative to a control (untreated cells). The primer sequences used in the qRT-PCR were:
Gapdh: Forward primer 5′-AGGTCGGTGTGAACGGATTTG-3′, Reverse primer 5′- TGTAGACCATGTAGTTGAGGTCA-3′
Il1b: Forward primer 5′-TCCAGGATGAGGACATGAGCAC-3′, Reverse primer 5′- GAACGTCACACACCAGCAGGTTA-3′
Western blot analysis
The total protein content was extracted from hippocampus tissue and BV2 cells by using lysis buffer containing protease inhibitors. The protein concentration was measured with the BCA-200 protein assay kit (Thermo, 23225). Equal amounts of protein were separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane. The membrane was blocked in TRIS-buffered saline (Sigma, T5912) with Tween 20 (Sigma, P9416) containing 5% nonfat dry milk for 2 h and probed with primary antibodies-NLRP3 (Abcam, ab4207), CASP1 (Abcam, ab108362), IL1B (Abcam, ab9722), BECN1 (Cell Signaling Technology, 3738), CTSB (Proteintech, 12216–1-AP), MAP1LC3B/LC3B (Cell Signaling Technology, 2775), ATG5 (Cell Signaling Technology, 12994S), SQSTM1 (Cell Signaling Technology, 8025S), and ACTB (Cell Signaling Technology, 12262) overnight at 4°C and then incubated for 2 h with a horseradish-peroxidase-conjugated anti-mouse IgG antibody or anti-rabbit IgG diluted 1: 2000 according to the primary antibodies. Protein bands were visualized on X-ray film by using an enhance chemiluminescence system (GE Healthcare, Buckinghamshire, UK). The relative protein expression intensities were quantified by densitometry with the Quantity One analysis software.
Immunofluorescence staining
Mice were perfused through the heart under deep anesthesia with 150 to 200 ml of 4% paraformaldehyde in phosphate buffer, pH 7.4. Hippocampus was dissected from brain, and then cryoprotected in 30% sucrose (Sigma, V900116) phosphate buffer (Sigma, 1805), pH 7.4. The tissues were frozen at −80°C, and sectioned serially (25 μm). BV2 cells were probed with anti-NLRP3 antibody. Three randomly chosen 25-μm-thick sections were probed with anti-NLRP3 antibody and anti-ITGAM (AbD Serotec, MCA275G, clone OX42) antibody. Cells were fixed with 4% paraformaldehyde for 30 min and then incubated in PBS containing 0.4% Triton X-100 (Sigma, T8787) for 10 min on ice. The cells and sections were blocked with bovine serum albumin (Sigma, V900933) for 60 min at 37°C. After the blocking step, the cells and sections were incubated with primary antibody at 4°C overnight; PBS was used as the negative control. The cells and sections were then washed with PBS and incubated for 1 h with the secondary antibodies, namely anti-rabbit IgG conjugated to fluorescein isothiocyanate (FITC; Cell Signaling Technology, 4412S) or anti-mouse IgG conjugated to rhodamine (Cell Signaling Technology, 8890S), each at 1:1000, at room temperature. After being rinsed with PBS, the sections and cells were examined under a Nikon A1R confocal microscope (Nikon, 2CE-SBTH-7, Takkuko, Nasu-Machi, Nasu-Gun, Tochigi, Japan).
ELISA
The production of IL1B (eBioscience, 88–7013) and IL18 (eBioscience, BMS618/2) in the culture supernatants was measured by ELISA as specified by the manufacturer, according to the manufacturer's instructions. BV2 cells plated in 12-well plates were cultured with Mn, LPS and sham for 6 h. The culture supernatants were aspirated and stored at −80°C until assayed by ELISA. The concentration of IL1B and IL18 was determined using a standard curve obtained with IL1B and IL18 protein.
Statistical analysis
Each experiment was performed at least 3 times, unless otherwise indicated. Data are reported as the mean ± SE (standard error) deviation from 3 independent experiments. Significant differences between the experimental and the control groups were determined by one-way analysis of variance (ANOVA) with Tukey-Kramer post-hoc test; Values of P<0.05 were considered statistically significant.
Abbreviations
- AD
Alzheimer disease
- ATG
autophagy related
- BAF
bafilomycin A1
- BECN1
Beclin 1 autophagy related
- CNS
central nervous system
- CTSB
cathepsin B
- CASP1
caspase 1
- IL1B
interleukin 1 β
- IL18
interleukin 18
- LPS
lipopolysaccharide
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 β
- Mn
manganese
- NLRP3 NLR family
NLR family pyrin domain containing 3
- NH4Cl
ammonium chloride
- PBS
phosphate-buffered saline
- PD
Parkinson disease
- TNF
tumor necrosis factor
- TEM
transmission electron microscopy
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by National Basic Research Program of China (973 Program, 2012CB525004); Key Project of Natural Science Foundation of China (81230063); National Natural Science Foundation of China (81673121, 81602815, 81372952 and 81673211); Program for Changjiang Scholars (T2011153) and Innovative Research Team in University (PCSIRT); Shaanxi science and technology coordinating innovative project (2011KTCL03–19); M.A was supported by NIEHS R01 ES 10563.
References
- [1].Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 297:353-6; PMID:12130773; http://dx.doi.org/ 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- [2].Heneka MT, O'Banion MK, Terwel D, Kummer MP. Neuroinflammatory processes in Alzheimer's disease. J Neural Transm 2010; 117:919-47; PMID:20632195; http://dx.doi.org/ 10.1007/s00702-010-0438-z [DOI] [PubMed] [Google Scholar]
- [3].Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer's disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol 1998; 8:65-72; PMID:9458167; http://dx.doi.org/ 10.1111/j.1750-3639.1998.tb00136.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rivera-Escalera F, Matousek SB, Ghosh S, Olschowka JA, O'Banion MK. Interleukin-1beta mediated amyloid plaque clearance is independent of CCR2 signaling in the APP/PS1 mouse model of Alzheimer's disease. Neurobiol Dis 2014; 69:124-33; PMID:24874542; http://dx.doi.org/ 10.1016/j.nbd.2014.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Erikson KM, Aschner M. Manganese neurotoxicity and glutamate-GABA interaction. Neurochem Int 2003; 43:475-80; PMID:12742094; http://dx.doi.org/ 10.1016/S0197-0186(03)00037-8 [DOI] [PubMed] [Google Scholar]
- [6].Butterworth J. Changes in nine enzyme markers for neurons, glia, and endothelial cells in agonal state and Huntington's disease caudate nucleus. J Neurochem 1986; 47:583-7; PMID:2874190; http://dx.doi.org/ 10.1111/j.1471-4159.1986.tb04539.x [DOI] [PubMed] [Google Scholar]
- [7].Hudnell HK. Effects from environmental Mn exposures: a review of the evidence from non-occupational exposure studies. Neurotoxicology 1999; 20:379-97; PMID:10385898 [PubMed] [Google Scholar]
- [8].Iregren A. Manganese neurotoxicity in industrial exposures: proof of effects, critical exposure level, and sensitive tests. Neurotoxicology 1999; 20:315-23; PMID:10385893 [PubMed] [Google Scholar]
- [9].Tuschl K, Mills PB, Clayton PT. Manganese and the brain. Int Rev Neurobiol 2013; 110:277-312; PMID:24209443 [DOI] [PubMed] [Google Scholar]
- [10].Butler PD, Wang Z, Ly DP, Longaker MT, Koong AC, Yang GP. Unfolded protein response regulation in keloid cells. J Surg Res 2011; 167:151-7; PMID:19631342; http://dx.doi.org/ 10.1016/j.jss.2009.04.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Banta RG, Markesbery WR. Elevated manganese levels associated with dementia and extrapyramidal signs. Neurology 1977; 27:213-6; PMID:557755; http://dx.doi.org/ 10.1212/WNL.27.3.213 [DOI] [PubMed] [Google Scholar]
- [12].Guilarte TR, Burton NC, Verina T, Prabhu VV, Becker KG, Syversen T, Schneider JS. Increased APLP1 expression and neurodegeneration in the frontal cortex of manganese-exposed non-human primates. J Neurochem 2008; 105:1948-59; PMID:18284614; http://dx.doi.org/ 10.1111/j.1471-4159.2008.05295.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Guilarte TR. APLP1, Alzheimer's-like pathology and neurodegeneration in the frontal cortex of manganese-exposed non-human primates. Neurotoxicology 2010; 31:572-4; PMID:20188756; http://dx.doi.org/ 10.1016/j.neuro.2010.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson DW, Bandyopadhyay U, Jiang Y, et al.. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy 2011; 7:788-9; PMID:21464620; http://dx.doi.org/ 10.4161/auto.7.7.15596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nat Rev Neurosci 2014; 15:84-97; PMID:24399084; http://dx.doi.org/ 10.1038/nrn3638 [DOI] [PubMed] [Google Scholar]
- [16].Zhao F, Cai T, Liu M, Zheng G, Luo W, Chen J. Manganese induces dopaminergic neurodegeneration via microglial activation in a rat model of manganism. Toxicol Sci 2009; 107:156-64; PMID:18836210; http://dx.doi.org/ 10.1093/toxsci/kfn213 [DOI] [PubMed] [Google Scholar]
- [17].Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140:821-32; PMID:20303873; http://dx.doi.org/ 10.1016/j.cell.2010.01.040 [DOI] [PubMed] [Google Scholar]
- [18].Jiang W, Lv H, Wang H, Wang D, Sun S, Jia Q, Wang P, Song B, Ni L. Activation of the NLRP3/caspase-1 inflammasome in human dental pulp tissue and human dental pulp fibroblasts. Cell Tissue Res 2015; 361:541-55; PMID:25684031; http://dx.doi.org/ 10.1007/s00441-015-2118-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Turk B, Turk D, Turk V. Lysosomal cysteine proteases: more than scavengers. Biochim Biophys Acta 2000; 1477:98-111; PMID:10708852; http://dx.doi.org/ 10.1016/S0167-4838(99)00263-0 [DOI] [PubMed] [Google Scholar]
- [22].Chiu HW, Chen CH, Chang JN, Chen CH, Hsu YH. Far-infrared promotes burn wound healing by suppressing NLRP3 inflammasome caused by enhanced autophagy. J Mol Med (Berl) 2016; 94(7):809-19; PMID:26864306; http://dx.doi.org/26568474 10.1007/s00109-016-1436-x [DOI] [PubMed] [Google Scholar]
- [23].Dhanavade MJ, Parulekar RS, Kamble SA, Sonawane KD. Molecular modeling approach to explore the role of cathepsin B from Hordeum vulgare in the degradation of Abeta peptides. Mol biosyst 2016; 12:162-8; PMID:26568474; http://dx.doi.org/ 10.1039/C5MB00718F [DOI] [PubMed] [Google Scholar]
- [24].Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 2008; 9:857-65; PMID:18604209; http://dx.doi.org/ 10.1038/ni.1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008; 9:847-56; PMID:18604214; http://dx.doi.org/ 10.1038/ni.1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bahr BA, Wisniewski ML, Butler D. Positive lysosomal modulation as a unique strategy to treat age-related protein accumulation diseases. Rejuvenat Res 2012; 15:189-97; PMID:22533430; http://dx.doi.org/ 10.1089/rej.2011.1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang J, Cao R, Cai T, Aschner M, Zhao F, Yao T, Chen Y, Cao Z, Luo W, Chen J. The role of autophagy dysregulation in manganese-induced dopaminergic neurodegeneration. Neurotox Res 2013; 24:478-90; PMID:23604964; http://dx.doi.org/ 10.1007/s12640-013-9392-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cassidy LD, Narita M. CELL BIOLOGY. GATA get a hold on senescence. Science 2015; 349:1448-9. [DOI] [PubMed] [Google Scholar]
- [30].Bowman AB, Kwakye GF, Herrero Hernandez E, Aschner M. Role of manganese in neurodegenerative diseases. J Trace Elem Med Biol 2011; 25:191-203; PMID:21963226; http://dx.doi.org/ 10.1016/j.jtemb.2011.08.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Aschner M, Aschner JL. Manganese neurotoxicity: cellular effects and blood-brain barrier transport. Neurosci Biobehav Rev 1991; 15:333-40; PMID:1956602; http://dx.doi.org/ 10.1016/S0149-7634(05)80026-0 [DOI] [PubMed] [Google Scholar]
- [32].Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med 2010; 362:329-44; PMID:20107219; http://dx.doi.org/ 10.1056/NEJMra0909142 [DOI] [PubMed] [Google Scholar]
- [33].Guilarte TR, McGlothan JL, Degaonkar M, Chen MK, Barker PB, Syversen T, Schneider JS. Evidence for cortical dysfunction and widespread manganese accumulation in the nonhuman primate brain following chronic manganese exposure: a 1H-MRS and MRI study. Toxicol Sci 2006; 94:351-8; PMID:16968886; http://dx.doi.org/ 10.1093/toxsci/kfl106 [DOI] [PubMed] [Google Scholar]
- [34].Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging 1999; 20:581-9; PMID:10674423; http://dx.doi.org/ 10.1016/S0197-4580(99)00065-2 [DOI] [PubMed] [Google Scholar]
- [35].Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, et al.. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci 2000; 20:5709-14; PMID:10908610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Griffin WS, Nicoll JA, Grimaldi LM, Sheng JG, Mrak RE. The pervasiveness of interleukin-1 in alzheimer pathogenesis: a role for specific polymorphisms in disease risk. Exp Gerontol 2000; 35:481-7; PMID:10959036; http://dx.doi.org/ 10.1016/S0531-5565(00)00110-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol 2014; 14:463-77; PMID:24962261; http://dx.doi.org/ 10.1038/nri3705 [DOI] [PubMed] [Google Scholar]
- [38].Kaushik DK, Gupta M, Kumawat KL, Basu A. NLRP3 inflammasome: key mediator of neuroinflammation in murine Japanese encephalitis. PLoS One 2012; 7:e32270; PMID:22393394; http://dx.doi.org/ 10.1371/journal.pone.0032270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, Sodhi K, Treuting PM, Busch MP, Norris PJ, et al.. IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog 2012; 8:e1003039; PMID:23209411; http://dx.doi.org/ 10.1371/journal.ppat.1003039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kumar M, Roe K, Orillo B, Muruve DA, Nerurkar VR, Gale M Jr., Verma S. Inflammasome adaptor protein Apoptosis-associated speck-like protein containing CARD (ASC) is critical for the immune response and survival in west Nile virus encephalitis. J Virol 2013; 87:3655-67; PMID:23302887; http://dx.doi.org/ 10.1128/JVI.02667-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hoegen T, Tremel N, Klein M, Angele B, Wagner H, Kirschning C, Pfister HW, Fontana A, Hammerschmidt S, Koedel U. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J Immunol 2011; 187:5440-51; PMID:22003197; http://dx.doi.org/ 10.4049/jimmunol.1100790 [DOI] [PubMed] [Google Scholar]
- [42].Mitchell AJ, Yau B, McQuillan JA, Ball HJ, Too LK, Abtin A, Hertzog P, Leib SL, Jones CA, Gerega SK, et al.. Inflammasome-dependent IFN-gamma drives pathogenesis in Streptococcus pneumoniae meningitis. J Immunol 2012; 189:4970-80; PMID:23071286; http://dx.doi.org/ 10.4049/jimmunol.1201687 [DOI] [PubMed] [Google Scholar]
- [43].Hanamsagar R, Torres V, Kielian T. Inflammasome activation and IL-1beta/IL-18 processing are influenced by distinct pathways in microglia. J Neurochem 2011; 119:736-48; PMID:21913925; http://dx.doi.org/ 10.1111/j.1471-4159.2011.07481.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et al.. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2013; 493:674-8; PMID:23254930; http://dx.doi.org/ 10.1038/nature11729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS One 2013; 8:e55375; PMID:23383169; http://dx.doi.org/ 10.1371/journal.pone.0055375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shi F, Yang L, Kouadir M, Yang Y, Wang J, Zhou X, Wang J, Zhou X, Yin X, Zhao D. The NALP3 inflammasome is involved in neurotoxic prion peptide-induced microglial activation. J Neuroinflammation 2012; 9:73; PMID:22531291; http://dx.doi.org/ 10.1186/1742-2094-9-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hafner-Bratkovic I, Bencina M, Fitzgerald KA, Golenbock D, Jerala R. NLRP3 inflammasome activation in macrophage cell lines by prion protein fibrils as the source of IL-1beta and neuronal toxicity. Cell Mol Life Sci 2012; 69:4215-28; PMID:22926439; http://dx.doi.org/ 10.1007/s00018-012-1140-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, Wang X, Yu G, Esposito L, Mucke L, et al.. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron 2006; 51:703-14; PMID:16982417; http://dx.doi.org/ 10.1016/j.neuron.2006.07.027 [DOI] [PubMed] [Google Scholar]
- [49].Ni BB, Li B, Yang YH, Chen JW, Chen K, Jiang SD, Jiang LS. The effect of transforming growth factor beta1 on the crosstalk between autophagy and apoptosis in the annulus fibrosus cells under serum deprivation. Cytokine 2014; 70:87-96; PMID:25127907; http://dx.doi.org/ 10.1016/j.cyto.2014.07.249 [DOI] [PubMed] [Google Scholar]
- [50].Yamagishi T, Sahni S, Sharp DM, Arvind A, Jansson PJ, Richardson DR. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J Biol Chem 2013; 288:31761-71; PMID:24062304; http://dx.doi.org/ 10.1074/jbc.M113.514091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Khalid M, Aoun RA, Mathews TA. Altered striatal dopamine release following a sub-acute exposure to manganese. J Neurosci Methods 2011; 202:182-91; PMID:21740928; http://dx.doi.org/ 10.1016/j.jneumeth.2011.06.019 [DOI] [PubMed] [Google Scholar]
- [52].Dodd CA, Ward DL, Klein BG. Basal Ganglia accumulation and motor assessment following manganese chloride exposure in the C57BL/6 mouse. Int J Toxicol 2005; 24:389-97; PMID:16393931; http://dx.doi.org/ 10.1080/10915810500366500 [DOI] [PubMed] [Google Scholar]
- [53].Wehner JM, Radcliffe RA. Cued and contextual fear conditioning in mice. Curr Protoc Neurosci 2004; Chapter 8:Unit 8 5C; PMID:18428608 [DOI] [PubMed] [Google Scholar]