

Abstract
The hallmarks of cancer described by Hanahan and Weinberg have proved seminal in our understanding of cancer’s common traits and in rational drug design. Not free of critique and with understanding of different aspects of tumorigenesis coming into clearer focus in the recent years, we attempt to draw a more organized and updated picture of the cancer hallmarks. We define seven hallmarks of cancer: selective growth and proliferative advantage, altered stress response favoring overall survival, vascularization, invasion and metastasis, metabolic rewiring, an abetting microenvironment, and immune modulation, while highlighting some considerations for the future of the field.
Keywords: Cancer, malignancy, tumor, hallmarks, cancer biology
Introduction
In the year 2000, Hanahan and Weinberg published their influential review: the hallmarks of cancer [1] (henceforth termed Hallmarks I) where they attempted to organize the dense complexities of cancer biology into six major hallmarks: self-sufficiency in growth signals, insensitivity to anti-growth signals, evading apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis. A decade later, an updating review [2] (henceforth termed Hallmarks II) added two emerging hallmarks: reprogramming energy metabolism and evading immune response, and two enabling traits: genome instability and mutation, and tumor-promoting inflammation. Although in Hallmarks I the authors anticipated that over the following decade cancer research would undergo a process of simplification where less layers of complexity are added, Hallmarks II arrived at such a decade mark with conversely daunting complexities. We have seemingly come full circle: from overwhelming complexity to anticipated simplicity, back again to substantial complexity-a perspective that was highlighted by Weinberg himself [3]. Nevertheless, the reviews have managed to persist at the core of the cancer biology literature, serving as blueprints for understanding the core traits of cancer.
Challenging arguments have not only brought about the inclusion\exclusion of specific hallmarks into question, but the bigger question of: what defines a cancer hallmark? In his essay [4] Lazebnik argued that “cancer” is often used to refer to malignant tumors and a “hallmark” is a distinguishing feature, then pointed out that five of the six initial hallmarks (all except invasion and metastasis) are shared by both benign and malignant growths and are thus rather indistinctive of “cancer” over non-malignant controls. We would add that even invasion and dissemination are properties of certain non-malignant conditions, an example of which is endometriosis, a relatively-common condition among females in which endometrial cells migrate to extra-anatomical sites and invade new tissues, all while maintaining a benign histological appearance [5]. We argue, however, that the goal of the hallmarks is providing an organizational framework of cellular properties uncovered during the transformation of (phenotypically) normal cells-whether such a transformation stops at a benign growth stage or continues in the direction of a more evolved and threatening malignancy. Therefore, we choose an evolutionary perspective on the mutation theory in which carcinogenesis is a dynamic process that might initiate (and terminate) within cells’ life-spans, with manifesting cancer hallmarks emerging throughout such a journey [6].
Critique has also been directed towards the reductionist adoption of the somatic mutation theory (SMT)-the ‘bottom-up’ approach to studying carcinogenesis, merely dealing with cancer as a disease of ‘genes gone awry’, with proposal of more organicistic, system-or tissue-disorganization, views [7-9]. It is clear that even the initial hallmarks list contains tissue-relevant rather than cancer-cell-specific components (e.g. angiogenesis) [1] and Hallmarks II has a section dealing with the tumor microenvironment (TME) [2]. More recently, phenotypically normal cells were shown to carry a high burden of non-silent somatic mutations that are positively selected upon without the formation of cancer [10-12]. Furthermore, the stromal component of tumors has proved to be an occasional initiator [13,14] and an indispensible accomplice [15] of the cancer process. We do believe that continuing to expand on SMT to encompass newer realizations while having a concurrent tissue view rather than abandoning it is best for the time being-at least until inevitable progress in systems biology presents us with a more comprehensive model. The attempts at classification of somatic mutations into driver and passenger has helped such conceptual expansion [16]. We are also beginning to see beyond the genes through the appreciation of the significant role of epigenetics in the cancer process [17]. Finally, the stromal compartment of tumors has been a subject of great dissection in the recent years [15]. Thus, a step back in time would bring us to the single malignant ‘renegade’ cell solely carrying the blame for cancer development [18], but where we are heading is a step back in perspective towards a concurrent holistic view, in which reductionism and organicism are complementary rather than competing [19].
A final critical point would be whether the description of the hallmarks has led to translational benefit in the clinic [6,7]. In Hallmarks II, various drug categories were linked to their targeted hallmark, but many have proved to only be effective for a limited time or within limited settings. There is referral to the important concept of shifting hallmark dependence during therapy [2]. Indeed, viewing the hallmarks as individual, segregated, and static targets is insufficient; the complementarity of the hallmarks, their codependence, and the evolutionary dynamics governing them are essential considerations [6].
Taking into account all the aforementioned points, we propose a more precise definition of cancer hallmarks as: acquired evolutionary-advantageous characteristics that complementarily promote transformation of phenotypically normal cells into malignant ones, and promote progression of malignant cells while sacrificing/exploiting host tissue (Figure 1). And with this current work, we aim to draw a more organized, robust, and updated picture of such hallmarks.
Figure 1.
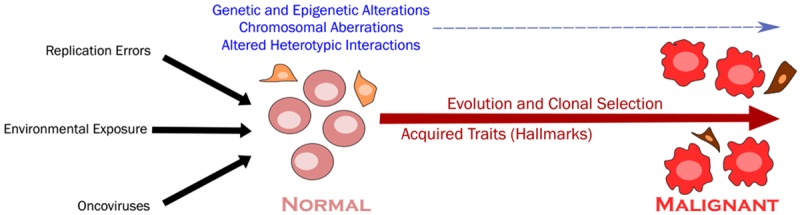
The transformation process. Different insults continuously act on cells leading to transformative alterations in (epi) genetics, chromosomal numbers and arrangements, and heterotypic interactions which, along the path towards malignancy, undergo cycles of evolutionary clonal selection leading to the acquisition of cancer-competent traits, the hallmarks of cancer.
Selective growth and proliferative advantage
Normal cells depend on growth signaling of a tightly-regulated cell cycle to controllably proliferate and maintain tissue homeostasis-this is disrupted in case of cancer [20,21]. It is currently appreciated that in cancer cells, the growth and proliferative signaling pathways harbor one or more driving alterations within their compartments giving them a survival edge [16]. Those compartments include growth ligands, their receptors or the cytosolic signaling molecules.
A simple depiction of growth ligands in a tumor setting would be “growth-promoting”, expected to be aberrantly produced in high levels by epithelial or stromal cells acting in an autocrine or paracrine fashion to promote tumor progression, and “growth-inhibiting”, expected to be shutdown to allow the tumor’s escape from braking signals [20]. While often the case, the distinction remains a virtual one. Take transforming growth factor-β (TGF-β) as an example, this conventional anti-growth ligand has been conversely shown to be implicated in tumor-progression both by stimulating cancer-cell de-differentiation [22] and reshaping the TME [23]. Such pleiotropic signaling demonstrates the continuous pressure on cancer cells that selectively survive and dominate to find the “right balance” (for each stage and environment) of growth ligands.
The receptors binding these ligands may also be altered in various manners, e.g. 1) receptors may be overexpressed through gene amplification, 2) somatic mutations may result in constitutive receptor activation, 3) chromosomal translocations may lead to fusion proteins and aberrant signaling, or 4) receptor recycling and degradation machinery may be impaired [24-26]. But perhaps the most common alterations are encountered in the downstream compartment. Such signaling networks are complex and still under investigation. An illustrative example is the RAS protein, serving as a “signaling hub” at the center of this network; it is chronically active in 30% of cancers and in over 90% of pancreatic carcinomas, often via missense mutations in its gene or inactivating mutations in one of its negative regulators (e.g. NF1) [27,28]. Downstream cascades mediate RAS functions, prominently the RAF-MEK-ERK and the PI3K-AKT-mTOR networks, the components of which could independently be mutated in a similar fashion in various types of cancer. Importantly, mutations in RAS, its regulatory proteins, or its downstream networks result in a plethora of effects beyond enhanced growth and proliferation that include suppression of apoptosis, rewiring of metabolism, promoting angiogenesis, and immune evasion, and thus reflect the fact that a single signaling cascade could be implicated in multiple hallmarks of cancer [29].
Even in the presence of growth signaling, tight regulation of the cell cycle via regulatory proteins keeps the division cycles in check. For cancer cells to grow, deregulation of the cell cycle and checkpoint disruption are crucial [21]. One key regulator is the retinoblastoma (RB) protein, commonly inactivated in a multitude of malignancies [30,31]. As is the case with RAS, it has been shown that the RB family are not limited to proliferative control, but are involved in multiple roles-impinging on various other hallmarks-that include maintenance of genomic stability, regulation of apoptosis, cell metabolism, senescence, angiogenesis, and suppression of invasion and metastasis [30,32]. Another key regulator is the p53, whose gene is the most commonly mutated causally-implicated cancer gene, mutated in over 50% of sequenced tumors [33,34]. P53 acts as a stress detector and responder, being sensitive to a variety of stresses that include genotoxic stress, excessive signaling, nutrient deprivation, and hypoxia. Arresting further proliferation, p53 is then involved in initiating repair mechanisms, or if the damage is beyond repair, initiating cell death or terminal differentiation states [35,36]. The molecule has also been linked to metabolic rewiring, regulation of autophagy, and redox homeostasis [35].
The description above makes it appealing-once again-to label cancer-cellular-components as “growth-permissive”, turned on by activated oncogenes (jammed throttles), and “growth-restraining”, tampered with by inactivated tumor-suppressor genes (failing brakes), and to conclude that the collaboration between both is a must for cancer to develop. This, also again, is not representative of the full picture where, for example, a great percentage of one tumor type (e.g. pancreatic adenocarcinoma) could carry driving mutation(s) in the labeled oncogene compartment alone [16]. Furthermore, such compartmental labeling is merely artificial; in reality, a context-dependent interchange of functions is seen in different tumor settings and at different stages of tumorigenesis [37-39].
Altered stress response favoring overall survival
On their journey towards full-scale malignancy, cancer cells face a wide range of stresses that include excessive signaling, DNA damage, hypoxia, nutrient scarcity, and even anticancer therapy. Physiologically, cells adopt a variety of responses to adapt to the stress if possible or, when the stress is overwhelming, altruistically be eliminated for the sake of healthy whole tissue. These mechanisms are subverted in cancer cells for the outcome of their overall survival and propagation (Figure 2). Metabolic response is elaborately discussed under Hallmark E, and we will discuss some of the other stress responses relevant to the cancer setting.
Figure 2.
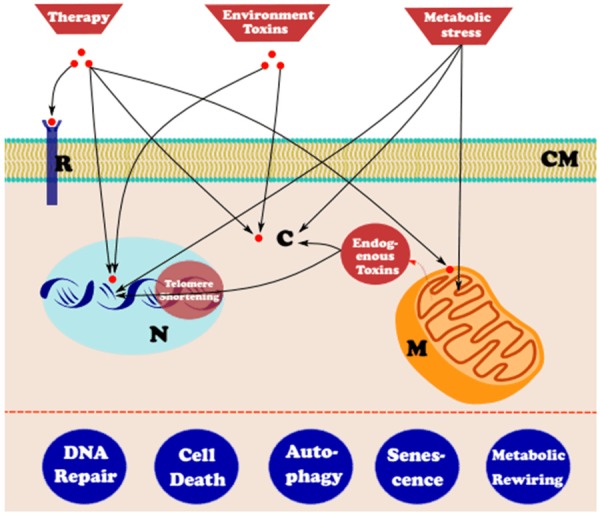
Altered stress response. Different stressors act on different compartments of normal and (to a higher degree) cancer cells. Some of the stress responses are subverted or hijacked for overall survival on the road towards malignancy. (CM: Cell Membrane, R: Receptor, C: Cytosol, N: Nucleus containing DNA, M: Mitochondria).
DNA repair
The cell employs a multitude of repair pathways (as part of the wider DNA damage response), each composed of a variety of molecules working cooperatively to amend a specific type of DNA lesion. Although not absolutely redundant, a pathway could be seen working in place of another were the latter to be defective (a concept exploited for targeting compensating pathways in repair-deficient tumors, “synthetic lethality”) [40,41].
The need for cumulative, driving, unrepaired genetic insults (persisting mutations), together with the unstable genetic makeup of most tumors, logically implicate defects in repair machinery in the story of tumorigenesis. Some of the most notable familial cancer syndromes entail germline mutations involving DNA repair genes [42-44]. Acquired cancers have also been shown to exhibit defects in repair pathways as they evolve [45], and polymorphisms involving repair genes have been correlated to cancer risk; those are probably further responsible for variability of therapeutic response (since most anti-cancer therapies function by inducing DNA damage) [46,47].
Perhaps counterintuitive is that overexpression of various repair proteins has also been observed in tumors; for example, upregulation of RAD51 has been demonstrated in leukemia, breast, and pancreatic cancers [48]. Such upregulation is linked to increased therapeutic resistance and post-treatment relapse [48,49]. Thus, DNA repair pathways are best considered multidimensional regulators in the cancer context, the disruption of which (over-reliance on an upregulated pathway or defects in another) serves overall tumor survival and progression, but may also represent a vulnerability that could be exploited for therapeutic purposes [40,49].
Apoptosis
Another response to transformation-associated stress, including irreparable DNA damage, uncontrolled proliferation, or matrix detachment, involves apoptotic cell death [50,51]. In cancer, the balance between cell proliferation and cell death that normally maintains healthy tissue homeostasis is disturbed [51].
The extrinsic pathway to apoptosis involves the interaction of cell surface receptors with their ligands. The more-relevant intrinsic pathway involves sensing of internal stress levels compatible with apoptosis, a shift of balance favoring pro-apoptotic over anti-apoptotic proteins, and mitochondrial outer membrane permeabilization (MOMP) with release of activating molecules from the intermembrane space. Both pathways converge on activation of caspases, cleavage proteins efficiently completing the task of cell death within minutes [51-53].
Cancer cells are able to surpass apoptotic response by various mechanisms [51,53]. Inactivating mutations involving p53 render the cell insensitive to many apoptotic stimuli (although, and fortunately, p53-independent cell death pathways exist). Upregulation of anti-apoptotic or loss of pro-apoptotic proteins through (epi) genetic means occur in various tumors. Cancer cells also occasionally inhibit caspase activity even with the occurrence of MOMP. It is important to note that, although the net outcome of those alterations is enhanced tumor survival, this does not mean that all cells within a tumor are insensitive to apoptotic signals. If this were the case, most of today’s anti-cancer therapies-effective by inducing cell death-would be useless, and tumors would grow to absurd sizes [54]. Contrarily, tumor cells are likely to be more sensitive to apoptotic stimulation than normal cells and apoptosis is continuously occurring in growing tumors [55,56]. In fact, apoptosis could serve an evolutionary role under conditions of selective pressure by eliminating less-fit lineages, evacuating a niche for predominance of better-suited clones and contributing to cancer progression [54].
Autophagy
Autophagy (macroautophagy, the most widely described type) is a recycling process of intracellular components that physiologically serves a quality-control function, operating at a low basal rate, removing pathologic long-lived or misfolded proteins and damaged organelles [57], and may also be involved in unconventional protein secretion [58]. It is upregulated as a protective response to a variety of stresses that include pathogenic, metabolic, and genotoxic ones [59].
The tumor-suppressor aspects of autophagy were described early on [60]. Engineered mice with homozygous loss of the beclin (encoding an essential autophagy protein) died during embryogenesis, while those haploinsufficient developed spontaneous tumors with ageing [61,62]. The gene is mono-allelically deleted in 40-75% of human sporadic breast, ovarian and prostate cancer cases [61]. The mechanisms by which autophagy suppresses tumorigenesis are still elusive, but may include selective elimination of damaged mitochondria during periods of stress reducing the burden of reactive oxygen species (ROS), metabolic homeostasis, degradation of overexpressed proteins, sharing in immune surveillance, and serving as a defense line against potentially-carcinogenic bacteria and viruses [59,63,64].
On the other hand, evidence has implicated autophagy in tumor survival and progression; upregulated autophagy (and even autophagy dependence) is a feature of many malignancies [63]. Autophagy may conversely enable cancer cells to survive harsh metabolic stress and hypoxia, evade immune-surveillance, acquire invasive and metastatic characters, or secrete TME-reshaping proteins [58,59,65], and thus represents another stress response with context-dependent roles in different tumor settings and stages, liable for hijacking for overall tumor survival [63].
Senescence
Another defense mechanism against cellular stress is senescence: the process of irreversible exit from the cell cycle [66]. On critical shortening of telomeres after exhaustion of replication potential, induction of senescence occurs; successive divisions would lead to cycles of chromosomal fusion and breakage, resulting in genomic instability and allowing accumulation of (potentially-transforming) alterations [66,67]. Short telomeric length is almost a universal feature of halted benign and pre-malignant lesions [68,69]. Consequently, such halt needs to be bypassed in fully transformed cells. This is mainly by an enzyme, telomerase, upregulated in 85-90% of human tumors and capable of reconstituting the telomeric ends, maintaining sufficient length for further replication [69,70]. (Of note, 10-20% of human tumors utilize alternative pathways for telomere lengthening, but their exact mechanisms are still subject of ongoing research) [70]. A variety of other stresses can also induce senescence: non-telomeric DNA damage (e.g. that caused by ROS or anti-cancer therapy), strong, long-lasting, or unbalanced mitogenic signaling, and activation of tumor-suppressors, all consistent with the cytoprotective role of senescence [66,68].
The passive role of senescent cells has, however, been challenged in the recent years, mainly through the characterization of senescence-associated secretory phenotype (SASP): soluble signaling factors, insoluble proteins and matrix components, and proteases released by senescent cells, capable of reshaping the microenvironment surrounding them [66,71]. Contrasting beneficial (e.g. autocrine tumor-suppressive effects, enhancing immune infiltration and clearance of senescent/transformed cells, and tissue repair) and detrimental (promoting inflammation, stimulation of angiogenesis, and contributing to metastasis) roles have been ascribed to SASP, obliterating the simplistic view of unidirectional tumor-suppressive senescence response, and rendering senescence a confusing yet-to-be-explored target for therapeutic design [71,72].
Vascularization
Tumors cannot grow beyond 2-3 mm3 nor metastasize without new vasculature [73]. Although angiogenesis is the most discussed, various other modes of tumor vascularization exist with redundancy in usage, partly explaining resistance towards antagonizing a single mode [74]. There exists a confusion in the literature where on occasions “angiogenesis” encompasses all forms of neo-vascularization, while on others it refers to the classic vascular sprouting with other modes treated as separate entities. We choose to adopt the latter terminology.
Angiogenesis
Angiogenesis is the process of sprouting, cell division, migration and assembly of endothelial cells (ECs) from pre-existing vessels [75]. It is utilized during embryogenesis for expansion and remodeling of primitive vascular networks, and is part of postnatal events including wound healing, the female reproductive cycle, and chronic inflammation [76]. In these events, however, angiogenesis is turned off or may be prolonged but self-limiting, unlike in case of malignancies where the process is continuously activated. Regulation of angiogenesis involves pro- and anti-angiogenic factors; their balance determines the status of the “angiogenic switch”. Only when a trigger tips the balance towards pro-angiogenic factors (as in case of malignancy) is the switch turned on and do vascular-quiescent tissues show signs of angiogenesis [77].
The most important trigger of angiogenesis is hypoxia. ECs possess a number of oxygen-sensing mechanisms, chiefly those interfacing with the hypoxia-inducible transcription factor (HIF) family, regulating the expression of a multitude of genes not only involved in angiogenesis, but in cell survival, metabolism, and inflammation as well. Responding to hypoxia, stabilized HIF initiates an adaptive transcriptional response, many products of which are factors involved in turning on the angiogenic switch [78,79]. With hypoxia being a feature of tumors, it is not surprising that HIF levels are higher in many cancers, correlating with poor clinical prognosis [80]. Other angiogenic switch triggers in tumors include metabolic rewiring of ECs creating an acidic TME, alterations in genes controlling production of angiogenic regulators, mechanical stress, and inflammatory cell infiltrate [77,79]. These triggers may be tumor- and tissue-specific and may alternate during various stages of tumor development [81].
The effectors include a plethora of pro-angiogenic molecules, the VEGF signaling pathway is the most potent of which [82,83]. Controlled by HIF activity, and also directly by growth signaling, VEGF is overexpressed in a multitude of malignancies [84] and its activated signaling leads to EC proliferation, survival, migration and differentiation, and mediation of vascular permeability [82,85]. A number of VEGF-independent effectors exist; those may work complementarily, independently, or compensatively for VEGF signaling [83,86]. A growing list of opposing anti-angiogenic factors stand on the other end of the angiogenic balance, susceptible to sabotage in tumor settings [87].
The sprouting tumor vasculature differs from normal one [77,88]. Dilated and tortuous vessels with ECs not forming regular monolayers, and resting on a basement membrane of variable thickness, and pericytes forming abnormally loose associations with ECs, all lead to leakiness. The blood flow is chaotic with resulting areas of hypoxia and acidosis; these stressful conditions have a number of effects including potentiating angiogenesis, lowering therapeutic effectiveness, and allowing resistant clonal expansion.
Other modes of tumor vascularization
Several modes of non-angiogenic vascularization of tumors exist and, although receiving less attention, constitute important players in understanding the complex tumor vascularity and in rational drug design.
Vascular co-option is the process by which tumor cells obtain their needed blood supply by hijacking existing vasculature [89]. It has been proposed that both angiogenesis and vascular co-option play distinct roles at the initiation of microtumors, and although tumor exponential growth is dependent on the former, the latter is an alternative but essential choice for survival of cells [90]. Co-opting cells are hypothesized to be refractory to anti-angiogenic therapy [89,91,92]. Although VEGF and angiopoietins are proposed to be the principal mediators [93], the exact pathways governing the process, its relation to angiogenesis, and necessity to tumors all need further investigation.
Intussusceptive microvascular growth (IMG, or “splitting angiogenesis”) is the process by which existing vessels split into daughter ones allowing the capillary network to expand within itself [94]. IMG is reported in a variety of tumor types [95], and with faster, more energy-conserving, and more physiological permeability levels than sprouting, the shift to IMG could serve an adaptive response to restore hemodynamic homeostasis to tumor vasculature or an escape mechanism from anti-angiogenic therapy [95,96]. To date, no molecule has been directly linked to IMG, although shear stress and blood flow seem to play some role [95].
Vasculogenic mimicry (VM, or “vascular mimicry”) describes the functional plasticity of aggressive tumor cells in expressing a stem-cell-like phenotype forming de-novo vascular networks [94,97]. VM has been described in almost all major types of malignancies [97,98], and correlates with poor prognosis [99]. The latter implies a functional advantage provided by VM, promoting survival of the aggressive tumor cell phenotype; experiments show physiological flow within VM networks, and anticoagulant properties of cancer cells lining VM channels facilitating perfusion [100]. Hypoxia seems to be an important driver of VM, but many molecules and signaling pathways relevant to VM are being pursued [97].
Invasion and metastasis
The defining feature of malignancy entails the ability to invade surrounding tissue and seed distant sites to form secondary growths (metastases). Metastatic disease is responsible for over 90% of cancer-related deaths [101], and involves a series of events, the “invasion-metastasis cascade”. For cancer cells to occur at distant sites they must 1) invade through the extracellular matrix (ECM), including the basement membrane, and stromal cells, 2) intravasate into tumor vasculature, 3) survive transport in circulation, 4) extravasate at parenchyma of distant organs, and 5) survive and manipulate foreign microenvironments forming micrometastases, that may later 6) grow into clinically-relevant macrometastases, a rate-limiting step termed “colonization” [102,103].
Invasion, intravasation, and circulation
Growing evidence supports a collective route for invasion resulting in polyclonal metastases [104]. Although most human carcinoma cells migrate collectively, most cells in in vivo and in vitro studies migrate individually; this could be related to the TME evolving alongside the former group [105].
Epithelial cells are immotile and tightly adherent to one another and to the surrounding matrix. Overcoming such barriers is linked to epithelial-mesenchymal transition (EMT) [106], the reversible biochemical changes that allow a polarized epithelial cell to acquire a mesenchymal phenotype, normally utilized during embryogenesis, and adult physiological (e.g. wound healing) and pathological (e.g. organ fibrosis) conditions [107]. Originally described as transformative between two binary-like states: full-epithelial and full-mesenchymal, this view has been broadened by demonstrating a spectrum of intermediary phases (commonly referred to as “partial EMT”) that could progress, revert, or exist as a final state, reflecting a more fluid phenomenon [108]. Context-dependent (tissue- or tumor-specific) triggers include a number of growth factors [22], signaling pathways [109], tissue hypoxia [110], metabolic and mechanical stress, and matrix stiffness [111]. Triggering often converges on the level of one of the master EMT transcription factors, repressing epithelial genes and activating mesenchymal ones [103]. Regulators of EMT also include epigenetic modifiers demonstrating non-linear fine-tunable control of the process [108]. Extensive evidence supports a role for EMT in cancer-cell invasion and migration [106,112]. Yet recently-emerging evidence has brought such contribution into question. Prominently, two recent studies [113,114] have demonstrated a dispensable role for EMT in metastasis, albeit contributing to chemo-resistance. It is worth mentioning that both reports have received their share of critique [108,112], but we currently hold insufficient evidence to include\exclude EMT in driving cancer progression; more experimental work (especially intravital studies) would prove useful to the debate [115].
Integral to local invasion is degradation of the ECM, a key player in which is the matrix metalloproteinases (MMPs) [116]. These endopeptidases not only contribute to invasion, but are involved in cell proliferation, survival, immune response, and angiogenesis as well [117,118]. MMPs are upregulated in almost every type of human cancer [119], and the expression of specific members has been linked to poor prognosis [120].
Intravasation is the entry of invasive cells into the lumina of vessels [103]. This could be active or passive depending on the tumor type, TME, and vasculature [121]. The process has been difficult to model in vitro limiting our understanding [122], but a recent microfluidic model may provide insight [123].
Once in circulation, circulating tumor cells (CTCs) are exposed to harsh selective conditions and must devise adaptive techniques [103,124]. Examples include platelet coats shielding from shear forces and immune-clearance [102,125], and metabolic rewiring blunting oxidant stress [126]. Serving as a “liquid biopsy”, isolated CTCs could provide means for cancer screening, estimation of metastatic relapse risk, identification of targetable components, exploring tumor heterogeneity, and monitoring therapeutic response [124,127]. Multiple challenges still stand in the way and will need to be addressed before clinical utilization [128].
Extravasation and organ predilection
As CTCs roam the circulation, they become entrapped in small capillaries where they either initiate growth leading to microvessel rupture, or engage in extravasation [102]. Some organs (like the liver and bone marrow) possess highly-permeable sinusoidal vessels and this may explain the high rates of liver and bone metastasis, but in most other organs, ECs form a tightly-adjacent lining enforced by a basement membrane and pericytes [129]. This is in contrast to the leaky neo-vasculature in the primary tumor, and since secondary sites lack the fostering TME that facilitates intravasation, it is clear that extravasation is a distinct (and likely more difficult) process from intravasation [103]. Factors involved in extravasation inlcude ligand-receptor interactions, chemokines, and circulating non-tumor cells, although our understanding of the process is incomplete [122,124,125].
Passive arrest (the “anatomical-mechanical” hypothesis) only manages to explain part of the metastatic predilection of human tumors. Cancer subsets favor organ subsets, the so called organotropism [103,129]. This was first proposed by Stephen Paget as the “seed and soil” hypothesis [130], and has held true in later observations where it has been especially studied in breast cancer [129,131]. A cooperation between genes mediating organ-specific metastasis [132] and adaptive programs working in the receptive tissue [129,133] seem to be at play. In conclusion, our current understanding would suggest that both the “seed and soil” and the “anatomical-mechanical” hypotheses are not mutually exclusive and reflect two mechanisms that serve together in a context-dependent manner to varying degrees [102,130].
Micrometastasis, dromancy, and colonization
The new microenvironment is foreign to the cells with harsh conditions challenging their survival [103]. Various systemic signals are implicated in preparation of secondary sites for reception of cancer cells. These may even be in effect long before dissemination, creating the so called “pre-metastatic niche” [134,135]. A number of tumor-derived secreted factors, and bone-marrow-derived cells are known stimulators of niche formation [135,136]. Growing evidence also supports a role for exosomes in the process [137,138].
Nevertheless, delayed adaptation at the niche results in entry of surviving cancer cells into dormancy. Dormant cancer cells are observed in the bone marrow of many cancer survivors long after radical removal of their primary tumors, possibly being responsible for late relapses [139]. Three (non-mutually-exclusive) types of dormancy are: 1) cellular dormancy in which cancer cells enter into a state of quiescence (similar to senescence but reversible), 2) angiogenic dormancy in which a balance is achieved between dividing and dying (vascular-lacking) cancer cells, and 3) immune-mediated dormancy where the tumor mass is kept constant by continuous cytotoxic activity of immune cells [140]. Exactly how cancer cells escape dormancy (“awaken”) and proceed to form overt metastasis (colonization) is poorly understood [139]; this most likely occurs in an organ-specific manner [102]. Of relevance, however, is that colonizing cells would need to have a high capacity for self-renewal [103]. This has supported the notion that a rare and unique subpopulation of cells (“metastasis-initiating cells”) possess advantageous stem-cell-like traits, outcompeting other cells and beating the challenging barriers to metastasis [141]. Although appealing, this idea needs further experimental validation.
Metabolic rewiring
Rewired metabolism uniquely provides a selective advantage during initiation and progression of tumors [142,143]. Pavlova and Thompson recently organized cancer metabolic alterations into six hallmarks: deregulated uptake of glucose and amino acids, opportunistic modes of nutrient acquisition, utilizing glycolysis and TCA cycle intermediates, increased nitrogen demand, alterations in metabolite-driven gene regulation, and metabolic interactions with the microenvironment [144]. These may exist fully or partly in different tumor settings and are likely to be revisited in the next years.
Nutrient uptake
Two principal nutrients absolutely required by growing cells are glucose and glutamine. Both provide a pool of carbon intermediates for assembly of macromolecules, and-through controlled oxidation-reducing intermediates used in ATP generation or maintaining redox balance [144]. Extra functions exist for glutamine, mainly due to its nitrogen-donation capacity; those include supporting the amino acid pool and being the obligate nitrogen donor for nucleotide biosynthesis [145,146]. This is in addition to being involved in cell-signaling and gene expression [146].
The discovery that cancer cells over-utilize glucose even in the presence of oxygen, the “Warburg effect” [147], was applied decades later in the [18F]fluorodeoxyglucose positron emission tomography scanners [148]. An elevated level of glucose transporter (GLUT) proteins is seen in many cancers [149], and has been associated with poor outcomes in some specific types [150-152]. Multiple mechanisms and signaling pathways in cancer cells promote glucose uptake [144,149,153]. HIF increases the expression of GLUT and hexokinase (trapping the inflowing glucose), and aberrant PI3K/Akt signaling increases expression of GLUT1 and protein translocation to the cellular membrane, and so do oncogenic KRAS and BRAF.
Conversely, the studies of glutamine in tumor-context have been scarce until recently [146]. Once pursued, its increased uptake and vast roles in transformed cells were appreciated. The regulators of glutamine uptake are less characterized: MYC enhances the expression of glutamine transporters and its utilization [145], and RB can negatively regulate glutamine synthesis [154]. Tumor cells may utilize alternative pathways other than transport for acquisition of glutamine (and other amino acids), those include macropinocytosis of extracellular proteins in periods of nutrient-deprivation, engulfment and digestion of living cells (“entosis”), or phagocytosis of neighboring apoptotic products [144,145]. Thus, deregulated nutrient uptake extends beyond glucose and glutamine, although those have been the most characterized overactive and rewired pathways. The acquisition of lipids or fatty acids, amino acids other than glutamine, and extracellular acetate, and their contribution and necessity to tumorigenesis are subjects of recent research [144,155-157].
Rewiring metabolic pathways
The metabolic aberration in tumor cells extends to the flow of nutrients into different pathways. Perhaps the prototypical example is the evolution of our understanding of the Warburg effect. While an initial explanation to the phenomenon was defective oxidative phosphorylation [148], it is now established that cancer cells (generally) have normally-functioning mitochondria capable of oxidative phosphorylation; in fact targeting mitochondrial DNA reduces tumorigenic potential of cells both in vitro and in vivo [153,158]. Alternatively, the emerging rationalization is that preferential glycolysis provides means for continuous glucose shunting into intermediary pathways (whose steps are frequently upregulated in cancer), producing precursors or reducing equivalents indispensible to the tumor cell [143,144]. Oxidative phosphorylation thus serves alongside glycolysis to fulfill the high anabolic demands of tumor cells [153], and the cells must devise ways to balance the high rate of alternative-pathway-shunting while maintaining some mitochondrial flow [144,158], mainly achieved by controlled influx into the TCA cycle via tight enzymatic regulation [159,160].
Despite regulation, oversaturation of the TCA cycle occurs with production of ROS [143]. Moderate amounts of ROS are useful to cancer: they stimulate stress signaling, add to the mutation burden, and promote cancer progression and spread [161]. However, high levels of ROS are detrimental to the cells, and thus tight control is crucial. A proposed model has emerged where initiation of tumors results in increased metabolic activity and ROS production, lending a hand to tumorigenesis, but as tumors progress and some cells face harsh hypoxic nutrient-deprived microenvironments, and to prevent toxic accumulation of ROS, these cells increase antioxidant capacity to maintain ROS in the stimulatory non-lethal levels [158].
Glutamine is also captured by the mitochondria and converted into glutamate; this may be utilized directly or further converted to α-ketoglutarate (α-KG) that could act as a glucose alternative to produce citrate or share in fatty acid synthesis under unfavorable conditions [145]. In fact, cancer cells are characterized by a dramatic increase in lipid production with frequent upregulation of all major components of fatty acyl chain synthesis [155]. This may be advantageous to proliferating tumor cells in the formation of lipid bilayers, and also in alteration of membrane composition towards an increasing percentage of oxidative-damage-resistant saturated fatty acids [144].
Contribution of metabolites to tumorigenesis
Rewired metabolism is not merely a passive consequence to tumorigenesis, but could be an active contributor. We have discussed some of the metabolite-induced influences on cell behavior and tumor progression within the context of other hallmarks. Epigenetic regulation in the form of the addition or removal of acetyl and methyl marks also depends greatly on metabolites’ availability [144]. But perhaps the most-characterized example is the production of the “oncometabolite” 2-HG. Point mutations in isocitrate dehydrogenase (IDH) genes, mainly in gliomas and leukemias, confer neomorphic enzymatic activity allowing the enzymes to act on α-KG instead of isocitrate to produce 2-HG, a structurally similar and competitive inhibitor of α-KG. This subsequently inhibits dioxygenases with hypermethylation silencing of an array of genes that may contribute to cancer progression [162-164]. 2-HG accumulation may also occur in settings of absent IDH mutations, as was recently demonstrated in triple-negative breast cancers [165].
An abetting microenvironment
Continuous paracrine communication between cancerous and stromal cells creates a rich and dynamic microenvironment during all stages of carcinogenesis. Several of the original hallmarks, and those described in our presentation, involve various stromal compartments. This section does not aim to cover details of the TME (reviewed in Hallmarks II [2] and the subsequent work of Hanahan and Coussens [15]). But just to demonstrate how components of the TME integrate with other hallmarks we select the cancer-associated fibroblasts. Those alone may share in providing a selective growth and proliferative advantage via growth signaling [166,167] or checkpoint inhibition [168,169], in stress response alteration, e.g. survival signal provision to resist cell death [170], in angiogenesis and various stages of the invasion-metastasis cascade [171], as well as metabolic rewiring of cancer cells [144].
A turning point in understanding the powerful TME’s role in carcinogenesis was demonstration of TME-related transformation. The integrity of normal hematopoietic progenitors seeded on osteolineage cells carrying Dicer-1 deletion was disrupted and myelodysplasia-related changes occurred [14]. Moreover, it is known that more than 5% of acute myeloid leukemia allograft recipients show complete-chimerism relapse with original leukemic cells, strengthening the argument for TME as a driver of leukemogenesis [172]. Such evidence pushed adopters of the “tissue organization field theory” to argue that the disruption “tissue disorganization” of the cellular-stromal signaling is the starting point of all cancers [9]. Backed up by fragmented evidence, it is currently unfounded to make such generalization.
TME studies face difficulty in modeling of tumor-associated stroma which consists of a large variety of cellular types (spatial heterogeneity) at various stages of differentiation (temporal heterogeneity). Spatial mapping of the TME [173], together with its molecular imaging [174], will prove useful in capturing the full picture. For temporal heterogeneity things are trickier. Previous studies focus on mature stromal cells, the origin of which remains unclear [175]. This begets the goal of pursuing the origin of stromal cells in the TME (the first settlers of the abetting neighborhood). Those may be derived from the mesoderm, potentially being the mesenchymal stem cells [176]. Recent studies have shown the latter to be under neuro-vegetative [177-179], endocrine [180,181], and signaling-pathway [182,183] control, and to undergo metabolic reprogramming [184].
Furthermore, different cancer-cell subpopulations may act as the abetting neighbors (or TME in a sense) of one another, the concept of intratumoral heterogeneity. Ablating such neighborhood by monoclonal transplantation leads to failure of reproducing parental tumor behavior [185]. This proves another challenge for many xenograft assays that rely on single-cell transplantation methods.
A final point to be made is our grown appreciation for “cancer-organ”, rather than “cancer-cellular”, therapeutic response [186]. Cancer-associated fibroblasts that contribute to therapeutic resistance, vascular-mediated resistance, and immune-mediated resistance are examples of recognized aspects that need to be considered in therapeutic planning [187-190]. Note that these are not unidirectional towards chemo-resistance [191,192].
Immune modulation
The theory of ‘cancer immune surveillance’ was supported early on by experimental data showing strong immune-mediated rejection of transplanted tumors in mice [193]. The fact that primary and acquired immunodeficiency (e.g. secondary to HIV infections) in humans and mice are associated with higher susceptibility of tumorigenesis further corroborated the existence of immune surveillance.
Despite immune surveillance, tumors continue to develop in bodies with intact immune systems. ‘Cancer immunoediting’ is the process by which the immune system eliminates and shapes malignant disease, and encompasses three phases: ‘elimination’, ‘equilibrium’ and ‘escape’. Elimination is the hallmark of the original concept in cancer immune surveillance whereby both the innate and adaptive immune responses cooperate to eradicate developing tumors. Dunn et al [194] proposed four phases of elimination: (a) recognition of tumor cells by innate immune cells and their limited killing, (b) maturation and migration of antigen-presenting cells and cross-priming of T-lymphocytes, (c) generation of tumor-antigen-specific T-lymphocytes and activation of cytotoxic mechanisms, and (d) homing of tumor-antigen-specific T-lymphocytes to the tumor site and elimination of tumor cells. This is followed by the equilibrium phase that involves continuous sculpting of tumor cells and selection of those with reduced immunogenicity, promoting the production of resistant variants. Cancer cells can enter quiescence or a slow-cycling state and remain latent for extended periods of time [195]. During the equilibrium phase, the longest of the three phases occurring over a period of many years, new variants emerge that harbor different mutations that increase resistance to immune pressure.
Despite cancer cells going through constant immune selection pressure, some clones emerge and ‘escape’ immune surveillance, which is the final phase in cancer immunoediting. During this phase tumor variants that are able to evade immune detection and elimination through genetic and epigenetic alterations grow to an uncontrolled manner forming a clinically detectable tumor. Several escape mechanisms have been described that fall under three major principles; (a) lack of tumor-antigen recognition medicated by alteration of either tumor or effector cells, (b) resistance to cell death, and (c) induction of immunological ignorance and tolerance particularly through immunosuppressive factors secreted by tumor cells [196]. Cancer cells can paralyze the cytotoxic components of immune system through secretion of immunosuppressive factors or recruitment of immunosuppressive inflammatory cells [197]. In addition, stromal cells secrete immunosuppressive factors, upregulating expression of other immunosupressive molecules [198]. Tumor-derived exosomes also convey signals that suppress the function of immune cells [199]. Metabolic rewiring [200] and altered microbiome [201] are two other relevant factors.
In summary, the immune modulation observed in tumors is currently recognized as a key player during cancer initiation and progression, and as a promising field for therapeutic manipulation. It remains difficult, however, to accurately model tumor immunology, restricting our ability to fully grasp the mechanisms at play [202].
Outlook
We have attempted to reimagine the hallmarks of cancer into seven hallmarks (Figure 3): selective growth and proliferative advantage, altered stress response favoring overall survival, vascularization, invasion and metastasis, metabolic rewiring, an abetting microenvironment, and immune modulation.
Figure 3.
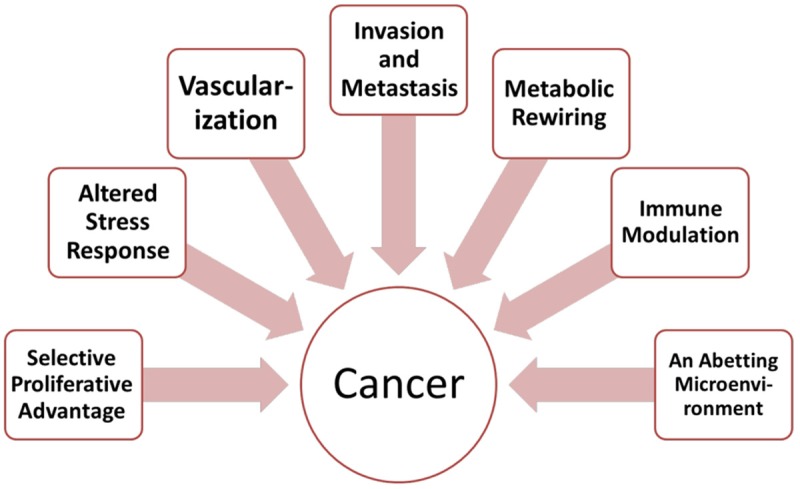
The hallmarks of cancer revisited.
The first two originally-proposed hallmarks were self-sufficiency in growth signals and insensitivity to antigrowth signals [1,2]. As our understanding grows, it is apparent that there is considerable overlap there [23,37,38]. Our dichotomous assignment of cancer-regulatory genes into oncogenes and tumor-suppressors (seemingly fit with the two original hallmarks, respectively) may even hamper our understanding of cancer biology at times [39]. We thus choose to view growth promoters and suppressors in an evolutionary context (Hallmark A), where cells compete and those that manipulate such factors, with the most acquired growth and proliferative advantage, are selected upon [6,203].
The third and fourth original hallmarks were evading apoptosis and limitless replicative potential [1,2]. The word-choice is rather misleading as it paints a picture of tumors full of immortal cells capable of cheating death. This is not the real picture; apoptosis occurs continuously in cancer cells [55] and may even serve an evolutionary role in tumor progression [54], and not all tumor cells are capable of bypassing senescence and crisis responses [68]. Consequently, we choose to view apoptotic and senescence pressures in the context of stresses facing cancer cells (Hallmark B), again positively selecting on cells that exhibit better adaptation, or those that could even subvert them to their own advantage.
We choose to maintain the fifth and sixth hallmarks in the original reviews [1,2]. Vascularization is certainly a hallmark of cancer development, however, we committed to using the term “angiogenesis” when we referred to sprouting, to denote the existence of other important forms of tumor vascularization (Hallmark C). Our understanding of the invasion-metastasis cascade has also evolved in the recent years (Hallmark D). Finally, two “enabling characteristics” (with no term-definition) and two emerging hallmarks were featured in Hallmarks II [2]. For genomic instability, we choose to view it as a stress (Hallmark B) allowed-and frequently utilized-by cancer cells where DNA damage response is altered [204,205]. For tumor-promoting inflammation, we disagree to its inclusion as a separate entity. The inflammatory cell components of tumors are no other than active members of the immune system [206], and are thus better viewed in the context of immune response (Hallmark G). A long-recognized association exists between chronic inflammation and cancer [207], but the aim of the hallmarks is describing commonality not causality (Figure 1); thus this would be equivalent to including viral transformation as a hallmark of cancer on the basis of its causal linkage to the disease. Finally, a tumor-promoting role for inflammation is not always the case; inflammation may be tumor-suppressive in settings [208], and may even favor response to immunotherapy [209].
The emerging hallmarks have managed to become established ones. Recognition of the importance of the immune system within the tumor context (Hallmark G) has come with an eruption in cancer immunotherapy developments [210]. We also continue to unravel the metabolic alterations observed in cancer cells, their causes and their consequences (Hallmark E). Lastly, our understanding of the TME has shifted through the years from passive components surrounding the cancerous cells, to active contributors abetting their initiation, progression, heterogeneity and survival (Hallmark F).
Although the idea of the hallmarks entails commonality, it is still important to keep in mind that cancer is not a single disease and that dependence on-and consequences of utilizing-the shared pathways are variable among different tumor types. For example, vascularization is an important hallmark of cancer, but some tumors are weakly-vascular and may depend less on such a hallmark [77]. Another example is the tissue-specific utilization of branched-chain amino acids in cancer cells sharing the same driving mutation [211]. We imagine cancer to continue being a diverse composition of diseases, each with a different agenda exploiting the hallmarks to different degrees, and consequently requiring therapeutic tailoring.
Finally, although the hallmarks model provides excellent description of what goes wrong in cancer cells, it does not answer the question of why those alterations are undertaken in the first place. In one of the most critically acclaimed cancer papers of the modern times [212]. Tomasetti and Vogelstein proposed that the variation in cancer risk among different tissues is grossly attributable to the different rates of stem-cell division, increasing the risk of progression by time. This is backed up by findings such as clonal hematopoiesis in aged individuals [213] and high mutational burden in aged skin cells [10], both harboring driver mutations of malignant transformation, perhaps just a few steps away. This might seem frustrating (depicting cancer as being a disease of ageing or chance for a great part), especially since our understanding of the cancer hallmarks has helped us gain insights into therapeutic targets, but not into prevention of the disease occurrence; this fact may explain why, since the original introduction of the cancer hallmarks, a translation into marked subverted cancer-related deaths is still absent [7]. Perhaps the key to cancer prevention would prove to be in the hands of understanding the hallmarks of normal cells and those of ageing, linking them to the hallmarks of cancer, and attempting to break the link.
Acknowledgements
This work originated as an assignment in Harvard Medical School’s Cancer Biology and Therapeutics program class of 2016. The authors would thus like to deeply thank the program directors for the inspiration and guidance. The authors also acknowledge the input of their program teammates: Ahmed El Ghamrawy, Mariam AlMuftah, and Amira Aljabiry.
Disclosure of conflict of interest
None.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Weinberg RA. Coming full circle-from endless complexity to simplicity and back again. Cell. 2014;157:267–271. doi: 10.1016/j.cell.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Lazebnik Y. What are the hallmarks of cancer? Nat Rev Cancer. 2010;10:232–233. doi: 10.1038/nrc2827. [DOI] [PubMed] [Google Scholar]
- 5.Wilbur MA, Shih IM, Segars JH, Fader AN. Cancer implications for patients with endometriosis. Semin Reprod Med. 2017;35:110–116. doi: 10.1055/s-0036-1597120. [DOI] [PubMed] [Google Scholar]
- 6.Horne SD, Pollick SA, Heng HH. Evolutionary mechanism unifies the hallmarks of cancer. Int J Cancer. 2015;136:2012–2021. doi: 10.1002/ijc.29031. [DOI] [PubMed] [Google Scholar]
- 7.Sonnenschein C, Soto AM. The aging of the 2000 and 2011 Hallmarks of Cancer reviews : a critique. J Biosci. 2013;38:651–663. doi: 10.1007/s12038-013-9335-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bizzarri M, Cucina A, Conti F, D’Anselmi F. Beyond the oncogene paradigm: understanding complexity in cancerogenesis. Acta Biotheor. 2008;56:173–196. doi: 10.1007/s10441-008-9047-8. [DOI] [PubMed] [Google Scholar]
- 9.Sonnenschein C SA, Rangarajan A, Kulkarni P. Competing views on cancer. J Biosci. 2014;39:281–302. doi: 10.1007/s12038-013-9403-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, Stratton MR, Jones PH, Campbell PJ. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, Hassimi M, Socci N, Bhatt PK, Gonen M, Mason CE, Melnick A, Godley LA, Brennan CW, Abdel-Wahab O, Levine RL. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44:1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, Ozenberger BA, Welch JS, Link DC, Walter MJ, Mardis ER, Dipersio JF, Chen F, Wilson RK, Ley TJ, Ding L. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R. Leukaemogenesis induced by an activating [bgr] -catenin mutation in osteoblasts. Nature. 2014;506:240–244. doi: 10.1038/nature12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raaijmakers MH. Myelodysplastic syndromes: revisiting the role of the bone marrow microenvironment in disease pathogenesis. Int J Hematol. 2012;95:17–25. doi: 10.1007/s12185-011-1001-x. [DOI] [PubMed] [Google Scholar]
- 15.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 16.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinberg RA. One renegade cell. Basic Books; 1998. [Google Scholar]
- 19.Malaterre C. Organicism and reductionism in cancer research: towards a systemic approach. International Studies in the Philosophy of Science. 2007;21:57–73. [Google Scholar]
- 20.Witsch E, Sela M, Yarden Y. Roles for growth factors in cancer progression. Physiology. 2010;25:85–101. doi: 10.1152/physiol.00045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 22.Katsuno Y, Lamouille S, Derynck R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25:76–84. doi: 10.1097/CCO.0b013e32835b6371. [DOI] [PubMed] [Google Scholar]
- 23.Pickup M, Novitskiy S, Moses HL. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer. 2013;13:788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 26.Bache KG, Slagsvold T, Stenmark H. Defective downregulation of receptor tyrosine kinases in cancer. EMBO J. 2004;23:2707–2712. doi: 10.1038/sj.emboj.7600292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015;15:290–301. doi: 10.1038/nrc3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Csermely P, Korcsmaros T, Nussinov R. Intracellular and intercellular signaling networks in cancer initiation, development and precision anti-cancer therapy: RAS acts as contextual signaling hub. Semin Cell Dev Biol. 2016;58:55–59. doi: 10.1016/j.semcdb.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 32.Indovina P, Marcelli E, Casini N, Rizzo V, Giordano A. Emerging roles of RB family: new defense mechanisms against tumor progression. J Cell Physiol. 2013;228:525–535. doi: 10.1002/jcp.24170. [DOI] [PubMed] [Google Scholar]
- 33.Kim MP, Zhang Y, Lozano G. Mutant p53: multiple mechanisms define biologic activity in cancer. Front Oncol. 2015;5:249. doi: 10.3389/fonc.2015.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stracquadanio G, Wang X, Wallace MD, Grawenda AM, Zhang P, Hewitt J, Zeron-Medina J, Castro-Giner F, Tomlinson IP, Goding CR. The importance of p53 pathway genetics in inherited and somatic cancer genomes. Nat Rev Cancer. 2016;16:251–265. doi: 10.1038/nrc.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393–405. doi: 10.1038/nrm4007. [DOI] [PubMed] [Google Scholar]
- 36.Haupt S, Raghu D, Haupt Y. Mutant p53 drives cancer by subverting multiple tumor suppression pathways. Front Oncol. 2016;6:12. doi: 10.3389/fonc.2016.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng GS. Conflicting roles of molecules in hepatocarcinogenesis: paradigm or paradox. Cancer Cell. 2012;21:150–154. doi: 10.1016/j.ccr.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shojaee S, Chan LN, Buchner M, Cazzaniga V, Cosgun KN, Geng H, Qiu YH, von Minden MD, Ernst T, Hochhaus A. PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. Nat Med. 2016;22:379–387. doi: 10.1038/nm.4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lou X, Zhang J, Liu S, Lou X, Liao DJ. The other side of the coin: the tumor-suppressive aspect of oncogenes and the oncogenic aspect of tumor-suppressive genes, such as those along the CCND-CDK4/6-RB axis. Cell Cycle. 2014;13:1677–1693. doi: 10.4161/cc.29082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelley MR, Logsdon D, Fishel ML. Targeting DNA repair pathways for cancer treatment: what’s new? Future Oncol. 2014;10:1215–1237. doi: 10.2217/fon.14.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60:547–560. doi: 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]
- 42.Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer: the xeroderma pigmentosum paradigm. Arch Dermatol. 1994;130:1018–1021. [PubMed] [Google Scholar]
- 43.Stoppa-Lyonnet D. The biological effects and clinical implications of BRCA mutations: where do we go from here? Eur J Hum Genet. 2016;24:S3–S9. doi: 10.1038/ejhg.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–932. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 45.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 46.Hung RJ, Hall J, Brennan P, Boffetta P. Genetic polymorphisms in the base excision repair pathway and cancer risk: a HuGE review. Am J Epidemiol. 2005;162:925–942. doi: 10.1093/aje/kwi318. [DOI] [PubMed] [Google Scholar]
- 47.Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. 2002;11:1513–1530. [PubMed] [Google Scholar]
- 48.Srivastava M, Raghavan SC. DNA double-strand break repair inhibitors as cancer therapeutics. Chem Biol. 2015;22:17–29. doi: 10.1016/j.chembiol.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 49.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801–817. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 50.Green DR, Evan GI. A matter of life and death. Cancer Cell. 2002;1:19–30. doi: 10.1016/s1535-6108(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 51.Lopez J, Tait SWG. Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer. 2015;112:957–962. doi: 10.1038/bjc.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Juin P, Geneste O, Gautier F, Depil S, Campone M. Decoding and unlocking the BCL-2 dependency of cancer cells. Nat Rev Cancer. 2013;13:455–465. doi: 10.1038/nrc3538. [DOI] [PubMed] [Google Scholar]
- 53.Koff JL, Ramachandiran S, Bernal-Mizrachi L. A time to kill: targeting apoptosis in cancer. Int J Mol Sci. 2015;16:2942–2955. doi: 10.3390/ijms16022942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Labi V, Erlacher M. How cell death shapes cancer. Cell Death Dis. 2015;6:e1675. doi: 10.1038/cddis.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev. 2011;21:12–20. doi: 10.1016/j.gde.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, Cantlon A, Fisch S, Golomb-Mello G, Ryan JA, Deng J, Jian B, Corbett C, Goldenberg M, Madsen JR, Liao R, Walsh D, Sedivy J, Murphy DJ, Carrasco DR, Robinson S, Moslehi J, Letai A. Developmental regulation of mitochondrial apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell. 2017;31:142–156. doi: 10.1016/j.ccell.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 58.Keulers TG, Schaaf MB, Rouschop KM. Autophagy-dependent secretion: contribution to tumor progression. Front Oncol. 2016;6:251. doi: 10.3389/fonc.2016.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–880. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edinger AL, Thompson CB. Defective autophagy leads to cancer. Cancer Cell. 2003;4:422–424. doi: 10.1016/s1535-6108(03)00306-4. [DOI] [PubMed] [Google Scholar]
- 61.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ozpolat B, Benbrook DM. Targeting autophagy in cancer management-strategies and developments. Cancer Manag Res. 2015;7:291–9. doi: 10.2147/CMAR.S34859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mowers EE, Sharifi MN, Macleod KF. Autophagy in cancer metastasis. Oncogene. 2017;36:1619–1630. doi: 10.1038/onc.2016.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martínez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer. 2011;11:161–176. doi: 10.1038/nrc3025. [DOI] [PubMed] [Google Scholar]
- 68.Perez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer. 2014;14:547–58. doi: 10.1038/nrc3773. [DOI] [PubMed] [Google Scholar]
- 69.Shay JW. Role of telomeres and telomerase in aging and cancer. Cancer Discov. 2016;6:584–593. doi: 10.1158/2159-8290.CD-16-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Günes C, Rudolph KL. The role of telomeres in stem cells and cancer. Cell. 2013;152:390–393. doi: 10.1016/j.cell.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 71.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tchkonia T, Zhu Y, Van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 74.Welti J, Loges S, Dimmeler S, Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–3200. doi: 10.1172/JCI70212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–396. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 76.Ribatti D, Nico B, Crivellato E. The development of the vascular system: a historical overview. Methods Mol Biol. 2015;1214:1–14. doi: 10.1007/978-1-4939-1462-3_1. [DOI] [PubMed] [Google Scholar]
- 77.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 78.Yang Y, Sun M, Wang L, Jiao B. HIFs, angiogenesis, and cancer. J Cell Biochem. 2013;114:967–974. doi: 10.1002/jcb.24438. [DOI] [PubMed] [Google Scholar]
- 79.Fraisl P, Mazzone M, Schmidt T, Carmeliet P. Regulation of angiogenesis by oxygen and metabolism. Dev Cell. 2009;16:167–179. doi: 10.1016/j.devcel.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 80.Hashimoto T, Shibasaki F. Hypoxia-inducible factor as an angiogenic master switch. Front Pediatr. 2015;3:33. doi: 10.3389/fped.2015.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Petrovic N. Targeting angiogenesis in cancer treatments: where do we Stand? J Pharm Pharm Sci. 2016;19:226–238. doi: 10.18433/jpps.v19i2.27608. [DOI] [PubMed] [Google Scholar]
- 82.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 83.Sakurai T, Kudo M. Signaling pathways governing tumor angiogenesis. Oncology. 2011;81(Suppl 1):24–29. doi: 10.1159/000333256. [DOI] [PubMed] [Google Scholar]
- 84.Kieran MW, Kalluri R, Cho YJ. The VEGF pathway in cancer and disease: responses, resistance, and the path forward. Cold Spring Harb Perspect Med. 2012;2:A006593. doi: 10.1101/cshperspect.a006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2005;23:1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 86.Zhao Y, Adjei AA. Targeting angiogenesis in cancer therapy: moving beyond vascular endothelial growth factor. Oncologist. 2015;20:660–673. doi: 10.1634/theoncologist.2014-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 88.Hida K, Maishi N, Torii C, Hida Y. Tumor angiogenesis-characteristics of tumor endothelial cells. Int J Clin Oncol. 2016;21:206–212. doi: 10.1007/s10147-016-0957-1. [DOI] [PubMed] [Google Scholar]
- 89.Donnem T, Hu J, Ferguson M, Adighibe O, Snell C, Harris AL, Gatter KC, Pezzella F. Vessel co-option in primary human tumors and metastases: an obstacle to effective anti-angiogenic treatment? Cancer Med. 2013;2:427–436. doi: 10.1002/cam4.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao C, Yang H, Shi H, Wang X, Chen X, Yuan Y, Lin S, Wei Y. Distinct contributions of angiogenesis and vascular co-option during the initiation of primary microtumors and micrometastases. Carcinogenesis. 2011;32:1143–1150. doi: 10.1093/carcin/bgr076. [DOI] [PubMed] [Google Scholar]
- 91.Leenders WPJ, Küsters B, de Waal RMW. Vessel co-option: how tumors obtain blood supply in the absence of sprouting angiogenesis. Endothelium. 2002;9:83–87. doi: 10.1080/10623320212006. [DOI] [PubMed] [Google Scholar]
- 92.Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, Shuman MA. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2:306–314. doi: 10.1038/sj.neo.7900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 94.Krishna Priya S, Nagare RP, Sneha VS, Sidhanth C, Bindhya S, Manasa P, Ganesan TS. Tumour angiogenesis-Origin of blood vessels. Int J Cancer. 2016;139:729–735. doi: 10.1002/ijc.30067. [DOI] [PubMed] [Google Scholar]
- 95.Ribatti D, Djonov V. Intussusceptive microvascular growth in tumors. Cancer Lett. 2012;316:126–131. doi: 10.1016/j.canlet.2011.10.040. [DOI] [PubMed] [Google Scholar]
- 96.Gianni-Barrera R, Trani M, Reginato S, Banfi A. To sprout or to split? VEGF, Notch and vascular morphogenesis. Biochem Soc Trans. 2011;39:1644–1648. doi: 10.1042/BST20110650. [DOI] [PubMed] [Google Scholar]
- 97.Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res. 2012;18:2726–2732. doi: 10.1158/1078-0432.CCR-11-3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Qiao L, Liang N, Zhang J, Xie J, Liu F, Xu D, Yu X, Tian Y. Advanced research on vasculogenic mimicry in cancer. J Cell Mol Med. 2015;19:315–326. doi: 10.1111/jcmm.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang JP, Liao YD, Mai DM, Xie P, Qiang YY, Zheng LS, Wang MY, Mei Y, Meng DF, Xu L, Cao L, Yang Q, Yang XX, Wang WB, Peng LX, Huang BJ, Qian CN. Tumor vasculogenic mimicry predicts poor prognosis in cancer patients: a meta-analysis. Angiogenesis. 2016;19:191–200. doi: 10.1007/s10456-016-9500-2. [DOI] [PubMed] [Google Scholar]
- 100.Seftor RE, Hess AR, Seftor EA, Kirschmann DA, Hardy KM, Margaryan NV, Hendrix MJ. Tumor cell vasculogenic mimicry: from controversy to therapeutic promise. Am J Pathol. 2012;181:1115–1125. doi: 10.1016/j.ajpath.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 102.Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi: 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cheung KJ, Ewald AJ. A collective route to metastasis: Seeding by tumor cell clusters. Science. 2016;352:167–169. doi: 10.1126/science.aaf6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Clark AG, Vignjevic DM. Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol. 2015;36:13–22. doi: 10.1016/j.ceb.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 106.Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25:675–686. doi: 10.1016/j.tcb.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 109.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 110.Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016;352:175–180. doi: 10.1126/science.aaf4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Diepenbruck M, Christofori G. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr Opin Cell Biol. 2016;43:7–13. doi: 10.1016/j.ceb.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 113.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, Choi H, El Rayes T, Ryu S, Troeger J. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–476. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li W, Kang Y. Probing the fifty shades of EMT in metastasis. Trends Cancer. 2016;2:65–67. doi: 10.1016/j.trecan.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Curran S, Murray GI. Matrix metalloproteinases: molecular aspects of their roles in tumour invasion and metastasis. Eur J Cancer. 2000;36:1621–1630. doi: 10.1016/s0959-8049(00)00156-8. [DOI] [PubMed] [Google Scholar]
- 117.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shuman Moss LA, Jensen-Taubman S, Stetler-Stevenson WG. Matrix metalloproteinases: changing roles in tumor progression and metastasis. Am J Pathol. 2012;181:1895–1899. doi: 10.1016/j.ajpath.2012.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 120.Hadler-Olsen E, Winberg JO, Uhlin-Hansen L. Matrix metalloproteinases in cancer: their value as diagnostic and prognostic markers and therapeutic targets. Tumor Biol. 2013;34:2041–2051. doi: 10.1007/s13277-013-0842-8. [DOI] [PubMed] [Google Scholar]
- 121.Bockhorn M, Jain RK, Munn LL. Active versus passive mechanisms in metastasis: do cancer cells crawl into vessels, or are they pushed? Lancet Oncol. 2007;8:444–448. doi: 10.1016/S1470-2045(07)70140-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reymond N, d’Água BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858–870. doi: 10.1038/nrc3628. [DOI] [PubMed] [Google Scholar]
- 123.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc Natl Acad Sci U S A. 2012;109:13515–13520. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Alix-Panabières C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6:479–491. doi: 10.1158/2159-8290.CD-15-1483. [DOI] [PubMed] [Google Scholar]
- 125.Stegner D, Dütting S, Nieswandt B. Mechanistic explanation for platelet contribution to cancer metastasis. Thrombosis Res. 2014;133:S149–S157. doi: 10.1016/S0049-3848(14)50025-4. [DOI] [PubMed] [Google Scholar]
- 126.Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, Dalin MG, Akyürek LM, Lindahl P, Nilsson J. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015;7:308re8. doi: 10.1126/scitranslmed.aad3740. [DOI] [PubMed] [Google Scholar]
- 127.Zhang C, Guan Y, Sun Y, Ai D, Guo Q. Tumor heterogeneity and circulating tumor cells. Cancer Lett. 2016;374:216–223. doi: 10.1016/j.canlet.2016.02.024. [DOI] [PubMed] [Google Scholar]
- 128.Alix-Panabières C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014;14:623–631. doi: 10.1038/nrc3820. [DOI] [PubMed] [Google Scholar]
- 129.Lorusso G, Rüegg C. New insights into the mechanisms of organ-specific breast cancer metastasis. Semin Cancer Biol. 2012;22:226–233. doi: 10.1016/j.semcancer.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 130.Langley RR, Fidler IJ. The seed and soil hypothesis revisited-The role of tumor-stroma interactions in metastasis to different organs. Int J Cancer. 2011;128:2527–2535. doi: 10.1002/ijc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, Ponomarev V, Gerald WL, Blasberg R, Massagué J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115:44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341–352. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- 133.Croucher PI, McDonald MM, Martin TJ. Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer. 2016;16:373–386. doi: 10.1038/nrc.2016.44. [DOI] [PubMed] [Google Scholar]
- 134.Psaila B, Kaplan RN, Port ER, Lyden D. Priming the ’soil’ for breast cancer metastasis: the pre-metastatic niche. Breast Dis. 2006;26:65–74. doi: 10.3233/bd-2007-26106. [DOI] [PubMed] [Google Scholar]
- 135.Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol. 2011;21:139–146. doi: 10.1016/j.semcancer.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 136.Sceneay J, Smyth MJ, Möller A. The pre-metastatic niche: finding common ground. Cancer Metastasis Rev. 2013;32:449–464. doi: 10.1007/s10555-013-9420-1. [DOI] [PubMed] [Google Scholar]
- 137.Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Ghajar CM. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–826. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell. 2013;155:750–764. doi: 10.1016/j.cell.2013.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14:611–622. doi: 10.1038/nrc3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Celià-Terrassa T, Kang Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016;30:892–908. doi: 10.1101/gad.277681.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cairns RA, Mak TW. The current state of cancer metabolism. Nature Reviews Cancer. 2016;16:613–614. [Google Scholar]
- 143.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619–634. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2009;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 149.Szablewski L. Expression of glucose transporters in cancers. Biochim Biophys Acta. 2013;1835:164–169. doi: 10.1016/j.bbcan.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 150.Younes M, Juarez D, Lechago LV, Lerner SP. Glut 1 expression in transitional cell carcinoma of the urinary bladder is associated with poor patient survival. Anticancer Res. 2000;21:575–578. [PubMed] [Google Scholar]
- 151.Cho H, Lee YS, Kim J, Chung JY, Kim JH. Overexpression of glucose transporter-1 (GLUT-1) predicts poor prognosis in epithelial ovarian cancer. Cancer Invest. 2013;31:607–615. doi: 10.3109/07357907.2013.849722. [DOI] [PubMed] [Google Scholar]
- 152.Kunkel M, Reichert TE, Benz P, Lehr HA, Jeong JH, Wieand S, Bartenstein P, Wagner W, Whiteside TL. Overexpression of Glut-1 and increased glucose metabolism in tumors are associated with a poor prognosis in patients with oral squamous cell carcinoma. Cancer. 2003;97:1015–1024. doi: 10.1002/cncr.11159. [DOI] [PubMed] [Google Scholar]
- 153.Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635–649. doi: 10.1038/nrc.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, Dean DC, Clem BF. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. 2014;33:556–566. doi: 10.1038/onc.2012.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. 2016;16:732–749. doi: 10.1038/nrc.2016.89. [DOI] [PubMed] [Google Scholar]
- 156.Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016;16:650–662. doi: 10.1038/nrc.2016.81. [DOI] [PubMed] [Google Scholar]
- 157.Schug ZT, Voorde JV, Gottlieb E. The metabolic fate of acetate in cancer. Nat Rev Cancer. 2016;16:708–717. doi: 10.1038/nrc.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166:555–566. doi: 10.1016/j.cell.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Iqbal MA, Gupta V, Gopinath P, Mazurek S, Bamezai RN. Pyruvate kinase M2 and cancer: an updated assessment. FEBS Lett. 2014;588:2685–2692. doi: 10.1016/j.febslet.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 160.Yang W, Lu Z. Regulation and function of pyruvate kinase M2 in cancer. Cancer Lett. 2013;339:153–158. doi: 10.1016/j.canlet.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14:709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gentric G, Mieulet V, Mechta-Grigoriou F. Heterogeneity in cancer metabolism: new concepts in an old field. Antioxid Redox Signal. 2017;26:462–485. doi: 10.1089/ars.2016.6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sullivan LB, Gui DY, Vander Heiden MG. Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer. 2016;16:680–693. doi: 10.1038/nrc.2016.85. [DOI] [PubMed] [Google Scholar]
- 164.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Terunuma A, Putluri N, Mishra P, Mathé EA, Dorsey TH, Yi M, Wallace TA, Issaq HJ, Zhou M, Killian JK, Stevenson HS, Karoly ED, Chan K, Samanta S, Prieto D, Hsu TYT, Kurley SJ, Putluri V, Sonavane R, Edelman DC, Wulff J, Starks AM, Yang Y, Kittles RA, Yfantis HG, Lee DH, Ioffe OB, Schiff R, Stephens RM, Meltzer PS, Veenstra TD, Westbrook TF, Sreekumar A, Ambs S. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest. 2014;124:398–412. doi: 10.1172/JCI71180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-ß signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 168.Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong JL. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–1091. doi: 10.1038/nature08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123:1001–1011. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 170.Franco OE, Shaw AK, Strand DW, Hayward SW. Cancer associated fibroblasts in cancer pathogenesis. Semin Cell Dev Biol. 2010;21:33–39. doi: 10.1016/j.semcdb.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wiseman DH. Donor cell leukemia: a review. Biol Blood Marrow Transplant. 2011;17:771–789. doi: 10.1016/j.bbmt.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 173.Yuan Y. Spatial heterogeneity in the tumor microenvironment. Cold Spring Harb Perspect Med. 2016:6. doi: 10.1101/cshperspect.a026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhou Z, Lu ZR. Molecular imaging of the tumor microenvironment. Adv Drug Deliv Rev. 2016 doi: 10.1016/j.addr.2016.07.012. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 175.Anderberg C, Pietras K. On the origin of cancer-associated fibroblasts. Cell Cycle. 2009;8:1461–1465. doi: 10.4161/cc.8.10.8557. [DOI] [PubMed] [Google Scholar]
- 176.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Méndez-Ferrer S, Battista M, Frenette PS. Cooperation of β2-and β3-adrenergic receptors in hematopoietic progenitor cell mobilization. Ann N Y Acad Sci. 2010;1192:139–144. doi: 10.1111/j.1749-6632.2010.05390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Arranz L, Sánchez-Aguilera A, Martín-Pérez D, Isern J, Langa X, Tzankov A, Lundberg P, Muntión S, Tzeng YS, Lai DM. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014;512:78–81. doi: 10.1038/nature13383. [DOI] [PubMed] [Google Scholar]
- 179.Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, Lacombe J, Armstrong SA, Dührsen U, Frenette PS. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell. 2014;15:365–375. doi: 10.1016/j.stem.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 181.Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, Purton LE. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor γ deficiency. Cell. 2007;129:1097–1110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Aanei CM, Flandrin P, Zugun Eloae F, Carasevici E, Guyotat D, Wattel E, Campos L. Intrinsic growth deficiencies of mesenchymal stromal cells in myelodysplastic syndromes. Stem Cells Dev. 2012;21:1604–1615. doi: 10.1089/scd.2011.0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Poitz DM, Augstein A, Gradehand C, Ende G, Schmeisser A, Strasser RH. Regulation of the Hif-system by micro-RNA 17 and 20a-role during monocyte-to-macrophage differentiation. Mol Immunol. 2013;56:442–451. doi: 10.1016/j.molimm.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 184.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tabassum DP, Polyak K. Tumorigenesis: it takes a village. Nat Rev Cancer. 2015;15:473–483. doi: 10.1038/nrc3971. [DOI] [PubMed] [Google Scholar]
- 186.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 187.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–1231. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Xu J, Escamilla J, Mok S, David J, Priceman S, West B, Bollag G, McBride W, Wu L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73:2782–2794. doi: 10.1158/0008-5472.CAN-12-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ohuchida K, Mizumoto K, Murakami M, Qian LW, Sato N, Nagai E, Matsumoto K, Nakamura T, Tanaka M. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. 2004;64:3215–3222. doi: 10.1158/0008-5472.can-03-2464. [DOI] [PubMed] [Google Scholar]
- 191.Cavallo F, De Giovanni C, Nanni P, Forni G, Lollini PL. 2011: the immune hallmarks of cancer. Cancer Immunol Immunother. 2011;60:319–326. doi: 10.1007/s00262-010-0968-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wang W, Kryczek I, Dostál L, Lin H, Tan L, Zhao L, Lu F, Wei S, Maj T, Peng D. Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell. 2016;165:1092–1105. doi: 10.1016/j.cell.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Burnet M. Cancer-a biological approach: I. The processes of control. II. The significance of somatic mutation. British Medical Journal. 1957;1:779. doi: 10.1136/bmj.1.5022.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 195.Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E, Massagué J. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. 2016;165:45–60. doi: 10.1016/j.cell.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Malmberg KJ. Effective immunotherapy against cancer. Cancer Immunol Immunother. 2004;53:879–892. doi: 10.1007/s00262-004-0577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, Holdman XB, Herschkowitz JI, Pond A, Xie G. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. 2016;18:632–644. doi: 10.1038/ncb3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jin P, Zhao Y, Liu H, Chen J, Ren J, Jin J, Bedognetti D, Liu S, Wang E, Marincola F. Interferon-γ and tumor necrosis factor-α polarize bone marrow stromal cells uniformly to a Th1 phenotype. Sci Rep. 2016;6:26345. doi: 10.1038/srep26345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Muller L, Mitsuhashi M, Simms P, Gooding WE, Whiteside TL. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci Rep. 2016;6:20254. doi: 10.1038/srep20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Buck MD, O’Sullivan D, Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166:63–76. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zitvogel L, Ayyoub M, Routy B, Kroemer G. Microbiome and anticancer immunosurveillance. Cell. 2016;165:276–287. doi: 10.1016/j.cell.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 202.Schreiber R. Tumor immunology. Academic Press; 2016. [Google Scholar]
- 203.Ewald PW, Ewald HA. Toward a general evolutionary theory of oncogenesis. Evol Appl. 2012;6:70–81. doi: 10.1111/eva.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FM. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer. 2015;15:166–180. doi: 10.1038/nrc3891. [DOI] [PubMed] [Google Scholar]
- 205.Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu Rev Pathol. 2015;10:425–448. doi: 10.1146/annurev-pathol-012414-040424. [DOI] [PubMed] [Google Scholar]
- 206.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chen R, Alvero AB, Silasi DA, Mor G. Inflammation, cancer and chemoresistance: taking advantage of the toll-like receptor signaling pathway. Am J Reprod Immunol. 2007;57:93–107. doi: 10.1111/j.1600-0897.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- 208.Kryvenko ON, Jankowski M, Chitale DA, Tang D, Rundle A, Trudeau S, Rybicki BA. Inflammation and preneoplastic lesions in benign prostate as risk factors for prostate cancer. Mod Pathol. 2012;25:1023–1032. doi: 10.1038/modpathol.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra232. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Couzin-Frankel J. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 211.Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, Lau AN, Ji BW, Dixit PD, Hosios AM. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science. 2016;353:1161–1165. doi: 10.1126/science.aaf5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tomasetti C, Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78–81. doi: 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert BL. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]