

Abstract
Cardiovascular diseases are among the leading causes of death worldwide. Reactive oxygen species (ROS) can act as damaging molecules but also represent central hubs in cellular signalling networks. Increasing evidence indicates that ROS play an important role in the pathogenesis of cardiovascular diseases, although the underlying mechanisms and consequences of pathophysiologically elevated ROS in the cardiovascular system are still not completely resolved. More recently, alterations of the epigenetic landscape, which can affect DNA methylation, post‐translational histone modifications, ATP‐dependent alterations to chromatin and non‐coding RNA transcripts, have been considered to be of increasing importance in the pathogenesis of cardiovascular diseases. While it has long been accepted that epigenetic changes are imprinted during development or even inherited and are not changed after reaching the lineage‐specific expression profile, it becomes more and more clear that epigenetic modifications are highly dynamic. Thus, they might provide an important link between the actions of ROS and cardiovascular diseases. This review will provide an overview of the role of ROS in modulating the epigenetic landscape in the context of the cardiovascular system.
Linked Articles
This article is part of a themed section on Redox Biology and Oxidative Stress in Health and Disease. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.12/issuetoc
Abbreviations
- 5hmC
5‐hydroxymethylcytosine
- 5mC
5‐methylcytosine
- 8‐oxodG
8‐oxo‐2′‐deoxyguanosine
- BAF
Brg1‐associated factors
- BER
base excision repair
- BRG1
Brahma‐related gene 1
- BRM
Brahma
- CBP
CREB binding protein
- CHD
chromodomain helicase DNA‐binding
- CK2
casein kinase 2
- CpG
5‐C‐phosphate‐G‐3′
- Cys
cysteine
- DNMT
DNA methyltransferase
- DPF3a
double plant homeodomain (PHD) finger protein 3a
- E2F1
E2F transcription factor 1
- ETC
electron transport chain
- EZH2
enhancer of zeste 2 PRC2 subunit
- GCN5
general control nonderepressible 5
- GPX1
glutathione peroxidase
- HAT
histone acetyltransferases
- HDAC
histone deacetylase
- HDM
histone demethylase
- HIF1
hypoxia‐inducible factor 1
- HMT
histone methyltransferases
- ISWI
imitation switch
- KDM
histone demethylase
- LINE‐1
long interspersed nuclear element‐1
- lncRNA
long non‐coding RNA
- LSD1
lysine demethylase 1A
- miRNA
microRNA
- mtDNA
mitochondrial DNA
- nDNA
nuclear DNA
- NOX
NADPH oxidases
- OGG1
8‐oxoguanine DNA glycosylase
- OXPHOS
oxidative phosphorylation
- PARP
poly(ADP‐ribose)‐polymerase
- PGC‐1α
peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha
- PHD
prolyl hydroxylase
- PolG
polymerase γ
- PPARγ
peroxisome proliferator‐activated receptor gamma
- PRC
polycomb repressive complex
- PRMT
protein arginine N‐methyltransferase
- ROS
reactive oxygen species
- SAM
S‐adenosyl methionine
- SET
Su(var)3–9, Enhancer of Zeste, Trithorax
- SIRT
sirtuin
- SMYD1
SET and MYND domain‐containing protein 1
- SNF2H
sucrose nonfermentable 2 homologue
- SWI/SNF
switch defective/sucrose nonfermentable
- TDG
thymine‐DNA glycosylase
- TET
ten eleven translocation
- TFAM
mitochondrial transcription factor A
- tRNA
transfer RNA
Introduction
Cardiovascular diseases are a major health burden and among the leading causes of death worldwide, promoted by an ageing population as well as Western diet and lifestyle. Thus, important risk factors of cardiovascular diseases are unhealthy diet, physical inactivity, smoking and harmful use of alcohol, which contribute to hypertension, hyperglycaemia, hyperlipidaemia, obesity and diabetes (Murray and Lopez, 2013).
Reactive oxygen species (ROS), at physiological levels, are important signalling molecules regulating many processes in the cardiovascular system to maintain cardiovascular homeostasis. Pronounced increases in ROS levels have been linked to initiation, progression and clinical consequences of cardiovascular diseases, including atherosclerosis, hypoxia‐reoxygenation, ischaemia–reperfusion injury, diabetic heart and vascular disease, arrhythmia, myocardial infarction, cardiac hypertrophy, cardiomyopathy, heart failure and systemic and pulmonary hypertension (Zhang and Shah, 2014; Brown and Griendling, 2015). Thereby, common risk factors for cardiovascular diseases as exemplified above, including ageing and inflammation as well as exposure to external factors such as drugs, toxins and air pollutants, have been shown to increase the ROS load in the body (Zhang and Shah, 2014; Brown and Griendling, 2015; Gorlach et al., 2015) (Figure 1).
Figure 1.
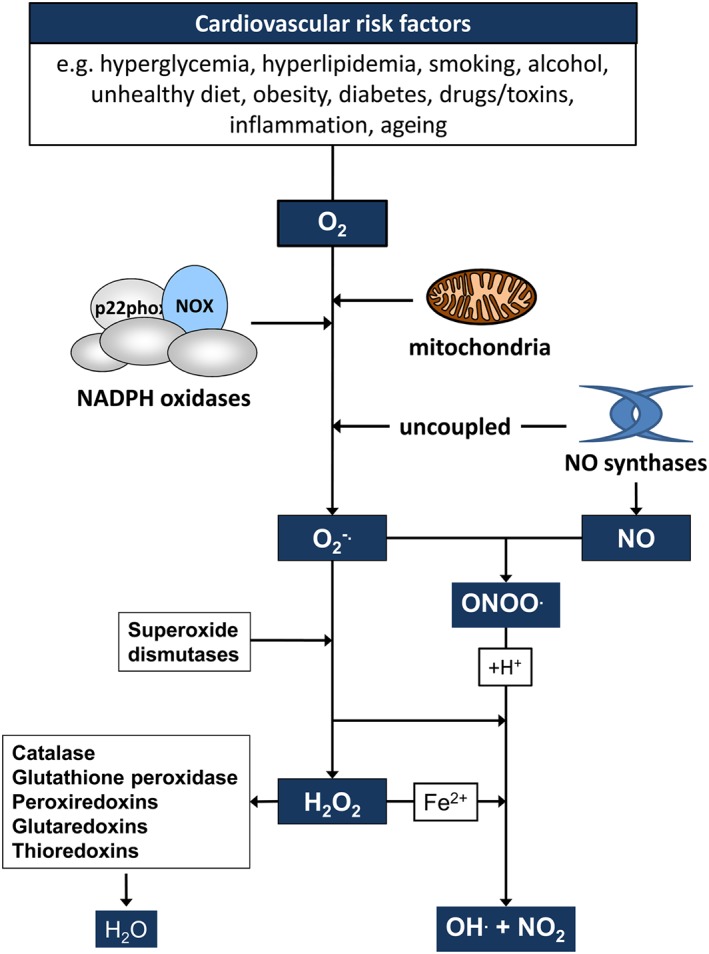
Cardiovascular risk factors promote the generation of ROS. Cardiovascular risk factors have been associated with increased generation of ROS. Superoxide anion radicals (O2 ·−) are generated from molecular oxygen via important sources such as NADPH oxidases, the mitochondrial electron transfer chain and uncoupled NO synthases. O2 ·− can be converted to H2O2 via superoxide dismutases (SOD) or in the presence of NO to peroxynitrite (ONOO−). H2O2 is decomposed or scavenged by catalase, glutathion peroxidase, glutaredoxins, peroxiredoxins or thioredoxins, respectively. O2 ·−, H2O2 and ONOO− can react via different reactions to form hydroxyl anion radicals (·OH) and nitrite (NO2).
Although the recent decades have seen great advances in the understanding of ROS biology and cardiovascular pathophysiology, a number of mechanisms underlying cardiovascular diseases and consequences of elevated ROS in the cardiovascular system are still not completely resolved.
Recently, epigenetic modifications of the genome have been added to the array of pathways leading to cardiovascular diseases (Kim et al., 2013; Friso et al., 2015; Uchida and Dimmeler, 2015; Keating et al., 2016). The term epigenetic roughly summarizes all changes at the nuclear and mitochondrial DNA (nDNA and mtDNA) or RNA level, which affect their structure or conformation, but not the DNA/RNA sequence. Epigenetic alterations commonly involve DNA methylation, post‐translational histone modifications, ATP‐dependent alterations to chromatin and non‐coding RNA transcripts (Li et al., 2007; Holoch and Moazed, 2015; Allis and Jenuwein, 2016).
Previously, epigenetic changes have been considered to be imprinted during development or even inherited and are not changed after reaching the lineage‐specific expression profile. Nowadays, it becomes increasingly clear that epigenetic modifications are as dynamic as transcription itself. Thus, they might provide an important link between the actions of ROS and cardiovascular diseases (Figure 2). This review will provide an overview of the involvement of ROS in modulating epigenetic pathways in the nuclear and mitochondrial genome with a specific emphasis on the cardiovascular system.
Figure 2.
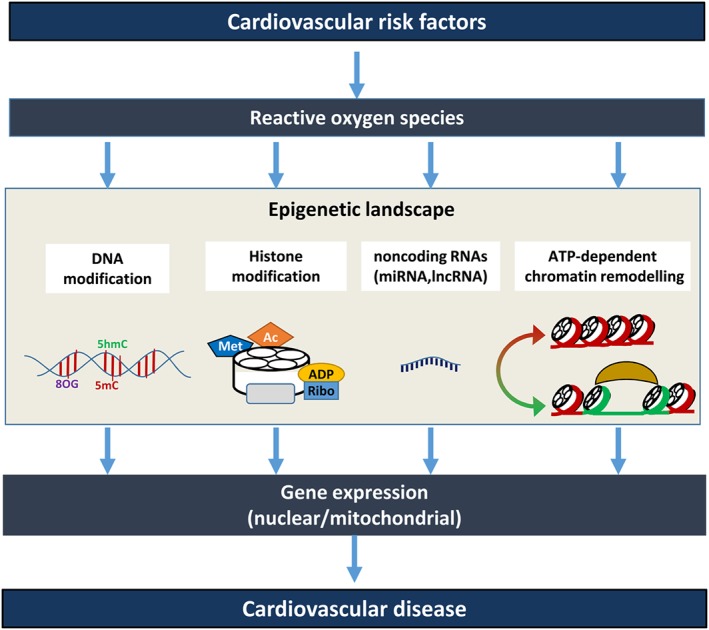
ROS modulate the epigenetic landscape contributing to the pathogenesis of cardiovascular diseases. Cardiovascular risk factors modulate the levels of ROS, which affect the epigenetic landscape by modulating histone modifications, DNA modifications, the expression of non‐coding RNAs and ATP‐dependent chromatin remodelling. This will subsequently affect gene expression patterns in the nucleus and mitochondria, which can contribute to cardiovascular diseases. DNA modifications include cytosine methylation (5mC), hydroxymethylation (5hmC) or 8‐oxo‐2′‐deoxyguanosine (8OG) formation. Histone modifications include methylation (Met), acetylation (Ac), ubiquitylation (Ubi), ADP‐ribosylation (ADP‐Ribo), SUMOylation (SUMO) and phosphorylation (P). Non‐coding RNAs include microRNAs (miRNA) and long non‐coding RNAs (lncRNA). ATP‐dependent chromatin remodelling includes moving and adding/removing nucleosomes by ATPase containing complexes (see text).
Generation of ROS
ROS are derived from molecular oxygen and include a number of free radicals and reactive molecules, which can modify DNA, RNA, proteins and lipids. Acquisition of an electron by molecular oxygen results in the formation of superoxide anion radicals (O2 ·−), which, via the family of superoxide dismutases (SOD) with its members Cu/ZnSOD (SOD1), mitochondrial MnSOD (SOD2) and extracellular (EC)SOD (SOD3), is converted to hydrogen peroxide (H2O2) (Figure 1). Catalase, as well as glutathione peroxidase (GPX), peroxiredoxins, glutaredoxins and thioredoxins can detoxify H2O2 to oxygen and water. Superoxide can react with Fe(III) to generate Fe(II) which in the presence of H2O2 produces the highly reactive hydroxyl radical (·OH) in the so‐called Fenton reaction. In the presence of NO, superoxide can, at a diffusion‐limited rate, lead to formation of peroxynitrite (ONOO−), another highly reactive ROS (Figure 1). High ROS levels can activate the transcription factor Nrf2 to increase the expression of antioxidant enzymes. Situations where the amount of ROS exceeds the antioxidant capacity are often referred to as oxidative stress and have been related to different aspects in the pathogenesis of cardiovascular diseases (Zhang and Shah, 2014; Brown and Griendling, 2015; Gorlach et al., 2015).
However, it is nowadays quite well known that ROS can also be produced in a regulated way in response to external or endogenous stimuli by different enzymic sources. At rather low levels, these ROS can act as signalling molecules in various cellular processes throughout the body (Petry et al., 2010; Samoylenko et al., 2013).
Among several sources of ROS, the NADPH oxidases (NOX) and mitochondria are of particular importance in the cardiovascular system (Zhang and Shah, 2014; Brown and Griendling, 2015; Gorlach et al., 2015) (Figure 1). NOX are multiprotein enzymes, which comprise seven members (NOX1 to NOX5, DUOX1/2). NOX are the only known class of enzymes whose sole purpose is to generate superoxide. In the cardiovascular system, in particular, NOX1, NOX2, NOX4 and NOX5 are expressed (Petry et al., 2010; Brown and Griendling, 2015). In mitochondria, O2 −· can be generated as a result of the premature univalent reduction of O2 during oxidative phosphorylation (OXPHOS) in the electron transport chain (ETC) and as byproducts of several enzyme reactions in the Krebs cycle (see below) (Murphy, 2009; Brand and Nicholls, 2011). Further sources of ROS with significance in the cardiovascular system include nitric oxide synthases (NOS) in their uncoupled state, xanthine oxidoreductase, cyclo‐ and lipoxygenases, but also members of the cytochrome P450 family and several peroxisomal oxidases (Petry et al., 2010; Samoylenko et al., 2013).
Cellular ROS levels themselves have been suggested to be the subject of epigenetic modulation. Thereby, ROS‐generating systems inside and outside the mitochondria, including different subunits of the NOX complex, as well as antioxidant enzymes such as SODs and catalase, have been reported to be epigenetically regulated by different mechanisms (see Cyr et al., 2013; Hayes and Knaus, 2013; Castegna et al., 2015; Manea et al., 2015; Mikhed et al., 2015). However, due to space limitations, we will concentrate in this review on ROS as modulators of epigenetic mechanisms.
Epigenetic mechanisms
DNA modifications
One of the first epigenetic modifications detected was methylation of cytosine. In mammals, primarily 5‐methylcytosine (5mC) is formed, but also adenine is known to be methylated. DNA methylation primarily occurs at 5‐C‐phosphate‐G‐3′ (CpG) sites. About 70–80% of CpG cytosines are methylated, which distinguishes between between newly synthesized and parent DNA during replication. CpG sites are often mutated due to spontaneous deamination of methylated cytosines into thymines, which can be at least in part counteracted by thymine‐DNA glycosylase (TDG). The presence of 5mC marks at gene regulatory regions is commonly associated with gene repression (Le and Fujimori, 2012).
Cytosine methylation is mainly processed by the DNA methyltransferases 1, 3A and 3B (DNMT1/3A/3B). All DNMTs use S‐adenosyl methionine (SAM) as methyl donor from which the methyl group is transferred to the 5‐carbon of the cytosine ring within DNA leading to the formation of S‐adenosylhomocysteine. This potent feedback inhibitor of SAM‐dependent methyltransferases is hydrolyzed to produce adenosine and homocysteine. The latter metabolite is critical for SAM regeneration via formation of methionine (Allis and Jenuwein, 2016).
Further, oxidation of 5mC to 5‐hydroxymethylcytosine (5hmC) has recently been shown to be an active process mediated by a group of dioxygenases named ten eleven translocation (TET) proteins. In mammals, there are three TET proteins known, which catalyse the conversion of 5mC to 5hmC, and further to 5‐formylcytosine and 5‐carboxylcytosine, which can subsequently undergo TDG‐mediated base excision and DNA base excision repair (BER) resulting in DNA demethylation (Branco et al., 2012; Rasmussen and Helin, 2016).
Thus, TET proteins seem to be important to avoid promoter hypermethylation and silencing of certain genes as seen with tumour suppressors during cancer development or in differentiation processes in the cardiovascular system (Greco et al., 2016).
While mass spectrometry identified 5hmC as a quite stable modification, typical methylation analysis methods, such as bisulfite sequencing, cannot discriminate between 5hmC and 5mC. Thus, cases where increased 5mC levels have been linked to increased gene expression make a concomitant analysis of 5hmC levels advisable (Branco et al., 2012).
Histone modifications
Modifications made on histones have an effect on chromatin organization and gene expression patterns. The core histones H3 and H4 are primary subjects for posttranslational modifications due to their accessible tails outside the globular nucleosome. The best known modifications are methylation and acetylation (see below), although they can comprise all types of available protein modifications, such as phosphorylation, ubiquitination, SUMOylation, ADP‐ribosylation or combinations of them, which appear to constitute the so‐called histone code (Li et al., 2007; Allis and Jenuwein, 2016). The following sections will give a short overview of the two types of modification most affected by ROS, histone methylation and acetylation.
Histone methylation
While the methylation of histones by histone methyltransferases (HMT) has been considered for a long time as a permanent epigenetic modification, the identification of histone demethylases (HDM) has shown that this chromatin modification is in fact dynamic (Bannister and Kouzarides, 2005; Cyr and Domann, 2011). Methylation occurs at lysine (K) and/or arginine (R) residues predominantly on histones H3 and H4. Dependent on genomic context and location within the histone protein, they can be mono‐, di‐ or tri‐methylated, leading to either transcriptional activation or repression (Allis and Jenuwein, 2016).
Two major types of HMTs exist, lysine‐specific and arginine‐specific, which both require SAM as methyl donor similar to DNMTs. The majority of lysine‐specific HMTs contains a SET domain as a catalytic core and methylates histones H3 and H4, while Dot1 HMTs do not contain a SET domain and methylate only histone H3 (Le and Fujimori, 2012). Protein arginine N‐methyltransferases (PRMT) act on H3 or H4, which can be monomethylated (Rme1), asymmetrically dimethylated (Rme2a) or symmetrically dimethylated (Rme2s) (Molina‐Serrano et al., 2013).
To reverse histone methylation, two families of HDMs have been identified: (a) The lysine‐specific demethylase 1A (KDM1 or LSD1) acts via a two‐step oxidation/reduction mechanism where the lysine's methylamine bond is oxidized to become an imine group and the cofactor FAD is reduced. The latter is then reoxidized by O2 to product H2O2, and the methyl group is removed from the unstable imine group as formaldehyde. (b) The jumonji‐C domain‐containing HDMs (JmjC KDM or JHDM) require Fe(II), α‐ketoglutarate and ascorbate to hydroxylate lysine's methyl group in the presence of O2. The subsequent oxidative demethylation releases formaldehyde, succinate and CO2. JmjC KDMs act on all three lysine methylation states whereas KDM1/LSD1 only acts on di‐ and mono‐methylation states (Cyr and Domann, 2011; Allis and Jenuwein, 2016).
Recently, a subset of JmjC KDMs has been shown to also act as arginine demethylases (Walport et al., 2016).
Histone acetylation
Histone acetylation results from the close interplay between histone acetyltransferases (HAT) and histone deacetylases (HDAC). HATs are either found in the nucleus (type A) or in the cytoplasm (type B). Type A HATs are further divided into five subclasses, which differ in sequence identity and structural features (Allis and Jenuwein, 2016). All HATs acetylate conserved lysine residues within histones by transferring the acetyl group from acetyl CoA to the ε‐amino group of the respective lysine. The lysine‐acetylated histones are involved in specific protein–protein interactions with transcription factors, which contain an acetyl‐lysine‐binding bromodomain. HATs are thus associated with euchromatin and linked to transcriptional activation and inhibition of DNA methylation (Wapenaar and Dekker, 2016).
HDACs, which are divided into four classes, reverse histone acetylation in higher eukaryotes, thus allowing tighter wrapping of DNA around histones and usually transcriptional inactivation (Allis and Jenuwein, 2016). Class I, II and IV HDACs use a redox‐active metal [Zn(II) or Fe(II)] to coordinate hydrolysis of acetate from lysine residues. Class III HDACs, which are also termed sirtuins (SIRT), couple lysine deacetylation with NAD+ hydrolysis. The latter reaction yields the deacetylated substrate, O‐acetyl‐ADP‐ribose, and nicotinamide. Both HATs and HDACs are also involved in the modulation of acetylation of non‐histone proteins such as transcription factors or nuclear receptors, thus explaining the wide range of effects of HDAC inhibitors on cardiovascular pathologies and other diseases.
Non‐coding RNA transcripts
A variety of differently sized RNA molecules, which are transcribed from DNA, but in general are not translated into proteins, have emerged as important modulators of the epigenetic landscape. These non‐coding RNAs (ncRNAs) are divided into small ncRNAs (<200 nts), among them the epigenetically relevant and mostly studied miRNA, and an increasing number of heterogeneous long non‐coding RNAs (lncRNA, >200 nts) (Li et al., 2007; Holoch and Moazed, 2015; Allis and Jenuwein, 2016). Small ncRNAs modify chromatin structure and silence transcription by guiding Argonaute‐containing complexes to complementary nascent RNA scaffolds and promote the recruitment of histone and DNA methyltransferases. Increasing evidence indicates that also lncRNAs are associated with chromatin complexes (Holoch and Moazed, 2015).
Moreover, the expression of ncRNAs can be subject to epigenetic regulation as well, thus helping to shape the epigenetic landscape via chromatin organization, heterochromatin formation, histone modifications and DNA methylation and enabling the detection and silencing of inappropriate transcription events (Li et al., 2007; Holoch and Moazed, 2015; Allis and Jenuwein, 2016).
ATP‐dependent chromatin remodelling
In addition to direct modulation of DNA and histones, the structure, organization and accessibility of chromatin is regulated by ATP‐dependent chromatin remodelers (Rosa‐Garrido et al., 2013). These large multi‐subunit complexes direct the localization of the nucleosomes on the chromatin to modify chromatin accessibility. They contain an evolutionary conserved Snf2‐like ATPase catalytic domain, which provides the energy to remodel nucleosomes by hydrolyzing ATP. Different associated subunits dictate the catalytic activity of the ATPase subunit and provide specificity to genome binding (Clapier and Cairns, 2009; Han et al., 2011). There are at least four main families of chromatin remodelling complexes known: switch defective/sucrose nonfermentable (SWI/SNF), initially identified in prokaryotes and yeast; imitation switch (ISWI), initially found in Drosophila; chromodomain helicase DNA‐binding (CHD), identified in mice, and INO80, initially found in yeast (Rosa‐Garrido et al., 2013). The different families have all important functions in development, since most, if not all, developmental transitions require chromatin regulation.
Effects of reactive oxygen species on epigenetic mechanisms
Modifications of DNA bases and histones by ROS: the direct attack
DNA bases can be directly modified by ROS. For example, hydroxyl radicals can lead to the formation of 5hmC from 5mC, initiated by abstraction of an H‐atom from the methyl group (Madugundu et al., 2014) (Figure 3). 5hmC has been proposed to interfere with DNMT1 to block the proper inheritance of methylation patterns, thus leading to indirect demethylation of CpG sites modification (Branco et al., 2012). Further, superoxide has been suggested to directly mediate cytosine methylation by deprotonating C5 followed by direct transfer of a methyl group from SAM without the need of a DNMT: however, a direct proof of this mechanism is pending (Afanas'ev, 2014).
Figure 3.
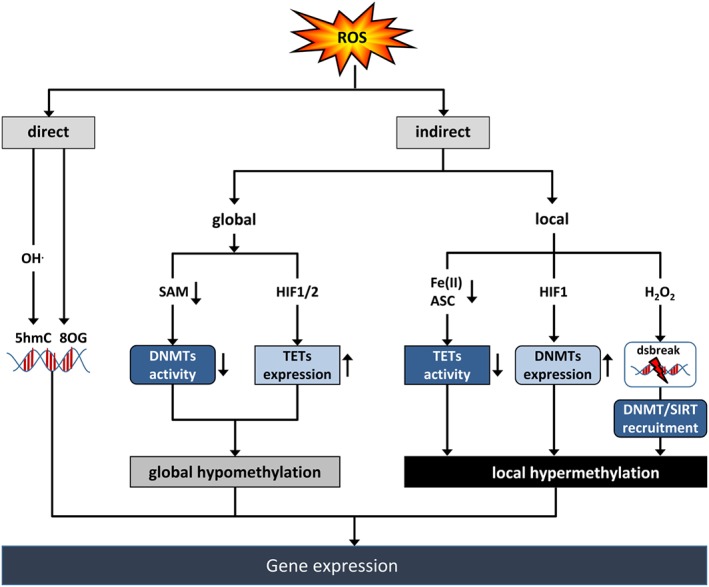
ROS affect DNA methylation. ROS can directly affect DNA by formation of 8‐oxo‐2′‐deoxyguanosine (8OG) or, via hydroxyl radicals (OH−), by formation of 5‐hydroxymethylcytosine (5hmC). ROS can also indirectly affect DNA methylation at the global or local level leading to modulation of gene expression. Reduction of the activity of DNA methyltransferases (DNMT) by reducing the availability of SAM or increasing the expression of TET proteins via the transcription factor HIF1 can lead to global hypomethylation. Decreasing TET activity by reducing Fe(II) or ascorbate (ASC) levels, or increasing DNMT expression via HIF1, or recruiting DNMT and the HDM SIRT1 containing complexes to H2O2‐induced DNA double strand breaks (dsbreak) can result in local hypermethylation.
ROS can also affect DNA methylation via oxidation of guanosine to 8‐oxo‐2′‐deoxyguanosine (8‐oxodG) (Figure 3). In order to prevent a mutagenic effect, the 8‐oxoguanine DNA glycosylase (OGG1) can remove the 8‐oxodG residues in most instances, and the gap is filled by base excision repair (BER) (Kreuz and Fischle, 2016). However, when 8‐oxodG persists, the adjacent cytosines cannot be methylated anymore, which results in hypomethylation and transcriptional activation (Le and Fujimori, 2012). Recruitment of OGG1 to the sites of 8‐oxodG formation can also promote DNA demethylation by interaction with TET1 (see below) (Zhou et al., 2016).
Interestingly, 8‐oxodG formation has been reported to preferentially take place at G‐rich sequences in promoters of putative oncogenes, such as VEGF, c‐MYC, KRAS, Bcl 2 and the transcription factor HIF1α (Balasubramanian et al., 2011; Pastukh et al., 2015). By promoting formation of G‐quadruplexes, 8‐oxodG formation has been suggested to contribute to transcriptional activation of these potential oncogenes, which could partially explain the preferential role of ROS in tumour progression (Balasubramanian et al., 2011). However, recent data show that such a process is not limited to tumours but is also present during inflammation in cells of the cardiovascular system (Balasubramanian et al., 2011; Ba et al., 2014). For example, 8‐oxodG formation promoted NFĸB‐dependent transcription of proinflammatory genes in response to TNFα (Pan et al., 2016). In addition, 8‐oxodG formation affected binding of HIF1 to the VEGF promoter and possibly other proangiogenic genes in hypoxic endothelial cells (Pastukh et al., 2015).
In a model of increased ROS due to removal of 2‐mercaptoethanol, 8‐oxodG formation resulted in activation of the transcription factor Tbx5 and enhanced differentiation of murine embryonic stem cells to cardiomyocytes (Park et al., 2016). High 8‐oxodG levels have been further found in atherosclerotic vessels and have been correlated with progression of disease (Nagayoshi et al., 2009). Two meta‐analyses recently indicated that 8‐oxodG levels in blood or urine samples are higher in patients with cardiovascular diseases such as atherosclerosis and heart failure than in controls (Kroese and Scheffer, 2014; Di Minno et al., 2016). However, larger prospective studies are needed to validate 8‐oxodG as a predictor of cardiovascular diseases.
In addition to DNA, ROS can also directly modify histones. Peroxynitrite has been shown to induce nitration and oxidation of H1, H2B and H3, which can lead to structural changes possibly protecting the packaged DNA, although it may have further implications in regulating chromatin structure and function as well as maintaining genome stability (Khan et al., 2016).
In the active phase of the cell cycle, ROS can form protein‐bound carbonyl groups by direct oxidation of mainly basic amino acid residues, including arginine and lysine, in histone H3, which may affect chromatin relaxation and accumulation of transcription factors. Moreover, H3, the only histone, which contains cysteines, has been shown to sense redox changes through S‐glutathionylation of Cys110, which leads to a more open chromatin structure. This modification is increased during cell proliferation and decreased during ageing (Garcia‐Gimenez et al., 2013). Lipid peroxidation products such as 4‐oxo‐2‐nonenal can form lysine adducts on H2, H3 and H4, for example, at H3K23 and H3K27, which were also detected in LPS‐stimulated macrophages, indicating that these modifications at histone acetylation and methylation sites may affect epigenetic patterning also in cardiovascular diseases (Galligan et al., 2014).
ROS effects on DNA and histone modifications: the indirect attack
ROS and DNA modifications
In general, increased levels of ROS have been associated with global DNA hypomethylation. This observation has been initially made in cancer, but recently also in cardiovascular diseases (Cyr and Domann, 2011; Byrne et al., 2014; Zhong et al., 2016). However, increasing evidence suggests that these observations cannot be generalized and that ROS rather differentially affect global and local DNA methylation.
DNA methyltransferases and ROS
One mechanism how ROS can affect DNA methylation is by acting on either activity or expression of DNMTs. For example, ROS can reduce the availability of the cofactor SAM, thus limiting the activity of DNMTs leading to DNA hypomethylation (Figure 3). This is achieved either by inhibiting methionine adenosyltransferase and thus SAM synthesis or by inhibiting methionine synthase and thus methionine regeneration. In addition, in conditions of oxidative stress, methionine is required for the synthesis of cysteine to produce the antioxidant glutathione and is thus depleted for SAM synthesis (Cyr and Domann, 2011). In support, long‐term exposure to H2O2 decreased SAM levels but increased glutathione levels, leading to hypomethylation of the long interspersed nuclear element‐1 (LINE‐1) (Kloypan et al., 2015). LINE‐1 hypomethylation as an indicator of global methylation status was found in blood from patients with ischaemic heart disease and stroke and has been related to higher risk for these diseases (Baccarelli et al., 2010).
In contrast, ROS can also induce DNA hypermethylation by increasing expression of DNMTs. Increased levels of DNMT1, DNMT3A and DNMT3B induced by the hypoxia‐inducible transcription factor HIF1α (Figure 3) were observed in several models of myocardial or cerebral ischaemia, as well as in pulmonary hypertension. This resulted either in global DNA hypermethylation and enhanced profibrotic gene expression or in specific hypermethylation of CpG islands in the SOD2 gene and subsequent loss of SOD2 expression, while DNMT inhibitors could alleviate ischaemia or oxidative stress‐induced injury (Kim et al., 2013; Watson et al., 2014; Wu et al., 2014). In isolated fetal rat hearts and cardiomyocytes, hypoxia‐induced hypermethylation of the PKCepsilon promoter has been associated with cardiac dysfunction, which was attenuated by antioxidants (Patterson et al., 2012). However, it remained open whether ROS were derived from the known HIF1α targets NOX2 (Diebold et al., 2012) or NOX4 (Diebold et al., 2010b). In fetal hearts exposed to noradrenaline, NOX1‐derived ROS were found to mediate hypermethylation of the PKCepsilon promoter, and DNMT inhibition prevented cardiac hypertrophy in this model (Xiong et al., 2012; Xiao et al., 2014).
Moreover, flow disturbances, which lead to increased ROS and endothelial dysfunction as a hallmark of atherogenesis, enhanced DNMT1 and DNMT3A expression as well as DNA hypermethylation of CpG islands of different genes involved in mechanotransduction. For example, hypermethylation of the gene encoding the transcription factor KLF4 decreased the expression of endothelial NOS under these conditions (Dunn et al., 2015). While these studies suggest that ROS can induce specific hypermethylation by up‐regulating DNMTs, several studies found that ROS can also affect DNA methylation by modulating the DNA recruitment of DNMTs without affecting DNMT expression (Lim et al., 2008; Zhao et al., 2016). For example, ROS induced Snail, which was required to recruit DNMT1 to the E‐cadherin promoter for hypermethylation (Lim et al., 2008).
Moreover, prolonged inflammation or exposure to H2O2 have been shown to recruit DNMT1 to damaged chromatin, forming an epigenetic silencing complex with DNMT3B, the HDAC SIRT1 and polycomb repressive complex (PRC) 4 members (O'Hagan et al., 2011) (Figure 3). This complex is enriched at GC‐rich areas of the genome, including CpG islands that become DNA hypermethylated and gain repressive histone marks leading to silencing of the associated genes. It has been proposed that such changes may persist and become permanent, which might explain aberrant DNA hypermethylation at distinct CpG islands in a context of DNA hypomethylation in tumours (O'Hagan et al., 2011), but possibly also in cardiovascular diseases. Recent findings in endothelial cells show that SIRT1 can affect DNA methylation at PRC target genes, a process which is also observed in ageing (Wakeling et al., 2015).
ROS, TETs and DNA demethylation
ROS have also been implicated to modulate DNA methylation by targeting expression and/or activity of the TET family proteins (Figure 3). Initially, ROS have been reported to induce nuclear TET1 protein levels and activity as well as to increase 5hmC but to decrease 5mC formation in hydroquinone‐stimulated HEK293 cells. This resulted in demethylation of LINE‐1 and several specific genes involved in ROS detoxification and cell cycle arrest (Coulter et al., 2013). Similarly, in a mouse model of cerebral ischaemia, global 5hmC abundance and TET2 levels were increased in the ischaemic regions, and these responses were diminished in TET2 knockout mice (Miao et al., 2015). Induction of TET1 and TET3 proteins concomitant with either global or local increases in 5hmC marks has also been observed under hypoxia in different tumour cell lines, involving the transcription factors HIF1 or HIF2 (Mariani et al., 2014; Tsai et al., 2014; Wu et al., 2015) (Figure 3). Since these transcription factors are also induced by ROS (BelAiba et al., 2004; Gorlach and Bonello, 2008; Diebold et al., 2010a), such a mechanism might be relevant also for increased TET protein levels in situations of oxidative stress.
As the TET proteins belong to the family of 2‐oxoglutarate (2OG) oxygenases, they require Fe(II), oxygen and ascorbate as cofactors. This enzyme family is particularly known for the HIF prolyl hydroxylases (PHDs) whose activity is substantially reduced by hypoxia and ROS (Bishop and Ratcliffe, 2015; Salminen et al., 2016), and ROS appear to reduce the activity of PHDs by decreasing the availability of Fe(II) and ascorbic acid (Diebold et al., 2010a). Similar to PHDs, ascorbate enhanced TET activity by acting on its catalytic domain, which was reflected by an increased 5hmC content in mouse embryonic stem cells (Chen et al., 2013).
Interestingly, a K M value of TET1/2 for O2 of 30 μM was reported, indicating that these enzymes, unlike the PHDs, can remain active even under hypoxic conditions (Laukka et al., 2016). Similar to PHDs, however, TET activity was also inhibited by succinate and fumarate, which are closely linked to ROS and mitochondrial metabolism (Tretter et al., 2016) (see below).
More recently, it was reported that hypoxia decreases, similar to the situation with PHDs, TET activity and 5hmC marks, thus leading to hypermethylation of hypoxic tumour areas (Thienpont et al., 2016). However, TET inhibition was only seen at oxygen concentrations of 2% O2 and below, confirming that TET activity is preserved under a wide range of oxygen concentrations. Interestingly, in a subset of cell lines, TET expression levels and 5hmC marks were increased under hypoxia, pointing to a cell type‐specific compensatory response, which is determined by oxygen availability and TET abundance (Thienpont et al., 2016).
This observation was not limited to tumours and hypoxia, since also in endothelial cells, H2O2 or hypoxia were shown to decrease TET activity and 5hmC base content (Niu et al., 2015; Sun et al., 2016) while DNMT3A, DNMT1 and 5mC levels were increased (Kalani et al., 2015) (Figure 3). In support, 5hmC levels were decreased in kidneys exposed to ischaemia/reperfusion (Huang et al., 2012) and in patients with preeclampsia and gestational diabetes mellitus (Sun et al., 2016) while in mice with combined knockout of the antioxidant enzymes GPX1, and −2 (Delatte et al., 2015), 5hmC levels were increased. Interestingly, in human atherosclerosis and in a mouse model of vascular injury, not only the 5hmC content but also the TET2 expression were reduced, which both contributed to a switch of vascular smooth muscle cells from a contractile to a proliferative phenotype (Liu et al., 2013).
Genome‐wide profiling in cell culture and mouse models of oxidative stress revealed a global decrease in 5hmC marks, although differentially hydroxymethylated regions were identified where a 5hmC enrichment was found for example in genes related to oxidative stress pathways (Delatte et al., 2015). Genome‐wide profiling of mouse brains after ischaemic stroke revealed a differential distribution of hydroxymethylated regions throughout the genome. 5hmC marks were reduced at intragenic CpG islands including transcription start sites, but were increased in exons, which promoted the expression of neuroprotective genes such as brain‐derived neurotrophic factor in ischaemic mouse brains (Miao et al., 2015).
Collectively, these data indicate that ROS are importantly involved in coordinating DNA methylation and demethylation.
ROS and histone modifications
ROS and histone methylation
Histone methylation is a highly diverse process with different sites and patterns, which can lead to open or closed chromatin and thus activate or repress the transcriptional activity of genes. Emerging evidence indicates that the histone methylation balance not only is critical in maintaining genome integrity, gene regulation and cancer evasion but also plays an important role in heart development and the pathogenesis of congenital heart defects and adult cardiovascular diseases (Zhang and Liu, 2015). ROS have been reported to modulate histone methylation marks, including activating marks such as H3K4me2/3 and repressing marks like H3K9me2/3 and H3K27me3 (Chervona and Costa, 2012; Niu et al., 2015) (Figure 4).
Figure 4.
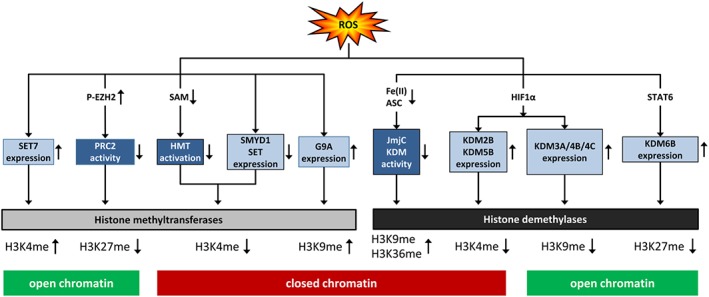
ROS affect histone lysine methylation. ROS affect histone lysine methylation via HMT or HDM either by diminishing their activity or modulating their expression, thus affecting either activating (H4K3me) or repressing (H3K9me, H3K27me, H3K36me) histone lysine methylation marks, subsequently resulting in open or closed chromatin. JmjC KDM, jumonji‐C domain‐containing HDM; STAT6, signal transducer and activator of transcription 6; p‐EZH2, enhancer of zeste 2 PRC2 subunit, phosphorylated.
Histone methyltransferases and ROS
ROS can affect the histone methylation status at the level of HMTs, either by affecting their activity or their expression. HMTs use, like DNMTs, SAM as a cofactor, thus depending on the status of methionine metabolism (Mentch et al., 2015), which can be affected by ROS as described above (Figure 4). In support, H3K4 methyltransferase activity was reduced with ageing in astrocytes following ischaemia, in conjunction with decreased H3K4me3 levels for example in the VEGF promoter, although neither ROS nor SAM levels were determined in this study (Chisholm et al., 2015).
Further, ROS decreased levels of the H3K4 methyltransferase SET and MYND domain‐containing protein 1 (SMYD1) in a model of cardiac pressure overload, and thioredoxin restored SMYD1 levels (Figure 4) and cardiac function indicating that HMTs might differ in their ROS sensitivity depending on the pathophysiological context although histone lysine methylation was not determined in this study (Liu et al., 2015b). As activation of SMYD1 has been considered to prevent cardiac hypertrophy and even heart failure, such a ROS‐related pathway might have important implications for further cardiovascular therapies (Franklin et al., 2016).
On the other hand, in different models of diabetes, hyperglycaemia and ROS have been shown to induce and/or activate the H3K4 methyltransferase SET7 (Figure 4) leading to the enrichment of H3K4me1 at various genes including Kelch‐like ECH‐associated protein, thus decreasing Nrf2 activity and antioxidant enzyme expression, or NFĸB‐65, resulting in pro‐inflammatory gene expression (Rajasekar et al., 2015; Yuan et al., 2016). Nrf2 has been shown to play an important role in cardiovascular diseases associated with increased ROS levels, such as atherosclerosis (Al‐Sawaf et al., 2015). It has thus been suggested that SET7, via ROS, would act as a sensor of vascular glucose (Keating and El‐Osta, 2013). Also, arsenite, which is known to induce ROS formation via NOX (Al Taleb et al., 2016), has been described to increase H3K4me3 levels (Zhou et al., 2008).
Further, arsenite or hypoxia up‐regulated the histone H3K9 methyltransferase G9a and the repressive mark H3K9me2 (Chervona and Costa, 2012) (Figure 4), a mechanism which has been associated with the adverse outcome of stroke (Zhao et al., 2016). Hypoxia also increased G9a‐mediated H3K9me2 levels in the promoters of neprilysin or dihydrofolate reductase (Chervona and Costa, 2012), mechanisms possibly involved in Alzheimer's disease (Wang et al., 2011b) or pulmonary hypertension (Chalupsky et al., 2015), respectively.
ROS‐dependent activation of Akt has been shown to phosphorylate the H3K27 methyltransferase EZH2, the catalytic component of PRC2, in response to different stimuli, including arsenite, thus weakening the association between EZH2 and other PRC2 subunits and decreasing the levels of the repressive mark H3K27me3 in the genome (Zhou et al., 2008; Li et al., 2014) (Figure 4). Suppression of EZH2 activity has been related to tumourigenesis, but also to impaired vascular function and atherosclerosis (Delgado‐Olguin et al., 2014), conditions where ROS‐dependent Akt activation, for example by NOX, is frequently observed (Djordjevic et al., 2005).
Furthermore, H2O2 has been shown to oxidize cysteine residues in several PRMT proteins to sulfenic acid, thus decreasing PRMT activity and histone arginine methylation (Morales et al., 2015). On the other hand, arsenite‐induced ROS promoted nuclear accumulation of PRMT1 and PRMT4 and methylation at H4R3 and H3R17 in the surrounding of antioxidant response elements, thus promoting binding of the transcription factor Nrf2 and antioxidant gene expression (Huang et al., 2013). In line, in a model of diabetic retinopathy, ROS‐increased expression of PRMT1 and PRMT4 was associated with reduced SIRT1 levels and increased cell damage in vitro and in vivo (Kim et al., 2015a).
Histone demethylases and ROS
ROS can also modulate histone methylation by affecting either expression or activity of HDMs. Increased levels of the lysine‐specific demethylase LSD1, leading to H3K4me1 or H3K4me0, have been found in cardiovascular diseases such as hypertension or diabetes (Friso et al., 2015), providing indirect evidence that ROS might affect LSD1 expression or activity. Similarly, natural polyphenols like resveratrol, curcumin and quercetin, which can decrease ROS levels (Hussain et al., 2016), inhibited LSD1 activity in C2C12 fibroblasts, thus decreasing myogenic expression and differentiation (Abdulla et al., 2013). However, direct experimental proof that ROS regulate either LSD1 expression or activity is still pending. Interestingly, LSD1 activity is accompanied by an H2O2 burst, which leads to 8‐oxodG formation and the recruitment of BER enzymes to repair the DNA (see above) (Li et al., 2013).
Broader evidence that ROS affect histone demethylation comes from the JmjC KDMs, which, like TET enzymes and PHDs, require molecular oxygen, α‐ketoglutarate, Fe(II) and ascorbate (Monfort and Wutz, 2013). Indeed, H2O2 or NO have been shown to inhibit HDM activity in a cell‐free system while increases in Fe(II) and ascorbate rescued this inhibition (Hickok et al., 2013; Niu et al., 2015) (Figure 4). Subsequently, in macrophages, ROS, NO and hypoxia or treatment with the pan‐dioxygenase inhibitor DMOG increased H3K9me2/me3 and H3K36me3 levels in specific promoter regions of chemokine genes, which is indicative for a loss of demethylase activity (Tausendschon et al., 2011).
In contrast, ROS, NO and hypoxia increased the expression of several KDMs (Hickok et al., 2013; He et al., 2016; Salminen et al., 2016). Thereby, HIF1α was found to up‐regulate expression of KDM3A, KDM4B, KDM4C and KDM6B, thus enhancing transcription by demethylating the repressive marks H3K9me2 and H3K27me3, and of KDM2B and KDM5B, thus repressing transcription by demethylating the activating marks H3K4me2/3 (Salminen et al., 2016) (Figure 4). Thus, similar to TET proteins, compensatory changes in KDM expression might counteract quenched KDM activity in response to hypoxia or ROS. Accordingly, increased H2O2 levels in the context of SOD1 overexpression promoted the expression of the H3K27 demethylase KDM6B/jmjd3 via STAT6, leading to macrophage M2 polarization and a profibrotic phenotype (He et al., 2016) (Figure 4). In line with these findings, the levels of H3K27me3 were decreased in atherosclerotic plaques (Greissel et al., 2015). Moreover, expression of KDM4A/JMJD2A was up‐regulated in hypertrophic cardiomyopathy in mouse and humans concomitant with a decrease in H3K9me2 (Zhang et al., 2011).
In summary, there is increasing evidence that ROS can serve to modulate the histone methylation balance at different levels and thus contribute to the dynamic adaptation of chromatin accessibility in response to various stimuli. Further studies will have to show how this pathway might specifically contribute to the pathogenesis of cardiovascular diseases.
ROS action on histone acetylation
Histone acetylation plays an important role in epigenetic regulation of gene expression. It is controlled on the one side by HATs, which induce transcriptional activation since adding acetyl groups weakens the association between histones and DNA. On the other side, HDACs repress transcription by removing those acetyl groups. Alterations in histone acetylation have been found associated with various cardiovascular disorders, including atherosclerosis, systemic and pulmonary hypertension, coronary heart disease, cardiomyopathy and heart failure (Kim et al., 2013; Wang et al., 2014; Matsushima and Sadoshima, 2015). Increased levels of ROS have also been widely associated with increased histone acetylation (Rajendrasozhan et al., 2008; Osoata et al., 2009; Santos et al., 2016), although opposing results have been reported (Afanas'ev, 2014). The following sections will give an overview of the different actions of ROS on HATs and HDACs.
Histone acetyltransferases and ROS
ROS increased the activity of HATs in different cell types, leading to increased acetylation of H3 or H4 histones (Gilmour et al., 2003; Tomita et al., 2003; Choudhury et al., 2010). In support, SOD2 overexpression leading to increased H2O2 levels promoted histone H3 acetylation and recruitment of the HAT p300/ CBP‐associated factor (p300/CBP) to the MMP‐1 promoter (Bartling et al., 2014), thus enhancing MMP‐1 expression, which has been associated with increased plaque instability in atherosclerosis (Lehrke et al., 2009).
Oxidative stress has also been linked to increased HAT activity of p300/CBP in conjunction with NFĸB DNA binding, thus promoting pro‐inflammatory gene expression (Sundar et al., 2013). Indirect evidence of a link between ROS and HATs comes also from studies, where histone H3 and H4 acetylation via p300/CBP was involved in disorders associated with increased ROS, such as inflammation, diabetes or cardiac hypertrophy (Rajendrasozhan et al., 2008; Usui et al., 2012; Li et al., 2016; Wapenaar and Dekker, 2016).
In a model of diabetes, enhanced expression of the HAT GCN5 and H3 acetylation in endothelial cells was associated with increased ROS levels (Paneni et al., 2012). Similarly, in hyperglycaemic adipocytes, insulin induced a fast increase in ROS, which was accompanied by increased histone H3 acetylation, an effect which was inhibited by catalase (Gupta and Tikoo, 2012). The HAT p300/CBP also mediated, by up‐regulating the kinases Syk and Btk, an Akt‐dependent survival pathway in response to H2O2 (Kikuchi et al., 2011b). On the other hand, it acetylated H3K9 at the NOX2 promoter, thus promoting ROS generation (Kikuchi et al., 2011a), indicating the complex involvement of these epigenetic modifications in ROS balance and response.
Moreover, SOD1 deficiency, which was associated with increased superoxide levels, decreased H3 acetylation at the peroxiredoxin1 gene promoter, while GPX1 deficiency associated with increased H2O2 levels up‐regulated these parameters (Wang et al., 2011a). Further studies have to show whether indeed superoxide and H2O2 might play, at least in part, opposite roles in the regulation of H3 acetylation in diabetes‐like and other cardiovascular disease conditions.
Histone deacetylation: ROS effects on class I/II HDACs
ROS‐mediated posttranslational modifications, including S‐glutathionylation, S‐nitrosylation, acetylation and phosphorylation, have been reported in both, class I HDACs, including HDAC1, HDAC2 and HDAC3, and class II HDACs such as HDAC4 and HDAC5 (Figure 5). In general, these modifications reduce enzymic activity or impair binding to targets or other regulatory protein complexes, thus leading to an open chromatin state (Rajendrasozhan et al., 2008; Cyr and Domann, 2011). In addition, lipid peroxides or 4‐hydroxynonenal can lead to tyrosine nitration or alkylation/carbonylation of HDAC1, HDAC2 and HDAC3 or to casein kinase 2 (CK2)‐mediated phosphorylation of HDAC2. This results in ubiquitination and proteasomal degradation and loss of HDAC function, increased acetylation of histones H3 and H4 in macrophages and other pro‐inflammatory cells and the release of proinflammatory cytokines (Adenuga et al., 2009; Osoata et al., 2009; Doyle and Fitzpatrick, 2010). Nitrated HDAC2 was also found in peripheral lung tissues of patients with chronic obstructive pulmonary disease (Osoata et al., 2009).
Figure 5.
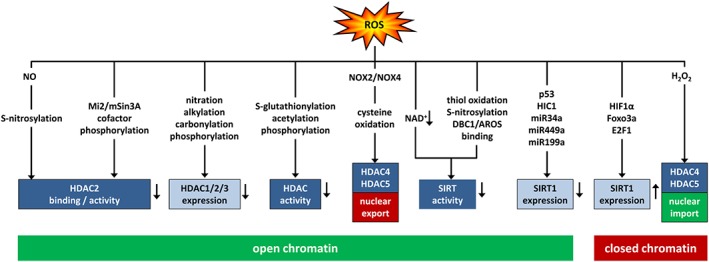
ROS shape histone acetylation by modulating histone deacetylases. ROS can affect histone acetylation by differentially modulating HDACs of class I (HDAC1/2/3), class II (HDAC4/5) and class III (SIRT). This can occur by affecting their activity or binding affinity, their expression or their nuclear localization. ROS can decrease expression or activity of class I HDACs due to posttranslational modifications or modulation of the cofactor mi2/mSin3a leading to increased histone acetylation and open chromatin. Similarly, ROS can promote nuclear export of oxidized class II HDACs or decrease activity of class III HDACs due to decreased availability of the cofactor NAD+ or posttranslational modifications or decreased expression of SIRT1 due to transcriptional or miRNA‐mediated repression leading to open chromatin. ROS can increase expression of SIRT1 or nuclear import of class II HDACs leading to decreased histone acetylation and closed chromatin. DBC1, deleted in breast cancer 1; AROS, active regulator of SIRT1; HIC1, hypermethylated in cancer 1; miR, microRNA; FOXO3a, Forkhead box O3a.
The outcome of oxidant‐dependent posttranslational modifications also seems to be dependent on the cellular context. While in neurons, NO‐dependent S‐nitrosylation of HDAC2 at Cys262/274 impaired HDAC2 binding to target DNA sequences (Nott et al., 2013), it reduced HDAC2 enzymic activity in C2C12 myoblasts (Colussi et al., 2008).
Oxidative stress has also been shown to lead to hypophosphorylation of an HDAC2 corepressor complex, Mi2/mSin3A, and may thereby reduce HDAC2 activity (Rajendrasozhan et al., 2008). In a mouse model of acute doxorubicin‐induced cardiotoxicity, increased levels of ROS associated with the NOX activator Rac1 decreased activity of HDACs, resulting in increased p53 acetylation associated with histone H2 phosphorylation (H2AX), cardiomyocyte death and cardiac dysfunction (Ma et al., 2013).
On the contrary, mitochondrial ROS have been reported to promote nuclear accumulation and activity of HDAC3 via c‐Src in LPS‐stimulated cardiomyocytes (Zhu et al., 2010), and HDAC2 activity was increased in an ROS‐dependent manner in kidneys of diabetic rats and obese mice as well as in H2O2 treated kidney cells (Li et al., 2016).
In contrast to class I HDACs, class II HDACs have a lower deacetylase activity, are expressed mainly in non‐proliferating cells and are regulated by nuclear‐cytoplasm shuttling, which derepresses gene expression (Yang et al., 2015). ROS‐induced oxidation of Cys667/669 in HDAC4 and Cys274/276 in its co‐regulator, DnaJb5, increased nuclear export of HDAC4, thus promoting NFAT‐regulated gene expression and cardiac hypertrophy (Ago et al., 2008) (Figure 5). NOX4 has been suggested as a source of ROS promoting oxidation and HDAC4 nuclear exit in mouse heart. Concomitantly, mice with heart‐specific NOX4 deficiency were protected against pressure overload‐induced cardiac hypertrophy (Matsushima and Sadoshima, 2015). However, a different study reported that deficiency of NOX4 leads to cardiac hypertrophy (Zhang et al., 2010). Thus, more detailed investigations are needed to elucidate the involvement of NOX4 in cardiac epigenetic processes. In the working skeletal muscle, NOX2‐dependent ROS generation has been reported to lead to nuclear efflux of HDAC4 and HDAC5 and muscle remodelling (Liu et al., 2012). Similarly, ROS increased nuclear exit of HDAC4/5 in angiotensin‐II‐induced vascular remodelling and cerebral ischaemic injury (Wang et al., 2014; Zhao et al., 2016) (Figure 5).
In contrast, treatment of neurons with H2O2 promoted HDAC4 translocation from the cytoplasm to the nucleus, leading to decreased PPARγ transcription, thus rendering neurons more vulnerable to H2O2 insult (Zhao et al., 2016). In models of redox‐associated pulmonary hypertension, increased nuclear accumulation of HDAC4 and HDAC5 was associated with decreased levels of miR‐424 and miR‐503, two miRNAs involved in maintenance of pulmonary vascular homeostasis, while application of a class II HDAC inhibitor prevented these responses (Kim et al., 2015b) (Figure 5). Increased levels of HDAC4/5 in response to ROS have also been shown in inflamed vessels contributing to hypertension and in fibrotic diabetic kidneys (Usui et al., 2012; Yan and Marsden, 2015; Li et al., 2016) while in ischaemic brain tissues decreased expression of HDAC4/5 has been observed, which could be restored by applying the antioxidant apocynin (Zhao et al., 2016).
Histone deacetylation: ROS effects on class III HDACs/sirtuins
While activation of class I/II HDACs does not require additional cofactors, the SIRT class III HDACs require NAD+, thus making this enzyme sensitive to metabolic and redox changes, and to a hub, which transmits metabolic alterations or cellular stress signals via modification of histones (and other proteins) to changes in gene expression (Hwang et al., 2013).
Oxidative stress has been related to reduced cellular levels of NAD+ and thus decreased SIRT1 activity in aged rat or human tissues (Braidy et al., 2011; Yoshino et al., 2011), but also in myocardial infarction and ischaemia/reperfusion (Matsushima and Sadoshima, 2015; Yamamoto et al., 2016) (Figure 5). Conditions of oxidative stress can activate PARPs, for example, to repair DNA, which will consume NAD+ on the expense of SIRT activity (Canto et al., 2013). In contrast, caloric restriction, an intervention shown to increase life span and to prevent metabolic syndrome, decreases oxidative stress which leads to increased NAD+ levels and SIRT3 activity and consequently to enhanced SOD2 activity and improved mitochondrial function (Qiu et al., 2010).
SIRT proteins further contain a highly conserved zinc tetra‐thiolate motif in the deacetylase domain, which is important for its activity. ROS or NO donors can induce thiol oxidation or S‐nitrosylation of these motifs in SIRT1 or SIRT3, leading to decreased deacetylation of target genes, such as endothelial NOS, and subsequently to endothelial dysfunction (Hwang et al., 2013; Santos et al., 2016) (Figure 5). Inhibition of SIRT1 has also been shown to increase expression of the NOX subunits p22phox and NOX4 and vascular superoxide production, which can further contribute to endothelial dysfunction (Zarzuelo et al., 2013). In line, SIRT1 levels are decreased in atherosclerosis in human and mouse models and have been associated with increased DNA damage, apoptosis and medial degeneration (Gorenne et al., 2013).
Similar to other HDACs, SIRTs can also be posttranslationally modified by ROS outside the zinc‐thiolate motif, leading to inactivation or proteasomal degradation (Hwang et al., 2013; Santos et al., 2016) (Figure 5). For example, oxidative stress decreased SIRT1 levels or activity in models of doxorubicin‐induced cardiotoxicity, oxidant stress‐induced diabetic retinopathy, myocardial infarction, stroke or in a setting of metabolic syndrome (de Kreutzenberg et al., 2010; Cattelan et al., 2015; Kim et al., 2015a; Ruan et al., 2015; Zhao et al., 2016)http://www.ncbi.nlm.nih.gov/pubmed/25863291. Finally, ROS can also alter SIRT binding to the regulatory proteins DBC1 and AROS (Santos et al., 2016), or lead to cytoplasmic sequestration and localization into caveolae, thus inhibiting deacetylase activity and promoting premature senescence (Volonte et al., 2015) (Figure 5).
Interestingly, while hypoxia decreased SIRT1 activity, attributable to decreased NAD+ levels, SIRT1 expression was increased dependent on HIF1α (Salminen et al., 2016) (Figure 5). Such a mechanism might aim to compensate decreased SIRT1 activity, not only under hypoxia but also in response to ROS, given that ROS can up‐regulate HIF1α (Bonello et al., 2007; Diebold et al., 2010a). In line, ROS have been shown to up‐regulate SIRT1 and SIRT2 expression and activity in vascular cells resulting in deacetylation of many genes involved in the cellular redox response, such as p53, FOXO3a, SOD2, GPX1, PPARγ coactivator 1‐α or NFκB (Santos et al., 2016). SIRT1 was also up‐regulated in several models of cardiac hypertrophy, heart failure and in aged hearts while it was either down‐ or up‐regulated in cardiac ischaemia/reperfusion (Matsushima and Sadoshima, 2015; Yamamoto et al., 2016). Of note, transcription of SIRT1 is reciprocally controlled by its targets p53 and FOXO3a. While p300/CBP‐dependent activation of p53 represses SIRT1 gene expression, activated FOXO3a can remove p53 from the SIRT1 promoter, thus activating SIRT1 transcription (Nemoto et al., 2004)http://dx.doi.org/10.1126/science.1101731 (Figure 5). As ROS generated by NOX4 can activate FOXO3a in vascular cells (Diebold et al., 2011), this mechanism might contribute to increased SIRT1 expression in ROS‐stimulated vascular cells (Santos et al., 2016). Furthermore, the transcription factors HIC1 and E2F1 have been identified to either repress or induce, respectively, SIRT1 transcription, under conditions of oxidative stress (Hwang et al., 2013) (Figure 5). Further, miR34a and miR449a, which are p53 and E2F1 transcriptional targets, respectively, and miR‐199a, which decreases HIF1α, have been shown to inhibit SIRT expression (Figure 5) (Rajendran et al., 2011). This tight regulatory network of ROS‐sensitive transcription factors and miRNAs (see below) might allow the fine‐tuning of SIRT expression dependent on the levels of ROS, the cell type and the stimulatory context and might counteract ROS‐mediated loss of SIRT activity. While less is known about expression regulation of other class HDACs, one might speculate that similar mechanisms might apply, thus explaining some seemingly contradictory observations.
Collectively, however, there is strong evidence that ROS are able to modulate chromatin accessibility by affecting the acetylation state of histones, via multiple modifications of HDAC expression and activity, which seem to have increasing importance in the cardiovascular system.
Non‐coding RNA transcripts and ROS
In recent years, evidence has been provided that ncRNAs, in particular miRNAs, but more recently also lncRNAs, play an important role in cardiovascular development and diseases (see Uchida and Dimmeler, 2015; Frank et al., 2016; Keating et al., 2016), while there is also evidence that miRNAs are critical regulators of the cellular stress response and thus responsive to ROS (Holoch and Moazed, 2015; Mikhed et al., 2015).
Various miRNAs are regulated by ROS (He and Jiang, 2016), among them several miRNAs, such as miR‐9, miR‐21, miR‐200 and miR‐210, which themselves can regulate ROS levels and thus have been termed ‘redoximiRs’ (Lin et al., 2009; Jajoo et al., 2013). Interestingly, the amount and time of exposure to ROS are important and can result in either up‐ or down‐regulation of miRNAs, as described for miR‐1 and its target myocardin in cardiomyocytes. Subsequently, in a model of chronic cardiac pressure overload, a miR‐1 mimic attenuated cardiac hypertrophy by suppressing increased myocardin, while application of anti‐miR‐1 ameliorated cardiac dysfunction upon acute myocardial infarction (Lee et al., 2015a).
Despite the complex interactions between ROS metabolism and miRNA levels, there seems to be a considerable overlap between ROS‐regulated miRNAs and miRNAs involved in cardiovascular pathologies. For example, a literature survey described that all 12 miRNAs, which have a confirmed role in atrial fibrillation, are modulated by ROS in cardiomyocytes or vascular cells, and target genes involved in electrical and/or structural cardiac remodelling (Lee et al., 2014). On an experimental level, a global miRNA profiling study in developing hearts showed that SOD1 overexpression could restore all miRNAs significantly altered by maternal pregestational diabetes mellitus, suggesting that oxidative stress might be responsible for dysregulation of miRNAs targeting cardiac development related pathways in offspring of diabetic mothers (Dong et al., 2016).
Mechanistically, ROS have been shown to be involved in various steps in miRNA biogenesis (He and Jiang, 2016). ROS can affect miRNA maturation by down‐regulating Dicer and modifying the argonaute RISC catalytic component 2 (Emde and Hornstein, 2014). During miRNA maturation, ROS can selectively stabilize the inactive, usually degraded miRNA star strand (miRNA*) of specific miRNAs (Bartel, 2009), leading, for example, to modulation of the NFκB pathway in oxidative stress‐responsive macrophages (Thulasingam et al., 2011).
Furthermore, ROS‐sensitive transcription factors such as NFκB, p53, Nrf2 or HIF1α can mediate ROS regulation of miRNA expression (Singh et al., 2013; Greco et al., 2014; Frank et al., 2016). In addition, ROS can act on miRNA genes by modulating epigenetic regulation such as DNA methylation or histone acetylation. For example, ROS increased DNMT1‐dependent methylation of miR‐199 and miR‐125 (He et al., 2012), thus allowing up‐regulation of their target HIF1α. Decreased levels of miR‐199 and increased levels of HIF1α are frequently found in cardiac ischaemia (Greco et al., 2014), while miR‐125 has been related to cardiac fibrosis (Nagpal et al., 2016). In contrast, ROS‐mediated inhibition of HDAC2 increased the levels of miR‐466 h‐5p, leading to apoptosis in hyperglycaemic conditions (Druz et al., 2012). Interestingly, genome‐wide profiling of mice with increased ROS load due to deficiency of GPX1 and 2 showed an unexpected high proportion of differentially hydroxymethylated miRNA‐encoding sequences. Of the major 20 miRNA genes found to be differentially hydroxymethylated, the majority of miRNAs, which lost 5hmC marks under oxidative stress, were related to cardiac dysfunction and cardiovascular diseases, and many targeted pathways were involved in oxidative stress (Delatte et al., 2015). Although these miRNAs did not include ‘redoximiRs’, they contained for example increased levels of miR‐137 or miR‐449a, which can target the HDM KDM5B (Denis et al., 2016) or the HDAC SIRT1 (Yamakuchi, 2012) respectively. They also contained decreased levels of miR‐30a, which has been shown to be down‐regulated in diabetic hearts (Costantino et al., 2016) and in doxorubicin‐induced cardiomyopathy where it was inversely correlated with ROS levels (Roca‐Alonso et al., 2015).
Increasing evidence suggests that not only miRNAs but also lncRNAs are related to increased ROS load. In a model of obesity and lipotoxicity, ROS were shown to increase the lncRNA gadd7 leading to further ROS‐induced endoplasmic reticulum stress and cell death (Wang et al., 2016). In a model of cardiac reperfusion injury, increased ROS load was associated with reduced expression of the lncRNA, UCA1, which was negatively correlated with p27 expression and mediated ROS‐induced cardiomyocyte apoptosis (Liu et al., 2015c). Another lncRNA termed necrosis‐related factor (NRF) was shown to mediate H2O2‐induced necrosis in cardiomyocytes by binding to miR‐873 and downregulating its targets, the RIPK1/3 family of kinases (Wang et al., 2016).
Intriguingly, it was recently shown that acute exposure to non‐toxic levels of H2O2 resulted in the down‐regulation of many protein‐coding genes as well as in the generation of thousands of lncRNAs, mostly with promoter‐associated antisense lncRNAs transcripts. These lncRNAs, which are associated with polysomes, might represent a novel component of the (early) cellular stress responses (Giannakakis et al., 2015). Further studies will have to show the implications of these lncRNAs for the acute and possibly also prolonged cardiovascular response to ROS‐related stress conditions.
ROS and ATP‐dependent chromatin remodelling
Members of ATP‐dependent chromatin remodelling complexes contribute to cardiac development and function, congenital heart defects and/or cardiovascular diseases such as cardiomyopathy, heart failure, atherosclerosis and pulmonary hypertension (Clapier and Cairns, 2009; Han et al., 2011; Vallaster et al., 2012; Rosa‐Garrido et al., 2013; Hota and Bruneau, 2016). Increasing evidence suggests that ROS can affect the function of these complexes at different levels.
In particular, the ATPase Brahma (BRM)‐related gene 1 (BRG1) of the Brg1‐associated factors (BAF) complex, the vertebrate homologue of the SWI/SNF complex (Han et al., 2011), has been implicated in the response to oxidative stress since it is recruited by Nrf2 to the promoter of haem oxygenase‐1 (HO‐1), where it facilitates Z‐DNA formation and RNA polymerase II‐dependent expression of this antioxidant gene (Zhang et al., 2006). In the diabetic heart, ROS down‐regulated BRG1 concomitant with decreased HO‐1 expression (Gao et al., 2016) while treatment with antioxidants restored BRG1 levels and improved the function of the diabetic heart (Xu et al., 2013). Similar to antioxidants, adiponectin reduced cardiac oxidative stress, ameliorated cardiomyocyte hypertrophy and prevented left ventricular dysfunction in diabetes by concomitantly activating Nrf2 and BRG1 to facilitate HO‐1 induction (Li et al., 2015). Furthermore, in hypertrophic hearts, induction of BRG1 recruited DNMT3 and the HMT G9a/G9a‐like protein to the Myh6 gene. This led to Myh6 gene silencing, impaired cardiomyocyte contraction and cardiomyopathy (Han et al., 2016). Similarly, in cardiac hypertrophy, CK2 phosphorylated the BAF45c subunit double plant homeodomain (PHD) finger protein 3a (DPF3a), thereby releasing it from the transcriptional repressor HEY. This allowed BRG1 to bind to DPF3a and to promote expression of HEY genomic targets such as natriuretic peptide precursor A and GATA binding protein 4 (Cui et al., 2016). While a role of ROS has not been explicitly investigated in this setting, ROS have been shown to activate CK2 leading to Nrf2 activation and HO‐1 induction (Kim et al., 2012) further suggesting an important role of BRG1 in the adaptation to oxidative stress in the heart.
Genetic and tissue‐specific effects may also account for differently affected ATP‐dependent chromatin remodelling effects. For example, in Fanconi anaemia, oxidative stress promoted the formation of a complex containing Fanconi anaemia proteins and BRG1, which protected the promoters of antioxidant genes like GPX1 and thioredoxin reductase 1 from oxidative damage (Du et al., 2012). Interestingly in neuroblastoma cells, the alternate ATPase of the BAF complex, BRM, was transcriptionally down‐regulated upon H2O2 application (Fontana et al., 2016). Further, BRM was up‐regulated by NO due to HDAC2 nitrosylation and subsequent derepression of the BRM promoter in the developing brain (Nott et al., 2013). Thus, ROS appear to affect ATP‐dependent chromatin remodelling by recruitment and transcriptional regulation of BAF complex ATPases.
ROS have been further shown to up‐regulate the Cockayne syndrome group B protein (CSB), which belongs to the SWI/SNF family, and to increase its interaction with the long‐range chromatin structural regulator CCCTC‐binding factor, thus increasing promoter occupancy of genes involved in RNA and protein homeostasis, energy control, OXPHOS and ROS production in the mitochondria (Lake et al., 2016). Mutations in the CSB gene result in Cockayne syndrome, which is associated with numerous developmental and neurological defects, sun sensitivity, premature ageing and increased sensitivity to oxidative stress.
Moreover, members of the SWI/SNF complex have been shown to increase transcription of the redox‐sensitive Caenorhabditis elegans transcription factor DAF‐16/FOXO leading to increased stress resistance and longevity (Riedel et al., 2013). Since mammalian FOXO transcription factors are essentially involved in controlling redox homeostasis and are sensitive to ROS in the vasculature (Diebold et al., 2011), it will be interesting to see whether such a pathway also plays a role in mammalian adaptation to ROS.
SWI/SNF components such as BAF57 were also found associated with the promoters of HIF‐α genes thus enhancing HIF transactivation under hypoxia (Kenneth et al., 2009). Further studies are required to elucidate whether SWI/SNF also plays a role in the regulation of HIF‐α by ROS. On the contrary, the active compounds of ISWI, hSNF2h and hSNF2l, have been found to depress HIF activity under hypoxia by increasing the levels of the HIF‐associated dioxygenase FIH, thereby acting as a hypoxia survival factor (Melvin et al., 2011). Since depletion of SNF2h leads to very early embryonic lethality, its role in cardiac development is not yet understood (Han et al., 2011). However, recent data indicate that in response to H2O2, SNF2h interacts with the XRCC1 protein, phosphorylated by CK2, to initiate DNA repair (Kubota et al., 2016). In summary, there is increasing evidence that ROS can affect ATP‐dependent chromatin remodelling complexes. Further studies are required to elucidate underlying mechanisms and their implications for the cardiovascular system.
Epigenetic mechanisms, mitochondria and ROS: old organelles, new players?
While epigenetic mechanisms have been widely related to nDNA, recent years have provided new evidence that mitochondria can also become targets of epigenetic mechanisms (see Chinnery et al., 2012; Manev and Dzitoyeva, 2013; Castegna et al., 2015; van der Wijst and Rots, 2015).
Mitochondria maintain their own genome that is encoded in the mtDNA. In mammals, the mt genome consists of 13 protein encoding genes, two ribosomal RNA and 22 transfer RNA (tRNA) genes. The 13 mitochondrial encoded proteins are essential subunits of the OXPHOS complexes I, III, IV and V. According to conservative estimates, mitochondrial functions require about 1100 or more proteins, which are encoded in genes of the nDNA and imported into mitochondria (Calvo et al., 2016).
Any damage to subunits of the OXPHOS system but also to the plethora of additional mitochondrial functions, such as the Krebs cycle, protein biosynthesis and fatty acid metabolism, leading to badly or non‐functioning mitochondria is generally referred to as mitochondrial dysfunction (Murphy, 2009; Brand and Nicholls, 2011). Mitochondrial dysfunction will affect ROS generation, but also mitochondrial metabolites like ATP, NADH/NAD+, SAM, acetyl‐CoA or the Krebs cycle intermediates succinate and fumarate with consequences for nuclear epigenetics (Wallace and Fan, 2010; Tretter et al., 2016) (see also above).
Epigenetic mechanisms involving mitochondria can roughly be divided into five classes: (i) nDNA methylation influencing nuclear‐encoded mitochondrial genes/protein‐expression; (ii) mtDNA copy‐numbers influencing nDNA methylation patterns; (iii) mitochondrial haplotypes influencing nDNA methylation patterns; (iv) intra‐mitochondrial epigenetics through mtDNA methylation; and (v) miRNA and lncRNA influencing mitochondrial metabolism (see Smiraglia et al., 2008; Bellizzi et al., 2012; Kelly et al., 2013; Castegna et al., 2015; Lee et al., 2015b).
Since a large number of mitochondrial proteins are encoded by nDNA, virtually all previously discussed aspects of epigenetic control mechanisms (DNA methylation, chromatin remodelling, histone modifications, ncRNA expression) can be involved in epigenetic regulation of mitochondrial functions (Wallace and Fan, 2010) and might thus be sensitive to ROS.
In mammalian mitochondria, mtDNA is always present as DNA‐protein complexes, so called nucleoids, and is packed mainly by the major DNA‐binding and bending protein mitochondrial transcription factor A (TFAM). TFAM is nDNA encoded, and emerging evidence suggests that its expression is regulated by nDNA promoter methylation (Li and Yang, 2015). Being the major DNA binding protein in mitochondria, TFAM also has a strong influence on mtDNA copy numbers (Pohjoismaki et al., 2006). Redox agents such as H2S decreased TFAM promoter methylation, most likely by inhibiting DNMT3A expression, thereby maintaining mtDNA copy numbers in vascular cells (Li and Yang, 2015). Several other studies have reported that ROS can elevate mtDNA copy numbers (Lee and Wei, 2005; Hori et al., 2009; Marine et al., 2014), although this effect may be time‐ and dose‐dependent (Al‐Kafaji et al., 2016) and possibly also dependent on the type of redox modulation. For example, in mice exposed to cigarette smoke extracts, TFAM promoter methylation was increased resulting in strongly reduced TFAM expression and endothelial cell apoptosis (Zhang et al., 2013). Although mtDNA copy numbers were not specifically assessed, the expression of mtDNA‐encoded COX2 was reduced, hinting to a reduction of mtDNA copy numbers. Accordingly, in patients with chronic obstructive pulmonary disease, a disorder associated with oxidative stress, leukocyte mtDNA copy numbers were lower than in healthy controls (Liu et al., 2015a).
Lowered mtDNA copy numbers are associated with the risk of heart failure in humans (Huang et al., 2016). In line with these pieces of evidence for a link between ROS, mtDNA copy numbers and cardiovascular disease are the recent findings in a mouse model that over‐expression of TFAM or the mtDNA helicase Twinkle leads to increased mtDNA copy numbers and prevents increased ROS generation, thus acting cardio‐protectively in mice that otherwise suffered from volume‐overload‐induced heart failure (Ikeda et al., 2015).
Recently, it was shown that singlet oxygen can facilitate mtDNA replication involving not only TFAM but also mtDNA polymerase γ (PolG) (Zhou et al., 2015). PolG, which consists of one catalytic PolG1 and two regulatory PolG2 subunits, replicates mtDNA and, through its DNA‐synthesizing function, also influences mtDNA copy numbers. Also, a POLG1 mutation (Y955C) caused a reduction of mtDNA copy numbers, oxidative stress and cardiomyopathy in mice (Lewis et al., 2007). Further, mtDNA copy numbers and POLG1 expression levels inversely correlated with the POLG1 gene methylation status at exon 2 (Tewari et al., 2012; Lee et al., 2015b). Although these findings imply a link between ROS and POLG1 function, further studies are needed to show whether ROS indeed modulate methylation of POLG1.
As with nDNA, mtDNA methylation comprises also 5mC and is catalysed by mtDNMT1, which is a mitochondrially targeted variant of nuclear DNMT1 (Shock et al., 2011). DNMT3A was also described in mitochondria in neurons, but a mitochondrial targeting sequence has yet to be identified (Castegna et al., 2015). In conditions of redox stress, expression of mtDNMT1 was found increased via the transcription factors NRF1 and PGC1‐α, suggesting a regulatory role for mtDNMT1 during oxidative stress (Shock et al., 2011). Further, 5hmC was found in mtDNA, and TET1 and TET2 were identified in mitochondrial protein fractions indicating their significance for mtDNA modification (Bellizzi et al., 2013). More so, since a major substrate not only for TET protein function but also for JmjC KDMs is α‐ketoglutarate, two other carboxylic acids, succinate and fumarate, are competitive inhibitors of these enzymes (see above). All three are intermediates of the mitochondrial Krebs cycle, and succinate is a potent substrate of mitochondrial ROS generation. Thus, mitochondrial (dys)function can affect the methylation status of mtDNA as well as nDNA not only via increased ROS levels but also by modulating Krebs cycle intermediates or the allocation of SAM to either the nucleus or mitochondria, thus modulating the function of DNMTs and TET proteins (Castegna et al., 2015).
Moreover, mtDNA shows non‐random methylation patterns, especially in a non‐coding control region known as the D‐loop in particular at sites where transcription and replication elements are located (Shock et al., 2011). Hypermethylation of this region has been found in diabetic patients leading to aberrant transcription of mitochondrial genes and increased generation of superoxide promoting diabetic retinopathy (Mishra and Kowluru, 2015).
Interestingly, it was recently reported that the nuclear MYST family acetyl transferase MOF is also present in mitochondria where it binds mtDNA and regulates expression of OXPHOS genes from both, nDNA and mtDNA (Chatterjee et al., 2016). MOF deficiency resulted not only in mitochondrial dysfunction but also in hypertrophic cardiomyopathy and cardiac failure (Chatterjee et al., 2016). Consistent with these data, MOF expression was down‐regulated in human failing hearts and in murine cardiac hypertrophy while overexpression of MOF protected against cardiac hypertrophy in response to pressure overload (Qiao et al., 2014). While no direct evidence for ROS regulation of MOF has been provided in this study, findings that MOF can control the expression of catalase, SOD2 and NOX4 (Qiao et al., 2014; Sanders et al., 2015) suggest that MOF is a dual transcriptional regulator of nuclear and mitochondrial genomes connecting epigenetics and ROS metabolism in the heart.
Like with nDNA, also non‐coding regulatory mitochondrial RNAs have been described, which undergo cell‐ and tissue‐specific regulation. While there are reports that miRNAs are encoded in the nucleus, processed in the cytoplasm and then translocated into mitochondria (Das et al., 2014), it is not clear whether they can also be (partially) processed in the mitochondria (Rackham et al., 2011). These mitomiRs target either mitochondrial gene functions encoded by nDNA or even mtDNA encoded genes (Latronico and Condorelli, 2012). For example, miRNA‐181c targeted mt‐encoded COX1, but increased the expression of mt‐encoded COX2 and COX3, leading to remodelling and dysfunction of complex IV, with increased mitochondrial respiration and ROS generation. Interestingly, systemic application of miRNA‐181c resulted in cardiac dysfunction (Das et al., 2014). On the other hand, the redoximiR miRNA‐210 (see above) was associated with mitochondria where it down‐regulates expression of Fe‐S‐cluster assembly factors (ISCU1 and ISCU2), thereby decreasing mt oxygen consumption and increasing ETC activity, mt ROS and lactate production (Chan et al., 2009). Thus, mitochondrial ROS generation not only affects epigenetic regulatory mechanisms in particular in the nDNA but also is itself regulated by (ROS‐regulated) epigenetic mechanisms affecting nDNA and mtDNA.
While mitochondrial (dys‐)function is an important determinant in cardiovascular diseases closely associated with ROS dysbalance (Ballinger, 2005), the involvement of mitochondrial epigenetics in the course of cardiovascular diseases is just starting to be elucidated. An initial association study demonstrates that mtDNA methylation was elevated in mitochondrial genes encoding proteins of complexes IV, V and tRNA‐Leu1 in thrombocytes from patients with cardiovascular diseases (Baccarelli and Byun, 2015). Thus, more studies are needed to progress in this widely unexplored field.
Conclusion
ROS and oxidative stress are emerging as novel players, shaping the epigenetic landscape of the entire genome. The increasing amount of data unravelling epigenetic mechanisms underlying the pathophysiology of cardiovascular diseases allows us, now, to link these mechanisms with the concepts of cardiovascular ROS signalling (Figure 2). Although a number of processes are just beginning to be understood, it is obvious that the action of a wide variety of enzymes and mitochondrial metabolism can contribute to ROS generation in response to environmental and nutritional changes associated with increased cardiovascular risk. ROS, in turn, can by modulating DNA and histone modifications, ncRNA transcripts and chromatin remodelling affect the epigenetic landscape with important implications for gene expression in the nucleus and mitochondria and consequences for cardiovascular pathophysiology (Figure 2).
Novel techniques and more comprehensive mechanistic analyses are necessary to solve conflicting data, which may derive from technical limitations in older studies where no differentiation between methylation and hydroxymethylation could be made. This might also help to further elucidate the complex interactions between different epigenetic modifications in DNA and chromatin and to increase the understanding regarding the role of lncRNAs in ROS signalling in the cardiovascular system. Finally, the increasing amount of data pointing to epigenetic alterations in mitochondria will need additional mechanistic studies to characterize their crosstalk with epigenetic phenomena in the nucleus and to fully assess their effects on the cardiovascular system.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors dedicate this work to Prof. Lorenz Poellinger, a member of the Epiros network, who passed away too early. The authors are grateful to all researchers who contributed to the field, and we apologize to all those whose work could not be cited due to space limitations. The work of the authors was supported by grants from BMBF (Epiros), DZHK (German Centre for Vascular Research), DFG (GO709/4‐5), the Academy of Finland, Jane and Aatos Erkko Foundation, the Estonian Research Council grant PUT (PUT610) and the European Cooperation in Science and Technology organization (COST Action BM1203/EU‐ROS).
Kietzmann, T. , Petry, A. , Shvetsova, A. , Gerhold, J. M. , and Görlach, A. (2017) The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. British Journal of Pharmacology, 174: 1533–1554. doi: 10.1111/bph.13792.
References
- Abdulla A, Zhao X, Yang F (2013). Natural polyphenols inhibit lysine‐specific demethylase‐1 in vitro. J Biochem Pharmacol Res 1: 56–63. [PMC free article] [PubMed] [Google Scholar]
- Adenuga D, Yao H, March TH, Seagrave J, Rahman I (2009). Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am J Respir Cell Mol Biol 40: 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afanas'ev I (2014). New nucleophilic mechanisms of ros‐dependent epigenetic modifications: comparison of aging and cancer. Aging Dis 5: 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD et al. (2008). A redox‐dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 133: 978–993. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5734–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Kafaji G, Sabry MA, Skrypnyk C (2016). Time‐course effect of high‐glucose‐induced reactive oxygen species on mitochondrial biogenesis and function in human renal mesangial cells. Cell Biol Int 40: 36–48. [DOI] [PubMed] [Google Scholar]
- Allis CD, Jenuwein T (2016). The molecular hallmarks of epigenetic control. Nat Rev Genet 17: 487–500. [DOI] [PubMed] [Google Scholar]
- Al‐Sawaf O, Clarner T, Fragoulis A, Kan YW, Pufe T, Streetz K et al. (2015). Nrf2 in health and disease: current and future clinical implications. Clin Sci 129: 989–999. [DOI] [PubMed] [Google Scholar]
- Al Taleb Z, Petry A, Chi TF, Mennerich D, Gorlach A, Dimova EY et al. (2016). Differential transcriptional regulation of hypoxia‐inducible factor‐1alpha by arsenite under normoxia and hypoxia: involvement of Nrf2. J Mol Med 94: 1153–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba X, Aguilera‐Aguirre L, Rashid QT, Bacsi A, Radak Z, Sur S et al. (2014). The role of 8‐oxoguanine DNA glycosylase‐1 in inflammation. Int J Mol Sci 15: 16975–16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarelli A, Wright R, Bollati V, Litonjua A, Zanobetti A, Tarantini L et al. (2010). Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology 21: 819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarelli AA, Byun HM (2015). Platelet mitochondrial DNA methylation: a potential new marker of cardiovascular disease. Clin Epigenetics 7: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S, Hurley LH, Neidle S (2011). Targeting G‐quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov 10: 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger SW (2005). Mitochondrial dysfunction in cardiovascular disease. Free Radic Biol Med 38: 1278–1295. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T (2005). Reversing histone methylation. Nature 436: 1103–1106. [DOI] [PubMed] [Google Scholar]
- Bartel DP (2009). MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartling TR, Subbaram S, Clark RR, Chandrasekaran A, Kar S, Melendez JA (2014). Redox‐sensitive gene‐regulatory events controlling aberrant matrix metalloproteinase‐1 expression. Free Radic Biol Med 74: 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BelAiba RS, Djordjevic T, Bonello S, Flugel D, Hess J, Kietzmann T et al. (2004). Redox‐sensitive regulation of the HIF pathway under non‐hypoxic conditions in pulmonary artery smooth muscle cells. Biol Chem 385: 249–257. [DOI] [PubMed] [Google Scholar]
- Bellizzi D, D'Aquila P, Giordano M, Montesanto A, Passarino G (2012). Global DNA methylation levels are modulated by mitochondrial DNA variants. Epigenomics 4: 17–27. [DOI] [PubMed] [Google Scholar]
- Bellizzi D, D'Aquila P, Scafone T, Giordano M, Riso V, Riccio A et al. (2013). The control region of mitochondrial DNA shows an unusual CpG and non‐CpG methylation pattern. DNA Res 20: 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop T, Ratcliffe PJ (2015). HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ Res 117: 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C et al. (2007). Reactive oxygen species activate the HIF‐1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol 27: 755–761. [DOI] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan‐Ling T, Poljak A, Grant R (2011). Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One 6: e19194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Branco MR, Ficz G, Reik W (2012). Uncovering the role of 5‐hydroxymethylcytosine in the epigenome. Nat Rev Genet 13: 7–13. [DOI] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG (2011). Assessing mitochondrial dysfunction in cells. Biochem J 435: 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DI, Griendling KK (2015). Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 116: 531–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne MM, Murphy RT, Ryan AW (2014). Epigenetic modulation in the treatment of atherosclerotic disease. Front Genet 5: 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, Mootha VK (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44 (Database issue): D1251–D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Sauve AA, Bai P (2013). Crosstalk between poly(ADP‐ribose) polymerase and sirtuin enzymes. Mol Aspects Med 34: 1168–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castegna A, Iacobazzi V, Infantino V (2015). The mitochondrial side of epigenetics. Physiol Genomics 47: 299–307. [DOI] [PubMed] [Google Scholar]
- Cattelan A, Ceolotto G, Bova S, Albiero M, Kuppusamy M, De Martin S et al. (2015). NAD(+)‐dependent SIRT1 deactivation has a key role on ischemia‐reperfusion‐induced apoptosis. Vascul Pharmacol 70: 35–44. [DOI] [PubMed] [Google Scholar]
- Chalupsky K, Kracun D, Kanchev I, Bertram K, Gorlach A (2015). Folic acid promotes recycling of tetrahydrobiopterin and protects against hypoxia‐induced pulmonary hypertension by recoupling endothelial nitric oxide synthase. Antioxid Redox Signal 23: 1076–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J (2009). MicroRNA‐210 controls mitochondrial metabolism during hypoxia by repressing the iron‐sulfur cluster assembly proteins ISCU1/2. Cell Metab 10: 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Seyfferth J, Lucci J, Gilsbach R, Preissl S, Bottinger L et al. (2016). MOF acetyl transferase regulates transcription and respiration in mitochondria. Cell 167: 722–738 e723. [DOI] [PubMed] [Google Scholar]
- Chen J, Guo L, Zhang L, Wu H, Yang J, Liu H et al. (2013). Vitamin C modulates TET1 function during somatic cell reprogramming. Nat Genet 45: 1504–1509. [DOI] [PubMed] [Google Scholar]
- Chervona Y, Costa M (2012). The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic Biol Med 53: 1041–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, Elliott HR, Hudson G, Samuels DC, Relton CL (2012). Epigenetics, epidemiology and mitochondrial DNA diseases. Int J Epidemiol 41: 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm NC, Henderson ML, Selvamani A, Park MJ, Dindot S, Miranda RC et al. (2015). Histone methylation patterns in astrocytes are influenced by age following ischemia. Epigenetics 10: 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury M, Park PH, Jackson D, Shukla SD (2010). Evidence for the role of oxidative stress in the acetylation of histone H3 by ethanol in rat hepatocytes. Alcohol 44: 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR (2009). The biology of chromatin remodeling complexes. Annu Rev Biochem 78: 273–304. [DOI] [PubMed] [Google Scholar]
- Colussi C, Mozzetta C, Gurtner A, Illi B, Rosati J, Straino S et al. (2008). HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci U S A 105: 19183–19187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino S, Paneni F, Luscher TF, Cosentino F (2016). MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur Heart J 37: 572–576. [DOI] [PubMed] [Google Scholar]
- Coulter JB, O'Driscoll CM, Bressler JP (2013). Hydroquinone increases 5‐hydroxymethylcytosine formation through ten eleven translocation 1 (TET1) 5‐methylcytosine dioxygenase. J Biol Chem 288: 28792–28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Schlesinger J, Schoenhals S, Tonjes M, Dunkel I, Meierhofer D et al. (2016). Phosphorylation of the chromatin remodeling factor DPF3a induces cardiac hypertrophy through releasing HEY repressors from DNA. Nucleic Acids Res 44: 2538–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr AR, Domann FE (2011). The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal 15: 551–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr AR, Hitchler MJ, Domann FE (2013). Regulation of SOD2 in cancer by histone modifications and CpG methylation: closing the loop between redox biology and epigenetics. Antioxid Redox Signal 18: 1946–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Bedja D, Campbell N, Dunkerly B, Chenna V, Maitra A et al. (2014). miR‐181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo . PLoS One 9: e96820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kreutzenberg SV, Ceolotto G, Papparella I, Bortoluzzi A, Semplicini A, Dalla Man C et al. (2010). Downregulation of the longevity‐associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes 59: 1006–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delatte B, Jeschke J, Defrance M, Bachman M, Creppe C, Calonne E et al. (2015). Genome‐wide hydroxymethylcytosine pattern changes in response to oxidative stress. Sci Rep 5: 12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado‐Olguin P, Dang LT, He D, Thomas S, Chi L, Sukonnik T et al. (2014). Ezh2‐mediated repression of a transcriptional pathway upstream of Mmp9 maintains integrity of the developing vasculature. Development 141: 4610–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis H, Van Grembergen O, Delatte B, Dedeurwaerder S, Putmans P, Calonne E et al. (2016). MicroRNAs regulate KDM5 histone demethylases in breast cancer cells. Mol Biosyst 12: 404–413. [DOI] [PubMed] [Google Scholar]
- Di Minno A, Turnu L, Porro B, Squellerio I, Cavalca V, Tremoli E et al. (2016). 8‐Hydroxy‐2‐deoxyguanosine levels and cardiovascular disease: a systematic review and meta‐analysis of the literature. Antioxid Redox Signal 24: 548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold I, Flugel D, Becht S, Belaiba RS, Bonello S, Hess J et al. (2010a). The hypoxia‐inducible factor‐2alpha is stabilized by oxidative stress involving NOX4. Antioxid Redox Signal 13: 425–436. [DOI] [PubMed] [Google Scholar]
- Diebold I, Petry A, Burger M, Hess J, Gorlach A (2011). NOX4 mediates activation of FoxO3a and matrix metalloproteinase‐2 expression by urotensin‐II. Mol Biol Cell 22: 4424–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold I, Petry A, Hess J, Gorlach A (2010b). The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia‐inducible factor‐1. Mol Biol Cell 21: 2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold I, Petry A, Sabrane K, Djordjevic T, Hess J, Gorlach A (2012). The HIF1 target gene NOX2 promotes angiogenesis through urotensin‐II. J Cell Sci 125 (Pt 4): 956–964. [DOI] [PubMed] [Google Scholar]
- Djordjevic T, Pogrebniak A, BelAiba RS, Bonello S, Wotzlaw C, Acker H et al. (2005). The expression of the NADPH oxidase subunit p22phox is regulated by a redox‐sensitive pathway in endothelial cells. Free Radic Biol Med 38: 616–630. [DOI] [PubMed] [Google Scholar]
- Dong D, Zhang Y, Reece EA, Wang L, Harman CR, Yang P (2016). MicroRNA expression profiling and functional annotation analysis of their targets modulated by oxidative stress during embryonic heart development in diabetic mice. Reprod Toxicol 65: 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle K, Fitzpatrick FA (2010). Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J Biol Chem 285: 17417–17424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druz A, Betenbaugh M, Shiloach J (2012). Glucose depletion activates mmu‐miR‐466h‐5p expression through oxidative stress and inhibition of histone deacetylation. Nucleic Acids Res 40: 7291–7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Rani R, Sipple J, Schick J, Myers KC, Mehta P et al. (2012). The FA pathway counteracts oxidative stress through selective protection of antioxidant defense gene promoters. Blood 119: 4142–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn J, Simmons R, Thabet S, Jo H (2015). The role of epigenetics in the endothelial cell shear stress response and atherosclerosis. Int J Biochem Cell Biol 67: 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emde A, Hornstein E (2014). miRNAs at the interface of cellular stress and disease. EMBO J 33: 1428–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana GA, Rigamonti A, Lenzken SC, Filosa G, Alvarez R, Calogero R et al. (2016). Oxidative stress controls the choice of alternative last exons via a Brahma‐BRCA1‐CstF pathway. Nucleic Acids Res 45: 902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Aguirre A, Hescheler J, Kurian L (2016). A lncRNA perspective into (Re)building the heart. Front Cell Dev Biol 4: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin S, Kimball T, Rasmussen TL, Rosa‐Garrido M, Chen H, Tran T et al. (2016). The chromatin‐binding protein Smyd1 restricts adult mammalian heart growth. Am J Physiol Heart Circ Physiol 311: H1234–H1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friso S, Carvajal CA, Fardella CE, Olivieri O (2015). Epigenetics and arterial hypertension: the challenge of emerging evidence. Transl Res 165: 154–165. [DOI] [PubMed] [Google Scholar]
- Galligan JJ, Rose KL, Beavers WN, Hill S, Tallman KA, Tansey WP et al. (2014). Stable histone adduction by 4‐oxo‐2‐nonenal: a potential link between oxidative stress and epigenetics. J Am Chem Soc 136: 11864–11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Yang Z, Shi R, Xu D, Li H, Xia Z et al. (2016). Diabetes blocks the cardioprotective effects of sevoflurane postconditioning by impairing Nrf2/Brg1/HO‐1 signaling. Eur J Pharmacol 779: 111–121. [DOI] [PubMed] [Google Scholar]
- Garcia‐Gimenez JL, Olaso G, Hake SB, Bonisch C, Wiedemann SM, Markovic J et al. (2013). Histone h3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxid Redox Signal 19: 1305–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakakis A, Zhang J, Jenjaroenpun P, Nama S, Zainolabidin N, Aau MY et al. (2015). Contrasting expression patterns of coding and noncoding parts of the human genome upon oxidative stress. Sci Rep 5: 9737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour PS, Rahman I, Donaldson K, MacNee W (2003). Histone acetylation regulates epithelial IL‐8 release mediated by oxidative stress from environmental particles. Am J Physiol Lung Cell Mol Physiol 284: L533–L540. [DOI] [PubMed] [Google Scholar]
- Gorenne I, Kumar S, Gray K, Figg N, Yu H, Mercer J et al. (2013). Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation 127: 386–396. [DOI] [PubMed] [Google Scholar]
- Gorlach A, Bonello S (2008). The cross‐talk between NF‐kappaB and HIF‐1: further evidence for a significant liaison. Biochem J 412: e17–e19. [DOI] [PubMed] [Google Scholar]
- Gorlach A, Dimova EY, Petry A, Martinez‐Ruiz A, Hernansanz‐Agustin P, Rolo AP et al. (2015). Reactive oxygen species, nutrition, hypoxia and diseases: problems solved? Redox Biol 6: 372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco CM, Kunderfranco P, Rubino M, Larcher V, Carullo P, Anselmo A et al. (2016). DNA hydroxymethylation controls cardiomyocyte gene expression in development and hypertrophy. Nat Commun 7: 12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco S, Gaetano C, Martelli F (2014). HypoxamiR regulation and function in ischemic cardiovascular diseases. Antioxid Redox Signal 21: 1202–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greissel A, Culmes M, Napieralski R, Wagner E, Gebhard H, Schmitt M et al. (2015). Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thromb Haemost 114: 390–402. [DOI] [PubMed] [Google Scholar]
- Gupta J, Tikoo K (2012). Involvement of insulin‐induced reversible chromatin remodeling in altering the expression of oxidative stress‐responsive genes under hyperglycemia in 3T3‐L1 preadipocytes. Gene 504: 181–191. [DOI] [PubMed] [Google Scholar]
- Han P, Hang CT, Yang J, Chang CP (2011). Chromatin remodeling in cardiovascular development and physiology. Circ Res 108: 378–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Li W, Yang J, Shang C, Lin CH, Cheng W et al. (2016). Epigenetic response to environmental stress: assembly of BRG1‐G9a/GLP‐DNMT3 repressive chromatin complex on Myh6 promoter in pathologically stressed hearts. Biochim Biophys Acta 1863 (7 Pt B): 1772–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes P, Knaus UG (2013). Balancing reactive oxygen species in the epigenome: NADPH oxidases as target and perpetrator. Antioxid Redox Signal 18: 1937–1945. [DOI] [PubMed] [Google Scholar]
- He C, Larson‐Casey JL, Gu L, Ryan AJ, Murthy S, Carter AB (2016). Cu,Zn‐SOD‐mediated redox regulation of Jmjd3 modulates macrophage polarization and pulmonary fibrosis. Am J Respir Cell Mol Biol 55: 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Jiang BH (2016). Interplay between reactive oxygen species and microRNAs in cancer. Curr Pharmacol Rep 2: 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Xu Q, Jing Y, Agani F, Qian X, Carpenter R et al. (2012). Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR‐199a/125b and DNA methylation. EMBO Rep 13: 1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickok JR, Vasudevan D, Antholine WE, Thomas DD (2013). Nitric oxide modifies global histone methylation by inhibiting Jumonji C domain‐containing demethylases. J Biol Chem 288: 16004–16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, Moazed D (2015). RNA‐mediated epigenetic regulation of gene expression. Nat Rev Genet 16: 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori A, Yoshida M, Shibata T, Ling F (2009). Reactive oxygen species regulate DNA copy number in isolated yeast mitochondria by triggering recombination‐mediated replication. Nucleic Acids Res 37: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hota SK, Bruneau BG (2016). ATP‐dependent chromatin remodeling during mammalian development. Development 143: 2882–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang BW, Ray PD, Iwasaki K, Tsuji Y (2013). Transcriptional regulation of the human ferritin gene by coordinated regulation of Nrf2 and protein arginine methyltransferases PRMT1 and PRMT4. FASEB J 27: 3763–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Tan L, Shen R, Zhang L, Zuo H, Wang DW (2016). Decreased peripheral mitochondrial DNA copy number is associated with the risk of heart failure and long‐term outcomes. Medicine 95: e3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Tan L, Xue Z, Cang J, Wang H (2012). Reduction of DNA hydroxymethylation in the mouse kidney insulted by ischemia reperfusion. Biochem Biophys Res Commun 422: 697–702. [DOI] [PubMed] [Google Scholar]
- Hussain T, Tan B, Yin Y, Blachier F, Tossou MC, Rahu N (2016). Oxidative stress and inflammation: what polyphenols can do for us? Oxid Med Cell Longev 2016 7432797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JW, Yao H, Caito S, Sundar IK, Rahman I (2013). Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic Biol Med 61: 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Ide T, Fujino T, Arai S, Saku K, Kakino T et al. (2015). Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS One 10: e0119687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jajoo S, Mukherjea D, Kaur T, Sheehan KE, Sheth S, Borse V et al. (2013). Essential role of NADPH oxidase‐dependent reactive oxygen species generation in regulating microRNA‐21 expression and function in prostate cancer. Antioxid Redox Signal 19: 1863–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalani A, Kamat PK, Tyagi N (2015). Diabetic stroke severity: epigenetic remodeling and neuronal, glial, and vascular dysfunction. Diabetes 64: 4260–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating ST, El‐Osta A (2013). Transcriptional regulation by the Set7 lysine methyltransferase. Epigenetics 8: 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating ST, Plutzky J, El‐Osta A (2016). Epigenetic changes in diabetes and cardiovascular risk. Circ Res 118: 1706–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RD, Rodda AE, Dickinson A, Mahmud A, Nefzger CM, Lee W et al. (2013). Mitochondrial DNA haplotypes define gene expression patterns in pluripotent and differentiating embryonic stem cells. Stem Cells 31: 703–716. [DOI] [PubMed] [Google Scholar]
- Kenneth NS, Mudie S, van Uden P, Rocha S (2009). SWI/SNF regulates the cellular response to hypoxia. J Biol Chem 284: 4123–4131. [DOI] [PubMed] [Google Scholar]
- Khan MA, Alam K, Dixit K, Rizvi MM (2016). Role of peroxynitrite induced structural changes on H2B histone by physicochemical method. Int J Biol Macromol 82: 31–38. [DOI] [PubMed] [Google Scholar]
- Kikuchi H, Kuribayashi F, Kiwaki N, Takami Y, Nakayama T (2011a). GCN5 regulates the superoxide‐generating system in leukocytes via controlling gp91‐phox gene expression. J Immunol 186: 3015–3022. [DOI] [PubMed] [Google Scholar]
- Kikuchi H, Kuribayashi F, Takami Y, Imajoh‐Ohmi S, Nakayama T (2011b). GCN5 regulates the activation of PI3K/Akt survival pathway in B cells exposed to oxidative stress via controlling gene expressions of Syk and Btk. Biochem Biophys Res Commun 405: 657–661. [DOI] [PubMed] [Google Scholar]
- Kim DI, Park MJ, Choi JH, Kim IS, Han HJ, Yoon KC et al. (2015a). PRMT1 and PRMT4 regulate oxidative stress‐induced retinal pigment epithelial cell damage in SIRT1‐dependent and SIRT1‐independent manners. Oxid Med Cell Longev 2015: 617919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GH, Ryan JJ, Archer SL (2013). The role of redox signaling in epigenetics and cardiovascular disease. Antioxid Redox Signal 18: 1920–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hwangbo C, Hu X, Kang Y, Papangeli I, Mehrotra D et al. (2015b). Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 131: 190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KM, Song JD, Chung HT, Park YC (2012). Protein kinase CK2 mediates peroxynitrite‐induced heme oxygenase‐1 expression in articular chondrocytes. Int J Mol Med 29: 1039–1044. [DOI] [PubMed] [Google Scholar]
- Kloypan C, Srisa‐art M, Mutirangura A, Boonla C (2015). LINE‐1 hypomethylation induced by reactive oxygen species is mediated via depletion of S‐adenosylmethionine. Cell Biochem Funct 33: 375–385. [DOI] [PubMed] [Google Scholar]
- Kreuz S, Fischle W (2016). Oxidative stress signaling to chromatin in health and disease. Epigenomics 8: 843–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroese LJ, Scheffer PG (2014). 8‐hydroxy‐2′‐deoxyguanosine and cardiovascular disease: a systematic review. Curr Atheroscler Rep 16: 452. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Shimizu S, Yasuhira S, Horiuchi S (2016). SNF2H interacts with XRCC1 and is involved in repair of H2O2‐induced DNA damage. DNA Repair 43: 69–77. [DOI] [PubMed] [Google Scholar]
- Lake RJ, Boetefuer EL, Won KJ, Fan HY (2016). The CSB chromatin remodeler and CTCF architectural protein cooperate in response to oxidative stress. Nucleic Acids Res 44: 2125–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latronico MV, Condorelli G (2012). The might of microRNA in mitochondria. Circ Res 110: 1540–1542. [DOI] [PubMed] [Google Scholar]
- Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG Jr et al. (2016). Fumarate and succinate regulate expression of hypoxia‐inducible genes via TET enzymes. J Biol Chem 291: 4256–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DD, Fujimori DG (2012). Protein and nucleic acid methylating enzymes: mechanisms and regulation. Curr Opin Chem Biol 16: 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Wei YH (2005). Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol 37: 822–834. [DOI] [PubMed] [Google Scholar]
- Lee S, Choi E, Cha MJ, Hwang KC (2014). Looking into a conceptual framework of ROS‐miRNA‐atrial fibrillation. Int J Mol Sci 15: 21754–21776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lim S, Ham O, Lee SY, Lee CY, Park JH et al. (2015a). ROS‐mediated bidirectional regulation of miRNA results in distinct pathologic heart conditions. Biochem Biophys Res Commun 465: 349–355. [DOI] [PubMed] [Google Scholar]
- Lee W, Johnson J, Gough DJ, Donoghue J, Cagnone GL, Vaghjiani V et al. (2015b). Mitochondrial DNA copy number is regulated by DNA methylation and demethylation of POLGA in stem and cancer cells and their differentiated progeny. Cell Death Dis 6: e1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrke M, Greif M, Broedl UC, Lebherz C, Laubender RP, Becker A et al. (2009). MMP‐1 serum levels predict coronary atherosclerosis in humans. Cardiovasc Diabetol 8: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis W, Day BJ, Kohler JJ, Hosseini SH, Chan SSL, Green E et al. (2007). Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase γ *. Lab Invest 87: 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL (2007). The role of chromatin during transcription. Cell 128: 707–719. [DOI] [PubMed] [Google Scholar]
- Li H, Yao W, Irwin MG, Wang T, Wang S, Zhang L et al. (2015). Adiponectin ameliorates hyperglycemia‐induced cardiac hypertrophy and dysfunction by concomitantly activating Nrf2 and Brg1. Free Radic Biol Med 84: 311–321. [DOI] [PubMed] [Google Scholar]
- Li J, Braganza A, Sobol RW (2013). Base excision repair facilitates a functional relationship between guanine oxidation and histone demethylation. Antioxid Redox Signal 18: 2429–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Qiu P, Chen B, Lu Y, Wu K, Thakur C et al. (2014). Reactive oxygen species contribute to arsenic‐induced EZH2 phosphorylation in human bronchial epithelial cells and lung cancer cells. Toxicol Appl Pharmacol 276: 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Yang G (2015). Hydrogen sulfide maintains mitochondrial DNA replication via demethylation of TFAM. Antioxid Redox Signal 23: 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Li C, Sun G (2016). Histone acetylation and its modifiers in the pathogenesis of diabetic nephropathy. J Diabetes Res 2016 4065382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SO, Gu JM, Kim MS, Kim HS, Park YN, Park CK et al. (2008). Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E‐cadherin promoter. Gastroenterology 135: 2128–2140, 2140 e2121‐2128. [DOI] [PubMed] [Google Scholar]
- Lin Y, Liu X, Cheng Y, Yang J, Huo Y, Zhang C (2009). Involvement of microRNAs in hydrogen peroxide‐mediated gene regulation and cellular injury response in vascular smooth muscle cells. J Biol Chem 284: 7903–7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G et al. (2013). Ten‐eleven translocation‐2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation 128: 2047–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SF, Kuo HC, Tseng CW, Huang HT, Chen YC, Tseng CC et al. (2015a). Leukocyte mitochondrial DNA copy number is associated with chronic obstructive pulmonary disease. PLoS One 10: e0138716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Wu C, Jain MR, Nagarajan N, Yan L, Dai H et al. (2015b). Master redox regulator Trx1 upregulates SMYD1 & modulates lysine methylation. Biochim Biophys Acta 1854: 1816–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Hernandez‐Ochoa EO, Randall WR, Schneider MF (2012). NOX2‐dependent ROS is required for HDAC5 nuclear efflux and contributes to HDAC4 nuclear efflux during intense repetitive activity of fast skeletal muscle fibers. Am J Physiol Cell Physiol 303: C334–C347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou D, Li G, Ming X, Tu Y, Tian J et al. (2015c). Long non coding RNA‐UCA1 contributes to cardiomyocyte apoptosis by suppression of p27 expression. Cell Physiol Biochem 35: 1986–1998. [DOI] [PubMed] [Google Scholar]
- Ma J, Wang Y, Zheng D, Wei M, Xu H, Peng T (2013). Rac1 signalling mediates doxorubicin‐induced cardiotoxicity through both reactive oxygen species‐dependent and ‐independent pathways. Cardiovasc Res 97: 77–87. [DOI] [PubMed] [Google Scholar]
- Madugundu GS, Cadet J, Wagner JR (2014). Hydroxyl‐radical‐induced oxidation of 5‐methylcytosine in isolated and cellular DNA. Nucleic Acids Res 42: 7450–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manea SA, Constantin A, Manda G, Sasson S, Manea A (2015). Regulation of Nox enzymes expression in vascular pathophysiology: focusing on transcription factors and epigenetic mechanisms. Redox Biol 5: 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manev H, Dzitoyeva S (2013). Progress in mitochondrial epigenetics. Biomol Concepts 4: 381–389. [DOI] [PubMed] [Google Scholar]
- Mariani CJ, Vasanthakumar A, Madzo J, Yesilkanal A, Bhagat T, Yu Y et al. (2014). TET1‐mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep 7: 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine A, Krager KJ, Aykin‐Burns N, Macmillan‐Crow LA (2014). Peroxynitrite induced mitochondrial biogenesis following MnSOD knockdown in normal rat kidney (NRK) cells. Redox Biol 2: 348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima S, Sadoshima J (2015). The role of sirtuins in cardiac disease. Am J Physiol Heart Circ Physiol 309: H1375–H1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melvin A, Mudie S, Rocha S (2011). The chromatin remodeler ISWI regulates the cellular response to hypoxia: role of FIH. Mol Biol Cell 22: 4171–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D et al. (2015). Histone methylation dynamics and gene regulation occur through the sensing of one‐carbon metabolism. Cell Metab 22: 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Z, He Y, Xin N, Sun M, Chen L, Lin L et al. (2015). Altering 5‐hydroxymethylcytosine modification impacts ischemic brain injury. Hum Mol Genet 24: 5855–5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhed Y, Gorlach A, Knaus UG, Daiber A (2015). Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol 5: 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra M, Kowluru RA (2015). Epigenetic modification of mitochondrial DNA in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci 56: 5133–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina‐Serrano D, Schiza V, Kirmizis A (2013). Cross‐talk among epigenetic modifications: lessons from histone arginine methylation. Biochem Soc Trans 41: 751–759. [DOI] [PubMed] [Google Scholar]
- Monfort A, Wutz A (2013). Breathing‐in epigenetic change with vitamin C. EMBO Rep 14: 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales Y, Nitzel DV, Price OM, Gui S, Li J, Qu J et al. (2015). Redox control of protein arginine methyltransferase 1 (PRMT1) activity. J Biol Chem 290: 14915–14926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CJ, Lopez AD (2013). Measuring the global burden of disease. N Engl J Med 369: 448–457. [DOI] [PubMed] [Google Scholar]
- Nagayoshi Y, Kawano H, Hokamaki J, Uemura T, Soejima H, Kaikita K et al. (2009). Differences in oxidative stress markers based on the aetiology of heart failure: comparison of oxidative stress in patients with and without coronary artery disease. Free Radic Res 43: 1159–1166. [DOI] [PubMed] [Google Scholar]
- Nagpal V, Rai R, Place AT, Murphy SB, Verma SK, Ghosh AK et al. (2016). MiR‐125b Is critical for fibroblast‐to‐myofibroblast transition and cardiac fibrosis. Circulation 133: 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T (2004). Nutrient availability regulates SIRT1 through a forkhead‐dependent pathway. Science 306: 2105–2108. [DOI] [PubMed] [Google Scholar]
- Niu Y, DesMarais TL, Tong Z, Yao Y, Costa M (2015). Oxidative stress alters global histone modification and DNA methylation. Free Radic Biol Med 82: 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Nitarska J, Veenvliet JV, Schacke S, Derijck AA, Sirko P et al. (2013). S‐nitrosylation of HDAC2 regulates the expression of the chromatin‐remodeling factor Brm during radial neuron migration. Proc Natl Acad Sci U S A 110: 3113–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW et al. (2011). Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell 20: 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osoata GO, Yamamura S, Ito M, Vuppusetty C, Adcock IM, Barnes PJ et al. (2009). Nitration of distinct tyrosine residues causes inactivation of histone deacetylase 2. Biochem Biophys Res Commun 384: 366–371. [DOI] [PubMed] [Google Scholar]
- Pan L, Zhu B, Hao W, Zeng X, Vlahopoulos SA, Hazra TK et al. (2016). Oxidized guanine base lesions function in 8‐oxoguanine DNA glycosylase‐1‐mediated epigenetic regulation of nuclear factor kappaB‐driven gene expression. J Biol Chem 291: 25553–25566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M et al. (2012). Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res 111: 278–289. [DOI] [PubMed] [Google Scholar]
- Park J, Park JW, Oh H, Maria FS, Kang J, Tian X (2016). Gene‐specific assessment of guanine oxidation as an epigenetic modulator for cardiac specification of mouse embryonic stem cells. PLoS One 11: e0155792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastukh V, Roberts JT, Clark DW, Bardwell GC, Patel M, Al‐Mehdi AB et al. (2015). An oxidative DNA “damage” and repair mechanism localized in the VEGF promoter is important for hypoxia‐induced VEGF mRNA expression. Am J Physiol Lung Cell Mol Physiol 309: L1367–L1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson AJ, Xiao D, Xiong F, Dixon B, Zhang L (2012). Hypoxia‐derived oxidative stress mediates epigenetic repression of PKCepsilon gene in foetal rat hearts. Cardiovasc Res 93: 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petry A, Weitnauer M, Gorlach A (2010). Receptor activation of NADPH oxidases. Antioxid Redox Signal 13: 467–487. [DOI] [PubMed] [Google Scholar]
- Pohjoismaki JL, Wanrooij S, Hyvarinen AK, Goffart S, Holt IJ, Spelbrink JN et al. (2006). Alterations to the expression level of mitochondrial transcription factor A, TFAM, modify the mode of mitochondrial DNA replication in cultured human cells. Nucleic Acids Res 34: 5815–5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao W, Zhang W, Gai Y, Zhao L, Fan J (2014). The histone acetyltransferase MOF overexpression blunts cardiac hypertrophy by targeting ROS in mice. Biochem Biophys Res Commun 448: 379–384. [DOI] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D (2010). Calorie restriction reduces oxidative stress by SIRT3‐mediated SOD2 activation. Cell Metab 12: 662–667. [DOI] [PubMed] [Google Scholar]
- Rackham O, Shearwood AM, Mercer TR, Davies SM, Mattick JS, Filipovska A (2011). Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear‐encoded proteins. RNA 17: 2085–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekar P, O'Neill CL, Eeles L, Stitt AW, Medina RJ (2015). Epigenetic changes in endothelial progenitors as a possible cellular basis for glycemic memory in diabetic vascular complications. J Diabetes Res 2015: 436879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran R, Garva R, Krstic‐Demonacos M, Demonacos C (2011). Sirtuins: molecular traffic lights in the crossroad of oxidative stress, chromatin remodeling, and transcription. J Biomed Biotechnol 2011: 368276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendrasozhan S, Yang SR, Edirisinghe I, Yao H, Adenuga D, Rahman I (2008). Deacetylases and NF‐kappaB in redox regulation of cigarette smoke‐induced lung inflammation: epigenetics in pathogenesis of COPD. Antioxid Redox Signal 10: 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen KD, Helin K (2016). Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 30: 733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel CG, Dowen RH, Lourenco GF, Kirienko NV, Heimbucher T, West JA et al. (2013). DAF‐16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat Cell Biol 15: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca‐Alonso L, Castellano L, Mills A, Dabrowska AF, Sikkel MB, Pellegrino L et al. (2015). Myocardial MiR‐30 downregulation triggered by doxorubicin drives alterations in beta‐adrenergic signaling and enhances apoptosis. Cell Death Dis 6: e1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa‐Garrido M, Karbassi E, Monte E, Vondriska TM (2013). Regulation of chromatin structure in the cardiovascular system. Circ J 77: 1389–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Y, Dong C, Patel J, Duan C, Wang X, Wu X et al. (2015). SIRT1 suppresses doxorubicin‐induced cardiotoxicity by regulating the oxidative stress and p38MAPK pathways. Cell Physiol Biochem 35: 1116–1124. [DOI] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K, Kauppinen A (2016). Hypoxia‐inducible histone lysine demethylases: impact on the aging process and age‐related diseases. Aging Dis 7: 180–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samoylenko A, Hossain JA, Mennerich D, Kellokumpu S, Hiltunen JK, Kietzmann T (2013). Nutritional countermeasures targeting reactive oxygen species in cancer: from mechanisms to biomarkers and clinical evidence. Antioxid Redox Signal 19: 2157–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders YY, Liu H, Liu G, Thannickal VJ (2015). Epigenetic mechanisms regulate NADPH oxidase‐4 expression in cellular senescence. Free Radic Biol Med 79: 197–205. [DOI] [PubMed] [Google Scholar]
- Santos L, Escande C, Denicola A (2016). Potential modulation of sirtuins by oxidative stress. Oxid Med Cell Longev 2016 9831825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM (2011). DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A 108: 3630–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Happel C, Manna SK, Acquaah‐Mensah G, Carrerero J, Kumar S et al. (2013). Transcription factor NRF2 regulates miR‐1 and miR‐206 to drive tumorigenesis. J Clin Invest 123: 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiraglia DJ, Kulawiec M, Bistulfi GL, Gupta SG, Singh KK (2008). A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol Ther 7: 1182–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44 (D1): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Song MM, Wei B, Gao Q, Li L, Yao B et al. (2016). 5‐Hydroxymethylcytosine‐mediated alteration of transposon activity associated with the exposure to adverse in utero environments in human. Hum Mol Genet 25: 2208–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar IK, Yao H, Rahman I (2013). Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking‐related diseases. Antioxid Redox Signal 18: 1956–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tausendschon M, Dehne N, Brune B (2011). Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine 53: 256–262. [DOI] [PubMed] [Google Scholar]
- Tewari S, Zhong Q, Santos JM, Kowluru RA (2012). Mitochondria DNA replication and DNA methylation in the metabolic memory associated with continued progression of diabetic retinopathy. Invest Ophthalmol Vis Sci 53: 4881–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thienpont B, Steinbacher J, Zhao H, D'Anna F, Kuchnio A, Ploumakis A et al. (2016). Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 537: 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thulasingam S, Massilamany C, Gangaplara A, Dai H, Yarbaeva S, Subramaniam S et al. (2011). miR‐27b*, an oxidative stress‐responsive microRNA modulates nuclear factor‐kB pathway in RAW 264.7 cells. Mol Cell Biochem 352: 181–188. [DOI] [PubMed] [Google Scholar]
- Tomita K, Barnes PJ, Adcock IM (2003). The effect of oxidative stress on histone acetylation and IL‐8 release. Biochem Biophys Res Commun 301: 572–577. [DOI] [PubMed] [Google Scholar]
- Tretter L, Patocs A, Chinopoulos C (2016). Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim Biophys Acta 1857: 1086–1101. [DOI] [PubMed] [Google Scholar]
- Tsai YP, Chen HF, Chen SY, Cheng WC, Wang HW, Shen ZJ et al. (2014). TET1 regulates hypoxia‐induced epithelial‐mesenchymal transition by acting as a co‐activator. Genome Biol 15: 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S, Dimmeler S (2015). Long noncoding RNAs in cardiovascular diseases. Circ Res 116: 737–750. [DOI] [PubMed] [Google Scholar]
- Usui T, Okada M, Mizuno W, Oda M, Ide N, Morita T et al. (2012). HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am J Physiol Heart Circ Physiol 302: H1894–H1904. [DOI] [PubMed] [Google Scholar]
- Vallaster M, Vallaster CD, Wu SM (2012). Epigenetic mechanisms in cardiac development and disease. Acta Biochim Biophys Sin 44: 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wijst MG, Rots MG (2015). Mitochondrial epigenetics: an overlooked layer of regulation? Trends Genet 31: 353–356. [DOI] [PubMed] [Google Scholar]
- Volonte D, Zou H, Bartholomew JN, Liu Z, Morel PA, Galbiati F (2015). Oxidative stress‐induced inhibition of Sirt1 by caveolin‐1 promotes p53‐dependent premature senescence and stimulates the secretion of interleukin 6 (IL‐6). J Biol Chem 290: 4202–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeling LA, Ions LJ, Escolme SM, Cockell SJ, Su T, Dey M et al. (2015). SIRT1 affects DNA methylation of polycomb group protein target genes, a hotspot of the epigenetic shift observed in ageing. Hum Genomics 9: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Fan W (2010). Energetics, epigenetics, mitochondrial genetics. Mitochondrion 10: 12–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walport LJ, Hopkinson RJ, Chowdhury R, Schiller R, Ge W, Kawamura A et al. (2016). Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat Commun 7: 11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Liu F, Liu CY, An T, Zhang J, Zhou LY et al. (2016). The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR‐873. Cell Death Differ 23: 1394–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Vatamaniuk MZ, Roneker CA, Pepper MP, Hu LG, Simmons RA et al. (2011a). Knockouts of SOD1 and GPX1 exert different impacts on murine islet function and pancreatic integrity. Antioxid Redox Signal 14: 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Miao X, Liu Y, Li F, Liu Q, Sun J et al. (2014). Dysregulation of histone acetyltransferases and deacetylases in cardiovascular diseases. Oxid Med Cell Longev 2014: 641979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yang D, Zhang X, Li T, Li J, Tang Y et al. (2011b). Hypoxia‐induced down‐regulation of neprilysin by histone modification in mouse primary cortical and hippocampal neurons. PLoS One 6: e19229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapenaar H, Dekker FJ (2016). Histone acetyltransferases: challenges in targeting bi‐substrate enzymes. Clin Epigenetics 8: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CJ, Collier P, Tea I, Neary R, Watson JA, Robinson C et al. (2014). Hypoxia‐induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast‐like phenotype. Hum Mol Genet 23: 2176–2188. [DOI] [PubMed] [Google Scholar]
- Wu MZ, Chen SF, Nieh S, Benner C, Ger LP, Jan CI et al. (2015). Hypoxia drives breast tumor malignancy through a TET‐TNFalpha‐p38‐MAPK signaling axis. Cancer Res 75: 3912–3924. [DOI] [PubMed] [Google Scholar]
- Wu X, Sun J, Zhang X, Li X, Liu Z, Yang Q et al. (2014). Epigenetic signature of chronic cerebral hypoperfusion and beneficial effects of S‐adenosylmethionine in rats. Mol Neurobiol 50: 839–851. [DOI] [PubMed] [Google Scholar]
- Xiao D, Dasgupta C, Chen M, Zhang K, Buchholz J, Xu Z et al. (2014). Inhibition of DNA methylation reverses norepinephrine‐induced cardiac hypertrophy in rats. Cardiovasc Res 101: 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong F, Xiao D, Zhang L (2012). Norepinephrine causes epigenetic repression of PKCepsilon gene in rodent hearts by activating Nox1‐dependent reactive oxygen species production. FASEB J 26: 2753–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lei S, Liu Y, Gao X, Irwin MG, Xia ZY et al. (2013). Antioxidant N‐acetylcysteine attenuates the reduction of Brg1 protein expression in the myocardium of type 1 diabetic rats. J Diabetes Res 2013: 716219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakuchi M (2012). MicroRNA regulation of SIRT1. Front Physiol 3: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Tamaki K, Shirakawa K, Ito K, Yan X, Katsumata Y et al. (2016). Cardiac Sirt1 mediates the cardioprotective effect of caloric restriction by suppressing local complement system activation after ischemia‐reperfusion. Am J Physiol Heart Circ Physiol 310: H1003–H1014. [DOI] [PubMed] [Google Scholar]
- Yan MS, Marsden PA (2015). Epigenetics in the vascular endothelium: looking from a different perspective in the epigenomics era. Arterioscler Thromb Vasc Biol 35: 2297–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Wang Q, Wang W, Zeng LF (2015). Histone deacetylases and cardiovascular cell lineage commitment. World J Stem Cells 7: 852–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S (2011). Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet‐ and age‐induced diabetes in mice. Cell Metab 14: 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Reddy MA, Deshpande S, Jia Y, Park JT, Lanting LL et al. (2016). Epigenetic histone modifications involved in profibrotic gene regulation by 12/15‐lipoxygenase and its oxidized lipid products in diabetic nephropathy. Antioxid Redox Signal 24: 361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarzuelo MJ, Lopez‐Sepulveda R, Sanchez M, Romero M, Gomez‐Guzman M, Ungvary Z et al. (2013). SIRT1 inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: implications for vascular aging. Biochem Pharmacol 85: 1288–1296. [DOI] [PubMed] [Google Scholar]
- Zhang H, Chen P, Zeng H, Zhang Y, Peng H, Chen Y et al. (2013). Protective effect of demethylation treatment on cigarette smoke extract‐induced mouse emphysema model. J Pharmacol Sci 123: 159–166. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ohta T, Maruyama A, Hosoya T, Nishikawa K, Maher JM et al. (2006). BRG1 interacts with Nrf2 to selectively mediate HO‐1 induction in response to oxidative stress. Mol Cell Biol 26: 7942–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M et al. (2010). NADPH oxidase‐4 mediates protection against chronic load‐induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A 107: 18121–18126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Shah AM (2014). ROS signalling between endothelial cells and cardiac cells. Cardiovasc Res 102: 249–257. [DOI] [PubMed] [Google Scholar]
- Zhang QJ, Chen HZ, Wang L, Liu DP, Hill JA, Liu ZP (2011). The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J Clin Invest 121: 2447–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QJ, Liu ZP (2015). Histone methylations in heart development, congenital and adult heart diseases. Epigenomics 7: 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Han Z, Ji X, Luo Y (2016). Epigenetic regulation of oxidative stress in ischemic stroke. Aging Dis 7: 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Agha G, Baccarelli AA (2016). The role of DNA methylation in cardiovascular risk and disease: methodological aspects, study design, and data analysis for epidemiological studies. Circ Res 118: 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Sun H, Ellen TP, Chen H, Costa M (2008). Arsenite alters global histone H3 methylation. Carcinogenesis 29: 1831–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang Y, Si J, Zhou R, Gan L, Di C et al. (2015). Laser controlled singlet oxygen generation in mitochondria to promote mitochondrial DNA replication in vitro. Sci Rep 5: 16925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Zhuang Z, Wang W, He L, Wu H, Cao Y et al. (2016). OGG1 is essential in oxidative stress induced DNA demethylation. Cell Signal 28: 1163–1171. [DOI] [PubMed] [Google Scholar]
- Zhu H, Shan L, Schiller PW, Mai A, Peng T (2010). Histone deacetylase‐3 activation promotes tumor necrosis factor‐alpha (TNF‐alpha) expression in cardiomyocytes during lipopolysaccharide stimulation. J Biol Chem 285: 9429–9436. [DOI] [PMC free article] [PubMed] [Google Scholar]