

Abstract
Purpose
The expanding number of targeted therapeutics for non-small cell lung cancer (NSCLC) necessitates real-time tumor genotyping, yet tissue biopsies are difficult to perform serially and often yield inadequate DNA for next-generation sequencing (NGS). We evaluated the feasibility of using cell-free circulating tumor DNA (ctDNA) NGS as a complement or alternative to tissue NGS.
Experimental Design
112 plasma samples obtained from a consecutive study of 102 prospectively enrolled patients with advanced NSCLC were subjected to ultra-deep sequencing of up to 70 genes and matched with tissue samples, when possible.
Results
We detected 275 alterations in 45 genes, and at least one alteration in the ctDNA for 86 of 102 patients (84%), with EGFR variants being most common. ctDNA NGS detected 50 driver and 12 resistance mutations, and mutations in 22 additional genes for which experimental therapies, including clinical trials, are available. While ctDNA NGS was completed for 102 consecutive patients, tissue sequencing was only successful for 50 patients (49%). Actionable EGFR mutations were detected in 24 tissue and 19 ctDNA samples, yielding concordance of 79%, with a shorter time interval between tissue and blood collection associated with increased concordance (p=0.038). ctDNA sequencing identified 8 patients harboring a resistance mutation who developed progressive disease while on targeted therapy, and for whom tissue sequencing wasn’t possible.
Conclusions
Therapeutically targetable driver and resistance mutations can be detected by ctDNA NGS, even when tissue is unavailable, thus allowing more accurate diagnosis, improved patient management, and serial sampling to monitor disease progression and clonal evolution.
Keywords: Next-generation sequencing, circulating tumor DNA, lung cancer, personalized medicine, liquid biopsy
INTRODUCTION
Tumor genotyping to identify actionable oncogenic driver mutations and mechanisms of resistance to targeted therapeutics has become increasingly important in the management of cancers. In non-small cell lung cancer (NSCLC), detection of activating epidermal growth factor receptor (EGFR) mutations, and anaplastic lymphoma kinase (ALK) fusions has expanded treatment options, and targeting these mutations has improved progression-free survival in patients with metastatic disease (1, 2). In addition, several other promising somatic genomic targets are recommended in the National Comprehensive Cancer Network (NCCN) NSCLC guidelines for existing targeted therapies including ROS1 and RET fusions, MET exon 14 skipping, and ERBB2 and BRAF mutations (3), as well as numerous other genetic alterations that are the focus of active clinical trials (4).
EGFR mutations are some of the most common variants detected in NSCLC, present in 10–40% of cases depending on patient demographics and smoking status (5, 6), with ALK rearrangements detected less frequently (1). The presence of a sensitizing EGFR mutation usually indicates a high likelihood of response to first-generation tyrosine kinase inhibitors (TKIs), but most patients will develop resistance within 12–24 months of treatment initiation (7–9). Mechanisms of primary and acquired resistance to EGFR-TKI therapy have been identified including amplifications in MET and ERBB2, deletions or point mutations in EGFR, KRAS, BRAF, and PIK3CA, and loss or inactivation of PTEN (10, 11). Multiple molecular resistance mechanisms to ALK-TKIs have also been identified (9). The accurate identification of such tumor molecular evolution has important clinical implications as second- and third-generation TKIs are now available for tumors harboring certain EGFR and ALK resistance mutations (12, 13).
Tumor tissue has been the preferred source for mutational analysis, and the NCCN guidelines now recommend repeat tissue biopsies to identify resistance mechanisms in patients whose cancers progress on first-line targeted therapeutics (14). However, tissue specimens may prove inadequate for testing in a significant proportion of cases. In addition, a small needle biopsy of a single lesion may fail to reflect the true underlying intra- and inter-tumor genetic heterogeneity (15–17). As a result, a non-invasive approach to accurately detect actionable driver and resistance mechanisms offers significant clinical utility.
There is growing interest in utilizing circulating tumor DNA (ctDNA) for the non-invasive molecular profiling of tumors. ctDNA consists of short double-stranded DNA fragments shed into the bloodstream by tumor cells undergoing apoptosis or necrosis (18, 19). ctDNA can be readily detected in advanced NSCLC patients, and several studies have shown that highly sensitive genotyping assays can detect mutations in ctDNA (20–26). Digital polymerase chain reaction assays have been developed with high sensitivity and specificity for detection of variants in ctDNA (24, 27), however, these approaches are typically limited to hotspot mutations in a few genes, and cannot interrogate the full spectrum of mutations that may emerge in the setting of acquired resistance during targeted therapy (28). Targeted next-generation sequencing (NGS) of ctDNA offers the ability to profile a much broader scope of genetic alterations on a single platform.
In this study, massively parallel digital sequencing of NSCLC patient plasma ctDNA was performed in a Clinical Laboratory Improvement Amendments (CLIA)-certified, College of American Pathology (CAP)-accredited laboratory (29, 30). The test detects single nucleotide variants in up to 70 genes, fusions in 6 genes, and insertion/deletions (indels) in 3 genes. We assessed the feasibility of using ctDNA NGS to identify actionable mutations in a cohort of NSCLC patients, including those for whom tissue sequencing could not be conducted. We also evaluated the concordance in genomic alterations and the ability to detect clinically actionable mutations between commercially available NGS gene-panels in paired tumor tissue biopsies and ctDNA.
MATERIALS AND METHODS
Patients
This single-center, observational study was conducted at the Hospital of the University of Pennsylvania between February 2015 and March 2016, enrolling a consecutive blood sample of 102 prospectively enrolled subjects. This included both new patients referred to our institution and existing patients. The clinical indications and inclusion criteria for this study were that the patient must have a diagnosis of NSCLC or suspected NSCLC (by pathology) seen in our Thoracic Oncology Group, and had blood samples sent for ctDNA NGS as part of their routine clinical care. This study did not require a specific course of treatment. Because our primary objective was to evaluate the feasibility of the ctDNA test for detecting actionable mutations, consecutive patients who fulfilled the inclusion criteria were enrolled, with a target sample size of 100 patients. Blood draws for commercial ctDNA NGS were ordered as clinically indicated by the primary oncologist. Clinical variables and results from solid tumor sequencing were determined by chart review. The study was approved by the University of Pennsylvania Internal Review Board (IRB).
Blood samples and circulating tumor DNA isolation and sequencing
Blood was collected in two 10 mL Streck tubes (Streck) and shipped overnight at ambient temperature to Guardant Health, Inc, a CLIA-certified, CAP-accredited laboratory facility. Cell-free DNA (cfDNA) was extracted from plasma and the amount of cfDNA in the sample cohort was quantified using electrophoretic separation in a massively parallel capillary array system allowing for post-extraction high-throughput, high-resolution fragment size-specific data acquisition for each sample processed. The cfDNA was then analyzed by paired-end sequencing by synthesis utilizing an Illumina Hi-Seq 2500 platform and hg19 as the reference genome as described (29). Digital sequences were reconstructed using Guardant Health’s proprietary bioinformatics algorithms, allowing the detection of 1–2 mutant fragments in 10 mL of blood with an analytic specificity greater than 99.9999%. The detection of somatic alterations in cfDNA was used to confirm the presence of ctDNA. Clinically significant somatic alterations were distinguished from germline polymorphisms by referencing multiple SNP databases and by their annotation in COSMIC (Catalogue of Somatic Mutations in Cancer; http://cancer.sanger.ac.uk/cosmic). Single nucleotide variants, indels, and fusions were quantitatively reported as the allelic fraction (AF), calculated as the total number of molecules with a given mutation divided by the total number of mutant plus wild-type molecules (29).
The current 70-gene Guardant360 panel (Supplemental Table 1) includes complete exons for 30 genes, and critical exons (those reported as having a somatic mutation in COSMIC) of 40 additional genes resulting in a 146,000 bp (146KB) target region. The platform detects fusions in 6 genes, and multiple indels in 3 genes. The average coverage depth was 10,000X. During this study, the Guardant platform expanded from a 68- to a 70-gene panel. This expansion added ERBB2 and MET indels, full exon coverage of RB1, and critical exon coverage of TSC1. Samples from 36 patients were sequenced on the 68-gene panel and samples from the remaining 66 patients on the 70-gene panel.
Tumor tissue DNA sequencing
All tissue samples were processed at the CAP/CLIA-certified University of Pennsylvania Center for Personalized Diagnostics clinical laboratory. 38 patient samples were processed using the Illumina TruSeq Amplicon – Cancer Panel (TSACP, FC-130-1008; Illumina) to sequence hotspots or exonic regions for 47 genes, as recently described (31, 32). For the remaining 12 patients, the DNA extracted from submitted tissue was of insufficient quantity for the full 47-gene panel; these samples were assessed using the Penn Precision Panel for mutations in a smaller panel of 20 commonly mutated genes (Supplemental Table 2). Genes covered on the ctDNA panel but not the tissue panel or vice versa are noted in Supplemental Tables 1 and 2. Libraries were prepared and sequenced on an Illumina MiSeq instrument, and always included a no template negative control. A clinically validated bioinformatics pipeline was used for tumor DNA (tDNA) sequencing analysis (33), with reads mapped to the hg19 genome build. The level of detection for the tissue NGS panel is 4.0%.
Statistical analysis and concordance analysis of ctDNA and tDNA
Tissue and plasma based platforms were compared using the Integrative Genome Viewer (https://www.broadinstitute.org/igv/) and variants covered and reported by both platforms were included in the concordance analysis. Although tissue testing was intended for all 102 patients, it was only successful for 50 (see Figure 1); post hoc calculations indicate that 50 paired samples provide precision for estimation of a 95% confidence interval around concordance with a maximum width of approximately 0.3. For patients with matched tDNA and ctDNA, we defined a concordant result as detection of the same variant in each sample, or in neither sample. The percent concordance was calculated for each patient, and then averaged over the matched tissue cohort (n=50) to determine overall concordance. To test whether concordance varied by elapsed time between tissue biopsy and blood draw, we applied the non-parametric trend test. We used paired t-tests to compare allelic fraction with each assay. We used independent-group t-tests to compare the number of variants and the concentration of cfDNA measured for patients who survived and did not. All t-test results were confirmed with their non-parametric equivalents. Overall survival was estimated using the Kaplan-Meier method and with Cox proportional hazards models. We evaluated the association of number of variants (categorized as <3 or ≥3 mutations) and cfDNA concentration (amount of cfDNA in ng/μL), with overall survival using the log-rank test. All significance tests were two-sided. Statistical analysis was conducted using Stata v 14 (StataCorp).
Figure 1.
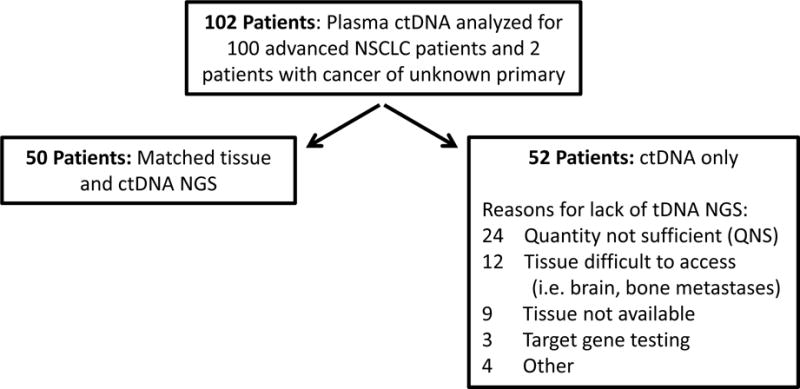
Overview of study for the detection of therapeutically targetable mutations in the tissue and plasma of patients with advanced lung cancer by next generation sequencing.
RESULTS
Patient characteristics and study design
Digital ctDNA sequencing was performed for 112 samples obtained from 102 total patients, including 100 patients with advanced NSCLC plus two patients (patients 37 and 51) with cancer of unknown primary who were treated at the University of Pennsylvania Abramson Cancer Center between February 2015 and March 2016 (Table 1). Most were women (68%) with adenocarcinoma (81%) and stage IV disease (96%). Only 4% of patients had a diagnosis of squamous cell carcinoma (SqCC) as compared to 15–20% in the overall lung cancer population (4, 34), likely due to the anticipated low frequency of actionable molecular abnormalities in SqCC, and also a competing SqCC trial at our institution at the time of this study. The female preponderance and significant proportion of never-smokers is likely a reflection of providers sending plasma samples for ctDNA analysis in a patient population most likely to harbor targetable mutations. In fact, the most common reason for ordering the ctDNA test was for initial detection of targetable mutations for 52 patients who either had no previous therapy (n=27), chemotherapy (n=18), or chemotherapy and immunotherapy (n=7). One patient who received chemotherapy also received erlotinib as second-line therapy according to the FDA label for unselected patients in whom EGFR mutation status had not been previously determined. Other reasons for ordering the ctDNA test included: detection of resistance mutations to targeted therapy (36 patients), identification of actionable mutations in patients with progressive disease (12 patients), and tracking a mutation identified in tDNA to monitor response to therapy (2 patients). While ctDNA analysis was successfully completed for all 112 samples obtained from 102 consecutively enrolled patients, tissue NGS could only be completed for 50 (49.0%) patients. Of the 52 patients with only ctDNA results, a tissue biopsy was either unobtainable, or yielded DNA of insufficient quantity or quality for 45 (86.5%) of 52 patients. (Figure 1). Thus, for over half our patients, ctDNA sequencing was the only option for detection of therapeutically targetable variants.
Table 1.
Patient characteristics
| All Patients (n = 102) |
Patients with Matched Tissue NGS (n = 50) |
Mutation Detected in ctDNA (n = 102) |
||||
|---|---|---|---|---|---|---|
| Number | % | Number | % | Number | % | |
| Median age (range) | 63 (32–88) | – | 64 (34–85) | – | ||
| Sex | ||||||
| Male | 33 | 32 | 17 | 34 | 27 | 82 |
| Female | 69 | 68 | 33 | 66 | 59 | 86 |
| Smoking | ||||||
| Former/Active | 65 | 64 | 25 | 50 | 55 | 85 |
| Never | 37 | 36 | 25 | 50 | 31 | 84 |
| Malignancy | ||||||
| NSCLC | 100 | 98 | 49 | 98 | 84 | 84 |
| Cancer of Unknown Primary | 2 | 2 | 1 | 2 | 2 | 100 |
| Histology | ||||||
| Adenocarcinoma | 83 | 81 | 39 | 78 | 70 | 84 |
| Squamous | 4 | 4 | 2 | 4 | 4 | 100 |
| Poorly differentiated carcinoma | 12 | 12 | 7 | 14 | 11 | 92 |
| Other | 3 | 3 | 2 | 4 | 1 | 33 |
| Disease stage at blood draw | ||||||
| II | 2 | 2 | 1 | 2 | 1 | 50 |
| III | 2 | 2 | 0 | 0 | 1 | 50 |
| IV | 98 | 96 | 49 | 98 | 84 | 86 |
| ECOG Performance Status | ||||||
| 0 | 26 | 25 | 14 | 28 | 22 | 85 |
| 1 | 59 | 58 | 30 | 60 | 50 | 85 |
| ≥ 2 | 17 | 17 | 6 | 12 | 14 | 82 |
| Number of Metastasesa | ||||||
| 0 | 4 | 3 | 1 | 2 | 2 | 50 |
| 1–2 | 72 | 71 | 36 | 72 | 61 | 85 |
| > 2 | 26 | 26 | 13 | 26 | 23 | 88 |
| Number of Previous Chemotherapeutic Regimens | ||||||
| 0 | 27 | 26 | 11 | 22 | 23 | 85 |
| 1–2 | 52 | 51 | 25 | 50 | 41 | 79 |
| ≥ 3 | 23 | 23 | 14 | 28 | 22 | 96 |
| On TKI Prior to Blood Draw | ||||||
| Yes | 39 | 38 | 27 | 54 | 34 | 87 |
| No | 63 | 62 | 23 | 46 | 52 | 83 |
| Intent of ctDNA Testing | ||||||
| Screen for actionable mutations | 52 | 51 | 17 | 34 | 43 | 83 |
| Detect resistance mutations | 36 | 35 | 23 | 46 | 33 | 92 |
| Identify potential actionable mutations following progression on chemotherapy | 12 | 12 | 8 | 16 | 9 | 75 |
| Track mutation identified in tDNA | 2 | 2 | 2 | 4 | 1 | 50 |
Number of organs with metastases
Therapeutically targetable driver and resistance mutations detected in ctDNA
In total, 275 variants were detected in the ctDNA of 86 (84.3%) of 102 patients, including SNVs, indels, and fusions. Sufficient input cfDNA was obtained and sequenced but no somatic variants, and thus no ctDNA, detected for 16 patients. Variants were detected in 45 genes with EGFR mutations the most prevalent at 20% of total variants (Figure 2, Supplemental Table 3). Among the driver mutations with FDA-approved therapies, and as previously reported (35), EGFR Exon 19 deletions were more frequently reported than L858R mutations, in 16 (15.7%) and 10 (9.8%) patients respectively (Supplemental Table 4). Ten ERBB2 mutations were detected, including 5 exon 20 insertions shown to confer sensitivity to ERBB2-targeted therapies (36). The EML4-ALK fusion, which confers sensitivity to the ALK TKI crizotinib (1), was detected in 2 samples.
Figure 2.
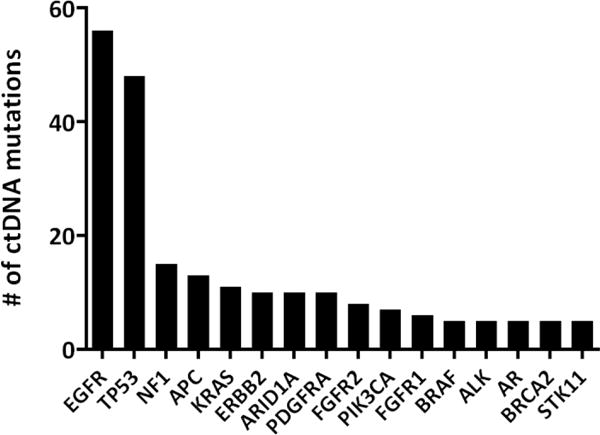
Variant detection in ctDNA. 275 total mutations were detected in the ctDNA of 86 out of 102 patients. Shown here is the frequency of variants by gene for the 219 variants detected in the 16 most commonly mutated genes.
The EGFR T790M resistance mutation was identified in the ctDNA of 10 (31%) of 32 patients receiving EGFR-TKI therapy, but the less common secondary resistance mutations EGFR D761Y, T854A, and L747S (37) were not detected in any samples. The original EGFR-activating mutation was detected in all 10 ctDNA samples harboring a T790M mutation. Objective progression of disease while on EGFR-directed therapy had been documented by cross-sectional imaging in 10/10 (100%) T790M-positive patients. Importantly, a T790M mutation was detected in the ctDNA of eight patients for whom tissue sequencing could not be performed. In addition, the BRAF D594G mutation, which has been associated with resistance to EGFR-targeted therapy (35), was detected for one patient. The PIK3CA E545K resistance mutation was not detected in any ctDNA samples. An ALK G1202R resistance mutation (9) was co-expressed with EML4-ALK for one patient whose cancer had progressed while receiving an ALK TKI. ROS1 or RET rearrangements were not detected. Eleven KRAS mutations were detected, including the G12C, G12D, and G12H variants shown to be associated with primary resistance to EGFR inhibitors in lung cancer (38). Consistent with prior reports, we found no co-expression of EGFR and KRAS mutations in any plasma samples (23, 39).
To further understand the potential clinical actionability of ctDNA NGS, all detected variants were cross-referenced against available FDA-approved, off-label, or investigational therapies. The majority of patients (70%) were determined to have a relevant clinical trial available, 56 (55%) patients had an off-label targeted therapy that could potentially be used, and 32 (31%) patients had an FDA-approved therapy available to target the detected variant. The mutations associated with available trials and therapies are detailed in Supplemental Table 5. Taken together, these data suggest that ctDNA analysis for NSCLC patients can yield results with high clinical relevance, including detection of therapeutically targetable mutations in EGFR, ALK, and other genes.
Comparison of tissue and plasma results
We next compared variant calls for the 50 patients who had both tDNA and ctDNA NGS performed. Of the 50 patients with matched tissue and plasma samples, 39 (78%) patients had ≥1 alteration in tDNA, and 42 (84%) patients had ≥1 mutation detected in ctDNA. Not surprisingly given the broader coverage of the ctDNA versus the tDNA panels (Supplemental Tables 1 and 2) the mean number of variants detected per patient by ctDNA was 2.8, compared to 1.5 using tDNA. When only the variants covered by both panels were considered, the difference narrowed to 55 and 67 variants respectively, while 41 mutations were detected by both (Figure 3A). The overall concordance for all variants covered and detected by both platforms was 60%. When wild-type calls, i.e., genes for which no variants were detected, are considered, the overall concordance was 97.5%. As one might expect given that DNA may be more dilute in the blood than tissue, there was a significant difference between the allelic fraction (AF) of variants detected in tDNA versus ctDNA for the 50 patients with matched tissue and ctDNA (tDNA mean AF 30.6% vs 6.2% for ctDNA; p<0.001; Figure 3B). 60% of variants detected in ctDNA were at an AF less than 4.0%, which is below the threshold at which calls can be made for the tDNA sequencing platform (detailed AF results for 50 patients with matched ctDNA and tissue shown in Supplemental Table 6). For the 14 variants detected in ctDNA but not tDNA, all 14 (100%) had an AF below this 4.0% cutoff. While overall concordance between ctDNA and tDNA was higher when tDNA was obtained for metastatic (79%) versus primary tumor tissue (51%), this difference was not statistically significant (p=0.063; Figure 3C).
Figure 3.
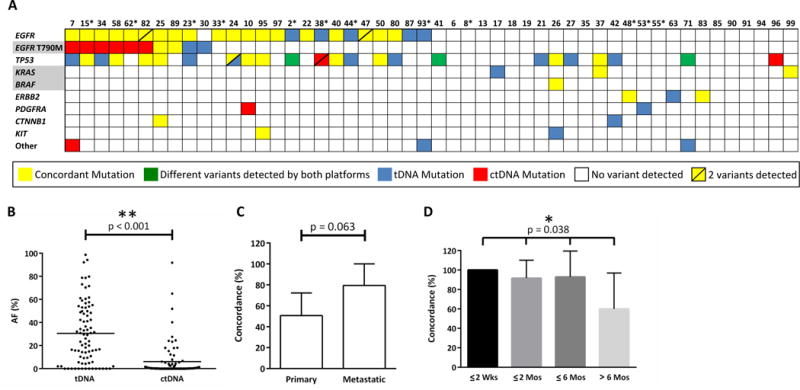
Mutational analysis for 50 patients for whom both plasma and tissue samples were obtained and sequenced. A. Detailed summary of all variants covered and called by both the tDNA and ctDNA platforms. The EGFR row includes the variants: L858R, G719A, L861Q, Exon 19 deletions, and Exon 20 insertions. Gene names with a grey background are known resistance mutations to EGFR-targeted therapy. Tumor DNA was sequenced using the 47-gene TSACP panel, except for those patient numbers with an asterisk indicating that the 20-gene panel was used due to low input DNA. Genes with two variants identified by either platform are denoted with a diagonal line. B. Allelic fractions for all variants detected in tDNA and ctDNA were compared. C. Concordance of all variant calls for tDNA and ctDNA by biopsy site (primary tumor, n=19; metastatic tumor, n=14) for samples obtained within 6 months. D. Concordance of actionable EGFR mutations detected in tDNA and ctDNA in relation to time interval between tissue biopsy and blood draw (≤ 2 weeks, n=9; ≤2 months, n=12; ≤6 months, n=14; > 6 months, n=10). AF (%) = Allelic Fraction.
Among the 50 patients with matched tDNA and ctDNA, 24 therapeutically targetable driver EGFR mutations were detected in tDNA and 19 in ctDNA samples, yielding an overall concordance of 79%. The EGFR T790M resistance mutation was identified in 4 (8%) tDNA samples and 8 (16%) ctDNA samples. Given the time interval between tissue and blood collection for our patients ranged from 0 days to >2 years, we next sought to understand whether tumor genetic evolution over time and under pressure of therapy might lead to a decrease in concordance when the time interval between plasma and tissue collection increased. To address this, we calculated the concordance between EGFR variants detected in tDNA and ctDNA for time intervals of ≤2 weeks, ≤2 months, ≤6 months, and >6 months and found the concordance rates (100%, 92%, 94%, and 60%, respectively) to be significantly correlated with time between tissue biopsy and blood draw (p=0.038; Figure 3D). There was no significant difference between the concordance for the 15 patients who received no treatment between ctDNA and tissue testing (76.7% concordance) and those (n=35) who received any treatment (52.4% concordance; p=0.11). This could be due to the wide variety of therapies patients received including chemotherapy (n=19), immunotherapy (n=6), targeted therapy (n=23), and 9 patients who received 2 or more types of therapy.
Among the 8 patients with discordant EGFR T790M calls (Supplemental Table 7), one patient (#23) had undergone tDNA and ctDNA testing 18 days apart, with the driver EGFR L858R mutation detected in both samples but T790M detected at a lower frequency in the tDNA but not the ctDNA. Upon review, it was determined that the total cfDNA yield was one of the lowest among our patients (14.5 ng versus a median of 32.1 ng (range 6–390 ng); Supplemental Figure 1), and the level of detection (LOD) for EGFR variants was therefore set at 0.2% rather than 0.1%. Since the L858R mutation was detected at 0.3% AF in the ctDNA, and EGFR resistance mutations are typically seen at a lower frequency in the blood compared to the originally detected EGFR driver mutations (24), it is possible that the T790M resistance mutation was present but below the LOD in the plasma. For patient #30, for whom EGFR T790M was detected in the tissue but not the plasma, 7 months had elapsed between the two tests and the patient had received afatinib during that time. It is well established that ctDNA levels may drop while on TKI therapy even with partial response (40). For the 6 patients for whom EGFR T790M was detected in the plasma but not the tumor, the ctDNA allelic fraction was well below the 4.0% level of detection for the tissue NGS panel, and 5 of the patients had received an EGFR-targeting therapy between tests, thus providing a possible explanation for the emergence of the T790M resistance mutation. The sixth patient, #15, in whom the variant was detected in plasma but not tDNA, received no EGFR-targeted therapy during the 7 days that elapsed between ctDNA and tDNA testing. Imaging in this patient showed multiple metastatic sites. If the T790M mutation had been present only in some metastases, but not the site that was biopsied, this tumor heterogeneity might provide an explanation for the discordant result.
Prognostic implications of molecular heterogeneity in ctDNA
While the main objective of this study was to determine the feasibility of ctDNA for detection of therapeutically targetable variants, others have shown that plasma-based testing can be used to predict patient outcome (22, 25, 41) and that ctDNA levels are associated with disease stage (23). Because molecular heterogeneity has also been associated with poor patient outcome (42, 43), we next sought to assess the prognostic significance of ctDNA molecular heterogeneity. For the 98 patients with metastatic disease, the median overall survival from time of metastatic diagnosis in patients with ≥3 variants detected in plasma was 46 months versus 62 months for those with <3 variants, although this result did not reach statistical significance (p=0.09; Figure 4A). Consistent with another recent report (30), we also found cfDNA level was associated with poor patient outcomes. The mean concentration of total cfDNA extracted from plasma was significantly higher in patients who died during the study period versus alive patients (4.0 vs. 1.6 ng/μL; p<0.001). Higher cfDNA concentrations were significantly associated with decreased overall survival from time of metastatic diagnosis, and a cfDNA concentration ≥3 ng/μL was associated with a median overall survival of 24 versus 46 months (log-rank. p<0.01; Figure 4B). This result remained significant when adjusted for age, performance status, EGFR mutation status, and number of metastatic sites.
Figure 4.
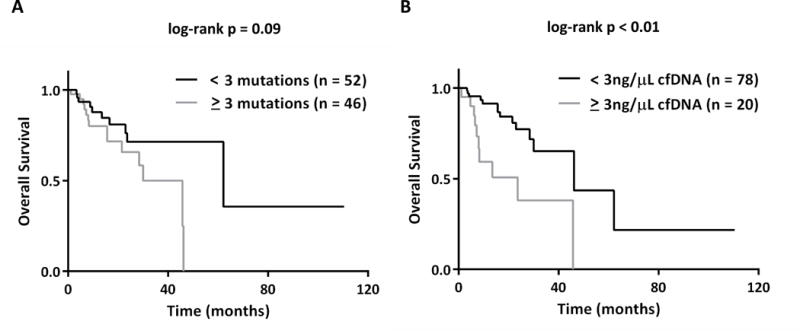
Use of ctDNA to predict survival. Patient survival was calculated as the number of months since date of metastatic diagnosis, and then compared to ctDNA measurements (n=98 patients with metastatic disease). A. Kaplan Meier survival curve and log-rank test dichotomized around a threshold of ≥ 3 mutations. B. Kaplan Meier survival curve and log-rank test dichotomized around a threshold of ≥ 3 ng/μL of cfDNA detected.
Real-time serial molecular monitoring of patients on therapy
As a non-invasive test, ctDNA NGS can be performed more frequently than tissue NGS, and thus offers the possibility of serial testing for molecular monitoring of disease. While this was not a primary objective of our study, serial ctDNA testing was performed on 6 patients as part of disease surveillance. For three of these patients, ctDNA testing did not reveal therapeutically targetable mutations, but could still be used to guide clinical decision-making. Patient #6 had newly diagnosed metastatic NSCLC. Initial tissue testing yielded insufficient DNA, therefore ctDNA was performed twice for initial mutation screening but no variants were detected, and the patient was put on chemotherapy. Subsequent tissue NGS confirmed the lack of detectable somatic variants. Patients #2 and #24 had metastatic EGFR mutation-positive NSCLC exhibiting progressive disease on an EGFR TKI. ctDNA analysis was performed at two timepoints for each patient to detect an EGFR resistance mutation as an indication to switch to a third-line TKI. However, no EGFR T790M mutation was detected for either timepoint or patient, so a third-line TKI was not prescribed.
In addition, we describe three patients for whom ctDNA did detect the emergence of therapeutically targetable variants over the course of therapy.
Patient #72 (Figure 5A) presented with metastatic disease at diagnosis with tissue biopsy detecting no variants in EGFR, ALK, or ROS1. Upon referral to our hospital, ctDNA testing revealed an EML4-ALK fusion (0.4% AF) and a TP53 variant (0. 8 % AF), and the patient was initiated on crizotinib monotherapy with significant improvement in symptoms. Two months following therapy, a repeat ctDNA test showed a decrease in the EML4-ALK AF to 0.1%, and the TP53 mutation was undetectable. A CT scan performed the same day demonstrated a decrease in size of the primary lung lesion as well as liver metastatic disease.
Figure 5. Serial ctDNA testing.
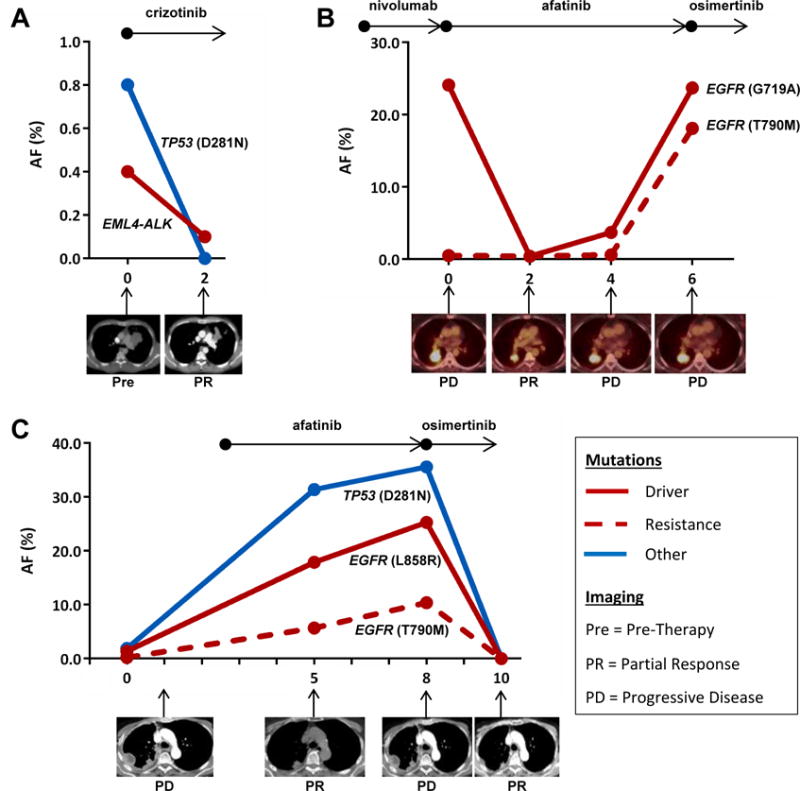
Real-time molecular monitoring of patients on therapy with time depicted on the x-axis as months since first plasma-based ctDNA analysis. The black arrows above each panel indicate targeted agents administered to patient over the indicated time periods. Below each panel are imaging results at the indicated time point. A. Patient #72 was treated with crizotinib following identification of EML4-ALK fusion in ctDNA but not tDNA. Imaging shows response to therapy correlating with decrease in plasma-based EML4-ALK AF. B. ctDNA analysis for Patient #82 confirmed EGFR (G719A) mutation and identified the emergence of EGFR (T790M) mutation that was not detected in tDNA several months prior. Serial ctDNA sampling shows AF of detected variants correlating with response to therapy. C. ctDNA analysis identified the presence of an EGFR (T790M) mutation at Month 0 that was not detected in a tDNA sample obtained 6 days prior. Following imaging- and ctDNA- confirmed disease progression, patient #15 was started on osimertinib with good clinical response. AF (%) = Allelic Fraction, Pre = Pre-Therapy, PD = Progressive Disease, PR = Partial Response.
Patient #82 (Figure 5B) developed progressive disease while receiving erlotinib with tumor sequencing at time of progression showing the EGFR G719A mutation (79% AF) but without a resistance mutation identified. Erlotinib was discontinued. A PET CT following three months of nivolumab (month 0 in Figure 5B) revealed further disease progression, and ctDNA analysis performed at that time confirmed EGFR G719A (24.1% AF), and also detected the emergence of a T790M mutation (0.5% AF). Afatanib was initiated and repeat ctDNA testing 2 months later showed a decrease in the AF of EGFR G719A to 0.4% and EGFR T790M to 0.4%, which correlated with imaging response. ctDNA performed two months later showed an increase in AF of EGFR G719A and EGFR T790M to 3.7% and 0.6%, respectively. This was associated with mild progression on PET CT, and afatinib was continued. ctDNA analysis two months later showed a further increase in the AF of EGFR G719A to 23.7% and EGFR T790M to 18.1% with clear progression on PET CT, and the patient was placed on osimertinib based on the EGFR T790M mutation detected only in blood. Interval imaging to monitor response to therapy had not yet been performed upon submission of this manuscript.
Patient #15 (Figure 5C) developed progressive disease soon after discontinuation of erlotinib due to toxicity. Tissue sampling revealed the EGFR L858R (16.5% AF) and TP53 D281N (15.8% AF), but no evidence of T790M mutation. ctDNA sequencing performed 7 days later identified EGFR L858R (1.4% AF), TP53 D281N (1.9% AF), and the emergence of EGFR T790M (0.2% AF). Afatinib was initiated and surveillance CT scan performed two months later showed a partial response. However, simultaneous ctDNA analysis noted a rising AF for all three variants: EGFR L858R 17.9%, EGFR T790M 5.7%, and TP53 D281N 31.4%. A CT scan three months later confirmed disease progression with simultaneous ctDNA sequencing showing a continued rise in AF for all three mutations: EGFR L858R 25.3%, EGFR T790M 10.4%, TP53 D281N 35.6%. Osimertinib was prescribed based on the EGFR T790M mutant detected only in the blood. A two-month follow up CT scan exhibited an interval response to therapy, and a simultaneous ctDNA test revealed that all 3 variants were now undetectable in the patient’s plasma.
DISCUSSION
Here we present evidence for the feasibility and clinical utility of liquid biopsies for the management of advanced NSCLC patients. The ever-expanding number of targeted therapies available for lung cancer patient treatment has been accompanied by a need for companion diagnostics for real-time detection of therapeutically targetable genetic lesions (44, 45). Treatment with first-line TKIs is facilitated by the identification at diagnosis of mutations such as EGFR L858R or exon 19 deletions, and can often be achieved through tissue biopsy. However, monitoring response to targeted therapy by assaying changes in the frequency of the targeted mutation, or identifying resistance mutations cannot be consistently achieved through repeat biopsy. Plasma-based ctDNA testing is a non-invasive means of patient monitoring and thus offers the advantage of testing without the risks associated with invasive biopsies. When applied to the blood of NSCLC patients, liquid biopsies can identify resistance mutations that allow for the treatment of patients with second- and third-line TKIs, or cytotoxic chemotherapy when no targetable mutation is identified.
In this prospective study, we have shown the successful next-generation sequencing of 102 consecutively obtained lung cancer patient plasma samples. The minimum input DNA (≥ 5 ng) was successfully extracted from all samples and libraries prepared. No mutations were detected in the cfDNA for 16 of 102 patients, suggesting the tumor was not actively shedding ctDNA, the patient’s disease was adequately controlled by therapy, or plasma-based somatic variants were either not covered by the 70-gene panel or below the 0.01% level of detection for the assay. In contrast, a tissue biopsy with sufficient quality and quantity of DNA for NGS was unobtainable or not obtained for 52 of 102 patients (51%). Similar to other reports for tissue NGS (46), quantity of tDNA was not sufficient for 24 of 52 (46.2%) patients. Thus, for over half our patients, a liquid biopsy was the only means of molecular monitoring. While circulating tumor cells (CTCs) are considered a form of liquid biopsy, and we (31) and others (47) have reported on approaches for molecular analysis of CTCs, NSCLC patient CTCs cannot always be reliably identified (44, 48, 49). Moreover, ctDNA is readily detectable in lung cancer patient blood (20, 27, 29, 30), and sensitivity of variant detection was recently shown to be higher in ctDNA than CTCs (25). Thus, ctDNA testing is well-suited for NSCLC patient molecular monitoring.
Droplet digital PCR (ddPCR) has been proposed as another means of liquid biopsy, and others have reported on the detection of driver and resistance mutations in a limited number of genes for NSCLC (23, 24) and other cancers (27, 50). While the results we have reported are based on a panel of up to 70 clinically relevant genes, including 30 with full exon coverage, ddPCR is typically used to detect hotspots in 3 to 5 genes. The EGFR T790M mutation accounts for more than 50% of cases of acquired resistance to EGFR-TKI therapy and would certainly be detected by many current ddPCR platforms (35). As the number of molecular mechanisms of resistance and associated approved therapies increases, however, NGS may emerge as the more clinically useful assay for identification of actionable targets. Digital sequencing of a large panel of genes (in this case 70) allows for detection of a large number of variants to aid clinical decision-making including: EGFR and ALK variants for first-line therapy; mutations in EGFR, ALK, BRAF, PIK3CA, and other genes associated with resistance to EGFR or ALK TKIs; KRAS variants associated with primary resistance to EGFR targeted therapy; and variants in other genes that may lead to the off-label use of FDA-approved therapies or enrollment to clinical trials of new therapeutic agents (see Supplemental Table 5). This approach may also facilitate the discovery of previously unreported resistance mutations and the emergence of low frequency subclones under pressure of therapy, which would have been undetectable in primary tumor tissue. Moreover, liquid biopsy may detect mutations that were either not present or undetectable in primary tissue or initial biopsy.
To our knowledge, this prospective study is the first of its kind to apply a comprehensive clinical NGS panel to ctDNA and matched tumor biopsies in advanced lung cancer patients, and demonstrate the feasibility and utility of plasma testing for a large subset of patients in whom matched tissue sequencing was not feasible. The logical extension of this work is to evaluate the test’s utility at diagnosis as a complement to tissue testing, and in the context of genetically heterogeneous metastatic disease. Here we reported on serial ctDNA testing for 6 patients, but larger scale studies will be required to further evaluate ctDNA monitoring for treatment selection, including patients for whom no therapeutically targetable mutations are detected who may be candidates for checkpoint inhibitors (34). While ctDNA testing alone will be insufficient to detect histological sources of therapy resistance, such as a small cell phenotype, a liquid biopsy may complement histological analysis by providing additional tumor molecular characterization. It may also be important to explore the clinical actionability of our finding that higher levels of cell-free DNA, irrespective of mutational profile, are associated with decreased survival. All the patients in our study either had active metastatic disease or had scans suspicious for progression; determining the feasibility of ctDNA-based disease monitoring in the context of minimal residual or early stage disease would broaden clinical utility. Adapting our approach to achieve the sensitivity and specificity necessary for nodule-positive patients at higher risk for the development of cancer, perhaps in conjunction with imaging, could greatly enhance early detection of tumors with a greater chance of achieving curative resection. In summary, this work demonstrates the promise of ctDNA testing for real-time molecular monitoring of patients with advanced lung cancer and other malignancies in clinical practice, and underscores the need for additional studies to further assess the biological evolution of metastatic disease and clinical utility of molecular non-invasive profiling.
Supplementary Material
TRANSLATIONAL RELEVANCE.
This study demonstrates the feasibility of conducting next-generation sequencing of a comprehensive gene panel for managing patients with advanced non-small cell lung cancer (NSCLC). Detection of driver and resistance mutations has never been more clinically important as the number of useful targeted agents continues to grow. Analyzable tissue samples are often difficult or impossible to obtain for this patient cohort, and serial biopsies are rarely possible. Here we demonstrate the use of a 70-gene cell-free circulating tumor DNA next-generation sequencing panel for the detection of clinically actionable variants in a study of 102 prospectively enrolled lung cancer patients. Matched tissue sequencing was successful for fewer than half the patients, mainly as a result of inaccessible tumor tissue or insufficient DNA for sequencing. These data demonstrate the feasibility and clinical utility of plasma-based liquid biopsy for personalized therapy of patients with advanced NSCLC.
Acknowledgments
The authors gratefully acknowledge Ms. Rebecca Nagy and Dr. Richard Lanman for helpful discussion, data analysis, and technical assistance. We also wish to thank the patients, their families, and any study staff involved in this study.
GRANT SUPPORT
This work was supported by NHLBI/NIH Training Grant 5T32HL007586-29 (to J.C. Thompson) and Abramson Cancer Center Translational Centers of Excellence (to E.L. Carpenter).
References
- 1.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167–77. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 2.Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16:141–51. doi: 10.1016/S1470-2045(14)71173-8. [DOI] [PubMed] [Google Scholar]
- 3.Ettinger DS, Wood DE, Akerley W, Bazhenova LA, Borghaei H, Camidge DR, et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 4.2016. J Natl Compr Canc Netw. 2016;14:255–64. doi: 10.6004/jnccn.2016.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. 2015;4:36–54. doi: 10.3978/j.issn.2218-6751.2014.05.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–67. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 6.Arrieta O, Cardona AF, Martin C, Mas-Lopez L, Corrales-Rodriguez L, Bramuglia G, et al. Updated Frequency of EGFR and KRAS Mutations in NonSmall-Cell Lung Cancer in Latin America: The Latin-American Consortium for the Investigation of Lung Cancer (CLICaP) J Thorac Oncol. 2015;10:838–43. doi: 10.1097/JTO.0000000000000481. [DOI] [PubMed] [Google Scholar]
- 7.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 8.Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–34. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 9.Awad MM, Shaw AT. ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clin Adv Hematol Oncol. 2014;12:429–39. [PMC free article] [PubMed] [Google Scholar]
- 10.Spaans JN, Goss GD. Drug resistance to molecular targeted therapy and its consequences for treatment decisions in non-small-cell lung cancer. Front Oncol. 2014;4:190. doi: 10.3389/fonc.2014.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol. 2010;7:493–507. doi: 10.1038/nrclinonc.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sequist LV, Rolfe L, Allen AR. Rociletinib in EGFR-Mutated Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:578–9. doi: 10.1056/NEJMc1506831. [DOI] [PubMed] [Google Scholar]
- 13.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–61. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ettinger DS, Wood DE, Akerley W, Bazhenova LA, Borghaei H, Camidge DR, et al. Non-Small Cell Lung Cancer, Version 6.2015. J Natl Compr Canc Netw. 2015;13:515–24. doi: 10.6004/jnccn.2015.0071. [DOI] [PubMed] [Google Scholar]
- 15.Sholl LM, Aisner DL, Varella-Garcia M, Berry LD, Dias-Santagata D, Wistuba II, et al. Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol. 2015;10:768–77. doi: 10.1097/JTO.0000000000000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z, et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci U S A. 2013;110:21083–8. doi: 10.1073/pnas.1320659110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–6. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 19.Chandrananda D, Thorne NP, Bahlo M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med Genomics. 2015;8:29. doi: 10.1186/s12920-015-0107-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548–54. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz LA, Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–40. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 23.Couraud S, Vaca-Paniagua F, Villar S, Oliver J, Schuster T, Blanche H, et al. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: a proof-of-concept study from BioCAST/IFCT-1002. Clin Cancer Res. 2014;20:4613–24. doi: 10.1158/1078-0432.CCR-13-3063. [DOI] [PubMed] [Google Scholar]
- 24.Oxnard GR, Paweletz CP, Kuang Y, Mach SL, O’Connell A, Messineo MM, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20:1698–705. doi: 10.1158/1078-0432.CCR-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Punnoose EA, Atwal S, Liu W, Raja R, Fine BM, Hughes BG, et al. Evaluation of circulating tumor cells and circulating tumor DNA in non-small cell lung cancer: association with clinical endpoints in a phase II clinical trial of pertuzumab and erlotinib. Clin Cancer Res. 2012;18:2391–401. doi: 10.1158/1078-0432.CCR-11-3148. [DOI] [PubMed] [Google Scholar]
- 26.Kuang Y, Rogers A, Yeap BY, Wang L, Makrigiorgos M, Vetrand K, et al. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin Cancer Res. 2009;15:2630–6. doi: 10.1158/1078-0432.CCR-08-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drilon A, Wang L, Arcila ME, Balasubramanian S, Greenbowe JR, Ross JS, et al. Broad, Hybrid Capture-Based Next-Generation Sequencing Identifies Actionable Genomic Alterations in Lung Adenocarcinomas Otherwise Negative for Such Alterations by Other Genomic Testing Approaches. Clin Cancer Res. 2015;21:3631–9. doi: 10.1158/1078-0432.CCR-14-2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanman RB, Mortimer SA, Zill OA, Sebisanovic D, Lopez R, Blau S, et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS One. 2015;10:e0140712. doi: 10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwaederle M, Husain H, Fanta PT, Piccioni DE, Kesari S, Schwab RB, et al. Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget. 2016;7:9707–17. doi: 10.18632/oncotarget.7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yee SS, Lieberman DB, Blanchard T, Rader J, Zhao J, Troxel AB, et al. A novel approach for next-generation sequencing of circulating tumor cells. Molecular Genetics & Genomic Medicine. 2016 doi: 10.1002/mgg3.210. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hiemenz MC, Kadauke S, Lieberman DB, Roth DB, Zhao J, Watt CD, et al. Building a Robust Tumor Profiling Program: Synergy between Next-Generation Sequencing and Targeted Single-Gene Testing. PLoS One. 2016;11:e0152851. doi: 10.1371/journal.pone.0152851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daber R, Sukhadia S, Morrissette JJ. Understanding the limitations of next generation sequencing informatics, an approach to clinical pipeline validation using artificial data sets. Cancer Genet. 2013;206:441–8. doi: 10.1016/j.cancergen.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 34.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 35.Stewart EL, Tan SZ, Liu G, Tsao MS. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl Lung Cancer Res. 2015;4:67–81. doi: 10.3978/j.issn.2218-6751.2014.11.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li BT, Ross DS, Aisner DL, Chaft JE, Hsu M, Kako SL, et al. HER2 Amplification and HER2 Mutation Are Distinct Molecular Targets in Lung Cancers. J Thorac Oncol. 2016;11:414–9. doi: 10.1016/j.jtho.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen KS, Kobayashi S, Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer. 2009;10:281–9. doi: 10.3816/CLC.2009.n.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson ML, Sima CS, Chaft J, Paik PK, Pao W, Kris MG, et al. Association of KRAS and EGFR mutations with survival in patients with advanced lung adenocarcinomas. Cancer. 2013;119:356–62. doi: 10.1002/cncr.27730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marchetti A, Palma JF, Felicioni L, De Pas TM, Chiari R, Del Grammastro M, et al. Early Prediction of Response to Tyrosine Kinase Inhibitors by Quantification of EGFR Mutations in Plasma of NSCLC Patients. J Thorac Oncol. 2015;10:1437–43. doi: 10.1097/JTO.0000000000000643. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 42.Mroz EA, Tward AD, Pickering CR, Myers JN, Ferris RL, Rocco JW. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer. 2013;119:3034–42. doi: 10.1002/cncr.28150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mahrooghy M, Ashraf AB, Daye D, McDonald ES, Rosen M, Mies C, et al. Pharmacokinetic Tumor Heterogeneity as a Prognostic Biomarker for Classifying Breast Cancer Recurrence Risk. IEEE Trans Biomed Eng. 2015;62:1585–94. doi: 10.1109/TBME.2015.2395812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neal JW, Gainor JF, Shaw AT. Developing biomarker-specific end points in lung cancer clinical trials. Nat Rev Clin Oncol. 2015;12:135–46. doi: 10.1038/nrclinonc.2014.222. [DOI] [PubMed] [Google Scholar]
- 45.Carpenter EL, Mosse YP. Targeting ALK in neuroblastoma–preclinical and clinical advancements. Nat Rev Clin Oncol. 2012;9:391–9. doi: 10.1038/nrclinonc.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zill OA, Greene C, Sebisanovic D, Siew LM, Leng J, Vu M, et al. Cell-Free DNA Next-Generation Sequencing in Pancreatobiliary Carcinomas. Cancer Discov. 2015;5:1040–8. doi: 10.1158/2159-8290.CD-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Polzer B, Medoro G, Pasch S, Fontana F, Zorzino L, Pestka A, et al. Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol Med. 2014;6:1371–86. doi: 10.15252/emmm.201404033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004;10:6897–904. doi: 10.1158/1078-0432.CCR-04-0378. [DOI] [PubMed] [Google Scholar]
- 49.Krebs MG, Hou JM, Sloane R, Lancashire L, Priest L, Nonaka D, et al. Analysis of circulating tumor cells in patients with non-small cell lung cancer using epithelial marker-dependent and -independent approaches. J Thorac Oncol. 2012;7:306–15. doi: 10.1097/JTO.0b013e31823c5c16. [DOI] [PubMed] [Google Scholar]
- 50.Arena S, Bellosillo B, Siravegna G, Martinez A, Canadas I, Lazzari L, et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin Cancer Res. 2015;21:2157–66. doi: 10.1158/1078-0432.CCR-14-2821. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.