

Abstract
TREK-1 (KCNK2 or K2P2.1) is a mechanosensitive K2P channel that is opened by membrane stretch as well as cell swelling. Here, we demonstrate that membrane phospholipids, including PIP2, control channel gating and transform TREK-1 into a leak K+ conductance. A carboxy-terminal positively charged cluster is the phospholipid-sensing domain that interacts with the plasma membrane. This region also encompasses the proton sensor E306 that is required for activation of TREK-1 by cytosolic acidosis. Protonation of E306 drastically tightens channel–phospholipid interaction and leads to TREK-1 opening at atmospheric pressure. The TREK-1–phospholipid interaction is critical for channel mechano-, pHi- and voltage-dependent gating.
Keywords: general anesthesia, KCNK, K2P, neuro-protection, stretch
Introduction
The mammalian K2P channel subunits have four transmembrane segments and two P domains in tandem that contribute to the K+-selective ionic pore (Lesage and Lazdunski, 2000; Patel and Honoré, 2001b; Kim, 2003; Talley et al, 2003; Yost, 2003). A functional K2P channel is a dimer of subunits (Lesage et al, 1996; Maingret et al, 2000a; Czirjak and Enyedi, 2001; Lauritzen et al, 2003; Berg et al, 2004; Kang et al, 2004). Two of the 15 human K2P subunits, TREK-1 and TREK-2, have particularly interesting functional properties. They form mechanosensitive K+ channels that are activated by membrane stretch as well as cell swelling (Patel et al, 1998; Maingret et al, 1999; Bang et al, 2000; Lesage et al, 2000; Honoré et al, 2002). These mechanosensitive K2P channels are also activated by polyunsaturated fatty acids, including arachidonic acid (AA) and lysophospholipids (Fink et al, 1998; Patel et al, 1998; Lesage et al, 2000; Maingret et al, 2000b). TREK-1 and TREK-2 are the targets of an important class of drugs, the volatile general anesthetics (Patel et al, 1999; Lesage et al, 2000; Heurteaux et al, 2004). Moreover, their opening is stimulated by the neuroprotective agent riluzole that is currently used to protect motorneurons in amyotrophic lateral sclerosis (Duprat et al, 2000). Neurotransmitters and hormones that stimulate the cAMP/PKA pathway strongly inhibit TREK-1 and TREK-2 channel activity (Patel et al, 1998; Lesage et al, 2000). Neurotransmitters that stimulate the Gq pathway, such as glutamate, decrease TREK-1 and TREK-2 activity via a direct diacylglycerol (DG) inhibition (Chemin et al, 2003). Mice in which the TREK-1 channel has been deleted have an increased vulnerability to both epileptic seizure and brain ischemia (Heurteaux et al, 2004). Moreover, they have lost neuroprotection by polyunsaturated fatty acids and are more resistant to volatile general anesthetics (Heurteaux et al, 2004).
A particular effort has been made to understand the gating mechanism of the TREK-1 channel (for a review, see Patel et al, 2001). The cytosolic carboxy-terminal domain of TREK-1 plays a key role in its function and regulation. A glutamate residue, E306, is a key element in the channel-gating machinery. Its protonation by cytosolic acidification leads to channel activation by drastically changing the pressure dependency in such a way as to render the channel active at atmospheric pressure (Maingret et al, 1999; Honoré et al, 2002). In the same region, the serine residue S333 is the target of PKA phosphorylation locking the channel in a closed state (Patel et al, 1998). The TREK-2 channel shares all the functional properties of TREK-1 (Lesage et al, 2000).
The phospholipid PIP2, although a minor component of the plasma membrane, is increasingly recognized as a key physiological regulator of ion channel activity (Hilgemann et al, 2001). PIP2 directly activates the inward rectifier IRK potassium channels and is essential for the Gβγ-protein activation of GIRK channels (Huang et al, 1998; Mirshahi et al, 2003). PIP2 controls nucleotide sensitivity of KATP channels and promotes channel opening (Baukrowitz et al, 1998; Shyng and Nichols, 1998). PIP2 also regulates both activation and inactivation processes of the voltage-gated K+ channels (Loussouarn et al, 2003; Oliver et al, 2004). Genetic alterations of K+ channel–PIP2 interactions can lead to channelopathies such as Andersen's and Bartter's syndromes (Lopes et al, 2002). Receptor-mediated PIP2 hydrolysis plays a key role in the regulation of several ion channel types (Kobrinsky et al, 2000; Chuang et al, 2001; Runnels et al, 2002; Suh and Hille, 2002; Zhang et al, 2003) and particularly in the inhibition of the TASK channel activity by hormones and neurotransmitters (Czirjak et al, 2001). This mechanism is involved in the downmodulation of the TASK-1/-3 K2P channels by stimulation of Gq-coupled type I metabotropic glutamate receptors (Chemin et al, 2003).
In the present report, we demonstrate that PIP2, but also other membrane phospholipids such as phosphatidyl serine (PS), are essential for TREK-1 channel activity. Phospholipids switch TREK-1 from a voltage-, stretch- and pH-activated channel into a leak K+ conductance. We identify the protein sequence elements that are involved in phospholipid sensing and provide a mechanistic model for channel gating.
Results
Cytosolic PIP2 stimulates TREK-1 channel activity
Both in situ hybridization and immunolocalization studies have shown that striatal neurons express high levels of TREK-1 (Fink et al, 1996; Maingret et al, 2000a; Talley et al, 2001). In striatal neurons, K+ channels (55.8±0.9 pS, n=6; physiological K+ gradient) that are opened by membrane stretch as well as AA were found to be absent in TREK-1 knockout (KO) mice (Heurteaux et al, 2004). Similarly, cytosolic acidosis as well as PIP2 stimulate the activity of this endogenous TREK-1 channel (Figure 1A–C). The single-channel current amplitude measured at 0 mV is not significantly altered by PIP2 (4.7±0.3 pA, n=5 versus 4.9±0.2 pA, n=6) and cytosolic acidosis (4.5±0.25 pA, n=5, at pHi 5.5), suggesting that NPo is increased in both conditions. Stimulation is maintained after PIP2 washout (Figure 1B). Addition of polylysine (pL) to the intracellular medium, a cationic molecule that is classically used to mask or even remove negatively charged membrane phospholipids such as PIP2 (Huang et al, 1998; Lopes et al, 2002), not only reverses PIP2 stimulation but also inhibits basal channel activity (Figure 1B and C). This PIP2-, pHi- and pL-sensitive 56 pS K+ channel is absent in striatal neurons isolated from TREK-1 KO mice (Figure 1C).
Figure 1.
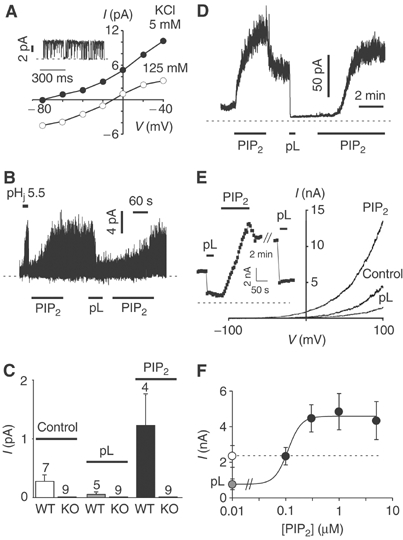
PIP2 stimulates native and cloned TREK-1 channel activity. (A) Endogenous K+ channel activity from cultured mouse striatal neurons was recorded in the presence of extracellular TEA, 4-AP and glibenclamide. A TREK-1-like channel stimulated by cytosolic acidosis to pH 5.5, membrane stretch and AA is recorded under these experimental conditions (Heurteaux et al, 2004). I–V curves (inside-out patch) were constructed in the presence of 5 mM (filled symbols) and 125 mM external K+ (empty symbols), with a pHi 5.5. The inset shows a current trace recorded at a holding potential of 0 mV in a physiological K+ gradient. (B) Effects of cytosolic acidosis (pHi 5.5), PIP2 (5 μM) and pL (30 μg/ml) on the endogenous striatal TREK-1 channel recorded in an inside-out patch held at 0 mV (physiological K+ gradient). (C) Histogram summarizing the effect of pL (30 μg/ml) and PIP2 (5 μM) on the mean TREK-1-like current amplitude of cultured striatal neurons from WT and KO mice. (D) Effect of PIP2 (5 μM) and pL (30 μg/ml) on the cloned TREK-1 channel transiently expressed in COS cells and measured in an inside-out patch held at 0 mV (physiological K+ gradient). (E) I–V curves elicited by voltage ramps showing the effect of pL and PIP2 in an inside-out patch. The inset shows current amplitude measured at 0 mV. (F) Dose–effect curve of PIP2 on TREK-1 channel activity after pL inhibition. The EC50 value is estimated to be 125 nM (n=10). In COS cells, we also occasionally observed inhibition of TREK-1 control currents at higher concentrations of PIP2 before pL treatment. In (A, B, D and E) inset, the dashed lines indicate the zero current. In (F), the dashed line indicates the control TREK-1 current (empty circles).
The same type of findings were obtained with the cloned TREK-1 channel transiently expressed in COS cells. Cytosolic PIP2 increases TREK-1 channel activity before pL treatment (37/81 patches) and again this effect is only partially reversible upon wash (Figure 1D). This PIP2/pHi-activated K+ current is absent in EYFP-transfected control COS cells (Supplementary Figure 1B). Addition of cytosolic pL (30 μg/ml) strongly and irreversibly inhibits basal channel activity (−78±2%, n=10) (Figure 1E and F). A similar inhibition is observed with the polyamine spermine (100 μM) (−55±5%, n=13) that is also used as a quencher of PIP2 (Huang et al, 1998). As expected, pL inhibition is fully reversed by the addition of the anionic molecule heparin (20 μg/ml) (n=8) that probably removes pL from its interaction with phospholipids (Supplementary Figure 1A and B). As described for the native TREK-1 channel, low concentrations of PIP2 not only restore the initial basal TREK-1 activity but also actually lead to a higher channel activity (95/95 patches) (Figure 1E and F). PIP2 (1 μM) leads to about a two-fold increase in current amplitude as compared to the control condition (n=10) (Figure 1F). Channel stimulation is independent of the level of expression of TREK-1 (5.1±0.8-fold increase, n=48, for currents of 200±16 pA amplitude and 4.1±0.7-fold increase, n=47, for currents of 2088±294 pA amplitude). Another addition of pL, after the PIP2-induced activation, again inhibits channel activity (Figure 1E). Similarly, PIP2 stimulation is observed in inside-out patches excised from TREK-1-expressing Xenopus oocytes (23/23 patches) (not shown).
PIP2 regulates TREK-1 gating
The cloned TREK-1 channel activity, like the endogenous striatal channel, is also reversibly stimulated by cytosolic acidification (Figures 1A, B and 2A). After a pL treatment that inhibits the basal current, cytosolic acidosis fails to activate TREK-1 (Figure 2A and C). When channel activity is restored (in fact at a higher level) by the addition of PIP2, the activating effect of cytosolic acidosis is recovered (n=18) (Figure 2A–C). However, the acidic stimulation after PIP2 addition becomes ‘irreversible' and the TREK-1 channel remains locked in a highly active form even when the cytosolic pH is returned to pH 7.2 (n=18) (Figure 2A–C). When TREK-1 is locked open by PIP2, a second cytosolic acidification actually produces a mild current inhibition (−32±4%, n=17) (Figure 2A), probably related to a decrease in single-channel conductance at low pHi (Maingret et al, 1999). Again, another addition of pL, after the co-stimulation with PIP2 and acidosis, inhibits channel activity, although only partially (n=12) (Figure 2A).
Figure 2.
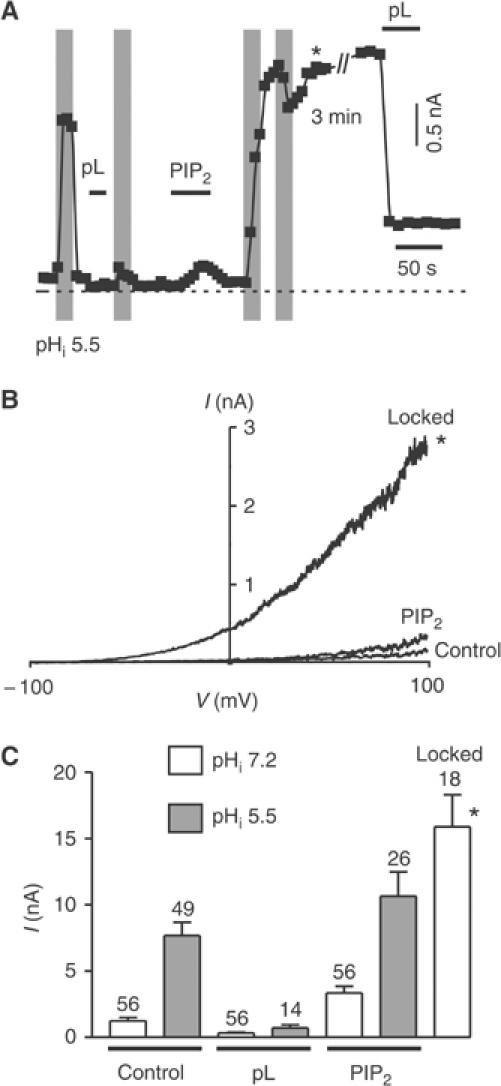
Acidic stimulation locks TREK-1 open in the presence of PIP2. (A) Effect of pHi 5.5 (indicated by gray shading) in the absence and the presence of pL (30 μg/ml) and PIP2 (5 μM) on TREK-1 recorded in an inside-out patch measured at 0 mV (physiological K+ gradient). (B) I–V curves of TREK-1 elicited by voltage ramps in control, in the presence of PIP2 (5 μM) and during open lock following cytosolic acidosis (pHi 5.5) after PIP2 (5 μM) washout (indicated by a star). (C) The histogram shows the effect of pHi 5.5 (gray columns) in the absence and the presence of pL (30 μg/ml) and PIP2 (5 μM).
pL also inhibits stretch activation of TREK-1 and the pressure effect curve is shifted towards more negative pressure values (n=6) (Figure 3A–C). Therefore, TREK-1 channel becomes more resistant to membrane stretch (Figure 3A and B). With a PIP2 treatment followed by a cytosolic acidification, the TREK-1 activity is high at atmospheric pressure and pressure stimulation only produces a modest further activating effect (n=6) (open lock indicated by a star; Figure 3A and B).
Figure 3.
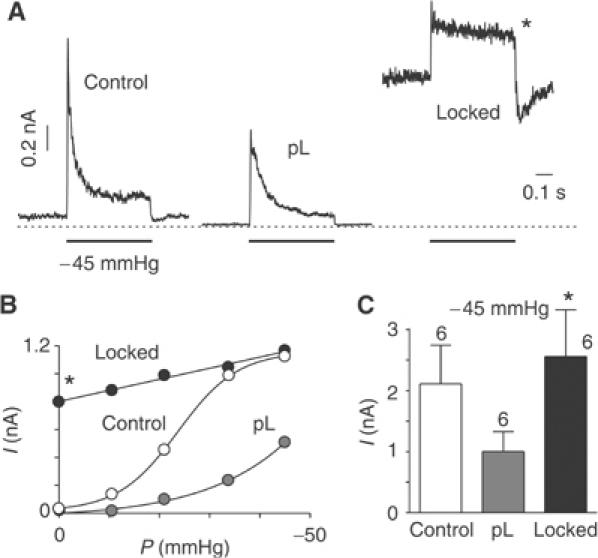
PIP2 affects TREK-1 mechanogating. (A) Effect of a membrane pressure of −45 mmHg on TREK-1 channel activity in an inside-out patch held at 0 mV (physiological K+ gradient). pL (30 μg/ml) and PIP2 (5 μM) were added intracellularly. After PIP2 addition (5 μM for 2 min), cytosolic pH was lowered from 7.2 to 5.5 for 30 s. Channels were locked open when returning to pHi 7.2 (indicated by a star). (B) Pressure–effect curve of TREK-1 in control, in the presence of pL (30 μg/ml), and following cytosolic acidosis (pHi 5.5) after PIP2 (5 μM) washout. The pressure–effect curves were fitted with Boltzmann relationships. The P0.5 values are −24 mmHg and estimated at −84 mmHg for control and pL, respectively. The slope factors are 6.0 and 15.2 for control and pL, respectively. (C) Mean current amplitude of TREK-1 at a pressure of −45 mmHg in control, pL (30 μg/ml) and following cytosolic acidosis (pHi 5.5) after PIP2 (5 μM) washout (open lock indicated by a star).
TREK-1 is known to display an intrinsic voltage dependency (Bockenhauer et al, 2001; Maylie and Adelman, 2001; Maingret et al, 2002). The basal TREK-1 current (at atmospheric pressure and at pHi 7.2) is composed of two distinct components (Maingret et al, 2002): (i) one K+-selective leak component that shows a modest outward rectification and that is instantaneous upon depolarization (Ito); (ii) one time-dependent outwardly rectifying component that slowly activates at depolarized potentials (above 40 mV) and then deactivates upon repolarization (Ist) (Figure 4C and Supplementary Figure 1C–F). Both current components are absent in control EYFP-transfected cells (Supplementary Figure 1C, E and F). pL (30 μg/ml) preferentially inhibits the time- and voltage-dependent component of the basal current (−96±2 and −52±8% for the voltage-dependent Ist and the leak component Ito, respectively; n=12) (Supplementary Figure 1C–F). When TREK-1 is locked open by PIP2, the I–V curve becomes symmetrical and the outward rectification disappears (Figure 4A and B). Furthermore, channel kinetics are altered, with activation becoming instantaneous followed by a partial inactivation (Figure 4A and D inset). PIP2, pH and stretch exert similar effects on TREK-1 voltage dependency (Figure 4B–D). For instance, when the membrane is progressively stretched, the outward rectification seen at atmospheric pressure gradually disappears (Figure 4C and D).
Figure 4.
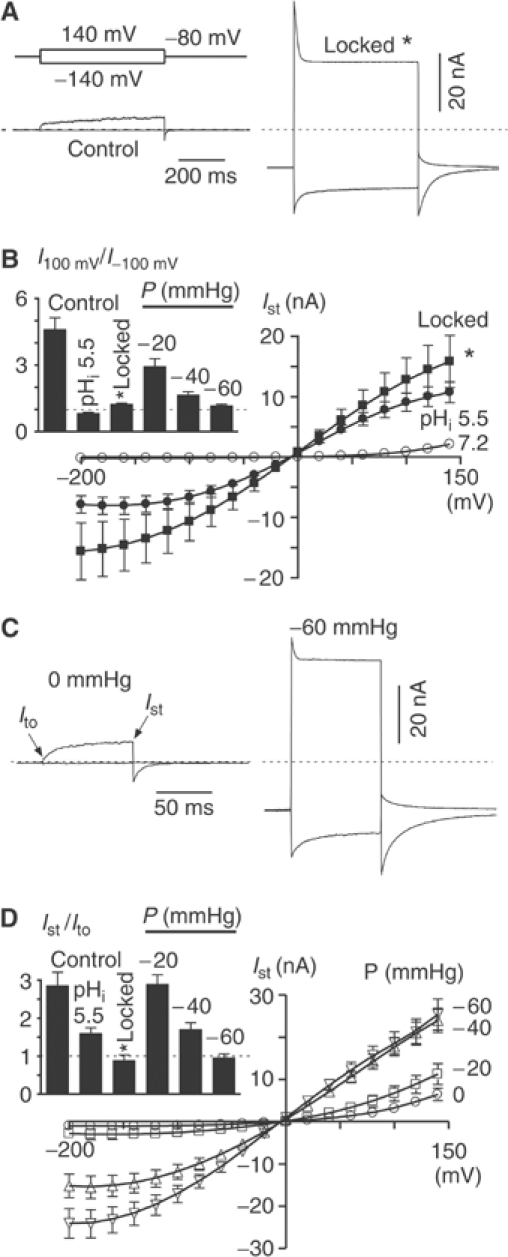
PIP2 affects TREK-1 voltage dependency. (A) Inside-out patches were excised in a symmetrical K+ gradient lacking divalent cations. The holding potential was −80 mV and voltage steps to −140 and 140 mV are illustrated. Currents were recorded in control and following cytosolic acidosis (pHi 5.5) after PIP2 (5 μM) washout (open lock indicated by a star). (B) I–V curves (Ist) in control (empty circles) (n=17), at pHi 5.5 (filled circles) (n=17) and following cytosolic acidosis (pHi 5.5) after PIP2 (5 μM) washout (filled squares) (n=5). The inset shows TREK-1 outward rectification determined by the ratio of current amplitude measured at −100 and 100 mV in control (pHi 7.2) (n=34), during cytosolic acidosis (pHi 5.5) (n=17), following cytosolic acidosis (pHi 5.5) after PIP2 (5 μM) washout (open lock indicated by a star) (n=5) and at increasing pressure (n=16, 17 and 7 for −20, −40 and −60 mmHg, respectively). (C) Effect of membrane stretch (−60 mmHg) on TREK-1 voltage dependency (symmetrical K+ gradient). The holding potential was −80 mV and voltage steps to −140 and 140 mV are illustrated. (D) I–V curves (Ist) were constructed at increasing pressure (indicated in mmHg) (0 mmHg, n=17; −20 mmHg, n=16; −40 mmHg, n=17 and −60 mmHg, n=10). The inset shows the time dependency of TREK-1 measured at 140 mV and expressed as the ratio of the steady state (Ist) over the initial (Ito) current amplitude in control (pHi 7.2) (n=34), during cytosolic acidosis (pHi 5.5) (n=17), following cytosolic acidosis (pHi 5.5) after PIP2 (5 μM) washout (open lock indicated by a star) (n=5) and at increasing pressure (n=16, 17 and 11 for −20, −40 and −60 mmHg, respectively).
Positive charges in the proximal carboxy-terminal domain of TREK-1 are critically required for PIP2 stimulation
To identify the channel region responsible for PIP2 stimulation of TREK-1, we generated truncated, chimeric as well as point mutants (Figure 5A and B). Complete deletion of the amino-terminal domain (ΔN) does not affect PIP2 stimulation (Figure 5A–C). Similarly, deletion of the last 100 amino acids in the carboxy-terminal domain of TREK-1 (ΔC100) has no significant effect on PIP2 effects (Figure 5A–C). A complete deletion of the carboxy-terminal domain (ΔC119) impairs membrane expression as well as channel activity (n=12) (Supplementary Figure 2A and B). However, a chimera between the whole carboxy-terminal domain of TASK-3 and the core domain of TREK-1 remains functional and is resistant to PIP2 (Figure 5A–C). Thus, the protein segment of the cytosolic carboxy-terminal domain of TREK-1 upstream of R311 is critical for PIP2 stimulation (Figure 5A). This region is characterized by the presence of several positively charged residues that appear to be excellent candidates for binding PIP2 (Figure 5A). We then engineered an internal deletion in this region (intraΔC) (Figure 5A–C). The intraΔC mutant is active and comparable to TREK-1 WT in current amplitude (Figure 5B), but, similar to the TREK-1/TASK-3 chimera, is not activated by cytosolic PIP2 (Figure 5C). A systematic substitution of the positively charged amino acids by alanines progressively leads to the suppression of TREK-1 stimulation by PIP2 (Figure 5A–C). When all five positive charges are removed simultaneously, the mutant 5+A remains active but becomes completely resistant to PIP2 activation (Figure 5B and C). Furthermore, it loses its capacity to be definitively locked open after a cytosolic acidic stimulation in the presence of PIP2 (n=9). Thus, this cationic cluster is critical for PIP2 stimulation of TREK-1.
Figure 5.
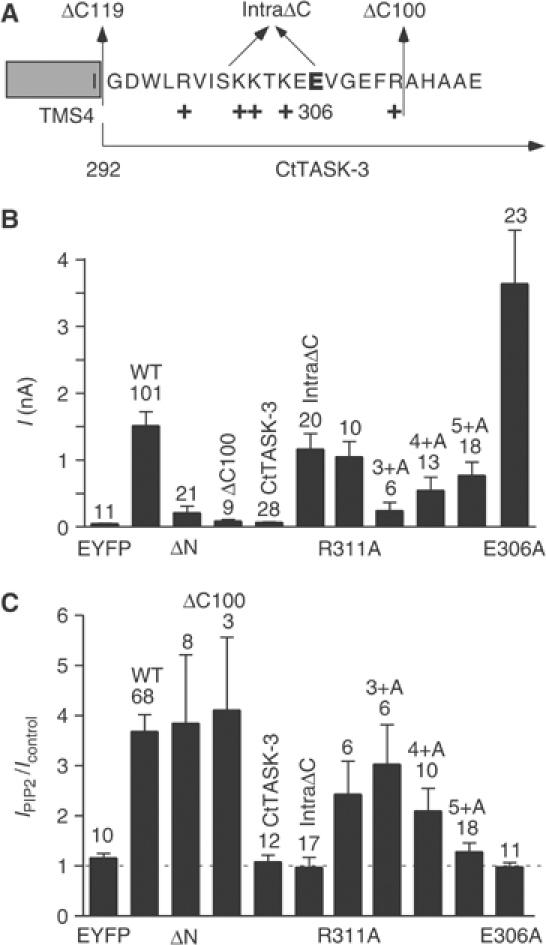
Positive charges in the carboxy-terminal domain of TREK-1 are critically required for PIP2 stimulation. (A) Sequence of the proximal carboxy-terminal domain of TREK-1 illustrating truncation, chimera, as well as point mutations. In the ΔN mutant, the whole amino-terminal domain was deleted (Met was introduced at position 46). In the intraΔC mutant, the region between K301 and E306 was deleted. The ΔC100 mutant was deleted after R311. The ΔC119 was deleted after I292. In the TREK-1/CtTASK-3 chimera, the whole carboxy-terminal domain of TASK-3 was fused to the core of TREK-1 (position 292). In the R311A mutant, arginine at position 311 is substituted by an alanine. In the 3+A mutant R297, K301 and K304 are substituted by an alanine. In the 4+A mutant R297, K301, K302 and K304 are substituted by an alanine. In the 5+A mutant, R297, K301, K302, K304 and R311 are substituted by an alanine. In the E306A mutant, glutamate at position 306 is substituted by an alanine. (B) The histogram illustrates the basal current amplitude of the TREK-1 mutants measured at 0 mV in the inside-out patch configuration. (C) The histogram illustrates the effect of PIP2 (5 μM) after pL (30 μg/ml) treatment.
Protonation of E306 increases TREK-1/PIP2 interaction
The positively charged cluster encompasses the negatively charged proton sensor E306, that is essential for cytosolic acidic stimulation of TREK-1 (Honoré et al, 2002) (Figure 5A). The E306A mutation mimics the effect of cytosolic acidosis in the stimulation of TREK-1 channel activity (Honoré et al, 2002). The E306A mutant is constitutively active and undergoes no further activation by cytosolic PIP2 (Figure 5A–C). There are two possible explanations for this observation. The first is that PIP2 is not required for E306A activity. The second one is that, on the contrary, the replacement of E306 by an alanine has drastically increased the capacity of TREK-1 to bind PIP2 in such a way that it becomes resistant to pL. Maximal channel inhibition is observed with about 0.3 μg/ml pL for TREK-1 WT, whereas only a small effect is seen for E306A under the same conditions (Figure 6A). Significant inhibition of the E306A mutant by pL can be observed, but at much higher concentrations (3–30 μg/ml) (Figure 6A). Also, while the time constant for TREK-1 WT inhibition by 0.3 μg/ml pL is about 15 s (Figure 6B and C), the time constant for inhibition of E306A by 0.3 μg/ml pL is much slower in the range of 45 s (Figure 6B and C). Since replacing E306 by an alanine is equivalent to protonating this particular glutamate, we would expect to find similar properties for TREK-1 at pHi 5.5. Indeed, TREK-1 WT becomes more resistant to pL inhibition at pHi 5.5 (Figure 6A–C). Thus, protonation of the negatively charged residue E306 at cytosolic acidic pH probably increases the affinity of TREK-1 (i.e. of the positively charged cluster) for PIP2, making it more resisant to pL inhibition, as also observed for E306A. An alternative explanation is that pL may directly inhibit TREK-1 by interacting with E306. However, the double mutant Δ100 TREK-1 E306A that is pHi-resistant but remains mechanogated (Honoré et al, 2002), unlike E306A which is locked open, is also sensitively inhibited by cytosolic pL (−63±6% with 0.3 mg/ml pL with a time constant of 17.1±3.9 s at 0 mV, n=6), suggesting that this possibility is unlikely.
Figure 6.
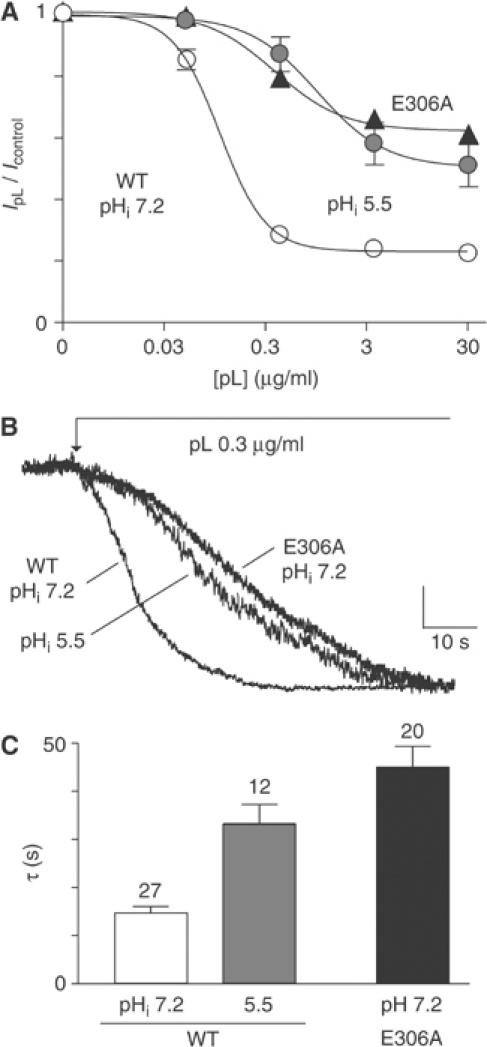
Cytosolic acidification increases TREK-1/PIP2 interaction. (A) Dose–effect curve of pL on TREK-1 WT at pHi 7.2 (n=8) and pH 5.5 (n=9), and on E306A at pHi 7.2 (n=8). EC50 values are 0.074, 0.774 and 0.285 μg/ml for TREK-1 WT at pH 7.2, pH 5.5 and E306A. Slope factors are 1.62, 1.33 and 1.26 for TREK-1 WT at pH 7.2, pH 5.5 and E306A. (B) Kinetic of current inhibition by 0.3 μg/ml pL. The vertical scale is 300, 200 and 250 pA for WT pH 7.2, WT pH 5.5 and E306A. (C) The histogram shows the mean time constant of current inhibition determined by mono-exponential fitting. In these experiments, inside-out patches were held at 0 mV (physiological K+ gradient). The time constants measured for TREK-1 WT at pHi 5.5 and for E306A at pHi 7.2 are significantly slower (P<0.01) as compared to TREK-1 WT measured at pHi 7.2.
Phospholipid specificity of TREK-1 stimulation
To determine whether PIP2 is specific in its action on TREK-1, various phospholipids were tested (Figure 7A–D). The phospholipid PI that lacks both phosphates on the inositol group similarly stimulates TREK-1 channel activity (Figure 7A and B). Moreover, PS and PE that do not belong to the phosphoinositide family, but are other inner leaflet phospholipids, also stimulate TREK-1 (n=16 and 5, respectively) (Figure 7A–D). Similar to PIP2, the various phospholipids lock TREK-1 in the open conformation during cytosolic acidosis (Figure 7C and D). The presence of a large polar head is not an absolute requirement since PA also produces a strong stimulation of TREK-1 after a previous pL treatment (n=6) (Figure 7A and B). However, the negative phosphate group at position 3 of the glycerol is critical as DG does not stimulate TREK-1 channel activity (n=5) (Figure 7A and B).
Figure 7.
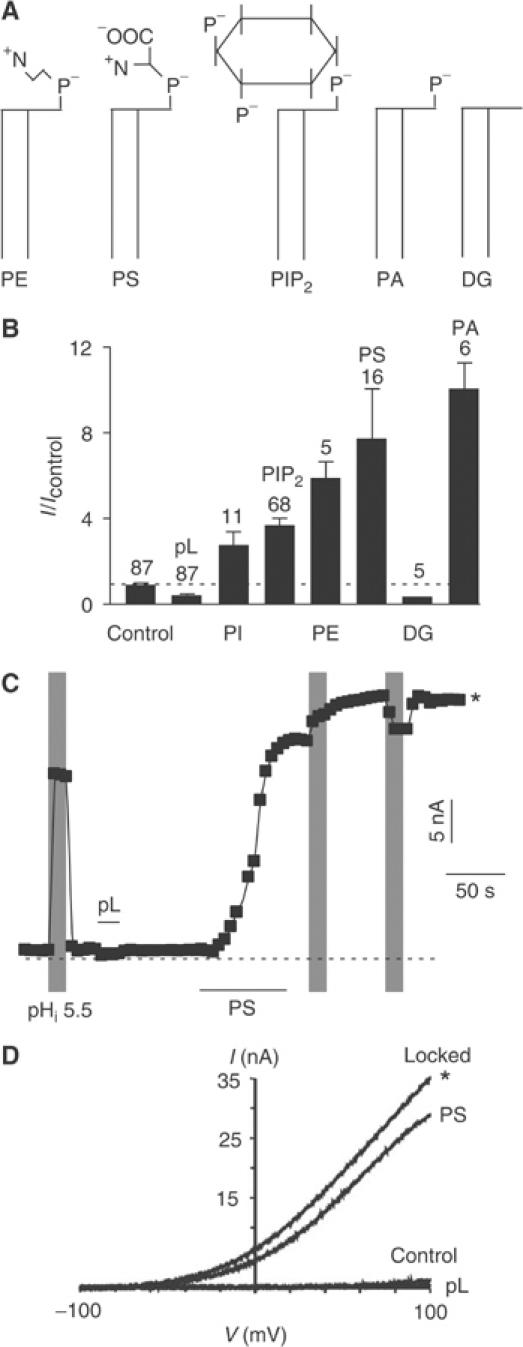
Phospholipid specificity of TREK-1 modulation. (A) Diagram of the different phospholipid molecules. (B) Histogram showing the effect of various phospholipids (5 μM) on TREK-1 currents after pL treatment (30 μg/ml), recorded in inside-out patches and measured at 0 mV (physiological K+ gradient). (C) Effect of cytosolic acidosis to pH 5.5 (indicated by gray shading), pL (30 μg/ml) and PS (5 μM) on TREK-1 recorded in an inside-out patch measured at 0 mV (physiological K+ gradient). (D) I–V curves elicited by voltage ramps illustrating the effect of pL (30 μg/ml) and PS (5 μM) before and after cytosolic acidosis to pH 5.5.
The carboxy-terminal domain of TREK-1 interacts with the plasma membrane
To demonstrate that the carboxy-terminal domain of TREK-1 interacts with the plasma membrane phospholipids, we took advantage of the SOS recruitment strategy in yeast (Aronheim, 2001). Briefly, the Saccharomyces cerevisiae cdc25-2 strain harbors a temperature-sensitive mutation in its guanyl exchange factor (GEF) cdc25, the yeast homolog of human SOS. This mutation prevents binding of GEF and activation of the small G protein Ras and consequently impairs cell growth at the nonpermissive temperature of 37°C (Figure 8A and B). However, Ras activation and cell growth occur at the permissive temperature of 25°C (Figure 8A and B). This system can be complemented by expressing the full-length hSOS (Aronheim, 2001). But, an amino-terminal fragment of hSOS (aa 1–1067) is unable to go to the membrane by itself and thus does not activate Ras (Figure 8A and B). Plasma membrane targeting of this hSOS deletion can be rescued by fusing it to a membrane-associated protein. Dimerization of Jun was used as a positive control (Figure 8B; see Materials and methods). Interestingly, the carboxy-terminal domain of TREK-1 brings the SOS fragment to the plasma membrane and allows cell growth at 37°C (Figure 8A and B). This result suggests that the cytosolic carboxy-terminal domain of TREK-1 is in close proximity with the plasma membrane. This effect is specific to TREK-1 as the fusion proteins SOS/TASK-1 or SOS/TWIK-2, two other K2P channels, do not allow growth at 37°C (Figure 8B). Deletion of the highly charged region (aa 293–307) in the 5′ end of the carboxy-terminal domain of TREK-1 (ΔCtTREK-1) impairs growth of transformed cdc25-2 cells at 37°C (Figure 8B). Substitution of the five positively charged residues required for PIP2 stimulation by an alanine (5+A mutant) similarly impairs cell growth, unlike the E306A mutation (Figure 8B).
Figure 8.
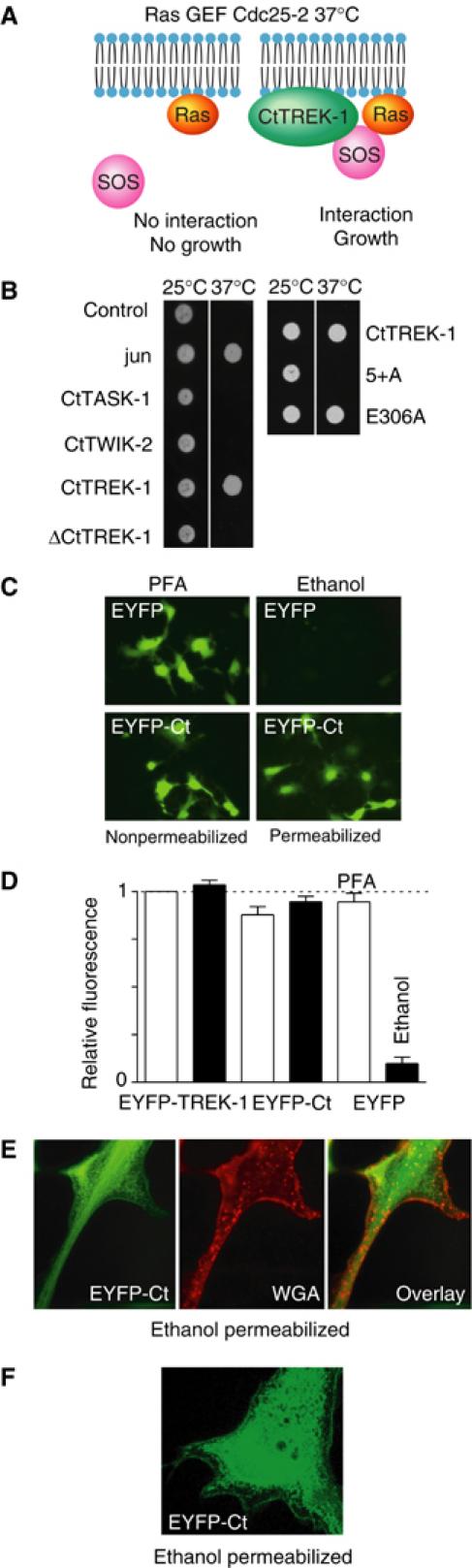
Interaction of the carboxy-terminal domain of TREK-1 with the plasma membrane. (A) The yeast SOS recruitment strategy was used to investigate the interaction of the carboxy-terminal domain of TREK-1 with the plasma membrane. Cdc25-2 cells are growing at the permissive temperature of 25°C and Jun dimerization is used as a positive control. At the nonpermissive temperature 37°C, cell growth is impaired. The cartoon illustrates the SOS strategy. (B) In these experiments, the rescue ability of the carboxy-terminal domain of various channels and mutants is illustrated by cell growth (left panel). Effects of the 5+A and E306A mutations on SOS recruitment (right panel). (C) Expression of the cytosolic EYFP and the fusion protein EYFP-CtTREK-1 in transiently transfected COS cells. Cells were fixed with either ethanol permeabilization or in the absence of permeabilization with PFA. Images were acquired with an × 25 oil immersion objective and an EYFP filter. (D) Quantification of the fluorescence in EYFP-TREK-1-, EYFP-Ct TREK-1- and EYFP-transfected COS cells fixed with (ethanol) or without permeabilization (PFA). (E) Cells were ethanol permeabilized and labelled with WGA. Images were collected with an × 63 oil immersion lens and filter sets for EYFP (green) and Cy3 (red). The overlay of EYFP and WGA fluorescence is illustrated in the right panel. (F) Confocal image of an ethanol-permeabilized EYFP-Ct TREK-1-transfected cell acquired with an × 100 oil immersion objective and laser excitation for FITC. The image corresponds to a section of 0.1 μm.
We next generated a fusion protein between the fluorescent protein EYFP and the carboxy-terminal domain of TREK-1 (EYFP-CtTREK-1). This construct was transiently expressed in COS cells that were either fixed without permeabilization by paraformaldehyde (PFA) or with permeabilization by ethanol (Figure 8C). In parallel experiments, COS cells expressing the cytosolic EYFP were fixed under the same conditions. Fixation with permeabilization leads to a loss of the fluorescence signal within less than 20 min in the control EYFP condition. On the contrary, the fluorescence signal remains very intense in the EYFP/CtTREK-1 condition even with a 12 h permeabilization step (Figure 8C and D). EYFP-CtTREK-1 is expressed at the membrane (visualized by wheat germ agglutinin fluorescence) of ethanol-permeabilized transfected cells (Figure 8E). Membrane localization was confirmed with confocal microscopy (Figure 8F). Deletion of this carboxy-terminal region in TREK-1 (ΔC119 TREK-1) impairs membrane localization and consequently channel activity (Supplementary Figure 2).
Discussion
TREK-1 is a polymodal K+ channel that is opened by both physical and chemical stimulations (Patel and Honoré, 2001a; 2001b; Patel et al, 2001). The channel has an unusual pharmacology and plays an important role in general anesthesia and neuroprotection (Heurteaux et al, 2004). TREK-1 is a mechanosensitive ion channel that is modulated by cytosolic acidosis (Patel et al, 1998; Maingret et al, 1999; Honoré et al, 2002). Cytosolic acidosis shifts the pressure–effect curve of TREK-1 towards more positive pressure values, leading to channel opening at atmospheric pressure (Maingret et al, 1999; Honoré et al, 2002). Cytosolic acidosis protonates E306 in the proximal carboxy-terminal domain of TREK-1 and is mimicked by the E306A mutation (Honoré et al, 2002).
In the present report, we demonstrate that both the cloned and endogenous neuronal TREK-1 channels are highly dependent for their activity on membrane phospholipids. The cationic molecules pL and spermine, that have a high affinity for phospholipids, inhibit TREK-1 channel activity at rest, but the activity is restored and even stimulated by PIP2 (Figure 9A). The inhibitory effect of polycationic molecules is thought to be due to the fact that they interact with the negative charges of essential membrane phospholipids, thus removing them from their electrostatic interaction with specific positively charged segments in the channel protein (Huang et al, 1998; Lopes et al, 2002). In the presence of intracellular pL or other polyamines, TREK-1 remains in an inactive state (Figure 9A). We have identified a cluster of five positive charges in the proximal carboxy-terminal domain of TREK-1 that is central to the effect of phospholipids (Figure 5A). When the positive charges are deleted, TREK-1 becomes resistant to the activation by PIP2. The upmodulation of TREK-1 channel activity by PIP2 or PA is not additive with AA stimulation (Chemin et al, 2003). By contrast, in the presence of AA, PA as well as DG inhibit TREK-1 activity (Chemin et al, 2003). Phospholipases control phospholipid levels. However, unlike the TASK-1/-3 channels, PIP2 hydrolysis via phosholipase C activation is not involved in TREK-1 inhibition by stimulation of metabotropic glutamate Gq-coupled receptors (Chemin et al, 2003). Rather, DG that is released after phospholipase C activation directly inhibits TREK-1 (Chemin et al, 2003).
Figure 9.
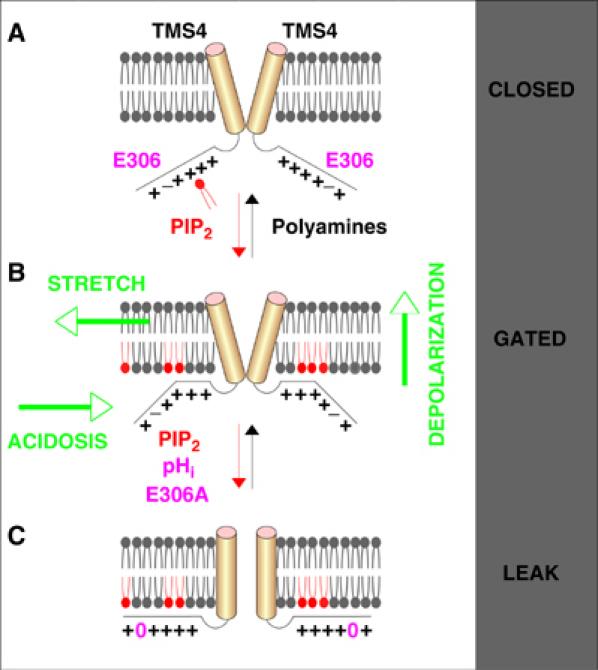
Model of TREK-1 gating. (A) In the presence of pL or endogenous polyamines, TREK-1 is in the closed state and not activable (closed state). (B) Phospholipids, including PIP2, electrostatically interact with the positively charged cluster (5+) in the cytosolic proximal carboxy-terminal domain of TREK-1. Insertion of PIP2 in the inner leaflet of the bilayer controls coupling of the carboxy-terminal domain of TREK-1 with the plasma membrane. When partially coupled, TREK-1 is in the closed state but activable by membrane stretch, depolarization and cytosolic acidosis (gated state). (C) This membrane interaction is favored when the negative charge of the proton sensor E306 is masked by either protonation at acidic pHi or by substitution with an alanine (E306A). The E306A mutant is locked open and behaves as a leak K+ channel. Similarly, in the presence of exogenous phospholipids, cytosolic acidosis irreversibly locks TREK-1 open (leak state).
Using a complementation yeast system designed to assay for membrane–protein interactions (Aronheim, 2001), we have shown that the carboxy-terminal domain of TREK-1 is in close proximity to the plasma membrane. Moreover, the positive charges in this cluster shown to be essential for PIP2 action are also crucial for this membrane interaction. These results were confirmed by a fluorescence assay also indicating that the carboxy-terminal domain of TREK-1 is probably closely associated with the plasma membrane.
A model presented in Figure 9 summarizes our results and our mechanistic interpretation concerning the effects of PIP2. When the positively charged segment cannot interact with the inner leaflet phospholipids, as for instance in the presence of polyamines, the TREK-1 channel is inactive and no stimulus such as stretch or internal acidification can promote channel activity (Figure 9A). When PIP2 interacts with the positively charged carboxy-terminal segment and inserts in the membrane, the channel becomes partially coupled to the membrane and activable by membrane stretch, depolarization and cytosolic acidification (Figure 9B). This model proposes that E306 may play an important regulatory role in the interaction between membrane phospholipids and the carboxy-terminal domain of TREK-1 (Figure 9B). When the side chain of E306 is protonated at cytosolic acidic pH, the positively charged character of the corresponding protein segment is increased, favoring a stronger electrostatic interaction with the negative charges of the inner leaflet phospholipid (Figure 9C). This interaction is so tight that application of PIP2 at acidic pH results not only in a very strong stimulation of TREK-1, but also an activity that is maintained even when the cytosolic pH is brought to normal. Channel locking in the open conformation is definitive. The TREK-1 channel becomes constitutively open. The same situation may be created when, instead of being protonated, E306 is replaced by an alanine. Although, TREK-1 becomes more resistant to pL at acidic pHi or when E306, the proton sensor, is substituted by an alanine, it remains to be directly demonstrated that protonation of this residue indeed results in a stronger PIP2 binding that locks the channel open. TREK-1 changes from a voltage-dependent to a leak mode when it is: (i) stimulated by PIP2; (ii) stimulated by cytosolic acidification; (iii) activated by membrane stretch. There is a tight link between TREK-1 stretch activation and membrane phospholipids. Although PIP2 is generally considered as the key phospholipid for regulation of many ion channels, we have shown in this work that other inner leaflet phospholipids such as PI, PE or PS can fulfill exactly the same key role for TREK-1 channel gating. In the basal condition, TREK-1 current is made up of a leak component and a voltage- and time-dependent component. Since the leak component is more resistant to inhibition by pL, it may therefore correspond to channels that are already fully coupled to the membrane phospholipids and thus locked in the open conformation (Figure 9C). The remaining voltage-dependent channels may be in a partially coupled conformation, thus activable by stretch, acidosis and depolarization (Figure 9B).
These findings provide a substantially better view of the gating mechanism of the TREK channels that were the first mammalian mechanosensitive ion channels to be cloned. They may also have a more general implication since many other types of mechanically activated ion channels (although most probably belonging to the TRP family of channels; Voets and Nilius, 2003) are known to be essential to sense pressure variations in mammalian cells (Sachs and Morris, 1998; Bode et al, 2001; Hamill and Martinac, 2001; Suchyna et al, 2004).
Materials and methods
Neuronal cell cultures
Primary culture of mouse striata was carried out as described previously (Heurteaux et al, 2004). Cells were plated in culture dishes previously coated with polyornithine and 50% fetal calf serum. Culture medium was DMEM (Invitrogen) with glucose (1.5 g/l) for 24 h and then B27 with uridine (2 μM) and 5-fluoro-2′-deoxyuridine (2 μM).
Electrophysiological recordings
Patch-clamp measurements of mouse striatal neurons were performed 2 or 3 days after plating. In inside-out configuration the internal solution contained (in mM): 155 KCl, 5 EGTA, 10 HEPES at pH 7.2 with KOH, and the external solution contained (in mM): 120 NaCl, 5 KCl, 3 MgCl2, 1 CaCl2 and 10 HEPES at pH 7.4 with NaOH. Tetra-ethylammonium chloride (10 mM), 3 mM 4-aminopyridine, 10 μM glibenclamide and 5 mM glucose were freshly added and pH adjusted with HCl. For COS cell experiments, culture, mutagenesis, transfection and the electrophysiological procedure have been detailed elsewhere (Patel et al, 1998; Maingret et al, 1999; 2002; Honoré et al, 2002). The long spliced form of mTREK-1, with an extension of 15 amino acids in the amino-terminal domain, was used in the present study (Bockenhauer et al, 2001) (mTREK-1 sequence submission). In some experiments, an amino-terminal EYFP-tagged mTREK-1 channel was used and no functional difference with untagged WT channel was observed, including sensitivity to phospholipids. For clarity and consistency with our previous work, amino-acid numbering is related to the short mTREK-1 splice variant (Fink et al, 1996). We routinely transfected between 0.1 and 0.5 μg DNA per 35 mm diameter plate containing 20 000 cells. The external medium contained (in mM): 150 NaCl, 5 KCl, 3 MgCl2, 1 CaCl2 and 10 HEPES at pH 7.4 with NaOH. To study the voltage dependency of TREK-1, we used an extracellular medium containing (in mM): 155 KCl, 5 EGTA, 5 EDTA and 10 HEPES at pH 7.4 with KOH. The cytosolic medium contained (in mM): 155 KCl, 5 EGTA, 10 HEPES at pH 7.2 with KOH. For Xenopus oocyte patch experiments, we used an extracellular medium containing (in mM): 96 NaCl, 2 KCl, 1 MgCl2, 1 CaCl2 and 5 HEPES at pH 7.4 with NaOH and an internal medium containing (in mM) 96 KCl, 5 EGTA and 10 HEPES at pH 7.2 with KOH. Patch pipettes of about 1.5 Mohm were used for excised inside-out patches. Patches were routinely held at a holding potential of −80 mV and stimulated by voltage ramps of 660 ms in duration from −100 to 100 mV every 5 s. Membrane stretch and cytosolic acidosis were applied as described previously (ALA fast pressure-clamp system) (Patel et al, 1998; Maingret et al, 1999; Honoré et al, 2002). Phospholipids (Sigma and Biomol TEBU) were dissolved in a mixture of chloroform/methanol/water/HCl (20/10/1/1) at a concentration of 5 mM. Aliquots were kept under argon at −80°C. Aliquots were daily evaporated with air and lipids were dissolved in the saline solution that was sonicated on ice for 15 min before use. pL was dissolved in water at a concentration of 30 mg/ml and stored at −20°C. Control experiments were carried out using the evaporated solvent alone. The Dunnet ANOVA test was used for statistical analysis. Differences were considered significant at P<0.05.
Yeast experiments
The yeast SOS recruitment strategy (Aronheim, 2001) was used to investigate the interaction of the carboxy-terminal domain of TREK-1 with the plasma membrane. We constructed the SOS (aa 1–1067)/CtTREK-1 (aa 293–411) fusion protein, the SOS/CtTASK-1 (aa 243–394) and the SOS/CtTWIK-2 (aa 244–313). We generated a truncated SOS/ΔCtTREK-1 mutant (aa 308–411). We introduced the E306A mutation in the SOS/CtTREK-1 as well as the 5+A mutant R297, K301, K302, K304 and R311. As a positive control, the following expression vectors were used: pADNS 5′SOS (aa 1–1066), JZ (aa 249–331) of c-Jun and pYES2 expresssing a myristilated c-Jun (249–331) (localization to the plasma membrane) (Aronheim, 2001). In isolation, these plasmids do not allow growth of transformed cdc25-2 cells at 37°C. However, when both plasmids are introduced into cdc25-2 cells, growth is obtained at the nonpermissive temperature of 37°C because of Jun dimerization. 5′SOS is now targeted to the membrane, can bind and activate Ras, thus rescuing cell growth at 37°C (Aronheim, 2001).
Fluorescence experiments
The cytosolic EYFP or the fusion protein EYFP/CtTREK-1 (starting at amino acid 293) was transiently expressed in COS cells as described previously. After 48 h of expression, cells were fixed for 20 min with paraformaldehyde (4% at room temperature) or with ethanol (70° on ice for 3–12 h). During ethanol fixation, cells are rapidly permeabilized. After fixation, cells were stained with fluorescent wheat germ agglutinin (Alexa Fluor-594 conjugated, 3 μg/ml, Molecular Probes) and DAPI (Vector Labs) for 5 min. After mounting, specimens were observed using an epifluorescence microscope (Axioplan 2, Carl Zeiss) equipped with × 25 and × 63 oil immersion objectives and appropriate combinations of filters. Images were recorded with a cooled CCD camera (Coolsnap HQ, Photometrics) driven by Metavue software. For confocal microscopy, cells were examined with a scanning laser confocal microscope (Leica) using an × 100 oil immersion objective. For fluorescence quantification, 20–40 independent fields from two separate experiments have been analyzed per experimental condition. Fluorescence was determined in blind and quantified as either strong, medium or absent. Mean values were normalized to the fluorescence of nonpermeabilized (PFA) EYFP-TREK-1-expressing COS cells. Similar results were obtained with three independent transfections.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Information
Acknowledgments
This work has been supported by the CNRS, the FRM, l'ARC and la Ligue contre le cancer and the Fondation Paul Hamel. We are grateful to Professor Michael Karin for providing reagents for the SOS experiments. We thank Bruno Antonny and Bruno Mesmin for their constructive comments on this work. Valérie Briet is acknowledged for preparing the manuscript. Martine Jodar is acknowledged for expert technical assistance.
References
- Aronheim A (2001) Ras signaling pathway for analysis of protein–protein interactions. Methods Enzymol 332: 260–270 [DOI] [PubMed] [Google Scholar]
- Bang H, Kim Y, Kim D (2000) TREK-2, a new member of the mechanosensitive tandem pore K+ channel family. J Biol Chem 275: 17412–17419 [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, Ruppersberg JP, Fakler B (1998) PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 282: 1141–1144 [DOI] [PubMed] [Google Scholar]
- Berg AP, Talley EM, Manger JP, Bayliss DA (2004) Motoneurons express heteromeric TWIK-related acid-sensitive K+ (TASK) channels containing TASK-1 (KCNK3) and TASK-3 (KCNK9) subunits. J Neurosci 24: 6693–6702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockenhauer D, Zilberberg N, Goldstein SA (2001) KCNK2: reversible conversion of a hippocampal potassium leak into a voltage-dependent channel. Nat Neurosci 4: 486–491 [DOI] [PubMed] [Google Scholar]
- Bode F, Sachs F, Franz MR (2001) Tarantula peptide inhibits atrial fibrillation. Nature 409: 35–36 [DOI] [PubMed] [Google Scholar]
- Chemin J, Girard C, Duprat F, Lesage F, Romey G, Lazdunski M (2003) Mechanisms underlying excitatory effects of group I metabotropic glutamate receptors via inhibition of 2P domain K+ channels. EMBO J 22: 5403–5411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411: 957–962 [DOI] [PubMed] [Google Scholar]
- Czirjak G, Enyedi P (2001) Formation of functional heterodimers between the TASK-1 and TASK-3 two pore domain potassium channel subunits. J Biol Chem 277: 5432–5436 [DOI] [PubMed] [Google Scholar]
- Czirjak G, Petheo GL, Spat A, Enyedi P (2001) Inhibition of TASK-1 potassium channel by phospholipase C. Am J Physiol Cell Physiol 281: C700–C708 [DOI] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Patel AJ, Fink M, Romey G, Lazdunski M (2000) The neuroprotective agent riluzole activates the two P domain K+ channels TREK-1 and TRAAK. Mol Pharmacol 57: 906–912 [PubMed] [Google Scholar]
- Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, Lazdunski M (1996) Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J 15: 6854–6862 [PMC free article] [PubMed] [Google Scholar]
- Fink M, Lesage F, Duprat F, Heurteaux C, Reyes R, Fosset M, Lazdunski M (1998) A neuronal two P domain K+ channel activated by arachidonic acid and polyunsaturated fatty acid. EMBO J 17: 3297–3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Martinac B (2001) Molecular basis of mechanotransduction in living cells. Physiol Rev 81: 685–740 [DOI] [PubMed] [Google Scholar]
- Heurteaux C, Guy N, Laigle C, Blondeau N, Duprat F, Mazzuca L, Lang-Lazdunski L, Widmann C, Zanzouri M, Romey G, Lazdunski M (2004) TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J 23: 2684–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann DW, Feng S, Nasuhoglu C (2001) The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE 2001: RE19. [DOI] [PubMed] [Google Scholar]
- Honoré E, Maingret F, Lazdunski M, Patel AJ (2002) An intracellular proton sensor commands lipid- and mechano-gating of the K+ channel TREK-1. EMBO J 21: 2968–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL, Feng S, Hilgemann DW (1998) Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 391: 803–806 [DOI] [PubMed] [Google Scholar]
- Kang D, Han J, Talley EM, Bayliss DA, Kim D (2004) Functional expression of TASK-1/TASK-3 heteromers in cerebellar granule cells. J Physiol 554: 64–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D (2003) Fatty acid-sensitive two-pore domain K+ channels. Trends Pharmacol Sci 24: 648–654 [DOI] [PubMed] [Google Scholar]
- Kobrinsky E, Mirshahi T, Zhang H, Jin T, Logothetis DE (2000) Receptor-mediated hydrolysis of plasma membrane messenger PIP2 leads to K+-current desensitization. Nat Cell Biol 2: 507–514 [DOI] [PubMed] [Google Scholar]
- Lauritzen I, Zanzouri M, Honoré E, Duprat F, Ehrengruber MU, Lazdunski M, Patel AJ (2003) K+-dependent cerebellar granule neuron apoptosis: role of TASK leak K+ channels. J Biol Chem 278: 32068–32076 [DOI] [PubMed] [Google Scholar]
- Lesage F, Lazdunski M (2000) Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol 279: F793–F801 [DOI] [PubMed] [Google Scholar]
- Lesage F, Reyes R, Fink M, Duprat F, Guillemare E, Lazdunski M (1996) Dimerization of TWIK-1 K+ channel subunits via a disulfide bridge. EMBO J 15: 6400–6407 [PMC free article] [PubMed] [Google Scholar]
- Lesage F, Terrenoire C, Romey G, Lazdunski M (2000) Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J Biol Chem 275: 28398–28405 [DOI] [PubMed] [Google Scholar]
- Lopes CM, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE (2002) Alterations in conserved Kir channel–PIP2 interactions underlie channelopathies. Neuron 34: 933–944 [DOI] [PubMed] [Google Scholar]
- Loussouarn G, Park KH, Bellocq C, Baro I, Charpentier F, Escande D (2003) Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J 22: 5412–5421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Honoré E, Lazdunski M, Patel AJ (2002) Molecular basis of the voltage-dependent gating of TREK-1, a mechano-sensitive K+ channel. Biochem Biophys Res Commun 292: 339–346 [DOI] [PubMed] [Google Scholar]
- Maingret F, Lauritzen I, Patel A, Heurteaux C, Reyes R, Lesage F, Lazdunski M, Honoré E (2000a) TREK-1 is a heat-activated background K+ channel. EMBO J 19: 2483–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E (1999) Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J Biol Chem 274: 26691–26696 [DOI] [PubMed] [Google Scholar]
- Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E (2000b) Lysophospholipids open the two P domain mechano-gated K+ channels TREK-1 and TRAAK. J Biol Chem 275: 10128–10133 [DOI] [PubMed] [Google Scholar]
- Maylie J, Adelman JP (2001) Beam me up, Scottie! TREK channels swing both ways. Nat Neurosci 4: 457–458 [DOI] [PubMed] [Google Scholar]
- Mirshahi T, Jin T, Logothetis DE (2003) G beta gamma and KACh: old story, new insights. Sci STKE 2003: PE32. [DOI] [PubMed] [Google Scholar]
- Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B (2004) Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science 304: 265–270 [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E (2001a) Anesthetic-sensitive 2P domain K+ channels. Anesthesiology 95: 1013–1025 [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E (2001b) Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci 24: 339–346 [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Lesage F, Fink M, Romey G, Lazdunski M (1999) Inhalational anaesthetics activate two-pore-domain background K+ channels. Nat Neurosci 2: 422–426 [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Maingret F, Lesage F, Fink M, Duprat F, Lazdunski M (1998) A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J 17: 4283–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AJ, Lazdunski M, Honoré E (2001) Lipid and mechano-gated 2P domain K+ channels. Curr Opin Cell Biol 13: 422–428 [DOI] [PubMed] [Google Scholar]
- Runnels LW, Yue L, Clapham DE (2002) The TRPM7 channel is inactivated by PIP2 hydrolysis. Nat Cell Biol 4: 329–336 [DOI] [PubMed] [Google Scholar]
- Sachs F, Morris CE (1998) Mechanosensitive ion channels in nonspecialized cells. Rev Physiol Biochem Pharmacol 132: 1–77 [DOI] [PubMed] [Google Scholar]
- Shyng SL, Nichols CG (1998) Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282: 1138–1141 [DOI] [PubMed] [Google Scholar]
- Suchyna TM, Tape SE, Koeppe RE II, Andersen OS, Sachs F, Gottlieb PA (2004) Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature 430: 235–240 [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B (2002) Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35: 507–520 [DOI] [PubMed] [Google Scholar]
- Talley EM, Sirois JE, Lei Q, Bayliss DA (2003) Two-pore-domain (KCNK) potassium channels: dynamic roles in neuronal function. Neuroscientist 9: 46–56 [DOI] [PubMed] [Google Scholar]
- Talley EM, Solorzano G, Lei Q, Kim D, Bayliss DA (2001) Cns distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci 21: 7491–7505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Nilius B (2003) TRPs make sense. J Membr Biol 192: 1–8 [DOI] [PubMed] [Google Scholar]
- Yost CS (2003) Update on tandem pore 2P domain K+ channels. Curr Drug Targets 4: 347–351 [DOI] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, Logothetis DE (2003) PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37: 963–975 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Information