


Abstract
Competing endogenous RNAs (ceRNAs) are RNA molecules that sequester shared microRNAs (miRNAs) thereby affecting the expression of other targets of the miRNAs. Whether genetic variants in ceRNA can affect its biological function and disease development is still an open question. Here we identified a large number of genetic variants that are associated with ceRNA's function using Geuvaids RNA-seq data for 462 individuals from the 1000 Genomes Project. We call these loci competing endogenous RNA expression quantitative trait loci or ‘cerQTL’, and found that a large number of them were unexplored in conventional eQTL mapping. We identified many cerQTLs that have undergone recent positive selection in different human populations, and showed that single nucleotide polymorphisms in gene 3΄UTRs at the miRNA seed binding regions can simultaneously regulate gene expression changes in both cis and trans by the ceRNA mechanism. We also discovered that cerQTLs are significantly enriched in traits/diseases associated variants reported from genome-wide association studies in the miRNA binding sites, suggesting that disease susceptibilities could be attributed to ceRNA regulation. Further in vitro functional experiments demonstrated that a cerQTL rs11540855 can regulate ceRNA function. These results provide a comprehensive catalog of functional non-coding regulatory variants that may be responsible for ceRNA crosstalk at the post-transcriptional level.
INTRODUCTION
RNA molecules can operate as competing endogenous RNAs (ceRNAs), which titrate away some of the active miRNAs and thereby indirectly regulate the expression of other transcripts targeted by the same set of miRNAs. Many classes of RNAs may act as ceRNAs, including mRNAs, pseudogene transcripts, long non-coding RNAs and circular RNAs (1), and play critical roles in cellular metabolism and disease development (2). Genetic alteration of ceRNA sequences has been associated with disease progression, which illustrates the potential importance of ceRNA to human health (3,4).
The ceRNA regulatory network is determined by miRNA response elements (MREs), which are the target sites of miRNAs. Genetic variants that perturb MREs can therefore change the dynamic equilibrium of all ceRNAs and miRNAs within the network (2,5,6). One example is the single nucleotide polymorphism (SNP) rs17228616, which disrupts the interaction between miR-608 and the mRNA product of the gene AChE. As a result, miR-608 released from the interaction is able to suppress other targets such as CDC42 and IL6 (7). Despite the fact that numerous examples of genetic variants that affect ceRNAs have been reported (8), no study to our knowledge has been conducted to systematically identify such variants at the genome-wide scale.
Recent advances in sequencing technologies have made it possible to identify genetic variants with molecular phenotypes, which are known as molecular quantitative trait loci (QTLs) (9). Previous studies have reported variants that affect miRNA gene expression (miRNA-eQTL) (9–13) which in turn affect the expression of coding genes whose 3΄ untranslated region are targeted by the miRNA (3΄UTR-eQTLs) (14,15). Here we use genotypes and RNA-seq data from the 1000 Genomes Project (16) and the Geuvadis Project (12) to better understand how genetic polymorphisms shape human ceRNA regulation. Our study is the first systematic investigation of genetic variants that are associated with ceRNA crosstalk.
MATERIALS AND METHODS
Expression data and genotype data
We used Geuvadis RNA sequencing data and small RNA sequencing data of 462 unrelated human lymphoblastoid cell line samples from the CEPH (CEU), Finns (FIN), British (GBR), Toscani (TSI) and Yoruba (YRI) populations in the 1000 Genomes Project (12,16).
Construct variant-miRNA-ceD-ceT unit
We extracted human 3΄UTR sequences using the GENCODE (17) V12 annotation, which was also used by Geuvadis for RNA-seq quantification, and obtained 714 Geuvadis-quantified miRNA sequences from miRBase (18). We mapped Geuvadis biallelic genotypes to 3΄UTR sequences to construct reference and alternative miRNA targets, and used TargetScan 6.2 (19) and ViennaRNA Package (20) to predict miRNA–target relationships. We then selected those ‘variant-miRNA–target’ units that show altered binding affinity between reference and alternative alleles. The miRNA targets that meet the above criteria are termed as putative ceRNA driver genes (ceDs), which were predicted to have differential binding affinity under reference and alternative alleles. For each selected variant-miRNA-ceD unit, we further searched for candidate ceRNA target genes (ceTs) under the control of the same miRNA as the ceD (on either reference or alternative 3΄UTR sequence) according to TargetScan predictions. Finally, we tested for association between genetic variants and their respective miRNA-ceD-ceT (details in Supplementary Methods).
Multivariate multiple regression and confounding factors
We sought to investigate whether a given variant regulates the expression of a ceD and a ceT in a reciprocal pattern due to the ceRNA competition. For each variant-miRNA-ceD-ceT unit, we considered gene expression, measured as the sum of all transcript RPKMs of the ceD (Yd) and the ceT (Yt), as two dependent variables and normalized to follow approximately a standard normal distribution. As predictors we include the individual's genotype (G) and the following factors which might influence the response variables: the PEER (21) residual of miRNA expression (Mr), the 10 PEER factors of ceD expression (PFd), the 10 PEER factors of ceT expression (PFt) and the first three principal components of individual genotype (PC). The multivariate multiple regression on the (ceD, ceT) pair is modeled as:
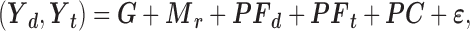 |
where we simultaneously observed two responses, Yd and Yt, and the same set of predictors on each sample unit. The error vector 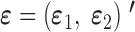 is assumed to follow a multivariate Gaussian distribution with expectation zero and an unknown covariance matrix (i.e. allowing for correlations and different variances for the two responses).
is assumed to follow a multivariate Gaussian distribution with expectation zero and an unknown covariance matrix (i.e. allowing for correlations and different variances for the two responses).
Thus, the variant effect for ceD and ceT can be estimated by the corresponding coefficients on genotype G (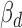 and
and 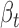 ), and the significance of the multivariate multiple regression model can be assessed by Pillai's trace test statistics (22). However, in our definition of cerQTLs, we required opposite expression trends between ceD and each of its ceTs accompanying the genotype change (AA = 0, AB = 1 and BB = 2), so we further filtered variants to only keep those with their
), and the significance of the multivariate multiple regression model can be assessed by Pillai's trace test statistics (22). However, in our definition of cerQTLs, we required opposite expression trends between ceD and each of its ceTs accompanying the genotype change (AA = 0, AB = 1 and BB = 2), so we further filtered variants to only keep those with their 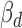 and
and 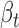 values having opposite signs (
values having opposite signs (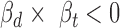 ). We finally reported the variant-miRNA-ceD-ceT units with a Benjamini-Hochberg false discovery rate (FDR) < 0.05 (23). To calibrate the effectiveness of model, we generated an empirical null by performing genotype permutation on each investigated locus by randomly switching the genotype among individuals and applying the model to the permuted data (details in Supplementary Methods).
). We finally reported the variant-miRNA-ceD-ceT units with a Benjamini-Hochberg false discovery rate (FDR) < 0.05 (23). To calibrate the effectiveness of model, we generated an empirical null by performing genotype permutation on each investigated locus by randomly switching the genotype among individuals and applying the model to the permuted data (details in Supplementary Methods).
cerQTL analysis
To be consistent with the Geuvadis RNA-seq quantification, we utilized GENCODE V12 gene annotation to investigate functional properties of ceDs and ceTs. We also used DAVID to find enriched gene annotations and pathways (24). We used SNVrap to annotate genetic variants (25,26), and grouped the genome-wide association study (GWAS) traits according to ontology mapping (human phenotype ontology and disease ontology) of GWASdb (27). We used six statistical measurements from dbPSHP (28), including difference of derived allele frequency (DDAF) (29), fixation Index (FST) (30), Tajima's D (31), integrated haplotype score (iHS) (32), cross-population extended haplotype homozygosity (XPEHH) (33) and cross-population composite likelihood ratio (XPCLR) (34), to evaluate signals of positive selection on each detected cerQTL SNP. To intuitively verify and visualize positive selection of the candidate cerQTLs, we applied hierarchical clustering on their derived allele frequencies (DAF) (details in Supplementary Methods).
GWAS enrichment
SNPs associated with GWAS traits were collected from GWASdb and NHGRI GWAS Catalog (34), and we obtained all linked SNPs with r2 > 0.8 in EUR for each GWAS lead SNP and identified cerQTLs overlapping with this expanded list. We used the hyper-geometric test to assess the enrichment of cerQTLs in GWAS signals (details in Supplementary Methods).
cerQTL experimental validation
We used in vitro assays to validate the functional role of a cerQTL unit (rs11540855-miR-4707-3p-ABHD8-AXIN1). First, we independently evaluated whether both ceD (ABHD8 A-allele or ABHD8 G-allele) and ceT (AXIN1) are biological targets of miR-4707-3p using dual luciferase reporter assay (35) and Western blot (details in Supplementary Methods). To investigate changes of ceRNA competition under different alleles, we then engineered the MCF116 breast cancer cells to stably express ABHD8 A-allele or ABHD8 G-allele and inspected the AXIN1 protein change upon miR-4707-3p agomir treatment. We further investigated ceRNA expression changes by miR-4707-3p agomir dose-dependent experiments (details in Supplementary Methods).
RESULTS
The logic for detecting genetic variants affecting ceRNA regulation
We assume that genetic variants in MREs, such as SNPs and indels, will perturb ceRNA regulation by titrating miRNA availability. Specifically, a variant in a gene's 3΄UTR can influence miRNA–target interaction, by creating, erasing, strengthening or weakening an MRE. We refer to genes with altered MREs as ceDs, and the interaction among miRNAs and ceRNAs as miRNA-ceRNA networks. Changes to ceDs can perturb the miRNA-ceRNA network and affect the expression of other ceRNAs. Considering the independent effect of one SNP or indel in a single MRE of a ceD, the variant will mostly affect the associated miRNA and its direct targets (ceTs) although other miRNAs and ceRNAs in the regulatory network can also be indirectly affected. To simplify the investigation of SNPs’ effects, we focused on each pair of ceD-ceT for a unique miRNA in this study, representing the minimum miRNA-ceRNA system (2,5,6,36). An SNP or indel that creates or strengthens an MRE will decrease the expression of its own host gene (the ceD), and thus increase the expression of the corresponding ceT, whereas a SNP or indel that erases or weakens an MRE will have the opposite effect (Figure 1A). We therefore termed these variants candidate competing endogenous RNA expression quantitative trait loci (cerQTLs).
Figure 1.

The schematic diagram of cerQTL detection for genetic variants affecting ceRNA regulation. (A) The model will investigate the variants effect of each ceD-ceT pair in local and unique miRNA-centered regulatory network (minimum miRNA-ceRNA system). If a mutation creates a gain-of-function MRE in the 3΄UTR of an mRNA (C1) targeted specifically by a miRNA (M1), C1 will be treated as a ceD and biologically participates in local network centered on M1. The original targets (including C2, T1 and T2) of M1 will naturally become ceRNA targets (ceTs) of C1. Alternatively, if a loss-of-function mutation erases an MRE of C1 which originally targeted by M1, then C1 will be released from M1-centered regulatory network. Therefore, both scenarios can initiate the perturbation of ceRNA regulation by mutation-driven redistribution of associated miRNA and ceRNAs. Ideally, assuming the concentration of miRNA is relatively stable among investigated individuals, one would observe reciprocal and coordinated expression trend between C1 and each of its ceTs (such as C2) under additive genetic effect. M: miRNA; T: target; C: investigated ceRNA; ceD: ceRNA driver; ceT: ceRNA target. (B) The pipeline first constructs variant-miRNA-ceD-ceT unit according to Geuvaids individual genotypes and TargetScan prediction. For each variant-miRNA-ceD-ceT unit, we test the genotype effect of ceRNA regulation using a multivariate multiple linear regression with two responses (ceD and ceT) by incorporating several confounding factors. Also, different function evidences area applied to prioritize the detected cerQTLs.
Genetic effects on ceRNA regulation in human populations
Summary of the detection pipeline
Using the genotype dataset of 462 Geuvadis individuals, we first identified genetic variants in the human genome and then examined the impact of these variants on miRNA binding according to TargetScan predictions (19). We identified 3544 candidate genetic variants (including 3263 SNPs and 281 indels) in seed binding sites of 2753 miRNA putative target genes (putative ceDs). These loci showed differential binding affinity to 439 miRNAs (out of 714 profiled by Geuvadis) between the reference and alternative alleles (see Supplementary Methods). For each putative ceD, we identified other genes (ceTs) that are targeted by the same miRNA and constructed a minimum variant-associated miRNA-ceRNA unit which we call variant-miRNA-ceD-ceT. To measure the genotype effect on ceRNA regulation, we applied a multivariate regression model to jointly model the genetic contribution on variability of ceRNA expression and considered several factors, such as miRNA concentration among individuals, covariates of RNA-seq quantification and population stratification (see Supplementary Methods and Figure 1B on whole detection pipeline). This multivariate regression model can simultaneously test for both ceD and ceT expressions and take advantage of the potentially correlated nature between ceD and ceT. At FDR 5%, we obtained the coefficients of ceD (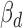 ) and ceT (
) and ceT (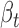 ) for the significant pairs which estimate the genotype effect on ceD and ceT, respectively. To capture the underlying miRNA sponge effect explained by the genotype change, we further filtered out units with
) for the significant pairs which estimate the genotype effect on ceD and ceT, respectively. To capture the underlying miRNA sponge effect explained by the genotype change, we further filtered out units with 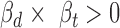 and only retained those showing opposite signs of association between genetic variants and gene expression of the two ceRNAs (details in Supplementary Methods).
and only retained those showing opposite signs of association between genetic variants and gene expression of the two ceRNAs (details in Supplementary Methods).
Genome-wide detection of cerQTLs in different populations
We applied our model and filtering strategy to five Geuvadis populations and detected genome-wide cerQTLs. We found 47 (YRI, 89 individuals), 66 (TSI, 93 individuals), 67 (CEU, 91 individuals), 97 (FIN, 95 individuals) and 106 (GBR, 94 individuals) significant cerQTLs at 5% FDR, respectively (Supplementary Table S1). To improve the detection power, we merged four European subpopulations (EUR, 373 individuals) and detected 387 significant cerQTLs and 1875 variant-miRNA-ceD-ceT units (Supplementary Table S2 and Figure 2). To assess whether the signals in our model differ significantly from the permuted null, we also applied our model to the EUR population with randomly permuted genotypes among individuals. The permutation result in the Q-Q plot shows little signal for these loci, indicating the effectiveness of model (Supplementary Figure S1). In the 387 cerQTLs associated with the EUR population, 344 are SNPs (out of 3263) and 43 are indels (out of 281) (Figure 3A), which suggests that indels are more likely to be cerQTLs than SNPs by accounting for all tested variants in each category (Chi-square test P = 0.04).
Figure 2.

Circos plot of all detected cerQTLs. Features or glyphs are displayed from the outer to the inner, include the number of chromosome, the chromosome ideograms, copy number variation hotspots (red region), Manhattan plot for cerQTLs with –log10(P-value), Manhattan plot for GWAS TASs in miRNA binding site predicted by TargetScan, genome variant density (red: dbSNP, black: 1000 Genomes, purple: HapMap 3), OMIM gene distribution and disease-susceptible region distribution.
Figure 3.

Genome-wide detection of cerQTLs. (A) The proportion of cerQTLs for SNP and indel. (B) The overlapping of cerQTLs in Geuvadis cis-eQTLs. (C) Venn diagram of cerQTLs in European populations. (D) Venn diagram of cerQTL between European and African populations.
cerQTLs are not only directly associated with their ceDs in cis, but also synchronously associated with their corresponding ceTs in trans through their common miRNA regulator. We found only six cerQTLs that overlap with the Geuvadis fine-mapped (‘the best’) cis-eQTL result from the EUR population, implying that most of these ‘best’ cis-eQTLs cannot be explained by ceRNA regulation. However, when we considered the Geuvadis ‘all mapped’ cis-eQTLs, the overlap with cerQTLs is significantly improved (43%, Figure 3B). This suggests that cerQTLs could explain many functional associations in the linked region of each Geuvadis ‘best’ cis-eQTL. Additionally, the cerQTL analysis has identified many additional gene expression-associated signals that were missed by conventional eQTL analyses.
Positive selection on ceRNA regulation
By simply overlapping the cerQTLs in different subpopulations, we can find many loci in common or specific to each subpopulation (Figure 3C and D). Similar patterns can also be observed for related miRNAs, ceDs and ceTs of cerQTLs as well (Supplementary Figure S2). Interestingly, the number of detected cerQTLs is different across subpopulations despite their similar sample sizes and expected statistical power. The number of detected cerQTLs is correlated with the subpopulation's distance from Africa, which suggests that ceRNA evolution may play a role in adaptation to new environments following migration (Supplementary Figure S3). To investigate whether cerQTLs are putative targets of recent positive selections, we screened cerQTLs using six statistical measures (including DDAF, FST, Tajima's D, iHS, XPEHH, XPCLR) for each subpopulation. We found 46 cerQTLs with positive selection signals for at least one of the measures according to their corresponding empirical thresholds (Supplementary Tables S3 and 4). To visualize loci with putatively positive selection, we clustered these 46 genetic variants according to their DAF in 5 investigated subpopulations. The clustering clearly recapitulates the population relationship and shows distinct patterns on individual locus (Supplementary Figure S4A). For example, one YRI-specific cerQTL rs1050286 (P = 8.11E-6) shows very different DAF in African (DAF of YRI: 0.89) and European populations (DAF of TSI: 0.44; CEU: 0.47; GBR: 0.54; FIN: 0.53) (Supplementary Figures S4B, C and 5).
Putative causality of cerQTLs
Properties of cerQTLs
Functional interpretation of cerQTLs is pivotal to our understanding of their underlying biological mechanisms and phenotype causality. Coefficients of ceD and ceT in our regression model can reflect the degree of gene expression perturbation under different genotypes. Using the 387 EUR cerQTLs, we found that majority of 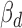 and
and 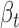 are small (<1) in the 1875 significant variant-miRNA-ceD-ceT units (Figure 4A and B), which indicates a moderate effect of these genetic variants on target gene expression and ceRNA regulation. To investigate whether the 298 cerQTL-associated ceDs and 1459 ceTs are engaged in important biological processes, we performed DAVID gene-annotation enrichment analysis of these two gene sets (24). We found that ceDs and ceTs were enriched in many transcriptional regulation and cell signaling processes (Supplementary Figures S6 and 7).
are small (<1) in the 1875 significant variant-miRNA-ceD-ceT units (Figure 4A and B), which indicates a moderate effect of these genetic variants on target gene expression and ceRNA regulation. To investigate whether the 298 cerQTL-associated ceDs and 1459 ceTs are engaged in important biological processes, we performed DAVID gene-annotation enrichment analysis of these two gene sets (24). We found that ceDs and ceTs were enriched in many transcriptional regulation and cell signaling processes (Supplementary Figures S6 and 7).
Figure 4.

The functional properties of cerQTLs for EUR population. (A) The distribution of 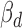 . (B) The distribution of
. (B) The distribution of 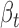 . (C) The direction concordance between association and functional prediction for all cerQTLs.
. (C) The direction concordance between association and functional prediction for all cerQTLs.
A variant's effect on miRNA–target interaction can be assessed by functional prediction algorithms that have been developed to estimate the change of binding affinity among different variant alleles (37). To evaluate whether the direction of association (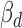 ) is concordant with computational prediction on ceD through variant effect in cis, we calculated two scores, the Δcontext+ score and ΔΔΔG, using TargetScan and an energy-based method (38) for the 387 cerQTLs in the EUR population. Intuitively, Δcontext+ score estimates the difference between alternative and reference alleles in binding affinity to the miRNA and ΔΔΔG reflects the difference between the two alleles in combined interaction energy with the miRNA. Overall, 53 and 59% of cerQTLs have
) is concordant with computational prediction on ceD through variant effect in cis, we calculated two scores, the Δcontext+ score and ΔΔΔG, using TargetScan and an energy-based method (38) for the 387 cerQTLs in the EUR population. Intuitively, Δcontext+ score estimates the difference between alternative and reference alleles in binding affinity to the miRNA and ΔΔΔG reflects the difference between the two alleles in combined interaction energy with the miRNA. Overall, 53 and 59% of cerQTLs have 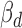 in a consistent direction with Δcontext+ score and ΔΔΔG in functional prediction, respectively, with a union of 75% (Figure 4C). These results suggest that many detected cerQTLs can be validated by functional predictions in their ceD locus.
in a consistent direction with Δcontext+ score and ΔΔΔG in functional prediction, respectively, with a union of 75% (Figure 4C). These results suggest that many detected cerQTLs can be validated by functional predictions in their ceD locus.
Functional prioritization of cerQTLs
To comprehensively evaluate the association between cerQTLs and ceRNA regulation, we rely not only on the statistical significance, but also on the magnitude of cerQTL function on titrating miRNA availability and ceRNA-dependent gene expression changes. Several factors have been reported to influence ceRNA effectiveness, including miRNA and ceRNA expression level, the binding affinity of MRE, as well as the positive correlation between ceRNA expression (3,5,39). We attempted to prioritize the functional cerQTLs according to these factors. Since our regression model has already accounted for factors from miRNA expression variation in the cerQTL calling step, we therefore only focused on ceRNA-centered factors in the prioritization. For 1875 significant variant-miRNA-ceD-ceT units, we first calculated and ranked the degree of gene expression change on ceD and ceT in different genotypes, which can be measured by the sum of 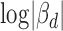 and
and 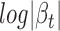 . We further required consistent direction between
. We further required consistent direction between 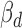 and the Δcontext+ functional prediction score from TargetScan. Finally, we required positive correlation (Pearson's r > 0.1) between ceD and ceT in the specific homozygous cerQTL genotypes, when ceD and ceT actively compete for miRNA binding. Based on these criteria, we identified 239 variant-miRNA-ceD-ceT units for 93 unique cerQTLs with sufficient functional evidences (Supplementary Table S5). The top variant rs3208409 creates a miR-940-3p binding site in the 3΄UTR of HLA-DRB1 gene, which competes with L3MBTL2 for miR-940-3p binding (P = 7.94E-27). The large effect of rs3208409 on the gene expression of ceD (
and the Δcontext+ functional prediction score from TargetScan. Finally, we required positive correlation (Pearson's r > 0.1) between ceD and ceT in the specific homozygous cerQTL genotypes, when ceD and ceT actively compete for miRNA binding. Based on these criteria, we identified 239 variant-miRNA-ceD-ceT units for 93 unique cerQTLs with sufficient functional evidences (Supplementary Table S5). The top variant rs3208409 creates a miR-940-3p binding site in the 3΄UTR of HLA-DRB1 gene, which competes with L3MBTL2 for miR-940-3p binding (P = 7.94E-27). The large effect of rs3208409 on the gene expression of ceD (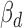 = -115.18) and ceT (
= -115.18) and ceT (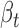 = 1.14), the consistent functional prediction (Δcontext+ score = −0.21), and the high correlation (r = 0.38) between ceD and ceT in homozygote individuals provides a strong support for the causality of this cerQTL (Supplementary Figure S8). We further overlapped this prioritized list with GWAS signals and identified 21 phenotype-associated variant-miRNA-ceD-ceT units (Supplementary Table S6). These results combine statistically significant association and functional evidences to support potential cerQTL causality.
= 1.14), the consistent functional prediction (Δcontext+ score = −0.21), and the high correlation (r = 0.38) between ceD and ceT in homozygote individuals provides a strong support for the causality of this cerQTL (Supplementary Figure S8). We further overlapped this prioritized list with GWAS signals and identified 21 phenotype-associated variant-miRNA-ceD-ceT units (Supplementary Table S6). These results combine statistically significant association and functional evidences to support potential cerQTL causality.
Illustration of cerQTL impact on ceRNA gene regulation
We use an example to illustrate how cerQTLs affect ceRNA regulation. SNP rs1056984 is predicted to affect the seed binding between hsa-miR-296-5p and the 3΄UTR of DIDO1. TargetScan predicted effective binding for the G allele (7mer-m8, context+ score: −0.236), but not for the A allele on this SNP. Furthermore, thermodynamic estimation confirms that allele G has a better binding stability to the miRNA (MFE of A allele: −26.7 kcal/mol; MFE of G allele: −30.5 kcal/mol). The simulated binding pattern also shows that allele G enhances the binding stability by creating a G:C match to position 8 of hsa-miR-296-5p (Figure 5A and B). Our model quantitatively identifies rs1056984 as a cerQTL (P = 4.76E-05) that affects the regulation of hsa-miR-296-5p to its targets ENSG00000101191 (DIDO1) and ENSG00000185361 (TNFAIP8L1). The 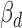 value of ceD DIDO1 is −1.34 (Figure 5C), and the
value of ceD DIDO1 is −1.34 (Figure 5C), and the 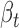 value of ceT TNFAIP8L1 is 0.20 (Figure 5D), with the opposite coefficient signs supporting the competing nature of ceRNA regulation. As the genotype of this variant changes from AA to AG to GG, the gradually enhanced sponge effect down-regulates DIDO1 expression (ceD) and up-regulates TNFAIP8L1 expression (ceT). When this locus is homozygous GG, we observed a significant positive correlation (r = 0.29, P = 0.01) between DIDO1 and TNFAIP8L1, further supporting the interaction between ceD and ceT through competition for hsa-miR-296-5p (Figure 5E).
value of ceT TNFAIP8L1 is 0.20 (Figure 5D), with the opposite coefficient signs supporting the competing nature of ceRNA regulation. As the genotype of this variant changes from AA to AG to GG, the gradually enhanced sponge effect down-regulates DIDO1 expression (ceD) and up-regulates TNFAIP8L1 expression (ceT). When this locus is homozygous GG, we observed a significant positive correlation (r = 0.29, P = 0.01) between DIDO1 and TNFAIP8L1, further supporting the interaction between ceD and ceT through competition for hsa-miR-296-5p (Figure 5E).
Figure 5.

The genetic effect of rs1056984 in ceRNA regulation for EUR population. (A) Hybridization pattern between miR-296-5p and binding site of DIDO1 on derived allele A. (B) Hybridization pattern between miR-296-5p and binding site of DIDO1 on ancestral allele G. (C) Boxplot of gene expression of DIDO1 on different genotype. (D) Boxplot of gene expression of TNFAIP8L1 on different genotypes. (E) The gene expression correlation of DIDO1 and TNFAIP8L1 under different genotypes. (F) Schematic diagram for rs1056984 affecting ceRNA competition under different alleles, it impacts the expression of tumor suppressor and oncogene in a reciprocal and coordinate manner. MFE: minimum free energy.
Previous studies on gene functions suggest the importance of DIDO1, TNFAIP8L1 and miR-296-5p in regulating cancer development. miR-296 has been characterized as ‘angiomiR,’ which can regulate angiogenesis (40). It is reported to have a specific role in promoting tumor angiogenesis by targeting HGS mRNA, resulting in the overexpression of VEGF receptors in angiogenic endothelial cells (41). MiR-296 may also contribute to carcinogenesis by dysregulating p53 (42). In this scenario, DIDO1 gene is a tyrosine-phosphorylated putative transcription factor, previously thought to induce apoptosis and mitotic division (43,44) and might be a tumor suppressor gene (45). In contrast, many publications reported TNFAIP8L1 to be an antiapoptotic molecule and oncogene in developing many cancers (10,46–48). Here, we predicted that cerQTL rs1056984 affect the ceRNA regulation by balancing the expression of the pro-apoptotic DIDO1 and the anti-apoptotic TNFAIP8L1 under different genotypes. Specifically, efficient miRNA competition occurs in the G allele of rs1056984, while the A allele has a protective effect in maintaining tumor suppressor DIDO1 expression and inhibiting oncogenic TNFAIP8L1 expression by shifting miR-296-5p binding from DIDO1 to TNFAIP8L1 (Figure 5F). Although there is no diseases/traits associated evidences for rs1056984 at the current stage, the cerQTL investigation could provide a disease-causal indication underlying this locus. Also, we found that African population has lower A allele frequency of rs1056984 (DAF of YRI is 0.28, DAF of CEU is 0.65) in the 1000 Genomes project (Supplementary Figure S9). Further calculation on FST (0.24) between CEU and YRI indicates that positive selection may drive the evolution of this locus.
cerQTLs explain GWAS traits and diseases associated signals in miRNA binding sites
To investigate whether cerQTL-affected gene expression changes contribute to human phenotypes, we connected cerQTLs in the EUR population to GWAS trait/disease-associated SNPs and found 8 of 387 cerQTLs to overlap with GWAS lead SNPs (Table 1). The top lead SNP is rs7294, which is located in the 3΄UTR region of VKORC1 and associated with warfarin maintenance dose in anticoagulant therapy (49–51) (Supplementary Figure S10A–D).
Table 1. The cerQTLs that overlap with GWAS leading TASs.
| Chr | Pos | SNP | Ref | Alt | cerQTL P-valuea | ceD | ceTs | miRNA | Effectb | GWAS P-valuec | GWAS traits |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 16 | 31102321 | rs7294 | C | T | 1.37E-06 | VKORC1 | EIF2B5, RPTOR, LRRFIP1 | miR-147a | Gain | 1.40E-45 | Warfarin and acenocoumarol maintenance dosage (68) |
| 11 | 18388128 | rs4596 | G | C | 1.15E-05 | GTF2H1 | TSTD2, TMOD1, TOB1, PPARGC1A, KLF5 | miR-642a-5p | Loss | 2.17E-35 | Amyloid A Levels (69), Colorectal cancer (70) |
| 12 | 56863770 | rs2657880 | G | C | 1.87E-51 | SPRYD4 | XKR6, AC004985.2 | miR-3157-5p | Loss | 2.28E-31 | Serum metabolite levels (71) |
| 10 | 100176869 | rs701801 | C | T | 4.70E-13 | HPS1 | TRIOBP, KLHL30 | miR-491-5p | Loss | 1.34E-25 | Serum metabolism (72) |
| 19 | 10397238 | rs281437 | C | T | 0.00019 | ICAM1 | C16orf54, FBXO41, SH2D4A | miR-3667-5p | Loss | 3.00E-10 | Soluble intercellular adhesion molecule 1 level (73) |
| 16 | 67708897 | rs12449157 | A | G | 7.69E-06 | GFOD2 | COASY, LMAN2L, VPS9D1 | miR-4792-5p | Gain | 2.00E-07 | Coronary artery disease (74) |
| 17 | 37921742 | rs907091 | C | T | 9.50E-05 | IKZF3 | RGL3, DDX11 | miR-330-5p | Gain | 3.38E-07 | Esophageal cancer (75) |
| 8 | 11643915 | rs804292 | G | A | 0.00024 | NEIL2 | ZNF583 | miR-143-3p | Loss | 2.00E-06 | Alcohol/nicotine dependence (76) |
athe best cerQTL P-value among all significant variant-miRNA-ceD-ceT units.
bthe predicted function effect of alternative allele for miRNA–target interaction.
cthe best GWAS P-value among all mapped GWAS traits.
Since GWAS lead SNPs may not be causal, we further scanned SNPs in high LD (r2 > 0.8) of the lead SNPs. We found 15.7% of 344 cerQTLs (SNPs only and not indels) to be associated with 145 GWAS hits (Supplementary Table S7), a significant enrichment of cerQTLs in GWAS hits compared with background SNPs for both the miRNA seed binding site (P = 7.54E-28, hypergeometric test) and differential miRNA binding signals (P = 3.30E-4, hypergeometric test) (Supplementary Table S8). Most of these 145 GWAS leading SNPs are located in introns or intergenic regions with poorly annotated functions and they are mostly related to autoimmune diseases and blood cell traits (Supplementary Table S9). Our analysis suggests that the causal SNPs for these leading SNPs might arise from cerQTLs which underly disease/trait-associated SNPs within GWAS LD proxies.
Functional effect of cerQTLs in ceRNA regulatory network
The altered expression of individual genes might affect the expression of many other genes in the ceRNA regulatory network by the miRNA sponge mechanism (52,53). We merged the 1875 significant variant-miRNA-ceD-ceT units our model identified and constructed the global ceRNA regulatory network under the control of 387 independent cerQTLs in the EUR population (Supplementary Figure S11). We also generated the network for 21 phenotype-associated variant-miRNA-ceD-ceT units (Supplementary Figure S12). The majority of ceDs can be associated with more than one ceT by a single genetic effect. For example, rs11540855 on ceD ABHD8 may influence the expression of two ceTs AXIN1 and RPRM through competing for binding to miR-4707-3p (Figure 6A) and the expression of ABHD8 and its two ceTs are positively correlated under the active genotype GG (Figure 6B–D). Interestingly, the cerQTL rs11540855 is located in the 3΄UTR of ABHD8 and has been recently reported to have the highest association with breast cancer risk (GWAS P = 1.65E-09) (54,55). In addition, AXIN1 and RPRM were recently reported as tumor suppressors in breast cancer development (56), and miR-4707-3p is highly expressed in breast cancer (57). These observations suggest that cerQTL rs11540855 might affect breast cancer risk by regulating tumor suppressors AXIN1 and RPRM through the ceRNA pathway. In addition to the aforementioned regulatory relationship, one ceD can also be regulated by multiple miRNAs and a single miRNA can regulate multiple ceDs and ceTs through different cerQTLs. These interactions highlight the complexity of genetic effect on ceRNA regulatory network.
Figure 6.

The genetic effect of rs11540855 in ceRNA regulation for EUR population. (A) Small ceRNA regulatory network driven by rs11540855, circle: ceD and ceTs; triangle: miRNA; suppression line with label: the miRNA-ceD regulation, G for gain-of-function mutation; other suppression lines: the miRNA-ceT regulation; arrow: ceD activate ceTs in gain-of-function situation (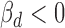 and
and 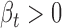 ). (B) Boxplot of gene expression for ABHD8 on different genotypes of Geuvaids individuals. (C) Boxplot of gene expression for AXIN1 and on different genotypes of Geuvaids individuals and the correlation with ABHD8 on genotype GG. (D) Boxplot of gene expression for RPRM and on different genotypes of Geuvaids individuals and the correlation with ABHD8 on genotype GG.
). (B) Boxplot of gene expression for ABHD8 on different genotypes of Geuvaids individuals. (C) Boxplot of gene expression for AXIN1 and on different genotypes of Geuvaids individuals and the correlation with ABHD8 on genotype GG. (D) Boxplot of gene expression for RPRM and on different genotypes of Geuvaids individuals and the correlation with ABHD8 on genotype GG.
In vitro cerQTL functional validation
To further illustrate cerQTL effect in the minimum miRNA-ceRNA system, we experimentally validated one breast cancer-associated cerQTL unit (rs11540855-miR-4707-3p-ABHD8-AXIN1) (Figure 7A) in the breast cancer cell line MCF116. We first attempted to independently validate whether both ceD and ceT were biological targets of miR-4707-3p, which was the premise of ceRNAs competition. Using dual luciferase reporter assay, we observed significantly reduced luciferase activity and mRNA expression level upon miR-4707-3p agomir treatment in the G-allele but not the A-allele in the 3΄UTR of ABHD8. This indicates that miR-4707-3p preferably binds to the 3΄UTR of ABHD8 G-allele and negative regulates ABHD8 protein level (Figure 7B and C). Moreover, miR-4707-3p agomir significantly inhibited the expression of AXIN1 in MCF116 (Figure 7D). We hence concluded that rs11540855 alters miR-4707-3p-ABHD8 interaction affinity in vitro, and both ceD (ABHD8 G-allele) and ceT (AXIN1) are valid targets of miR-4707-3p.
Figure 7.

In vitro experimental validation of rs11540855-miR-4707-3p-ABHD8-AXIN1. (A) Graphic representation of genetic effect on miR-4707-3p-ABHD8-AXIN1, circle: ceD and ceT; triangle: miRNA; suppression line with label: the miRNA-ceD regulation, G for gain-of-function mutation; other suppression lines: the miRNA-ceT regulation; arrow: ceD activate ceTs in gain-of-function situation (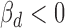 and
and 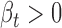 ). (B) Fold change of luciferase activity ratio for ABHD8 A-allele or ABHD8 G-allele (Firefly luciferase) and the internal control (Renilla luciferase) in response to miR-4707-3p agomir, n = 6 experiments; (C) Fold change of relative ABHD8 A-allele or ABHD8 G-allele mRNA expression level (with GAPDH) in response to miR-4707-3p agomir, n = 6 experiments; (D) A representative immunoblot and fold change of MCF116 native AXIN1 expression in response to miR-4707-3p agomir, n = 5 experiments; (E) A representative immunoblot and fold change of AXIN1 expression in MCF116 cells stably expressing the two ABHD8 alleles in response to miR-4707-3p agomir, n = 5 experiments; (F) Relative expression level of miR-4707-3p, ABHD8 and AXIN1, as a function of miR-4707-3p agomir dosage (range: 0, 5, 10, 15, 20 pmol/well), n = 6 experiments.
). (B) Fold change of luciferase activity ratio for ABHD8 A-allele or ABHD8 G-allele (Firefly luciferase) and the internal control (Renilla luciferase) in response to miR-4707-3p agomir, n = 6 experiments; (C) Fold change of relative ABHD8 A-allele or ABHD8 G-allele mRNA expression level (with GAPDH) in response to miR-4707-3p agomir, n = 6 experiments; (D) A representative immunoblot and fold change of MCF116 native AXIN1 expression in response to miR-4707-3p agomir, n = 5 experiments; (E) A representative immunoblot and fold change of AXIN1 expression in MCF116 cells stably expressing the two ABHD8 alleles in response to miR-4707-3p agomir, n = 5 experiments; (F) Relative expression level of miR-4707-3p, ABHD8 and AXIN1, as a function of miR-4707-3p agomir dosage (range: 0, 5, 10, 15, 20 pmol/well), n = 6 experiments.
Given that miR-4707-3p is associated with the 3΄UTR of both ABHD8 and AXIN1, we next aimed to evaluate the competitive effect of these two ceRNAs in different allele states of ABHD8. By constructing MCF116 cells that stably express ABHD8 A-allele or ABHD8 G-allele, we inspected the changes of ceRNA expression with the treatment of miR-4707-3p agomir. MCF116 cells carrying the 3΄UTR of ABHD8 A-allele suppressed the protein level of AXIN1 more than those carrying the G-allele (Figure 7E), suggesting stronger competition between ABHD8 G-allele and miR-4707-3p competing target AXIN1. Furthermore, dose-dependent experiments showed that increasing miR-4707-3p treatment suppresses the protein levels of both ceRNA genes. At the same level of miR-4707-3p treatment, AXIN1 level decreases less in MCF116 cells carrying the ABHD8 G-allele which binds stronger to miR-4707-3p than in MCF116 cells carrying the ABHD8 A-allele (Figure 7F). Taken together, these experiments support the genetic effect of cerQTL in the minimum miRNA-ceRNA system and motivate further functional evaluation of cerQTL cancer relevance.
DISCUSSION
In this study, we integrated the 1000 Genomes genotype and Geuvadis RNA sequencing data to investigate the association between human genetic variation and ceRNA regulation. Using a multivariate multiple regression model, we identified hundreds of cerQTLs and related ceD-ceTs genome-wide. We found evidence suggesting that recent natural selection has acted on many cerQTLs in different human populations. cerQTLs are significantly enriched in GWAS risk loci and are likely to be important for many biological processes. Furthermore, we prioritized these loci with their associated ceRNAs according to different criteria and evaluated their collective effect on the ceRNA regulatory network. We also experimentally validated a cerQTL by in vitro functional assays. Our study provides a novel angle to interpret genetic effect in post-transcriptional gene regulation and highlights the utility of ceRNA gene regulation in explaining disease susceptibility.
Although our regression model already considered many potential confounding factors, such as miRNA expression level, ceRNA expression variability, as well as population genetic structures, there may still be missing factors that impact the performance and statistical power in the cerQTL detection. One potential limitation is that we only treated each pair of ceD and ceT as an independent test unit in the local miRNA-centered regulatory network instead of modeling the whole ceRNA regulatory network. Studies have shown that a small perturbation of ceRNA expression usually shifts the equilibrium of ceRNA regulatory network especially when concentrations of miRNAs and targets are comparable (6). The cascade effect from miRNA redistribution and ceRNA competition at the global level (2,58,59) requires a much more complex mathematical model to accurately describe the full responses of the whole network.
Another limitation of our study is that current computational predictions of miRNA binding sites are far from perfect (60). In our study, we chose to use a strict context+ score threshold from TargetScan predictions and described the result based on this criterion. It is possible that our cerQTLs detection can both miss target sites not predicted by TargetScan and introduce false positives. Alternatively, one could consider the union or intersection of multiple miRNA–target prediction algorithms such as TargetScan, PITA (38), miRanda (61), etc. However, union might introduce many false positives and intersection might introduce false negatives and drastically reduce the number of valid variant-miRNA-ceD-ceT units (from 1875 to 331, Supplementary Table S10). Our current criteria may provide a reasonably reliable reference for further functional investigation. Future experiments combining CLIP-seq and Degradome-Seq could better capture global miRNA–-target interactions and improve our cerQTL inference.
Using genetic and transcriptomic data from different populations, we found many population-specific cerQTLs and identified loci that are putative targets of recent positive selections. These results represent a useful supplement to studies of recent natural selection of human miRNA targets (56,62,63) and significantly extend functional categories for positively selected loci (64). Besides, GWAS signals enrichment of our detected cerQTLs implies putative disease-causal mechanisms driven by ceRNA gene regulation, and we can connect cerQTLs to many human complex diseases and phenotypes. Importantly, we have experimentally validated a breast cancer-associated SNP rs11540855 that can regulate the competition of two ceRNAs for miR-4707-3p availability in vitro, including a tumor suppressor gene AXIN1. This may highlight that cerQTL detection could be an important addition to our current understanding of disease pathogenesis and trait development.
Since many genes contain multiple binding sites for the same miRNA, single MRE perturbations might be expected to have small effects on ceRNA expression and the downstream miRNA regulatory network. However, although many QTLs individually exert relatively small effects, in combination they might contribute substantially to complex traits (65). Theoretical simulations and quantitative experiments have demonstrated that perturbations on individual miRNA binding site can indeed affect the entire ceRNA regulatory network (2,36,66,67). Although our in vitro functional evaluation demonstrates the SNP effect on ceRNA competition in the minimum miRNA-ceRNA system, it still could not characterize the entire miRNA-ceRNA network. In summary, our cerQTL analyses on human populations suggest that DNA polymorphisms affecting ceRNA regulation might be a common phenomenon and likely contribute substantially to complex traits.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Graham McVicker and Russell J. H. Ryan for helpful discussions and suggestions. We also thank the associate editor and reviewers for constructive suggestions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Y S and Christabel Lung Postgraduate Scholarship from The University of Hong Kong and Talent Excellence Program from Tianjin Medical University (to M.J.L.); National Natural Science Foundation of China [81400239 to J.Z.]; Research Grants Council, Hong Kong SAR, China [17121414M to J.W.W.]; Startup funding from the Mayo Clinic Arizona and Mayo Clinic Center for Individualized Medicine (to J.W.W); National Institute of Health [ R01 GM113242-01 to J.S.L.]; National Institute of Health [U41 HG7000, U01 CA180980 to X.S.L.]. The open access publication charge for this paper has been waived by Oxford University Press - NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
REFERENCES
- 1. Tay Y., Rinn J., Pandolfi P.P.. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014; 505:344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ala U., Karreth F.A., Bosia C., Pagnani A., Taulli R., Leopold V., Tay Y., Provero P., Zecchina R., Pandolfi P.P.. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:7154–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P.. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?. Cell. 2011; 146:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li L., Wang D., Xue M., Mi X., Liang Y., Wang P.. 3΄UTR shortening identifies high-risk cancers with targeted dysregulation of the ceRNA network. Sci. Rep. 2014; 4:5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mukherji S., Ebert M.S., Zheng G.X., Tsang J.S., Sharp P.A., van Oudenaarden A.. MicroRNAs can generate thresholds in target gene expression. Nat. Genet. 2011; 43:854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levine E., Zhang Z., Kuhlman T., Hwa T.. Quantitative characteristics of gene regulation by small RNA. PLoS Biol. 2007; 5:e229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanin G., Shenhar-Tsarfaty S., Yayon N., Hoe Y.Y., Bennett E.R., Sklan E.H., Rao D.C., Rankinen T., Bouchard C., Geifman-Shochat S. et al. Competing targets of microRNA-608 affect anxiety and hypertension. Hum. Mol. Genet. 2014; 23:4569–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pan W., Liu L., Wei J., Ge Y., Zhang J., Chen H., Zhou L., Yuan Q., Zhou C., Yang M.. A functional lncRNA HOTAIR genetic variant contributes to gastric cancer susceptibility. Mol. Carcinog. 2015; 55:90–96. [DOI] [PubMed] [Google Scholar]
- 9. Siddle K.J., Deschamps M., Tailleux L., Nedelec Y., Pothlichet J., Lugo-Villarino G., Libri V., Gicquel B., Neyrolles O., Laval G. et al. A genomic portrait of the genetic architecture and regulatory impact of microRNA expression in response to infection. Genome Res. 2014; 24:850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gamazon E.R., Innocenti F., Wei R., Wang L., Zhang M., Mirkov S., Ramirez J., Huang R.S., Cox N.J., Ratain M.J. et al. A genome-wide integrative study of microRNAs in human liver. BMC Genomics. 2013; 14:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Civelek M., Hagopian R., Pan C., Che N., Yang W.P., Kayne P.S., Saleem N.K., Cederberg H., Kuusisto J., Gargalovic P.S. et al. Genetic regulation of human adipose microRNA expression and its consequences for metabolic traits. Hum. Mol. Genet. 2013; 22:3023–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lappalainen T., Sammeth M., Friedlander M.R., t Hoen P.A., Monlong J., Rivas M.A., Gonzalez-Porta M., Kurbatova N., Griebel T., Ferreira P.G. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013; 501:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huan T., Rong J., Liu C., Zhang X., Tanriverdi K., Joehanes R., Chen B.H., Murabito J.M., Yao C., Courchesne P. et al. Genome-wide identification of microRNA expression quantitative trait loci. Nat. Commun. 2015; 6:6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gamazon E.R., Ziliak D., Im H.K., LaCroix B., Park D.S., Cox N.J., Huang R.S.. Genetic architecture of microRNA expression: implications for the transcriptome and complex traits. Am. J. Hum. Genet. 2012; 90:1046–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu J., Clark A.G.. Impact of microRNA regulation on variation in human gene expression. Genome Res. 2012; 22:1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Genomes Project C., Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A.. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kozomara A., Griffiths-Jones S.. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014; 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lewis B.P., Burge C.B., Bartel D.P.. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120:15–20. [DOI] [PubMed] [Google Scholar]
- 20. Lorenz R., Bernhart S.H., Honer Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stegle O., Parts L., Piipari M., Winn J., Durbin R.. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 2012; 7:500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Constantine A.G. Some non-central distribution problems in multivariate-analysis. Ann. Math. Stat. 1963; 34:1270–1285. [Google Scholar]
- 23. Benjamini Y., Hochberg Y.. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. Met. 1995; 57:289–300. [Google Scholar]
- 24. Huang da W., Sherman B.T., Lempicki R.A.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 25. Bruno A.E., Li L., Kalabus J.L., Pan Y., Yu A., Hu Z.. miRdSNP: a database of disease-associated SNPs and microRNA target sites on 3΄UTRs of human genes. BMC Genomics. 2012; 13:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li M.J., Wang J.. Current trend of annotating single nucleotide variation in humans—a case study on SNVrap. Methods. 2014; 79-80:32–40. [DOI] [PubMed] [Google Scholar]
- 27. Li M.J., Wang P., Liu X., Lim E.L., Wang Z., Yeager M., Wong M.P., Sham P.C., Chanock S.J., Wang J.. GWASdb: a database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res. 2012; 40:D1047–D1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leslie R., O’Donnell C.J., Johnson A.D.. GRASP: analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics. 2014; 30:i185–i194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grossman S.R., Shlyakhter I., Karlsson E.K., Byrne E.H., Morales S., Frieden G., Hostetter E., Angelino E., Garber M., Zuk O. et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science. 2010; 327:883–886. [DOI] [PubMed] [Google Scholar]
- 30. Weir B.S., Hill W.G.. Estimating F-statistics. Annu. Rev. Genet. 2002; 36:721–750. [DOI] [PubMed] [Google Scholar]
- 31. Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989; 123:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Voight B.F., Kudaravalli S., Wen X., Pritchard J.K.. A map of recent positive selection in the human genome. PLoS Biol. 2006; 4:e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sabeti P.C., Varilly P., Fry B., Lohmueller J., Hostetter E., Cotsapas C., Xie X., Byrne E.H., McCarroll S.A., Gaudet R. et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007; 449:913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen H., Patterson N., Reich D.. Population differentiation as a test for selective sweeps. Genome Res. 2010; 20:393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bilan V., Allegra D., Kuchenbauer F., Mertens D.. In vivo processing assay based on a dual-luciferase reporter system to evaluate DROSHA enzymatic activity. Methods Mol. Biol. 2014; 1095:87–93. [DOI] [PubMed] [Google Scholar]
- 36. Yuan Y., Liu B., Xie P., Zhang M.Q., Li Y., Xie Z., Wang X.. Model-guided quantitative analysis of microRNA-mediated regulation on competing endogenous RNAs using a synthetic gene circuit. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:3158–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang G.L., Chen X.C., Chan L., Zhang M.X., Zhu B.H., Wang L.T., Zhu X.Y., Zhang J.Y., Zhou B.P., Wang J.W.. An SNP selection strategy identified IL-22 associating with susceptibility to tuberculosis in Chinese. Sci. Rep. 2011; 1:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kertesz M., Iovino N., Unnerstall U., Gaul U., Segal E.. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007; 39:1278–1284. [DOI] [PubMed] [Google Scholar]
- 39. Ebert M.S., Sharp P.A.. Emerging roles for natural microRNA sponges. Curr. Biol. 2010; 20:R858–R861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang S., Olson E.N.. AngiomiRs–key regulators of angiogenesis. Curr. Opin. Genet. Dev. 2009; 19:205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wurdinger T., Tannous B.A., Saydam O., Skog J., Grau S., Soutschek J., Weissleder R., Breakefield X.O., Krichevsky A.M.. miR-296 regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell. 2008; 14:382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yoon A.R., Gao R., Kaul Z., Choi I.K., Ryu J., Noble J.R., Kato Y., Saito S., Hirano T., Ishii T. et al. MicroRNA-296 is enriched in cancer cells and downregulates p21WAF1 mRNA expression via interaction with its 3΄ untranslated region. Nucleic Acids Res. 2011; 39:8078–8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Garcia-Domingo D., Leonardo E., Grandien A., Martinez P., Albar J.P., Izpisua-Belmonte J.C., Martinez A.C.. DIO-1 is a gene involved in onset of apoptosis in vitro, whose misexpression disrupts limb development. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:7992–7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stark A.L., Hause R.J. Jr, Gorsic L.K., Antao N.N., Wong S.S., Chung S.H., Gill D.F., Im H.K., Myers J.L., White K.P. et al. Protein quantitative trait loci identify novel candidates modulating cellular response to chemotherapy. PLoS Genet. 2014; 10:e1004192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Futterer A., Campanero M.R., Leonardo E., Criado L.M., Flores J.M., Hernandez J.M., San Miguel J.F., Martinez A.C.. Dido gene expression alterations are implicated in the induction of hematological myeloid neoplasms. J. Clin. Invest. 2005; 115:2351–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shi T.Y., Cheng X., Yu K.D., Sun M.H., Shao Z.M., Wang M.Y., Zhu M.L., He J., Li Q.X., Chen X.J. et al. Functional variants in TNFAIP8 associated with cervical cancer susceptibility and clinical outcomes. Carcinogenesis. 2013; 34:770–778. [DOI] [PubMed] [Google Scholar]
- 47. Kumar D., Whiteside T.L., Kasid U.. Identification of a novel tumor necrosis factor-alpha-inducible gene, SCC-S2, containing the consensus sequence of a death effector domain of fas-associated death domain-like interleukin- 1beta-converting enzyme-inhibitory protein. J. Biol. Chem. 2000; 275:2973–2978. [DOI] [PubMed] [Google Scholar]
- 48. Liu T., Gao H., Chen X., Lou G., Gu L., Yang M., Xia B., Yin H.. TNFAIP8 as a predictor of metastasis and a novel prognostic biomarker in patients with epithelial ovarian cancer. Br. J. Cancer. 2013; 109:1685–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scott S.A., Edelmann L., Kornreich R., Desnick R.J.. Warfarin pharmacogenetics: CYP2C9 and VKORC1 genotypes predict different sensitivity and resistance frequencies in the Ashkenazi and Sephardi Jewish populations. Am. J. Hum. Genet. 2008; 82:495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kringen M.K., Haug K.B., Grimholt R.M., Stormo C., Narum S., Opdal M.S., Fosen J.T., Piehler A.P., Johansen P.W., Seljeflot I. et al. Genetic variation of VKORC1 and CYP4F2 genes related to warfarin maintenance dose in patients with myocardial infarction. J. Biomed. Biotechnol. 2011; 2011:739751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Limdi N.A., Beasley T.M., Crowley M.R., Goldstein J.A., Rieder M.J., Flockhart D.A., Arnett D.K., Acton R.T., Liu N.. VKORC1 polymorphisms, haplotypes and haplotype groups on warfarin dose among African-Americans and European-Americans. Pharmacogenomics. 2008; 9:1445–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sumazin P., Yang X., Chiu H.S., Chung W.J., Iyer A., Llobet-Navas D., Rajbhandari P., Bansal M., Guarnieri P., Silva J. et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011; 147:370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bosia C., Pagnani A., Zecchina R.. Modelling competing endogenous RNA networks. PLoS One. 2013; 8:e66609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Antoniou A.C., Wang X., Fredericksen Z.S., McGuffog L., Tarrell R., Sinilnikova O.M., Healey S., Morrison J., Kartsonaki C., Lesnick T. et al. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat. Genet. 2010; 42:885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stevens K.N., Fredericksen Z., Vachon C.M., Wang X., Margolin S., Lindblom A., Nevanlinna H., Greco D., Aittomaki K., Blomqvist C. et al. 19p13.1 is a triple-negative-specific breast cancer susceptibility locus. Cancer Res. 2012; 72:1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li J., Liu Y., Xin X., Kim T.S., Cabeza E.A., Ren J., Nielsen R., Wrana J.L., Zhang Z.. Evidence for positive selection on a number of MicroRNA regulatory interactions during recent human evolution. PLoS Genet. 2012; 8:e1002578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Persson H., Kvist A., Rego N., Staaf J., Vallon-Christersson J., Luts L., Loman N., Jonsson G., Naya H., Hoglund M. et al. Identification of new microRNAs in paired normal and tumor breast tissue suggests a dual role for the ERBB2/Her2 gene. Cancer Res. 2011; 71:78–86. [DOI] [PubMed] [Google Scholar]
- 58. Seitz H. Redefining microRNA targets. Curr. Biol. 2009; 19:870–873. [DOI] [PubMed] [Google Scholar]
- 59. Thomson D.W., Dinger M.E.. Endogenous microRNA sponges: evidence and controversy. Nat. Rev. Genet. 2016; 17:272–283. [DOI] [PubMed] [Google Scholar]
- 60. Pasquinelli A.E. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012; 13:271–282. [DOI] [PubMed] [Google Scholar]
- 61. John B., Enright A.J., Aravin A., Tuschl T., Sander C., Marks D.S.. Human microRNA targets. PLoS Biol. 2004; 2:e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen K., Rajewsky N.. Natural selection on human microRNA binding sites inferred from SNP data. Nat. Genet. 2006; 38:1452–1456. [DOI] [PubMed] [Google Scholar]
- 63. Saunders M.A., Liang H., Li W.H.. Human polymorphism at microRNAs and microRNA target sites. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:3300–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Grossman S.R., Andersen K.G., Shlyakhter I., Tabrizi S., Winnicki S., Yen A., Park D.J., Griesemer D., Karlsson E.K., Wong S.H. et al. Identifying recent adaptations in large-scale genomic data. Cell. 2013; 152:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mackay T.F., Stone E.A., Ayroles J.F.. The genetics of quantitative traits: challenges and prospects. Nat. Rev. Genet. 2009; 10:565–577. [DOI] [PubMed] [Google Scholar]
- 66. Xie P., Liu Y., Li Y., Zhang M.Q., Wang X.. MIROR: a method for cell-type specific microRNA occupancy rate prediction. Mol. Biosyst. 2014; 10:1377–1384. [DOI] [PubMed] [Google Scholar]
- 67. Bosson A.D., Zamudio J.R., Sharp P.A.. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol. Cell. 2014; 56:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rieder M.J., Reiner A.P., Gage B.F., Nickerson D.A., Eby C.S., McLeod H.L., Blough D.K., Thummel K.E., Veenstra D.L., Rettie A.E.. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 2005; 352:2285–2293. [DOI] [PubMed] [Google Scholar]
- 69. Marzi C., Albrecht E., Hysi P.G., Lagou V., Waldenberger M., Tonjes A., Prokopenko I., Heim K., Blackburn H., Ried J.S. et al. Genome-wide association study identifies two novel regions at 11p15.5-p13 and 1p31 with major impact on acute-phase serum amyloid A. PLoS Genet. 2010; 6:e1001213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Naccarati A., Pardini B., Stefano L., Landi D., Slyskova J., Novotny J., Levy M., Polakova V., Lipska L., Vodicka P.. Polymorphisms in miRNA-binding sites of nucleotide excision repair genes and colorectal cancer risk. Carcinogenesis. 2012; 33:1346–1351. [DOI] [PubMed] [Google Scholar]
- 71. Inouye M., Ripatti S., Kettunen J., Lyytikainen L.P., Oksala N., Laurila P.P., Kangas A.J., Soininen P., Savolainen M.J., Viikari J. et al. Novel loci for metabolic networks and multi-tissue expression studies reveal genes for atherosclerosis. PLoS Genet. 2012; 8:e1002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hong M.G., Karlsson R., Magnusson P.K., Lewis M.R., Isaacs W., Zheng L.S., Xu J., Gronberg H., Ingelsson E., Pawitan Y. et al. A genome-wide assessment of variability in human serum metabolism. Hum. Mutat. 2013; 34:515–524. [DOI] [PubMed] [Google Scholar]
- 73. Pare G., Chasman D.I., Kellogg M., Zee R.Y., Rifai N., Badola S., Miletich J.P., Ridker P.M.. Novel association of ABO histo-blood group antigen with soluble ICAM-1: results of a genome-wide association study of 6,578 women. PLoS Genet. 2008; 4:e1000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Waterworth D.M., Ricketts S.L., Song K., Chen L., Zhao J.H., Ripatti S., Aulchenko Y.S., Zhang W., Yuan X., Lim N. et al. Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2010; 30:2264–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wu C., Kraft P., Zhai K., Chang J., Wang Z., Li Y., Hu Z., He Z., Jia W., Abnet C.C. et al. Genome-wide association analyses of esophageal squamous cell carcinoma in Chinese identify multiple susceptibility loci and gene-environment interactions. Nat. Genet. 2012; 44:1090–1097. [DOI] [PubMed] [Google Scholar]
- 76. McGue M., Zhang Y., Miller M.B., Basu S., Vrieze S., Hicks B., Malone S., Oetting W.S., Iacono W.G.. A genome-wide association study of behavioral disinhibition. Behav. Genet. 2013; 43:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.