

SUMMARY
Extensive transcriptional and ontogenetic diversity exists among normal tissue-resident macrophages, with unique transcriptional profiles endowing the cells with tissue-specific functions. However, it is unknown whether the origins of different macrophage populations affect their roles in malignancy. Given potential artifacts associated with irradiation-based lineage tracing, it remains unclear if bone marrow-derived macrophages (BMDM) are present in tumors of the brain, a tissue with no homeostatic involvement of BMDM. Here, we employed multiple models of murine brain malignancy and genetic lineage tracing to demonstrate that BMDM are abundant in primary and metastatic brain tumors. Our data indicate that distinct transcriptional networks in brain-resident microglia and recruited BMDM are associated with tumor-mediated education, yet are also influenced by chromatin landscapes established before tumor initiation. Furthermore, we demonstrate that microglia specifically repress Itga4 (CD49D), enabling its utility as a discriminatory marker between microglia and BMDM in primary and metastatic disease in mouse and human.
Keywords: Macrophage, microglia, tumor-associated macrophages, CD49D, glioma, brain metastasis
INTRODUCTION
Macrophages are a terminally differentiated cell of the myeloid lineage, with critical functions in tissue development and homeostasis (Okabe and Medzhitov, 2015). These cells serve as a nexus between adaptive and innate immunity, regulating responses to inflammation and wound healing (Mosser and Edwards, 2008). To facilitate these diverse functions, macrophages employ considerable plasticity in response to a range of cytokines. These responses fall within a spectrum of different phenotypes ranging from classically activated pro-inflammatory macrophages to alternatively activated anti-inflammatory macrophages (Xue et al., 2014). Macrophages also possess substantial diversity and plasticity, with recent studies revealing important insights into the developmental origins of tissue-resident macrophages and uncovering tissue-specific gene expression patterns and enhancer landscapes (Gautier et al., 2012; Ginhoux et al., 2010; Gomez Perdiguero et al., 2015; Lavin et al., 2014; Mass et al., 2016).
While the local tissue environment sculpts macrophage transcriptional profiles and epigenetic states in homeostasis (Lavin et al., 2014), it is unknown whether an inflammatory tissue environment may promote differences between macrophage populations of distinct ontogenies. This is particularly relevant in cancer, where tumor-associated macrophages (TAMs) are derived from monocytes and also potentially from tissue-resident macrophages (Du et al., 2008; Pyonteck et al., 2013; Solga et al., 2015).
Brain-resident macrophages, microglia (MG), develop from erythromyeloid precursors in the yolk sac (Gomez Perdiguero et al., 2015; Kierdorf et al., 2013a; Schulz et al., 2012). Unlike other tissue-resident macrophages, during homeostasis MG undergo self-renewal and their pool is not replenished by monocytes (Ajami et al., 2007). Microglia are also resistant to myeloablative irradiation (Kennedy and Abkowitz, 1997). Indeed this property has been used extensively in bone marrow transplantation (BMT) models to distinguish radio-resistant MG from BM-derived macrophages (BMDM) (Huang et al., 2014; Sedgwick et al., 1991). However, only under conditions of blood-brain barrier (BBB) disruption, e.g. via irradiation (IR) or chemical manipulation, does there appear to be a significant contribution of BMDM to the brain macrophage pool in a non-pathological context (Bruttger et al., 2015; Mildner et al., 2007). This is relevant to brain tumors such as gliomas where there is also disruption of the BBB with disease progression (Dubois et al., 2014). IR-BMT has shown BMDM abundance in murine CNS cancers (Biffi et al., 2004; De Palma et al., 2005; Huang et al., 2014; Muller et al., 2015; Pyonteck et al., 2013); however given the current lack of markers definitively distinguishing MG and BMDM, it remains unclear if BMDM recruitment indeed occurs in brain tumors in the absence of irradiation. The need for markers distinguishing these cells is especially critical in human disease, where lineage tracing is not possible.
Here, we utilize multiple genetic lineage tracing models to demonstrate that BMDM are indeed present in murine brain tumors. Gene expression profiling showed that while BMDM and MG share features of tumor education, they also exhibit distinct activation modes. Our data suggest these faculties are a result of inherent transcriptional networks poised before the onset of tumorigenesis, where ontogeny pre-biases cells to engage in distinct macrophage activation states. Lastly, we identify markers that distinguish MG and peripherally-derived macrophages under homeostasis, as well as in glioma and brain metastasis in both mice and humans.
RESULTS
Tumor-associated BMDM are present in mouse glioma models
To track the ontogeny of myeloid cells in murine gliomas we utilized a hematopoietic lineage tracing system, Flt3:Cre; Rosa26:mTmG, which has been used to show that peripheral myeloid cells develop from Flt3+ short term hematopoietic stem cells (ST-HSC), and are GFP+, while parenchymal MG develop independently of ST-HSC precursors, and are thus negative for the GFP reporter, remaining TdTomato+ (Boyer et al., 2011; Gomez Perdiguero et al., 2015). In non-tumor bearing mice, >98% of blood monocytes were GFP+, and <1% of MG showed recombination for the mTmG reporter (Figure 1A). The spleen was composed of GFP+ lymphocyte-rich follicles, surrounded by TdTomato+ stromal cells, while the brain parenchyma did not contain any detectable GFP+ cells (Figure S1A).
Figure 1. Lineage tracing systems demonstrate heterogeneity in TAM ontogeny in multiple models of glioma.
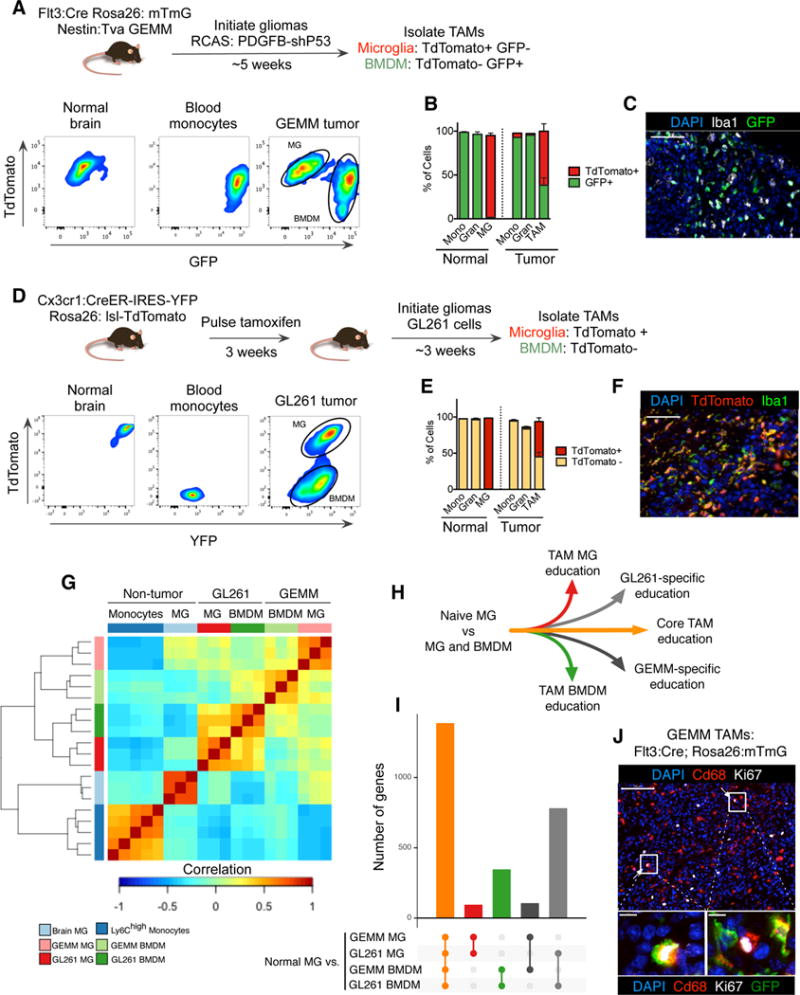
(A) Experimental scheme for the GEMM-shP53 model, see Supplemental Experimental Procedures for details. Representative flow cytometry panels for TdTomato and GFP are shown for Cd45+Cd11b+Ly6G−Ly6C− microglia (MG), Cd45+Cd11b+Ly6G−Ly6C+ monocytes, and Cd45+Cd11b+Ly6G−Ly6C−TAMs from GEMM-shP53 gliomas. (B) Quantitation of TdTomato+ and GFP+ monocytes (Mono) and granulocytes (Gran) in peripheral blood, MG in non-tumor bearing brain, and monocytes, granulocytes and TAMs in GEMM-shP53 gliomas as depicted in (A). Bars represent mean and s.e.m. n=3–5 for each group. (C) Representative immunofluorescence (IF) staining of Iba1 (white), GFP (green) and DAPI (blue) in a GEMM-shP53 tumor as depicted in (A). Scale bar: 50μm. Representative of n=5 tumors. (D) Experimental design for Cx3cr1 lineage tracing model, see methods for details. Monocytes, MG and TAMs were isolated as described in (A), and evaluated for TdTomato and YFP reporter expression. Representative of n=3 mice. (E) Flow cytometry quantitation of TdTomato+ and TdTomato− monocytes and granulocytes in peripheral blood, MG in non-tumor bearing brain, and monocytes, granulocytes and TAMs in GL261 gliomas as depicted in (D). Bars represent mean and s.e.m. n=3 for each group. (F) Representative IF staining for Iba1 (green), TdTomato (red) and DAPI (blue) in a GL261 tumor. Scale bar: 50μm. (G) Pairwise correlation matrix of normalized RNA-seq counts from monocytes (n=5), normal MG (n=3), and the four TAM populations from the different models: GEMM-shP53 TAM MG, GEMM-shP53 TAM BMDM, GL261 TAM MG, and GL261 TAM BMDM (n=3 for each group). (H) Diagram depicting different modules of TAM education compared to normal MG. (I) Differentially expressed genes between normal MG and the four TAM populations were tabulated. Barchart depicts the number of differentially expressed genes shared between the different groups, (J) Representative IF staining of Ki67+ TAM BMDM and TAM MG in the GEMM-shP53 model as depicted in (A). Ki67 (white), CD68 (red), DAPI (blue), GFP (green) (omitted from top panel). Scale bars: 100μm on upper panel, 10μm on lower panels. Representative of n=5 tumors.
We next bred this line to the nestin:Tva (nTva) line to trace myeloid cell ontogeny in a genetically engineered mouse model (GEMM) of glioma. We induced gliomas by intracranial injection of DF1 cells transfected with RCAS vectors encoding PDGFB and a short hairpin against P53 (Ozawa et al., 2014) (Figure 1A), termed GEMM-shP53 herein. Flow cytometry of end-stage gliomas demonstrated that all monocytes (Cd45+Cd11b+Ly6ChighLy6G−) and granulocytes (Cd45+Cd11b+Ly6ClowLy6Ghigh) in the tumor were GFP+ (Figure 1B), while the bulk TAM compartment (Cd45+Cd11b+Ly6C-Ly6G−) was composed of both GFP+ TAM BMDM and GFP− TAM MG (Figure 1A, B, Figure S1B), confirmed by tissue immunofluorescence (IF) co-staining with the pan-macrophage marker Iba1 (Figure 1C). By contrast, the contralateral, non-malignant brain contained only GFP− MG, demonstrating the specific abundance of TAM BMDM only within the tumor mass (Figure 1B).
We and others have utilized IR-BMT to show that TAM BMDM are recruited to murine gliomas (Huang et al., 2014; Pyonteck et al., 2013). However, IR can lead to ectopic recruitment of BMDM to the brain and thereby increase their relative abundance (Muller et al., 2015). We verified these findings in the orthotopic, syngeneic GL261 glioma model, and found the TAM compartment was composed of both TAM MG and TAM BMDM using both IR-BMT lineage tracing and IR-independent Flt3:Cre lineage tracing (Figure S1C, D). TAM BMDM abundance was significantly increased in the IR-BMT model compared to the Flt3:Cre model (Figure S1D), reinforcing previous reports that IR-BMT can skew the ratio of MG and BMDM. Critically, however, using Flt3:Cre lineage tracing we found that BMDM composed >35% of the bulk TAM population in gliomas without IR-preconditioning, demonstrating that BMDM infiltration into tumors is not solely an artifact of IR (Figure S1D).
To exclude the possibility that this finding was due to a subset of TAM MG spontaneously upregulating Flt3 expression, we utilized a complementary lineage tracing approach previously indicated to be specific for MG in the normal brain: Cx3cr1:CreER-IRES YFP; Rosa26:lsl-TdTomato (see Supplemental Experimental Procedures for details) (Parkhurst et al., 2013). At 3 days post tamoxifen-induced labeling, >99% of MG and circulating monocytes were TdTomato+ (Figure S1E). However, after 3 weeks blood monocytes no longer retained the TdTomato+ reporter, indicating their turnover and replenishment by tamoxifen ‘naïve’ monocytes (Figure S1E). By contrast, >99% of MG remained TdTomato+ (Figure S1E). We induced GL261 tumors in these mice, at 7 weeks of age, and observed both TdTomato+ TAM MG and TdTomato− TAM BMDM (Figure 1D, E). Meanwhile, all monocytes and granulocytes were TdTomato− in the tumor and periphery (Figure 1E). These findings were substantiated by IF co-staining of tissue sections with Iba1 (Figure 1F). Importantly, there was a gradient of eYFP reporter expression levels, with highest expression in TdTomato+ TAM MG, slightly lower levels in TdTomato− TAM BMDM, and lowest levels in monocytes (Figure S1F), demonstrating the capacity of TAM BMDM to express Cx3cr1 in brain tumors. Thus, Cx3cr1 expression alone cannot be used to strictly identify MG in gliomas. Together, these complementary genetic lineage-tracing models show that BMDM contribute to the TAM pool in several murine models of glioma, in the absence of IR.
RNA-sequencing reveals multimodal patterns of TAM education
We next analyzed the transcriptional profiles of TAM MG and TAM BMDM in gliomas. We performed RNA-sequencing on sorted populations of TAM MG and TAM BMDM from GEMM-shP53 and GL261 tumors using the Flt3-based and Cxc3cr1-based lineage tracing systems respectively. We also collected MG and Ly6Chigh blood monocytes from non-tumor bearing Flt3:Cre Rosa26:mTmG mice. Global correlation analyses revealed distinct clustering of all TAM populations from normal MG and monocytes, with further cell type-specific and tumor-specific clustering (Figure 1G). As expected, monocytes were enriched for Ly6c2 expression, while both TAM MG and TAM BMDM expressed higher levels of macrophage differentiation markers (e.g. Aif1, Mertk) compared to monocytes (Figure S1G). Normal MG and TAM MG expressed higher levels of MG-enriched genes (e.g. Cx3cr1, P2ry12, Tmem119) compared to monocytes and TAM BMDM (Figure S1G).
We next delineated cell type-specific, tumor-specific, and conserved patterns of tumor education among TAMs (Figure 1H). We identified differentially expressed genes between each TAM population from GEMM-shP53 and GL261 tumors compared to normal MG and monocytes (Figure 1I, Figure S1H, Table S1A). Using normal MG as the reference, we found 91 genes specifically upregulated in TAM MG from both GEMM-shP53 and GL261 models (Figure 1I, red bar), and 342 genes upregulated in TAM BMDM from both GEMM-shP53 and GL261 models (Figure 1I, green bar). We also identified genes that were specifically upregulated in TAM MG and TAM BMDM from either the GEMM-shP53 (n=102) or GL261 (n=778) models. The largest gene set (n=1383) was significantly upregulated in all TAM populations compared to normal MG (Figure 1I, orange bar). Similar patterns of expression were observed when monocytes were used as the reference population (Figure S1H, Table S1B).
Many cell cycle-related genes were upregulated, suggesting increased TAM proliferation compared to normal MG and monocytes (Table S1A,B). Indeed, we found Ki67+ cells in both Iba1+GFP+ TAM BMDM and Iba1+GFP− TAM MG in the Flt3-based lineage-tracing model (Figure 1J). Conserved upregulation of complement-related factors, extracellular matrix components, proteases, lipid metabolism mediators, and clotting factors were also evident in both TAM populations (Table S1A). In addition to these programmatic changes, compared to normal MG there was upregulation of growth factors (Igf1, Areg, Osm), chemokines and cytokines (Spp1, Ccl5, Cxcl9, Cxcl10), and other immune modulators including Cd274/PD-L1 and MHC class I molecules (H2-K1, H2-D1, B2m) (Table S1A). A similar distribution of differentially expressed genes was evident in comparing the TAM populations from both glioma models to blood monocytes (Figure S1H, Table S1B). Interestingly, we found several MG-enriched genes (e.g. Tmem119, Olfml3, Lag3, Jam2, Sparc) (Gautier et al., 2012) enriched in TAM BMDM in both GL261 and GEMM-shP53 models compared to monocytes (Table S1B). Despite this difference, there was still higher expression of MG-related genes in normal MG and TAM MG than in TAM BMDM. Meanwhile, other MG-enriched genes showed no such induction in TAM BMDM (P2ry12, Sall1, Mef2c). Collectively, these data are consistent with Cx3cr1 upregulation specifically in gliomas (Figure S1F), and the notion that macrophages acquire tissue-resident gene expression upon infiltration into a foreign tissue (Gosselin et al., 2014; Lavin et al., 2014).
TAM BMDM and TAM MG possess distinct education patterns
We investigated transcriptional differences between TAMs derived from BMDM versus MG and identified 378 differentially expressed genes enriched in TAM MG compared to TAM BMDM in both GEMM-shP53 and GL261 models and 485 genes enriched in TAM BMDM compared to TAM MG (Figure 2A, Table S2). As expected, among the 378 TAM MG genes, we found markers previously shown to be enriched in MG compared to other macrophage populations including P2ry12, Tmem119, Slc2a5, Pros1, and Sall1 (Figure 2A) (Gautier et al., 2012). Consistent with their tissue-specific functions, we found that normal MG and TAM MG were enriched for Jam2 and Ocln (Figure S2A, Table S2), integral components of the blood-brain barrier (Liu et al., 2012). Similarly, TAM MG expressed higher levels of classical complement factors C4b, C2, and Cfh (Figure S2A), a pathway important for MG function in synaptic pruning and host defense (Stephan et al., 2012).
Figure 2. TAM BMDM and TAM MG possess distinct gene expression patterns.
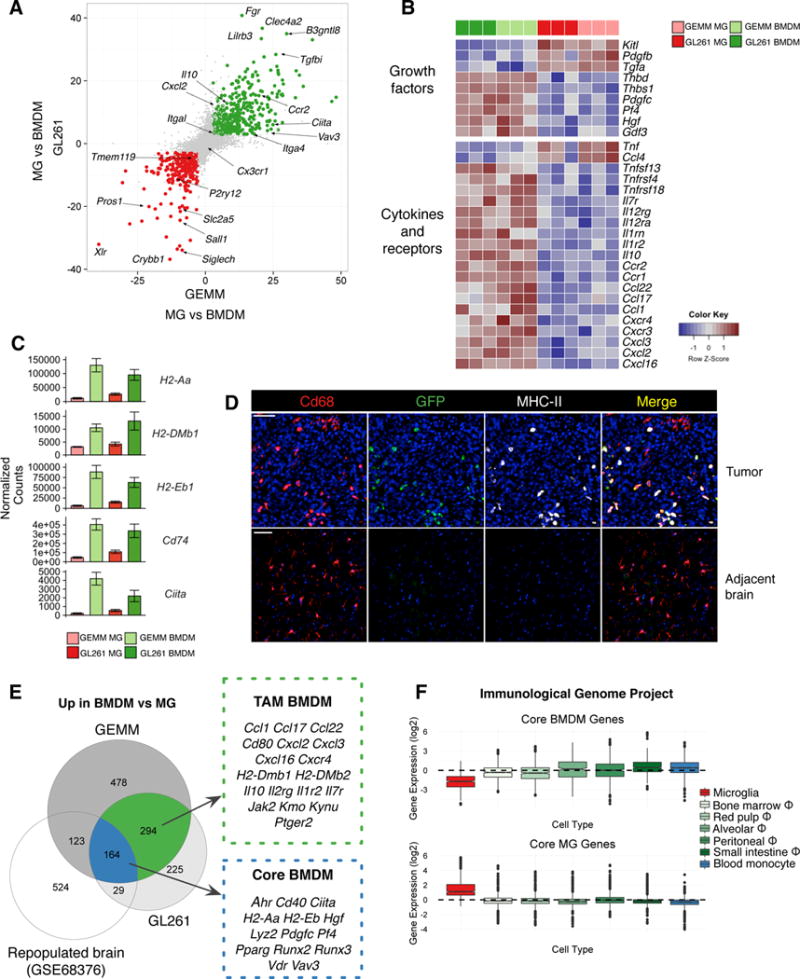
(A) Scatterplot depicting −log10(pvalue)*sign(fold change) between TAM BMDM and TAM MG in GEMM-shP53 gliomas (x-axis) and GL261 gliomas (y-axis). Significantly upregulated genes (log2 fold change > ±1 and FDR<1%) are in green for BMDM and red for MG. (B) Heatmap depicting row-normalized log2 gene expression values for indicated genes in GL261 TAM BMDM (dark green), GEMM-shP53 TAM BMDM (light green), GL261 TAM MG (dark red), and GEMM-shP53 TAM MG (light red). (C) Barplots depicting normalized gene expression values for indicated genes in these four different TAM populations. Bars represent mean ± s.e.m. (D) Representative IF staining in GEMM-shP53 Flt3:Cre Rosa26:mTmG gliomas and adjacent normal brain for Cd68 (red, AlexaFluor-594), GFP (green) and MHC-II (white). DAPI is shown in blue and TdTomato fluorescence is not shown. Scale bar: 100μm. Representative of n=5 tumors. (E) Venn diagram depicting significantly upregulated genes in BMDM vs MG in GL261 model, GEMM-shP53, and non-malignant brain (GSE68376 dataset). Select genes are listed. (F) Boxplot of Core BMDM genes (Figure 2E) and Core MG genes (Figure S2D) where each data point represents the z-scored expression of a gene across the indicated cell populations using available datasets from the Immunological Genome Project.
Meanwhile, TAM BMDM expressed high levels of alternative complement cascade components Cfb and Cfp (Figure S2A), and enrichment of many immune effectors including Cd40, Jak2, Ifitm1, Ifitm2, Tlr11, Tlr5, Tlr8, Mefv, and Fas (Figure S2A). In the GEMM-shP53 model IL-1 pathway ligands were differentially expressed, with Il1a enriched in TAM MG and Il1b in TAM BMDM, and similar trends observed in the GL261 model (Figure S2B). While Il1r1 levels did not significantly differ, TAM BMDM expressed higher levels of the IL-1 signaling antagonist Il1rn, and the IL-1 decoy receptor Il1r2 (Figure S2B). These results complement reports in non-cancer contexts demonstrating Il1a enrichment in MG compared to BMDM, where IL-1 signaling played a critical role in MG repopulation and maintenance (Bruttger et al., 2015).
We next interrogated chemokines, growth factors and immune modulators associated with different macrophage activation states. In addition to model-specific gene expression changes (Figure S2C, Table S3), we found in both GEMM and GL261 models that TAM BMDM were enriched for chemokines involved in wound healing including Ccl22, Ccl17, Cxcl2, Cxcl3, and Cxcl16 (Figure 2B) (Xue et al., 2014). Interestingly, TAM MG were enriched for expression of Ccl4 and Tnf, chemokines associated with a pro-inflammatory response (Xue et al., 2014). This difference in activation states was supported by a programmatic increase in antigen presentation centered around increased expression of the MHC-II master regulator Ciita (Reith et al., 2005), and its transcriptional targets H2-Aa, H2-DMb1, H2-Eb1 and Cd74 in TAM BMDM (Figure 2C). IF staining in Flt3:Cre; GEMM-shP53 tumors revealed a marked increase in MHC-II in tumors, compared to adjacent brain, restricted to GFP+ TAM BMDM (Figure 2D). In addition to this antigen presentation program, costimulatory molecules such as Cd80, Cd40, and Cd200r4 were increased (Figure S2A). These findings were further complemented by TAM BMDM-enriched expression of the Aryl-hydrocarbon receptor (Ahr), a transcription factor previously shown to mediate immune suppression (Murray et al., 2014; Opitz et al., 2011) (Figure S2A). Critically, we also found that the immunosuppressive cytokine Il10 was enriched in TAM BMDM compared to TAM MG (Figure 2B). Collectively, these results suggest that TAM BMDM engage in a chronic wound healing-like state reminiscent of an alternatively-activated macrophage (Mosser and Edwards, 2008). Similar phenotypes have been shown in models of oligodendrocyte cell death, where, despite high MHC-II expression, myeloid cells did not activate a robust T cell response (Locatelli et al., 2012), suggestive of a tolerogenic program.
We next asked if the differences in inflammatory mediators were an inherent feature of BMDM upon entry into the brain or rather a consequence of tumor education. Previous studies demonstrated that when MG are depleted and the brain preconditioned by IR, BMDM can seed the brain and contribute significantly to the brain macrophage pool (hereafter termed “ectopic BMDM” in a normal “repopulated brain”) (Bruttger et al., 2015). We used this dataset for comparative analyses with our TAM BMDM and TAM MG RNA-seq data to discriminate tumor education differences from ontogenetic, non-tumor associated differences. This juxtaposition allowed us to identify genes enriched in TAM MG vs TAM BMDM as well as normal MG vs “ectopic BMDM”. These “Core MG” genes included known MG markers such as Jam2, Siglech and P2ry12, but also complement factors C2, C4b, and Cfh, as well as the pro-inflammatory cytokines Ccl4 and Tnf (Figure S2D, n=245 genes, Table S4). We identified genes enriched in TAM BMDM (n=294) specifically in the context of a tumor, including Il10, Cxcl2, Cxcl3, Ccl17, Ccl22, and H2-Dmb1 (Figure 2E). In contrast to this tumor-specific expression profile, there were also 164 “Core BMDM” genes enriched in BMDM compared to MG, regardless of the presence or absence of a tumor, including Ciita, Ahr, Runx2, Runx3, Vav3 and Vdr (Figure 2E, Table S4). These data indicate some features distinguishing TAM BMDM and TAM MG are inherent to their differential ontogenies, while others are only acquired upon interaction with, and education by, the tumor microenvironment.
As many of the “Core BMDM” genes are central players in innate immunity, we queried the immunological genome project database to determine if these genes were overrepresented in any particular myeloid cell population. Interestingly, we found that these genes were actually repressed in MG compared to tissue-resident macrophages of the BM, spleen, lung, peritoneum, small intestine, and monocyte progenitors (Figure 2F). Meanwhile “Core MG” genes were indeed enriched in MG compared to other myeloid cells (Figure 2F). These data suggest that “Core BMDM” genes are neither specifically enriched in TAM BMDM nor macrophages in general, but rather are specifically repressed in MG.
Recent studies have highlighted extensive epigenetic diversity amongst tissue-resident macrophages (Lavin et al., 2014); thus we hypothesized that the MG-repressed genes may be epigenetically altered in MG compared to even the distantly related monocytes. Indeed, when we analyzed these published datasets, we observed increased H3K27 acetylation in the promoters of normal monocytes compared to normal MG for the “Core BMDM” genes (Figure S2E). Similarly, there was increased H3K27 acetylation in the promoters of “Core MG” genes in MG compared to monocytes (Figure S2E). Enhancer specification and epigenetic states in MG and other macrophage populations have been associated with differential PU.1 occupancy. Interrogating previously published data (Gosselin et al., 2014), we observed that several macrophage subsets (including BMDM) all showed increased PU.1 binding at the promoters of our “Core BMDM” genes compared to normal MG (Figure S2F). Meanwhile, variability in PU.1 occupancy was minimal at the promoters of “Core MG” genes (Figure S2F). Similar binding dynamics were evident in enhancer elements, where PU.1 occupancy in enhancer regions of “Core BMDM” genes was higher in BMDM than MG, with less pronounced differences present in “Core MG” genes (Figure S2G). Thus, epigenetic landscapes established before the development of a tumor may play a role in regulating differential activation patterns subsequently observed in malignancy.
Identification of transcription factor networks underlying TAM activation
Given the epigenetic differences in the non-malignant setting, we next determined if chromatin states also differed between TAM BMDM and TAM MG. We performed Assay for Transposase-Accessible Chromatin (ATAC)-sequencing (Buenrostro et al., 2013) to assess chromatin accessibility in TAM BMDM and TAM MG sorted from the GL261 model (Figure 1D). We found the ATAC-seq signal was associated with cell type-specific gene expression. In TAM BMDMs, the promoters of Core BMDM and TAM BMDM genes had higher ATAC-seq signal than Core MG and TAM MG genes, while TAM MG promoters of Core MG and TAM MG genes had a higher ATAC-seq signal than Core BMDM and TAM BMDM genes (Figure S3A). Within enhancers and intronic elements of these gene sets we identified 120 BMDM-specific peaks in TAM BMDM genes including Vav3, and 704 MG-specific peaks in TAM MG genes including P2ry12 and Sall1 (Figure 3A, Figure S3B,C, Table S5A).
Figure 3. Cell-specific transcription factor activities underlie differences between TAM BMDM and TAM MG.
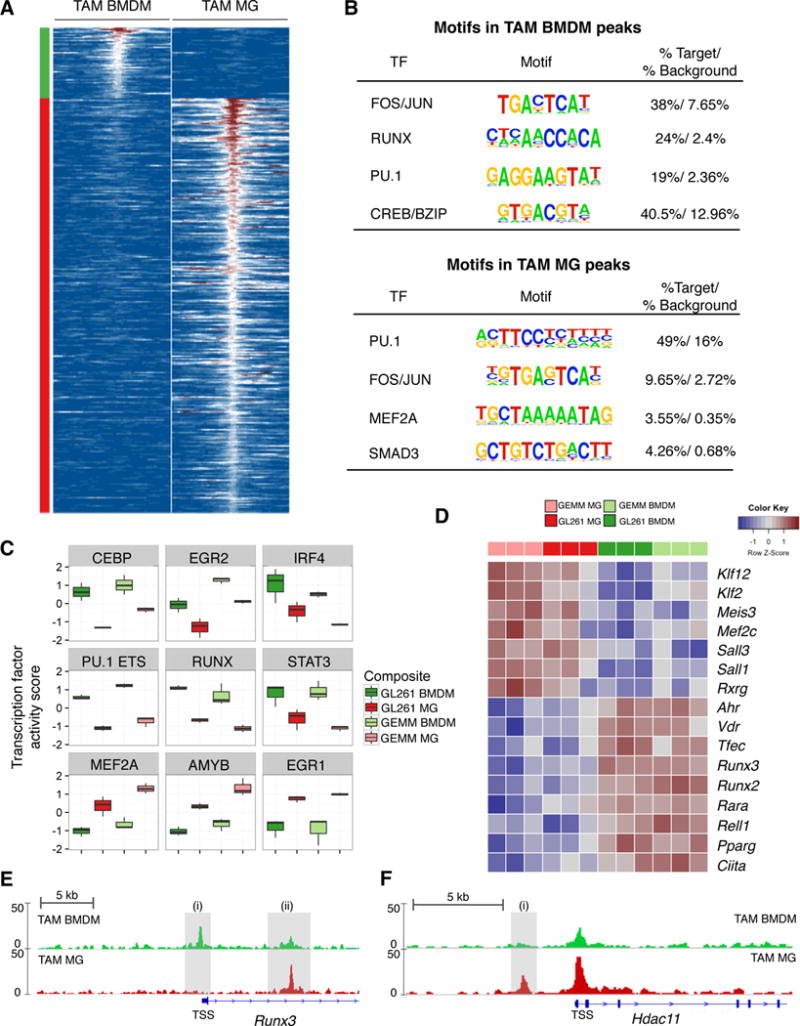
(A) Heatmap depicting ATAC-seq signal 1kb upstream and downstream of peaks specifically enriched in GL261 TAM BMDM (left panel) and GL261 TAM MG (right). Peaks were selected based on association with differentially expressed genes between TAM BMDM (top, green) and TAM MG (bottom, red). (B) Motifs identified by HOMER to be enriched in TAM BMDM and TAM MG peaks shown in (A). (C) Boxplots depicting normalized TF activity scores for indicated motifs across TAM BMDM and TAM MG from GL261 and GEMM-shP53 gliomas. (D) Heatmap depicting row-normalized log2 gene expression values for indicated genes in four different TAM populations. (E-F) ATAC-sequencing tracks from TAM BMDM (top, green) and TAM MG (bottom, red) from GL261 gliomas for (E) Runx3 and (F) Hdac11. Shaded grey regions indicate peaks specifically referenced in text. Y-axis values indicate tags per 10,000,000 with a range of 0–50. TSS denotes transcription start site.
We analyzed the transcription factor (TF) landscape underlying these different peaks, and performed de novo motif analysis (motifs are shown in all capital letters). Motif analysis of these peaks revealed an enrichment of FOS/JUN and PU.1 binding sites in both TAM BMDM and TAM MG peaks (Figure 3B, Table S5B), reinforcing previous analyses demonstrating the critical role of PU.1 in establishing specific enhancer landscapes in tissue-resident macrophages (Gosselin et al., 2014). Besides these shared enrichments, we found TAM BMDM peaks enriched for RUNX and CREB/bZIP motifs, while TAM MG peaks were enriched for SMAD3 and MEF2A motifs (Figure 3B).
To determine if these motifs reflected pathway activation of particular TFs we modeled the expression of their predicted downstream targets (see Supplemental Experimental Procedures). We identified TF families with enriched activity in TAM BMDM relative to TAM MG in the GEMM-shP53 and GL261 models (and vice versa) (Figure 3C, Figure S3D). Among a panel of different TFs, EGR1 and MEF2A were enriched in TAM MG (Figure 3C, Figure S3D, Table S5C). Interestingly, MEF2 is associated with MG identity (Lavin et al., 2014). In TAM BMDM, TF motifs involved in monocyte to macrophage differentiation were enriched including RUNX, CEBP, and PU.1 (Figure 3C, Figure S3D) (Alder et al., 2008). STAT3 and IRF4 were also enriched (Figure 3C), both of which have been associated with differential functions in macrophage activation (Mosser and Edwards, 2008; Ostuni and Natoli, 2011). We complemented these genome-wide TF activity analyses with motif enrichment analysis on the promoters of TAM BMDM-specific and TAM MG-specific genes using HOMER (Heinz et al., 2010) (Figure S3E). This also revealed an enrichment of MEF2 motifs in TAM MG, demonstrating the consistent role of tissue-specific transcriptional programs in TAM MG education. Meanwhile, TAM BMDM-specific genes were again enriched in PU.1, RUNX and CEBP motifs (Figure S3E). These findings were further corroborated by increased expression of brain-specific TFs (Mef2c, Sall1, Sall3) in TAM MG, while TAM BMDM were enriched for Ciita, Vdr, Ahr, and Runx family members (Figure 3D, Table S2A).
Given the consistent enrichment of RUNX activity in TAM BMDM, we next focused on examining the expression and chromatin state of Runx family members. Runx2 and Runx3 were enriched in TAM BMDM compared to TAM MG (Figure 3D). While no differences were found in the chromatin state of Runx1 or Runx2, in the first intron of Runx3 (Figure 3E-ii) we observed a peak present in TAM MG but reduced in TAM BMDM (Figure 3E). Meanwhile, the Runx3 promoter showed little open chromatin in TAM MG, and a distinct peak in TAM BMDM near the transcription start site (Figure 3E-i). Interestingly, both peaks have been shown to be TGFβ-responsive PU.1 binding sites associated with Runx3 expression (Chopin et al., 2013), indicating the same signal transduction pathway can produce distinct outputs in TAM BMDM and TAM MG.
We also identified enrichment of the epigenetic modifiers Hdac7 and Hdac9 in TAM BMDM, while Hdac11 was enriched in TAM MG (Figure S3F), the latter of which has been shown to repress Il10 expression in macrophages (Villagra et al., 2009). Interestingly, an upstream enhancer element in Hdac11 was significantly enriched in TAM MG compared to TAM BMDM, a peak that contained a SMAD-responsive element (Figure 3F-i). Collectively, these results suggest that differential genomic PU.1 occupancy underlies distinct open chromatin states in BMDM and MG, whereupon additional factors such as TGFβ/SMAD signaling and RUNX family members cooperate with PU.1 to enforce distinct transcriptional networks. Subsequent regulation of TFs and chromatin modifying factors, such as Hdac11, may explain the distinct cytokine expression patterns observed such as TAM BMDM expression of Il10, and TAM MG expression of Tnf.
Itga4/Cd49d distinguishes microglia and peripherally-derived macrophages in murine models of brain malignancy
We next sought to identify tools capable of distinguishing TAM BMDM and TAM MG in human disease, where genetic lineage tracing is not possible. Given that TAM BMDM in gliomas upregulated Cx3cr1 (Figure S1F), a proposed MG marker, we sought to identify TAM BMDM-specific markers that instead remained silent in TAM MG. From the 164 “Core BMDM” genes, we identified 40 candidate transmembrane proteins that might serve as useful markers for flow cytometry. Amongst these, the integrin subunit alpha 4, Itga4/Cd49d, emerged as a promising candidate, particularly given previous reports that it, along with the integrin subunit alpha L, Itgal/Cd11a, is regulated by RUNX family members, including Runx1 and Runx3 (Dominguez-Soto et al., 2005). Consistently, we found that Itga4 and Itgal were specifically repressed in MG compared to other macrophage populations (Figure S4A). This was confirmed by flow cytometry, where Cd49d expression in MG was negligible or absent compared to macrophages of the spleen, liver, lung, bone marrow and blood Ly6C+ monocytes (Figure 4A). Ly6G+ granulocytes were also Cd49d−, which, along with Cd49d+ lymphocytes in the CD45+Cd11b− gate, served as useful gating controls in subsequent experiments (Figure 4A).
Figure 4. Itga4/Cd49d distinguishes TAM BMDM and TAM MG in murine brain malignancy.
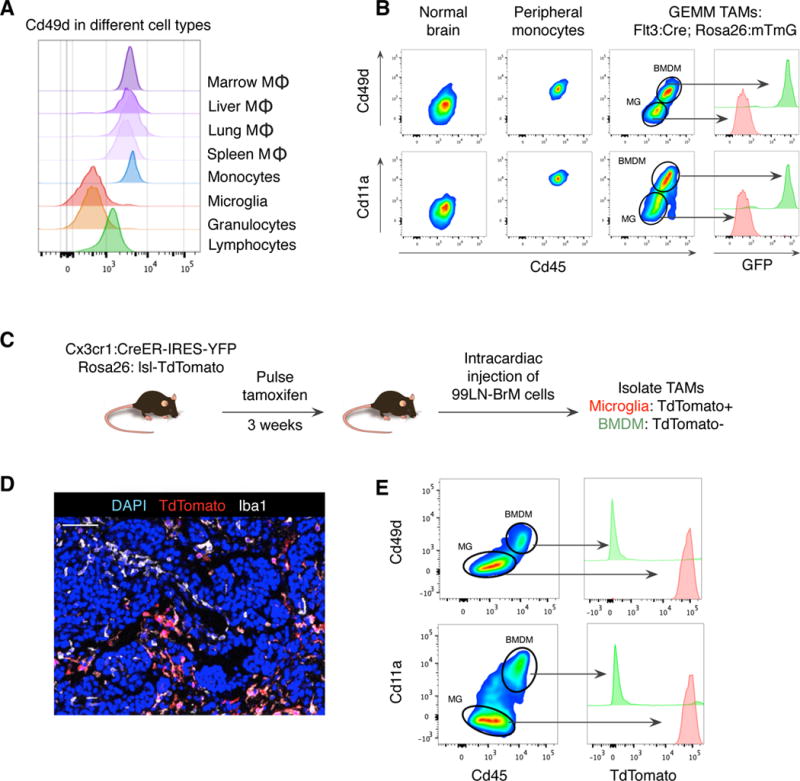
(A) Histogram of Cd49d expression for indicated populations from non-tumor bearing mice. (B) Flow cytometry for Cd45 and either Cd49d (top) or Cd11a (bottom) in normal blood monocytes, normal MG (from adjacent normal brain), or TAMs isolated from Flt3:Cre Rosa26:mTmG mice with GEMM-shP53 tumors. Adjacent histograms indicate GFP expression in indicated populations. (C) Experimental schematic for 99LN-BrM model in Cx3cr1-lineage tracing mice. (D) Representative IF staining of TdTomato (red), Iba1 (white) and DAPI (blue) 99LN-BrM tumors as depicted in (C). Scale bar: 50μm. (E) Flow cytometry as in (B) for 99LN-BrM model, with TdTomato expression indicated in the adjacent histogram. Flow plots are representative of n=5–8 mice.
We examined Cd49d and Cd11a expression in TAM BMDM and TAM MG using Flt3:Cre-based lineage tracing in the GEMM-shP53 model. After gating on Cd45+Cd11b+Ly6C−Ly6G− cells, the normal brain only contained Cd45lowCd49d− cells, and all peripheral monocytes were Cd45highCd49d+ (Figure 4B). In tumors we found two cell populations; Cd45lowCd49d− and Cd45highCd49d+, which contained GFP− TdTomato+ MG and GFP+ TdTomato− BMDM respectively (Figure 4B). Similar results were found for Cd11a (Figure 4B), and were replicated in the GL261 model using both Cx3cr1-based and Flt3:Cre lineage tracing strategies (Figure S4B, C). Lastly, we evaluated Cd49d expression in a Pten loss of function-PDGFB-driven glioma model (GEMM-PtenFl°x) where PtenFl°x/Fl°x; nTva+ mice were injected with RCAS vectors encoding PDGFB and Cre (Huse et al., 2009). Using IR-BMT for lineage tracing, we found that Cd49d distinguishes donor and host-derived cells, including in glioma models with extended latency (~12 weeks for the GEMM-PtenFl°x model) (Figure S4D).
To evaluate other models of brain malignancy, we utilized an intracardiac injection model of brain metastasis (BrM) colonization using a tumor cell line (99LN-BrM). 99LN-BrM cells were originally derived from the lymph node of a MMTV:PyMT breast cancer GEMM and subjected to in vivo selection. We used this syngeneic, immunocompetent BrM model in conjunction with Cx3cr1-based lineage tracing and found that BrM lesions contained both TdTomato+ Iba1+ and TdTomato− Iba1+ cells indicating recruitment of both TAM MG and TAM BMDM respectively (Figure 4C, D). We validated these findings by flow cytometry, where Cd49d and Cd11a served as reliable markers of BMDM as in the glioma models described above (Figure 4E). We again found that eYFP levels, a direct readout of Cx3cr1 expression, were similar between TAM BMDM and TAM MG in BrM, reinforcing the necessity of the Cx3cr1:CreER lineage tracing approach over that of the Cx3cr1 reporter (Figure S4E). Lastly, we confirmed these data in a well-established xenograft BrM model using brain homing MDA-MB-231 cells (Bos et al., 2009), in conjunction with IR-BMT lineage tracing using mRFP+ donor cells. In this model, we identified two cell populations; Cd45lowCd49d− MG and Cd45highCd49d+ BMDM. The mRFP+ donor cells were exclusively found within the Cd45highCd49d+ BMDM gate (Figure S4F).
Together, our results obtained in multiple models of brain malignancy with distinct lineage tracing approaches demonstrate that TAM BMDM accumulation is independent of BBB preconditioning by IR or intracranial injection. These data also thoroughly establish Cd49d as an efficient marker to distinguish resident MG and peripherally-derived macrophages in homeostasis as well as in primary and metastatic brain malignancies.
CD49D identifies microglia and macrophages in human brain malignancies
We next investigated whether CD49D could be used to discriminate MG and peripherally-derived macrophages in human brain tumors. We assessed CD49D expression by flow cytometry across a panel of surgical samples composed of nonmalignant normal brain (n=3), untreated high-grade glioma (GBM) (n=3), lung adenocarcinomas (n=6), and peripheral blood mononuclear cells (PBMCs) (n=6). Consistent with our data in mice, granulocytes (CD45+CD11B+CD66B+CD14lowCD16+) did not express CD49D, and were used as a reference guide for gating CD49D+ and CD49D− TAMs (Figure S5A). Importantly, we never identified CD49D− TAMs in primary lung tumors, or CD49D− monocytes in healthy donor PBMCs, indicating that, as predicted, low expression of CD49D is restricted to MG, and not a general phenotype of tissue-resident macrophages (Figure 5A). By contrast, the CD45+CD11B+CD66B−CD14+CD16− compartment in non-malignant brain was predominantly composed of CD49D− MG (Figure 5A). Critically, in each GBM sample we identified both CD49D+ and CD49D− TAMs, presumably representing BMDM and brain-resident MG respectively (Figure 5A).
Figure 5. CD49D discriminates TAM BMDM and TAM MG in human brain malignancy.
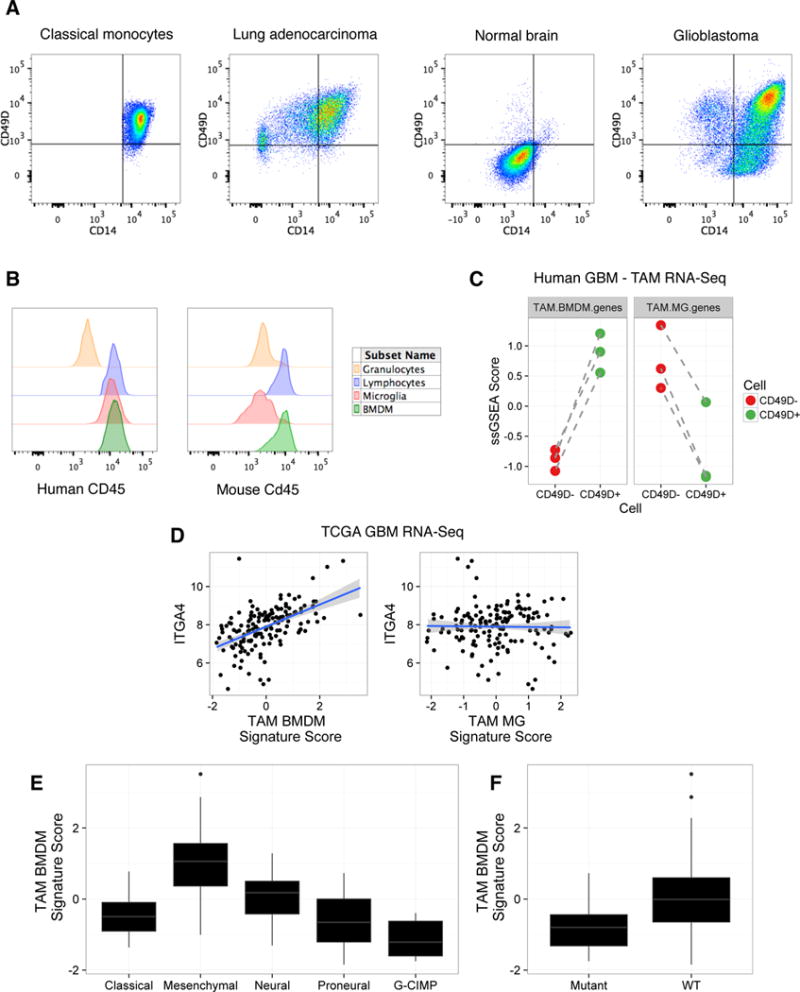
(A) Classical monocytes, MG, and TAMs were defined as CD45+CD11B+CD66B−CD14+CD16−. Gated cells are then shown for CD14 and CD49D in representative samples of human classical monocytes from peripheral blood (n=6), TAMs from a lung adenocarcinoma patient (n=6), MG from a non-malignant brain (n=3), and TAMs from a GBM patient (n=3). (B) Histogram of CD45 expression by flow cytometry in human (left) and mouse (right) samples. In human GBM, CD45 expression is shown for granulocytes (CD45+CD11B+CD66B+CD16+CD14low), lymphocytes (CD45+CD11B−), TAM MG (CD45+CD11B+CD66B−CD16−CD14+CD49D−) and TAM BMDM (CD45+CD11B+CD66B−CD16−CD14+CD49D+). In mouse, Cd45 expression is shown for granulocytes (Cd45+Cd11b+Ly6ClowLy6G+), lymphocytes (Cd45+Cd11b−), TAM MG (Cd45+Cd11b+Ly6C−Ly6G−Tomato+GFP−) and TAM BMDM (Cd45+Cd11b+Ly6C−Ly6G−Tomato−GFP+) from a Flt3:Cre Rosa26:mTmG GEMM-shP53 glioma. Representative of n=3 patients and n=5 mice. (C) Z-scored single sample gene set enrichment analysis scores for TAM BMDM genes (left, paired t-test, p≤5.01×10−3) and TAM MG genes (right, paired t-test, p≤7.78×10−3) in matched CD49D− and CD49D+ TAMs from GBM patients. Dashed lines indicate matched samples (n=3 patients). (D) Scatterplot of TAM BMDM signature score (x-axis, left, Spearman rho=0.564, p≤2.2×10−16) and TAM MG signature score (x-axis, right, Spearman rho=0.067, p≤0.411) and ITGA4 expression (y-axis) from TCGA-GBM RNA-seq data. Solid blue line indicates line of best fit with shaded areas depicting standard deviation confidence intervals. (E-F) Z-scored TAM BMDM signature scores across (E) GBM subtype (ANOVA p≤2.2×10−16), and (F) IDH1 mutation status (Student’s t-test p≤5.93×10−3).
Interestingly, in human samples we found no difference in CD45 expression between CD49D− and CD49D+ TAMs (Figure 5B), a marker previously suggested to be informative for distinguishing BMDM and MG in brain malignancy (Hussain et al., 2006; Parney et al., 2009; Sedgwick et al., 1991). Indeed, CD45 expression differed most prominently between granulocytes and TAMs, as opposed to MG and BMDM (Figure 5B). However, this lack of differential CD45 expression is not the case in mouse, where Cd45 adequately discriminates MG and BMDM in the models tested (Figure 5B). We next sorted paired CD49D− and CD49D+ TAMs from GBM patients to verify these populations indeed reflected TAM MG and TAM BMDM respectively. Using genes specific for TAM MG and TAM BMDM from our mouse models (Figure 2A), we found that CD49D− TAMs were indeed enriched for TAM MG genes (p≤7.78×10−3), while CD49D+ TAMs were enriched for TAM BMDM genes (p≤5.01×10−3) (Figure 5C).
Previous analyses of TAM expression in human gliomas have utilized bulk CD11B+ cells, a population likely composed of both TAM BMDM and TAM MG, as well as other myeloid populations. We queried one available RNA-seq dataset from bulk CD11B+ cells (Szulzewsky et al., 2016), which showed increased ITGA4/CD49D expression in purified CD11B+ cells in GBM compared to normal MG from either post-mortem samples, or resections from epileptic patients (Figure S5B). This was complemented by a relative decrease in the MG-enriched transcript P2RY12 in GBM compared to nonmalignant brain (Figure S5B). In querying an additional microarray-based dataset of purified CD11B+ cells (Gabrusiewicz et al., 2016), we observed that peripheral blood CD11B+ cells from GBM patients expressed similar levels of ITGA4 compared to GBM tumor samples, while there was higher P2RY12 expression in GBM samples compared to peripheral blood (Figure S5C), as we would have expected. We extended these analyses to whole tissue RNA-seq data from the TCGA-GBM cohort (Brennan et al., 2013), and observed that ITGA4 expression was significantly increased in GBM compared to normal brain (Figure S5D). Collectively, these analyses suggest that TAMs in GBM represent a heterogeneous population composed of both BMDM and MG, reinforcing the necessity of refined sorting strategies for accurate discrimination between these cells, and highlighting the utility of a CD49D-based gating approach.
We next assessed TAM BMDM and TAM MG gene set expression in the TCGA cohort as a whole. TAM BMDM genes and TAM MG genes showed high intra-gene set correlation, where TAM BMDM genes such as RUNX2, IL10, RUNX3, ITGA4 and VDR, showed significant pairwise correlations, and TAM MG genes such as MEF2C, P2RY12, RXRG, SALL1, KLF12, and SALL3 similarly showed significant pairwise correlations (Figure S5E). Moreover, ITGA4 showed a high correlation with a TAM BMDM gene signature score (p≤2.2×10−16), but not with a TAM MG signature score (Figure 5D), showing increased ITGA4 expression is specific to TAM BMDM abundance, and not TAMs as a whole.
Previous transcriptional and epigenetic analyses have identified distinct GBM subtypes (Noushmehr et al., 2010; Verhaak et al., 2010), where the Mesenchymal subtype was enriched for tumor stroma and inflammatory molecules. Here, we find TAM BMDM signature scores are significantly different among molecular subtypes of GBM (p≤2.2×10−16), with the highest scores in the Mesenchymal GBM subtype, and lowest in G-CIMP patients (Figure 5E). Correspondingly, TAM BMDM signature scores were lowest in patients with IDH1 mutations (Figure 5F, p≤5.93×10−3). By comparison, TAM MG signature scores displayed a blunted association with tumor subtype (p≤0.041) and no association with IDH1 mutation status (p≤0.153) (Figure S5F, G). These analyses reinforce our findings that TAM BMDM are a distinguishable immune cell population from TAM MG with distinct abundance and characteristics in specific subtypes of human GBM.
DISCUSSION
IR-BMT has been used widely in animal models to perform lineage tracing of TAMs in brain malignancy (Ajami et al., 2007; De Palma et al., 2005; Huang et al., 2014; Mildner et al., 2007; Muller et al., 2015), albeit with concerns regarding potential artifacts due to effects of IR on BBB disruption. Alternative chemical BMT approaches have been suggested, though similar effects on BBB permeability cannot be ignored (Alder et al., 2008; Kierdorf et al., 2013b). Here, we confirm that IR-BMT leads to increased TAM BMDM content in the GL261 glioma model, a finding that has been recently reported by juxtaposing IR-BMT with and without head-shielding (Muller et al., 2015). While IR-BMT may confound lineage-tracing studies, it remains to be seen if IR preconditioning before the onset of tumorigenesis significantly alters TAM activity in tumor development, or if the inflammatory environment of the tumor supersedes any antecedent effects of the IR-BMT protocol.
Other than IR-BMT, the most widely employed approach to discriminate MG and peripherally-derived macrophages relies upon Cd45 expression, with Cd45high cells considered BMDM and Cd45low cells considered MG (Gabrusiewicz et al., 2011; Sedgwick et al., 1991). While this marker seems adequate in the murine models we have employed here, cell type-specific CD45 expression appears to be different between mouse and human. Our data indicate that CD45 does not accurately discriminate MG and BMDM in patient samples, emphasizing the need for extensive flow cytometry panels to clearly distinguish these cells in both species. Additionally, our genetic lineage tracing models also show that expression of Cx3cr1, which is commonly used to trace normal MG, is subject to upregulation in BMDMs upon tumor education (Figure 2A, Figure S1F, Figure S4E) and thus cannot be used to discriminate MG versus BMDM in brain tumors.
Instead, we present Itga4 (Cd49d) as an effective, consistent marker that works in both mice and humans to distinguish MG and peripherally-derived macrophages in multiple brain malignancies. Cd49d may also prove a useful tool in determining the precise origin and kinetics of peripherally-derived macrophages in brain tumors. Recent efforts to understand the heterogeneity and origins of non-parenchymal myeloid cells in the brain (including perivascular, meningeal and choroid plexus macrophages) revealed that a subset of these cells are labeled using similar Flt3-Cre and Cx3cr1-CreER based lineage tracing systems as employed here (Goldmann et al., 2016). Thus it will be of interest to determine if any of these populations, in addition to monocytes, contribute to the TAM pool.
Our data supports the hypothesis that epigenetic states influence stimulus-dependent transcriptional induction, thus leading to differential TAM education between MG and BMDM. Differential genomic occupancy of PU.1 between MG and other macrophage populations in non-cancer contexts has been shown to dictate differential enhancer selection (Gosselin et al., 2014). Indeed, within this dataset we found that PU.1 binding sites at enhancers and promoters were already different between MG and BMDM for the genes we identified to be specific to their respective TAM populations. This suggests that TAM BMDM and TAM MG are poised to engage in different transcriptional networks based on initial enhancer selection. It is likely that differential expression of binding partners influences PU.1 genomic occupation. Cooperative binding is evident between PU.1 and CEBPβ to promote macrophage differentiation, and in B cell development where PU.1 occupancy is influenced by E2A expression (Heinz et al., 2010). Such a hypothesis has also been shown to account for MG-specific PU.1 binding in cooperation with TGFβ-induced SMAD activity (Gosselin et al., 2014). Similar dynamics may be at play in brain tumors, where binding partners that are absent in MG and expressed in BMDM can sculpt genomic PU.1 occupancy. For example, the RUNX family member Runx3 is one such candidate, which is enriched in TAM BMDM versus TAM MG and shows motif enrichment in promoters where PU.1 binds in BMDM, but not MG.
While our studies here focus predominantly on identifying recurrent signatures distinguishing TAM MG and TAM BMDM across multiple mouse models and patient samples, there were also tumor-specific gene expression patterns in TAM education (Figure 2E, Figure S2C,D), which may provide insights into how tumor-derived signals can generate inter-tumoral heterogeneity in TAM activation profiles. In addition, analysis of TCGA data showed that gene signatures associated with TAM BMDM were differentially enriched in the distinct tumor subtypes of GBM. Recent reports have identified mixed activation states in bulk TAM populations in glioma patients (Gabrusiewicz et al., 2016; Szulzewsky et al., 2016), and our data now shows that TAM MG and TAM BMDM possess distinct activation states, potentially resolving this mixed phenotype. Importantly, the identification of CD49D as a cell surface marker to discriminate between TAM MG and TAM BMDM in human disease will permit extensive interrogation of these cell populations in patient samples.
Collectively, the studies presented here definitively demonstrate that peripherally-derived macrophages are indeed present in multiple mouse and human brain malignancies, and have distinct transcriptional profiles from their brain-resident counterparts. We posit that while macrophages can acquire tissue-resident macrophage-like traits upon entry into a tissue (Lavin et al., 2014), an inflammatory microenvironment, such as in the context of cancer or neuroinflammation, may further amplify differences between the cells leading to diverse functional outcomes for tissue-resident and peripherally-derived macrophage populations.
EXPERIMENTAL PROCEDURES
Tumor and lineage tracing models
Mouse models of gliomagenesis and brain metastasis, cell line generation, and the use of lineage tracing models have been previously reported (Boyer et al., 2011; Parkhurst et al., 2013; Pyonteck et al., 2013; Sevenich et al., 2014) and are described in full in Supplemental Experimental Procedures.
Institutional Review Board (IRB) approval and patient information
All human specimens were collected from patients consented to MSKCC IRB protocols #06-107, #14-230. Glioma patients that presented with contrast-enhancing brain lesions and no prior history of brain malignancy or therapy were included. Tumor specimens were collected from the operating room and processed as described below. Pathological analyses confirmed grade IV GBM. Non-malignant normal brain samples were collected from two sources: non-malignant distant sites from low-grade disease, and post-mortem samples with no history of brain malignancy. Pathological analysis confirmed the absence of tumor. Samples from patients with primary lung tumors were included based on pathological analysis of lung adenocarcinoma, with no screening based on prior malignancy or therapy.
Flow cytometry and cell sorting
For blood analysis, mice were bled via either retro-orbital or submandibular routes under isoflurane anesthesia. For all other tissue analyses, mice were anesthetized with 1.25% avertin, and transcardially perfused with PBS. Single cell suspensions from spleen and BM were isolated by macrodissection and mechanical tissue dissociation. Liver, kidney, and lung were macrodissected and dissociated using the Mouse Tumor Dissociation Kit (mTDK, Miltenyi) and the OctoMACS dissociator. Mouse and human brain specimens were macrodissected and dissociated using the Brain Tumor Dissociation Kit (BTDK Miltenyi) and a single cell suspension generated using the OctoMACS dissociator. Human lung tumors were dissociated with the Human Tumor Dissociation Kit (hTDK Miltenyi.) All tissue suspensions were filtered through a 40 μM mesh filter and underwent red blood cell lysis (PharmLyse BD). Brain and brain tumor tissues were incubated with Myelin Removal Beads (Miltenyi). Single cell suspensions were FC blocked (BD #553141) for 15 mins at 4°C, then incubated with directly-conjugated antibody panels for 15 mins at 4°C. Cell suspensions were washed (PBS +2% fetal bovine serum) and resuspended in a DAPI solution. All flow cytometry analysis was completed on a BD Fortessa device and all sorting was performed on an Aria III. Cells were sorted directly into Trizol LS and snap frozen in liquid nitrogen. Antibodies and methods for immunohistochemistry can be found in Supplemental Experimental Procedures.
Statistical methods
RNA-sequencing, ATAC-sequencing and bioinformatics
RNA was isolated by chloroform extraction and isopropanol precipitation. RNA-sequencing libraries were generated with the SMART-Seq preparation kit (CloneTech). Single end, 100 base pair, sequencing was performed by GeneWiz (New Jersey, USA) on an Illumina HiSeq 2500. FASTQ files were mapped to the mouse genome (mm10) or the human genome (hg19) using STAR (version 2.5.0e) with default parameters (Dobin et al., 2013). Transcript abundance was quantified using STAR with a GTF file from iGenomes (Illumina). A count matrix was produced in R and differential gene expression was assessed with DESeq2 using a fold change cutoff of +/− 2 and a false discovery rate of 5% (Love et al., 2014). Gene ontology analysis was performed using DAVID with default parameters (Dennis et al., 2003). ATAC-Sequencing was performed as previously described (Buenrostro et al., 2013). Paired end, 50 base pair sequencing was performed on an Illumina HiSeq 2500 with an average read depth of ~35,000,000 reads per sample. Reads were mapped to mm10 using STAR (version 2.5.0e) using (– alignIntronMax 1 –alignEndsType EndToEnd). Peak calling, annotation, and differential peak identification was performed using HOMER.
Methods for analyzing external datasets, TF activity analysis, and additional statistical methods are described in Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank X. Chen for excellent technical support, Drs. D. Yan and N. Ben-Chetrit for experimental assistance, and members of the Joyce lab for insightful comments and discussion. We are grateful to Dr. B. Santomasso, N. Distefano and Dr. L. DeAngelis, MSKCC Neurosurgery, and Dr. C. Rudin, MSKCC Thoracic Oncology, for help in acquiring patient samples. We thank Drs. E. Holland and T. Ozawa for generously providing the RCAS vectors and Nestin-Tva mice, Dr. C. Forsberg for the Flt3:Cre Rosa26:mTmG mice, Drs. C. Sawyers and B. Carver for the PtenFlox/Flox mice, Drs. S. Coniglio and J. Segall for the GL261 cell line, and Dr. J. Massagué for the brain-homing MDA-MB-231 cell line. This research was supported by: R01CA181355 (J.A.J.), Ludwig Institute for Cancer Research (J.A.J.), MSKCC Center for Metastasis Research (J.A.J.), MSK Cancer Center Support Grant from NCI (P30 CA008748), Gerstner Sloan-Kettering Graduate School (R.L.B.), and fellowships from National Cancer Institute 5F31CA167863 (R.L.B.), Deutsche Forschungsgemeinschaft (F.K. (KL 2491/1-1) and L.S. (SE2234/1-1)), American Brain Tumor Association (L.A.), and Canadian Institutes of Health Research (D.F.Q.).
Footnotes
Accession numbers:
All gene expression data and ATAC-sequencing data generated in this study can be found under GEO accession number: GSE86573.
AUTHOR CONTRIBUTIONS
R.L.B. and J.A.J. conceived the study, designed and interpreted experiments, and wrote the manuscript. R.L.B., F.K., L.A., S.M.P., L.S., D.F.Q., S.D. and K.S. performed experiments and analyzed results. R.L.B. performed all computational analyses. E.E.G., C.I.D., C.W.B., V.T., P.H.G. provided patient samples. J.A.J. supervised the study. All authors commented on the manuscript.
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Alder JK, Georgantas RW, 3rd, Hildreth RL, Kaplan IM, Morisot S, Yu X, McDevitt M, Civin CI. Kruppel-like factor 4 is essential for inflammatory monocyte differentiation in vivo. J Immunol. 2008;180:5645–5652. doi: 10.4049/jimmunol.180.8.5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, De Palma M, Quattrini A, Del Carro U, Amadio S, Visigalli I, Sessa M, Fasano S, Brambilla R, Marchesini S, et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest. 2004;113:1118–1129. doi: 10.1172/JCI19205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, Massague J. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman RL, Joyce JA. Therapeutic targeting of tumor-associated macrophages and microglia in glioblastoma. Immunotherapy. 2014;6:663–666. doi: 10.2217/imt.14.48. [DOI] [PubMed] [Google Scholar]
- Boyer SW, Schroeder AV, Smith-Berdan S, Forsberg EC. All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell. 2011;9:64–73. doi: 10.1016/j.stem.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruttger J, Karram K, Wortge S, Regen T, Marini F, Hoppmann N, Klein M, Blank T, Yona S, Wolf Y, et al. Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity. 2015;43:92–106. doi: 10.1016/j.immuni.2015.06.012. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopin M, Seillet C, Chevrier S, Wu L, Wang H, Morse HC, 3rd, Belz GT, Nutt SL. Langerhans cells are generated by two distinct PU.1-dependent transcriptional networks. J Exp Med. 2013;210:2967–2980. doi: 10.1084/jem.20130930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, Naldini L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–226. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Soto A, Relloso M, Vega MA, Corbi AL, Puig-Kroger A. RUNX3 regulates the activity of the CD11a and CD49d integrin gene promoters. Immunobiology. 2005;210:133–139. doi: 10.1016/j.imbio.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois LG, Campanati L, Righy C, D’Andrea-Meira I, Spohr TC, Porto-Carreiro I, Pereira CM, Balca-Silva J, Kahn SA, DosSantos MF, et al. Gliomas and the vascular fragility of the blood brain barrier. Front Cell Neurosci. 2014;8:418. doi: 10.3389/fncel.2014.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrusiewicz K, Ellert-Miklaszewska A, Lipko M, Sielska M, Frankowska M, Kaminska B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PloS One. 2011;6:e23902. doi: 10.1371/journal.pone.0023902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, Thomas G, Zhou S, Wang Q, Elakkad A, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI insight 1. 2016 doi: 10.1172/jci.insight.85841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nature Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nature Immunol. 2016;17:797–805. doi: 10.1038/ni.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, Glass CK. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159:1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Hoffman C, Rajappa P, Kim JH, Hu W, Huse J, Tang Z, Li X, Weksler B, Bromberg J, et al. Oligodendrocyte progenitor cells promote neovascularization in glioma by disrupting the blood-brain barrier. Cancer Res. 2014;74:1011–1021. doi: 10.1158/0008-5472.CAN-13-1072. [DOI] [PubMed] [Google Scholar]
- Huse JT, Brennan C, Hambardzumyan D, Wee B, Pena J, Rouhanifard SH, Sohn-Lee C, le Sage C, Agami R, Tuschl T, Holland EC. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009;23:1327–1337. doi: 10.1101/gad.1777409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8:261–279. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DW, Abkowitz JL. Kinetics of central nervous system microglial and macrophage engraftment: analysis using a transgenic bone marrow transplantation model. Blood. 1997;90:986–993. [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013a;16:273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Katzmarski N, Haas CA, Prinz M. Bone marrow cell recruitment to the brain in the absence of irradiation or parabiosis bias. PloS One. 2013b;8:e58544. doi: 10.1371/journal.pone.0058544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WY, Wang ZB, Zhang LC, Wei X, Li L. Tight junction in blood-brain barrier: an overview of structure, regulation, and regulator substances. CNS Neurosci Ther. 2012;18:609–615. doi: 10.1111/j.1755-5949.2012.00340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locatelli G, Wortge S, Buch T, Ingold B, Frommer F, Sobottka B, Kruger M, Karram K, Buhlmann C, Bechmann I, et al. Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat Neurosci. 2012;15:543–550. doi: 10.1038/nn.3062. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, Jacome-Galarza CE, Handler K, Klughammer J, Kobayashi Y, et al. Specification of tissue-resident macrophages during organogenesis. Science. 2016;353:pii. doi: 10.1126/science.aaf4238. aaf4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Bruck W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller A, Brandenburg S, Turkowski K, Muller S, Vajkoczy P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int J Cancer. 2015;137:278–288. doi: 10.1002/ijc.29379. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe Y, Medzhitov R. Tissue biology perspective on macrophages. Nat immunol. 2015;17:9–17. doi: 10.1038/ni.3320. [DOI] [PubMed] [Google Scholar]
- Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- Ostuni R, Natoli G. Transcriptional control of macrophage diversity and specialization. Eur J Immunol. 2011;41:2486–2490. doi: 10.1002/eji.201141706. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parney IF, Waldron JS, Parsa AT. Flow cytometry and in vitro analysis of human glioma-associated macrophages. J Neurosurg. 2009;110:572–582. doi: 10.3171/2008.7.JNS08475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, Holland EC, Sutton JC, Joyce JA. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. 2016;352:aad3018. doi: 10.1126/science.aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith W, LeibundGut-Landmann S, Waldburger JM. Regulation of MHC class II gene expression by the class II transactivator. Nat Rev Immunol. 2005;5:793–806. doi: 10.1038/nri1708. [DOI] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, Brogi E, Brastianos PK, Hahn WC, Holsinger LJ, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol. 2014;16:876–888. doi: 10.1038/ncb3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solga AC, Pong WW, Kim KY, Cimino PJ, Toonen JA, Walker J, Wylie T, Magrini V, Griffith M, Griffith OL, et al. RNA sequencing of tumor-associated microglia reveals Ccl5 as a stromal chemokine critical for Neurofibromatosis-1 glioma growth. Neoplasia. 2015;17:776–788. doi: 10.1016/j.neo.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Ann Rev Neurosci. 2012;35:369–389. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- Szulzewsky F, Arora S, de Witte L, Ulas T, Markovic D, Schultze JL, Holland EC, Synowitz M, Wolf SA, Kettenmann H. Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia. 2016;64:1416–1436. doi: 10.1002/glia.23014. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, Nguyen D, Wright KL, Atadja PW, Bhalla K, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2009;10:92–100. doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.