

Abstract
Key points
Spontaneous activity of the sensory inner hair cells shapes maturation of the developing ascending (afferent) auditory system before hearing begins.
Just before the onset of hearing, descending (efferent) input from cholinergic neurons originating in the brainstem inhibit inner hair cell spontaneous activity and may further refine maturation.
We show that agonist activation of the group I metabotropic glutamate receptor mGluR1 increases the strength of this efferent inhibition by enhancing the presynaptic release of acetylcholine.
We further show that the endogenous release of glutamate from the inner hair cells may increase the strength of efferent inhibition via the activation of group I metabotropic glutamate receptors.
Thus, before the onset of hearing, metabotropic glutamate signalling establishes a local negative feedback loop that is positioned to regulate inner hair cell excitability and refine maturation of the auditory system.
Abstract
Just before the onset of hearing, the inner hair cells (IHCs) receive inhibitory efferent input from cholinergic medial olivocochlear (MOC) neurons originating in the brainstem. This input may serve a role in the maturation of the ascending (afferent) auditory system by inhibiting spontaneous activity of the IHCs. To investigate the molecular mechanisms regulating these IHC efferent synapses, we combined electrical stimulation of the efferent fibres with patch clamp recordings from the IHCs to measure efferent synaptic strength. By examining evoked responses, we show that activation of metabotropic glutamate receptors (mGluRs) by general and group I‐specific mGluR agonists enhances IHC efferent inhibition. This enhancement is blocked by application of a group I mGluR1‐specific antagonist, indicating that enhancement of IHC efferent inhibition is mediated by group I mGluRs and specifically by mGluR1s. By comparing spontaneous and evoked responses, we show that group I mGluR agonists act presynaptically to increase neurotransmitter release without affecting postsynaptic responsiveness. Moreover, endogenous glutamate released from the IHCs also enhances IHC efferent inhibition via the activation of group I mGluRs. Finally, immunofluorescence analysis indicates that the efferent terminals are sufficiently close to IHC glutamate release sites to allow activation of mGluRs on the efferent terminals by glutamate spillover. Together, these results suggest that glutamate released from the IHCs activates group I mGluRs (mGluR1s), probably present on the efferent terminals, which, in turn, enhances release of acetylcholine and inhibition of the IHCs. Thus, mGluRs establish a local negative feedback loop positioned to regulate IHC activity and maturation of the ascending auditory system in the developing cochlea.
Keywords: glutamate, medial olivocochlear nucleus, metabotropic glutamate receptors, synapse, quantal analysis
Key points
Spontaneous activity of the sensory inner hair cells shapes maturation of the developing ascending (afferent) auditory system before hearing begins.
Just before the onset of hearing, descending (efferent) input from cholinergic neurons originating in the brainstem inhibit inner hair cell spontaneous activity and may further refine maturation.
We show that agonist activation of the group I metabotropic glutamate receptor mGluR1 increases the strength of this efferent inhibition by enhancing the presynaptic release of acetylcholine.
We further show that the endogenous release of glutamate from the inner hair cells may increase the strength of efferent inhibition via the activation of group I metabotropic glutamate receptors.
Thus, before the onset of hearing, metabotropic glutamate signalling establishes a local negative feedback loop that is positioned to regulate inner hair cell excitability and refine maturation of the auditory system.
Abbreviations
- AP
action potential
- CNS
central nervous system
- CPCCOEt
7‐(hydroxyimino)cyclopropa[b]chromen‐1a‐carboxylate ethyl ester
- DHPG
(S)‐3,5‐dihydroxyphenylglycine
- eIPSC
evoked inhibitory postsynaptic current
- iGluR
ionotropic glutamate receptor
- IHC
inner hair cell
- mGluR
metabotropic glutamate receptor
- MOC
medial olivocochlear
- MPEP
2‐methyl‐6‐(phenylethynyl)pyridine hydrochloride
- OHC
outer hair cell
- P
postnatal day
- sIPSC
spontaneous inhibitory postsynaptic current
- SK2
small conductance, calcium‐activated potassium channel type 2
- t‐ACPD
(1S,3R)‐1‐aminocyclopentane‐1,3‐dicarboxylic acid
- TBOA
dl‐threo‐β‐benzyloxyaspartic acid
Introduction
Inner hair cells (IHCs), the sensory cells of the cochlea, relay information about sound to the central nervous system (CNS) via graded receptor potentials. In contrast, during a short developmental period, immature IHCs fire action potentials (APs; Kros et al. 1998; Marcotti et al. 2003; Sendin et al. 2014). This activity is believed to shape maturation of the ascending (afferent) auditory system (reviewed in Wang & Bergles, 2015). In turn, acetylcholine (ACh) inhibits IHC APs (Glowatzki & Fuchs, 2002; Marcotti et al. 2004), suggesting that efferent cholinergic input from the brainstem to the IHCs also serves to shape maturation of the auditory system (Clause et al. 2014). Medial olivocochlear (MOC) efferent inhibition of the IHCs relies on Ca2+ influx through a cholinergic receptor and subsequent activation of Ca2+‐dependent potassium (SK2) channels that hyperpolarize the IHCs (Glowatzki & Fuchs, 2000). These synapses are both plastic and transient (Katz & Elgoyhen, 2014), disappearing around the onset of hearing.
In both the immature and mature cochlea, IHCs release glutamate that activates ionotropic glutamate receptors (iGluRs) on the afferent dendrites of the spiral ganglion neurons (Grant et al. 2010). In addition to activating iGluRs (Traynelis et al. 2010), glutamate released from the IHCs may also activate metabotropic glutamate receptors (mGluRs). Unlike iGluRs, which are ligand‐gated cation channels, mGluRs modulate neuronal excitability and synaptic transmission via G‐protein‐coupled pathways and are not necessarily positioned postsynaptically (Niswender & Conn, 2010). mGluRs are known to be important regulators of nervous system development (reviewed in Maiese et al. 2005) and neuronal plasticity (reviewed in Anwyl, 1999). Although more widely studied in the CNS, mGluRs also regulate peripheral activity in a variety of sensory systems (Gerber, 2003; Montana & Gereau, 2011; Roper, 2013) including the cochlea (Kleinlogel et al. 1999; Peng et al. 2004; Doleviczenyi et al. 2005).
Using patch clamp electrophysiology and immunofluorescence, we provide evidence indicating that glutamate released from the IHCs enhances IHC efferent inhibition by activating group I mGluR1s present on the efferent presynaptic terminals. This work suggests that mGluR signalling contributes to the plasticity of these synapses and, therefore, may play an important role in the immature cochlea in modulating activity that shapes maturation of the developing auditory system.
Methods
Ethical approval
Animal protocols were approved by the Johns Hopkins University, University of North Carolina at Wilmington, and Instituto de Investigaciones en Ingeniería Genética y Biología Molecular Animal Care and Use Committees.
Animal procedures
Procedures similar to those published previously were used to prepare apical turns of the postnatal rat organ of Corti for electrophysiology (Goutman et al. 2005) or immunofluorescence (McLean et al. 2009). Animals were deeply anaesthetized by isoflurane or halothane inhalation and decapitated. For all experiments, Sprague–Dawley rat pups were used at postnatal days (P) 7–11. No differences in responses were observed within this developmental age range.
Electrophysiology
Whole‐cell voltage‐clamp recordings were made using intracellular solution containing (in mm): 150 KCl, 3.5 MgCl2, 0.1 CaCl2, 5 EGTA, 5 Hepes, 2.5 Na2ATP (290 mosmol l−1, pH 7.2 adjusted by KOH). The extracellular solution contained (in mm): 5.8 KCl, 155 NaCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 5.6 glucose, 10 Hepes, (300 mosmol l−1, pH 7.4 adjusted byNaOH). Recordings were made at room temperature (22–25°C). Extracellular solutions, including experimental drugs, were applied by gravity flow into the recording chamber at a rate of 2–3 ml min−1. (1S,3R)‐1‐Aminocyclopentane‐1,3‐dicarboxylic acid (t‐ACPD) and dl‐threo‐β‐benzyloxyaspartic acid (TBOA) were purchased from Tocris, and 7‐(hydroxyimino)cyclopropa[b]chromen‐1a‐carboxylate ethyl ester (CPCCOEt) and 2‐methyl‐6‐(phenylethynyl)pyridine hydrochloride (MPEP) were purchased from Tocris (Techne, Lille Cedex, France or Fisher Scientific, Pittsburgh, PA, USA) or Abcam Biochemicals (Cambridge, MA, USA). All other chemicals were purchased from Sigma (St. Louis, MO, USA). Drug doses for mGluR agonist and antagonists were based on previously published pharmacological characterization for t‐ACPD (Palmer et al. 1989), (S)‐3,5‐dihydroxyphenylglycine (DHPG) (Ito et al. 1992), CPCCOEt (Litschig et al. 1999) and MPEP (Gasparini et al. 1999).
Electrical stimulation of efferent axons was applied as described previously (Goutman et al. 2005). Currents and voltages were recorded with pCLAMP10 and an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA), low‐pass filtered at 10 kHz and digitized at 20 kHz with a Digidata 1322A board. Indicated holding potentials were not corrected for liquid junction potentials (approximately −4 mV). Because series resistance errors were calculated to be less than 5 mV, series resistance compensation was not used or corrected offline.
Spontaneous inhibitory postsynaptic current (sIPSC) amplitudes were calculated using MiniAnalysis (Synaptosoft, Chapel Hill, NC, USA) as described previously (Goutman et al. 2005). Evoked inhibitory postsynaptic current (eIPSC) amplitudes were calculated using Clampfit 10 Data Analysis Module (Molecular Devices, Sunnyvale, CA, USA). For quantal analyses, mean amplitudes of IPSCs were determined from the Gaussian means of amplitude distributions. The quantal content (m) was determined using either the ‘direct method,’ where m equals the ratio between the mean amplitudes of eIPSCs and the mean amplitude of sIPSCs, or the ‘method of failures,’ where m equals the natural log of the ratio between the total number of stimuli and the total number of failures (Del Castillo & Katz, 1954).
Immunofluorescence and microscopy
Immunofluorescent staining was performed using mouse monoclonal (IgG1) anti‐CTBP2 (612044, 1:300; BD Bioscience, San Jose, CA, USA) and rabbit polyclonal anti‐synapsin I (AB1543; 1:500; Millipore, Temecula, CA, USA). Secondary antibodies, AlexaFluor 488 goat anti‐mouse (A‐11029) and AlexaFluor 594 goat anti‐rabbit (A‐11037) were purchased from Life Technologies (Grand Island, NY, USA) and used diluted 1:500. Fluorescence images were acquired as described previously (Sadeghi et al. 2014) using an Olympus Fluoview FV1000 confocal microscope with a ×60 Olympus PlanApo oil‐immersion lens (NA 1.42) under the control of the Olympus FluoView FV1000 v2.1 software (Olympus, Center Valley, PA, USA). To determine their relative location (x, y and z coordinates), immunolabels were detected automatically using the Spots function in the Imaris 6.4 3‐D image visualization and analysis software (Bitplane, Zurich, Switzerland). The Euclidean distance between a given CTBP2 immunopunctum and its nearest synapsin immunopunctum was calculated using MATLAB.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 6 as described in the Results. For all analyses, P < 0.05 was used to establish statistical significance. Group results are reported as the mean ± SEM.
Results
Activation of mGluRs increases the amplitude of IHC efferent eIPSC amplitudes
Before the onset of hearing (approximately P12), electrical stimulation of the MOC efferent fibres just below the bases of the IHCs evokes efferent synaptic responses that can be recorded from the postsynaptic IHCs in the isolated rat (Goutman et al. 2005) and mouse (Zorrilla de San Martin et al. 2010) organ of Corti. In these experiments, efferent synaptic currents were evoked by electrically stimulating the efferent fibres using multiple sets of electrical stimuli applied at a rate of 1 stimulation s−1 (1 Hz). 1 Hz stimulation was chosen because it does not facilitate or suppress the efferent synaptic response (Goutman et al. 2005). Synaptic responses were recorded at −90 mV (below the K+ reversal potential of −82 mV in these recording conditions) and, therefore, include inward cholinergic and SK2 components. This recording potential was chosen to maximize detection of synaptic responses that otherwise would have been much smaller and harder to detect had they consisted of the cholinergic component alone. Importantly, amplitude distributions of synaptic responses recorded at −90 mV follow quantal steps (Goutman et al. 2005), indicating linear coupling between the cholinergic and SK2 channels in these conditions.
mGluRs are classified into three groups (group I: mGluR 1 and 5, group II: mGluR 2 and 3, and group III: mGluR 4, 6, 7 and 8), and several general and group‐specific agonists and antagonists have been identified (Niswender & Conn, 2010). To monitor the effect of a general mGluR agonist, t‐ACPD (100 μm), on eIPSCs, multiple sets of 100 stimuli were applied at a rate of 1 stimulation s−1 (1 Hz) every 2 min first in control solution, then during agonist application, and finally during wash out. The average eIPSC amplitude was calculated for each set of 100 stimuli (including failures). Example averaged eIPSCs from an individual IHC in control, agonist (t‐ACPD), and wash conditions are shown (Fig. 1 A). In this example, the amplitude increased from a mean value of 23 pA in control conditions to 56 pA in the presence of t‐ACPD. To examine possible changes in both the probability of release and the amplitude of individual EPSCs examined, averaged amplitudes included failures since failures provide a measure of the probability of release. To compare values across IHCs, average eIPSC amplitudes for each stimulus set were normalized to the mean of the averaged eIPSC amplitude over the last three sets of stimuli before application of t‐ACPD (here at 3, 5 and 7 min, grey filled circles, Fig. 1 C). The normalized eIPSC amplitudes had a mean value of 1.1 ± 0.1 in control conditions (at 7 min) and 2.2 ± 0.1 after t‐ACPD application (at 15 min or 7 min after agonist application, n = 5, Fig. 1 D).
Figure 1. IHC efferent eIPSC amplitudes are enhanced by activation of general and group I mGluRs.
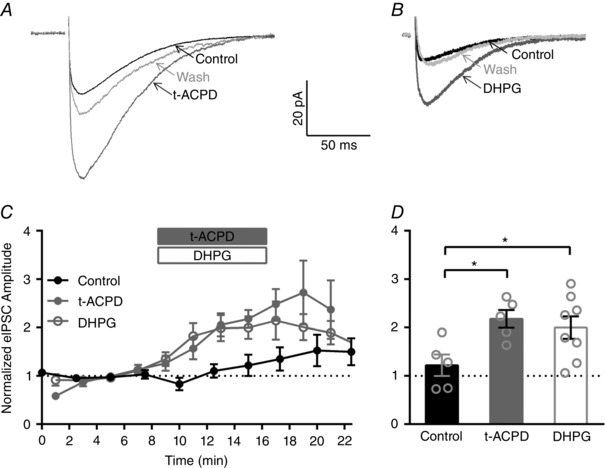
A, averaged eIPSCs from an individual IHC in control, general mGluR agonist (t‐ACPD; 15 min), and wash conditions (23 min). B, averaged eIPSCs from an individual IHC in control, group I mGluR agonist (DHPG; 13 min), and wash conditions (25 min). C, mean normalized eIPSC amplitudes across IHCs in control conditions (black filled circles) or before, during and after application of either t‐ACPD (grey filled circles) or DHPG (grey open circles). D, a significant increase in the mean normalized eIPSC amplitude (measured at 15 min or 7 min after agonist application) is observed after application of either t‐ACPD (grey filled bar) or DHPG (grey open bar) compared to control conditions (measured at 15 min, black filled bar).
Secondly, we tested the effect of a group I specific agonist, DHPG (50 μm), using a similar experimental design. Example averaged eIPSCs from individual IHCs in control, agonist (DHPG), and wash condition are shown in Fig. 1 B. In this example, the amplitude increased from a mean value of 9 pA in control conditions to 25 pA in the presence of DHPG. To compare across cells, average eIPSC amplitudes for each set of 100 stimuli were normalized to the mean eIPSC amplitude over the last three sets of stimuli before application of DHPG (at 3, 5 and 7 min, Fig. 1 C). The normalized eIPSC amplitudes had a mean value of 1.1 ± 0.06 in control conditions (at 7 min) and 2.0 ± 0.1 after DHPG application (at 15 min or 7 min after agonist application, n = 8, Fig. 1 D).
Although 1 Hz stimulation is not expected to facilitate the efferent synaptic response (Goutman et al. 2005), we also monitored eIPSC amplitudes in control conditions (absence of mGluR agonists). Multiple sets of 100 stimuli were applied at a rate of 1 stimulation s−1 (1 Hz) every 2.5 min, and the average eIPSC amplitudes for each set of 100 stimuli were normalized to the mean eIPSC amplitudes over the first three sets of stimuli (at 0, 2.5 and 5 min, Fig. 1 C). Although an increase in the average eIPSC amplitudes was seen in some cells, there were no significant differences in the mean normalized eIPSC amplitudes measured at 7.5, 15 or 22.5 min (ordinary one‐way ANOVA with Dunnett's multiple comparisons test). In control conditions, the normalized eIPSC amplitudes had a mean value of 1.0 ± 0.09 (averaged over 0, 2.5 and 5 min) and 1.2 ± 0.2 (at 15 min, n = 5, Fig. 1 D). Importantly, the mean normalized eIPSC amplitudes (at 15 min, 7 min after agonist application) were significantly larger in the presence of t‐ACPD (n = 5) and DHPG (n = 8) compared to those measured in control conditions (at 15 min, n = 5, ordinary one‐way ANOVA with Dunnett's multiple comparisons test). These data indicate that activation of group I mGluRs enhances the IHC efferent eIPSC amplitudes.
Activation of mGluR1 increases IHC efferent eIPSC amplitudes
To determine which group I mGluR, mGluR1 and/or mGluR5, is responsible for the enhancement of IHC efferent eIPSC amplitudes, we next examined the effects of mGluR1 and mGluR5 specific blockers on t‐ACPD‐mediated increases in eIPSC amplitudes (Fig. 2) using a similar experimental design (described in Fig. 1). Enhancement of eIPSC amplitudes persisted in the presence of MPEP (10 μm, mGluR5 antagonist) in individual IHCs (Fig. 2 A) and across cells (grey open circles, Fig. 2 C). In the example traces (Fig. 2 A), the amplitude increased from a mean value of 24 pA in control conditions to 37 pA in the presence of t‐ACPD/MPEP. Normalized eIPSC amplitudes had a mean value of 3.4 ± 1.0 in t‐ACPD/MPEP (at 21.5 min, 9.5 min after agonist application, n = 5, Fig. 2 D). In contrast, no increases in eIPSC amplitudes were observed in the presence of CPCCOEt (100 μm, mGluR1 antagonist) in either individual IHCs (Fig. 2 B) or across cells (black open circles, Fig. 2 C). In the example traces (Fig. 2 B), the amplitude had a mean value of 15 pA in control conditions and 12 pA in the presence of t‐ACPD/CPCCOEt. Normalized eIPSC amplitudes had a mean value of 0.6 ± 0.2 in t‐ACPD/CPCCOEt (at 21.5 min, 9.5 min after agonist application, n = 5, Fig. 2 D). Likewise, the effect of t‐ACPD was completely blocked during continuous application of both mGluR antagonists (MPEP and CPCCOEt). Normalized eIPSC amplitudes had a mean value of 1.1 ± 0.1 in the presence of t‐ACPD/CPCCOEt/MPEP (at 15 min, 7 min after agonist application, n = 4, Fig. 2 D). Importantly, the mean normalized eIPSC amplitudes were significantly larger in the presence of t‐ACPD/MPEP (after 9.5 min of agonist application, n = 5) compared to those measured in t‐ACPD/CPCCOEt (after 9.5 min of agonist application, n = 5, two‐tailed unpaired t test, Fig. 2 D). Likewise, the mean normalized eIPSC amplitudes were significantly larger in the presence of t‐ACPD agonist alone (7 min after application, n = 5, replotted from Fig. 1 D for reference) compared to those measured in the presence of t‐ACPD/MPEP/CPCCOEt (7 min after agonist application, n = 4, two‐tailed unpaired t test, Fig. 2 D). These data indicate that activation of mGluR1 and not mGluR5 enhances the IHC efferent eIPSC amplitudes.
Figure 2. IHC efferent eIPSC amplitudes are specifically enhanced by activation of mGluR1.
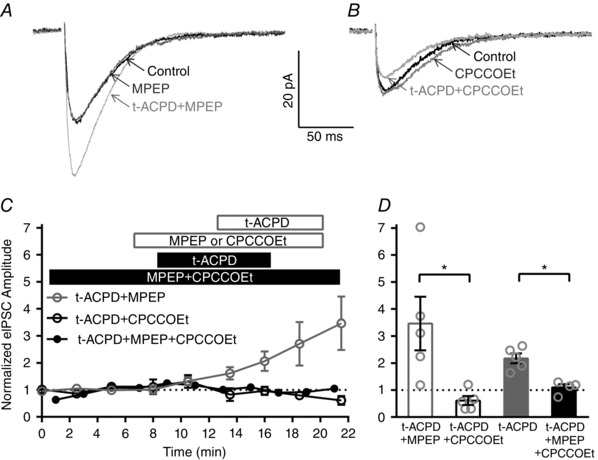
A, averaged eIPSCs from an individual IHC in control, mGluR5 antagonist (MPEP; 8 min), and mGluR5 antagonist with the general mGluR agonist (t‐ACPD; 16 min). B, averaged eIPSCs from an individual IHC in control (average of first files), mGluR1 antagonist (CPCCOEt; 8 min), and mGluR1 antagonist with the general mGluR agonist (t‐ACPD; 13.5 min). C, normalized eIPSC amplitudes across IHCs before, during and after general mGluR agonist (t‐ACPD) application in the presence of either the mGluR5 antagonist alone (MPEP, grey open circles), the mGluR1 antagonist alone (CPCCOEt, black open circles), or both antagonists (MPEP + CPCCOEt, black filled circles). D, a significant decrease in the mean normalized eIPSC amplitude is observed during either application of the mGluR1 antagonist alone (CPCCOEt, 9.5 min after t‐ACPD agonist application, filled grey bar) or application of both mGluR5 and mGluR1 antagonists (MPEP + CPCCOEt, 7 min after t‐ACPD agonist application, black filled bar) compared to application of the mGluR5 antagonist alone (MPEP, 9.5 min after t‐ACPD agonist application, grey open bar).
mGluR agonists act presynaptically to increase IHC efferent eIPSC amplitudes
Amplitudes of postsynaptic currents are regulated by processes that are both presynaptic, such as neurotransmitter release, and also postsynaptic, such as receptor sensitivity, number of receptors and receptive field size. To determine whether mGluR agonists target pre‐ or postsynaptic processes to enhance efferent eIPSC amplitudes, changes in the quantum size (q, the size of a single vesicle) and quantal content (m, the number of vesicles released per stimulation) were calculated. Changes in quantal content often indicate presynaptic mechanisms, whereas changes in quantum size often suggest postsynaptic mechanisms (McLachlan, 1978). Previous work in this preparation indicates that quantal content of efferent terminals is normally quite low: approximately 1 (Goutman et al. 2005; Zorrilla de San Martin et al. 2010).
sIPSCs and eIPSCs were recorded at −90 mV in the same IHCs in control conditions and after application of DHPG (50 μm). Amplitude distributions (shown for a single IHC in Fig. 3 A and B) were then analysed. In both control conditions (grey) and after agonist application (black), amplitude distributions of sIPSCs were well fitted by single Gaussian curves (Fig. 3 A), consistent with spontaneous events resulting from uniquantal (single vesicle) release events. Across cells, quantum sizes (q, average amplitudes of sIPSCs) were similar in control conditions (12.6 ± 0.8 pA) and after agonist application (13.3 ± 1.3 pA, n = 5, Fig. 3 C), suggesting that activation of group I mGluRs does not affect postsynaptic responsiveness. Compared to spontaneous events, amplitude distributions of eIPSCs in both control (grey) conditions and after agonist application (black) were shifted toward larger amplitudes (Fig. 3 B), probably reflecting the release of more than one vesicle upon stimulation. We also observed a decrease in failures (events with 0 pA amplitude) after agonist application (Fig. 3 B). Across cells, the mean amplitude of eIPSCs was 15.5 ± 1.3 pA in control conditions and 26.0 ± 3.8 pA in the presence of DHPG (n = 5, Fig. 3 C). As expected, the mean amplitude of eIPSCs increased significantly with the activation of group I mGluRs compared to control conditions (two‐tailed paired t test, Fig. 3 C). Although there was a trend toward increasing sIPSC frequency in the presence of DHPG application (Fig. 3 D), this trend was not significant (two‐tailed unpaired t test) probably because of the overall low frequency of spontaneous events.
Figure 3. Quantal content is increased by activation of group I mGluRs.
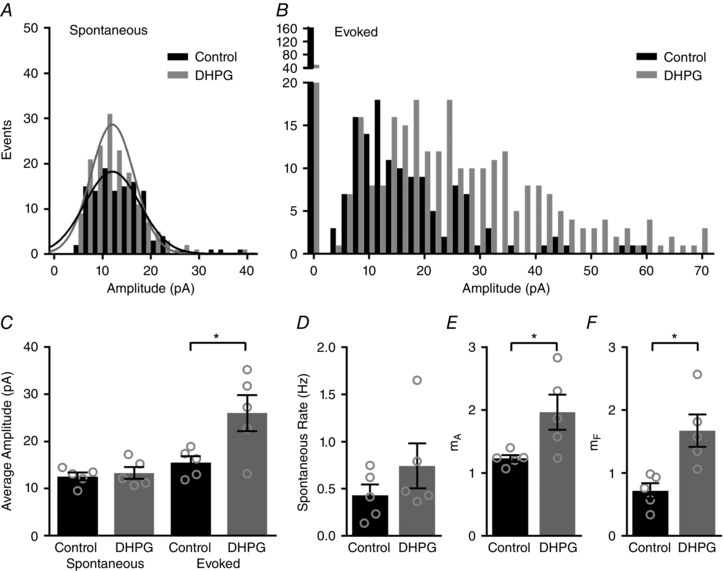
A and B, distributions of sIPSC (A) and eIPSC amplitudes (B) shown for an individual IHC in control conditions (black) and after application of a group I mGluR agonist (DHPG, grey). sIPSCs are fitted with single Gaussian curves (A). C, mean IPSC amplitudes across IHCs in the various conditions. D, mean sIPSC rate before and after after group I mGluR agonist (DHPG) application. E and F quantal content (m), calculated from changes in amplitude (E) or from changes in failures (F) increased significantly after group I mGluR agonist (DHPG) application.
To estimate the efficiency of vesicle release for each cell before and after activation of group I mGluRs, quantal content was determined in two ways. First, m A was calculated using the direct methods by dividing the mean amplitude of the eIPSCs (not including failures) by the mean amplitude of sIPSCs. In these experiments, m A increased significantly from 1.2 ± 0.05 in control conditions to 2.0 ± 0.3 after agonist application (two‐tailed paired t test, n = 5, Fig. 3 D). Second, m F was calculated using the method of failures. m F also increased significantly from 0.7 ± 0.3 in control conditions to 1.7 ± 0.3 after agonist application (two‐tailed paired t test, n = 5, Fig. 3 E), reflecting a significant decrease in the percentage of failures from 50.3 ± 6.3% in control conditions to 21.0 ± 4.5% after application of DHPG. In total, these data indicate that activation of group I mGluRs increases presynaptic glutamate release per stimulation without effecting postsynaptic responsiveness to neurotransmitter release.
Glutamate released from IHCs increases IHC efferent eIPSC amplitudes
To examine the endogenous activation of group I mGluRs, the enhancement of eIPSC amplitudes by glutamate released from the IHCs was tested. Conditions that maximized glutamate release from the IHCs and also promoted accumulation of glutamate were found to enhance IHC efferent eIPSCs. To maximize glutamate release from the IHCs, during every cycle of efferent stimulation, IHCs were depolarized in a series of five steps from −90 mV to −20 mV (Δ70 mV) for 50 ms each with an interstep interval of 100 ms. To facilitate glutamate accumulation extracellularly, glutamate transporters were blocked by bath application of 200 μm TBOA (Glowatzki et al. 2006). To enhance the stability of recordings, the efferent stimulation frequency was reduced to 0.5 Hz and delivered in sets of 50 (rather than 100) stimuli. Together, the entire protocol consisted of four sets of 50 stimuli applied in control solution, seven sets of 50 stimuli during application of TBOA with IHC depolarization, and three sets of 50 stimuli during wash out of TBOA. Efferent stimulation evoked inward eIPSCs, and IHC depolarization produced large outward currents through voltage‐activated K+ channels (Fig. 4 A).
Figure 4. Glutamate released from IHCs also enhances IHC efferent eIPSC amplitudes via activation of group I mGluRs.
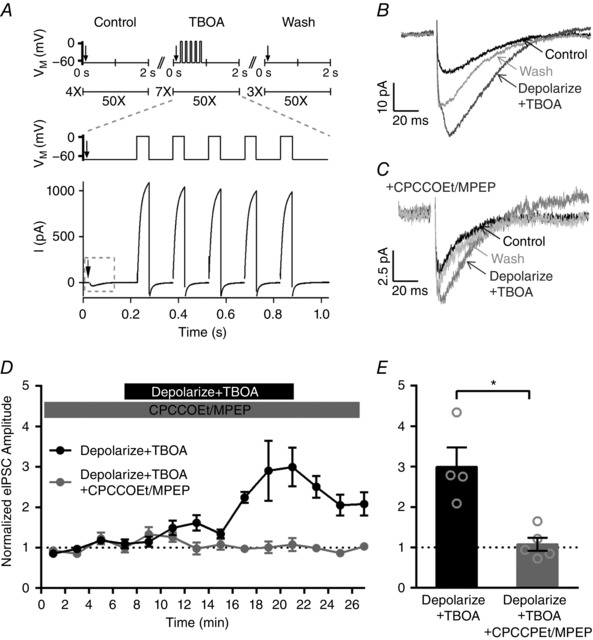
A, the stimulus protocol consists of efferent stimulation (arrows) that evoked inward eIPSCs and IHC depolarization (ΔV M) that produced large outward currents through voltage‐activated K+ channels. B, averaged eIPSCs from an individual IHC in control, stimulation (Depolarize + TBOA) and wash conditions. C, averaged eIPSCs from an individual IHC in control, stimulation (Depolarize + TBOA) and wash conditions in the continuous presence of group I mGluR antagonists (CPCCOEt/MPEP). D, normalized eIPSC amplitudes across IHCs before, during and after depolarization with TBOA application in the absence (filled black circles) and continuous presence (filled grey circles) of group I mGluR antagonists (CPCCOEt/MPEP). E, the increase in the mean normalized eIPSC amplitude observed during depolarization with TBOA application (21 min, filled black bar) is blocked by the continuous presence of group I mGluR antagonists (CPCCOEt/MPEP, 21 min, filled grey bar).
Example averaged eIPSCs from an individual cell in control, depolarization with TBOA, and wash conditions are shown (Fig. 4 B). In this example, the amplitude increased from a mean value of 14 pA in control conditions to 37 pA upon depolarization with TBOA application. To compare changes in eIPSC amplitudes across cells, average eIPSC amplitudes for each stimuli set were normalized to the average eIPSC amplitude over the first three sets of stimuli (at 1, 3 and 5 min, black, Fig. 4 D). Across cells, depolarization with TBOA application caused an increase in the mean normalized eIPSC amplitude to 3.0 ± 0.2 (at 21 min, n = 4, Fig. 4 E). Importantly, the effect of depolarization with TBOA application could be completely blocked by the continuous application of the group I mGluR antagonists CPCCOEt (100 μm) and MPEP (10 μm). Example averaged eIPSCs from an individual cell in the continuous application of CPCCOEt/MPEP before, during and after depolarization with TBOA are shown (Fig. 4 C). In this example, the amplitude was 5.5 pA before depolarization with TBOA and 8.0 pA upon depolarization with TBOA application (Fig. 4 C). To compare changes in eIPSC amplitudes across cells, average eIPSC amplitudes for each stimuli set were normalized to the average eIPSC amplitude over the first three sets of stimuli (at 1, 3 and 5 min, grey, Fig. 4 D). Across cells, depolarization with TBOA application in the continuous presence of CPCCOEt/MPEP was 1.1 ± 0.07 (at 21 min, n = 5, Fig. 4 E) and significantly less than those measured in the absence of antagonist (two‐tailed unpaired t test). These data provide evidence that activation of group I mGluRs by glutamate released from IHCs is able to enhance the efferent synaptic response. In initial experiments that had been performed in the absence of TBOA and with long step depolarizations (500 ms), no increase in the eIPSC amplitudes was observed (n = 4, data not shown). These data suggest that in our experimental conditions depolarization alone does not cause sufficient glutamate accumulation to activate mGluRs. In these in vitro experimental conditions, glutamate release is elicited from a single clamped IHC, and release from multiple IHCs may be necessary to active presynaptic efferent mGluRs in vivo. Additionally, the micro‐environment surrounding the IHC becomes damaged when performing whole cell recordings and could reduce the local glutamate concentration surrounding synapses to the IHCs. Despite differences between the in vitro conditions of these recordings and in vivo conditions in the developing cochlea, these experiments are nevertheless consistent with the hypothesis that inner hair cells are the source of glutamate required for mGluR activation.
Glutamate release sites and efferent presynaptic terminals are closely apposed
Quantal analysis (Fig. 3) suggests that group I mGluRs might be physically localized to the efferent presynaptic terminals to induce an increase in neurotransmitter (ACh) release. The anatomical localization of mGluRs was investigated using immunofluorescence with various antibodies against group I mGluRs and mGluR1 without consistent results. Instead, the proximity of IHC afferent synapses (the sites of glutamate release) and efferent presynaptic terminals was quantified to determine if, in principle, spillover of glutamate released from the IHCs could activate mGluRs on presynaptic efferent terminals. The relative locations of the IHC afferent ribbons and the efferent presynaptic terminals were assessed using a monoclonal antibody against CTBP2 (green) and a rabbit polyclonal antibody against synapsin (red). A single optical section (Fig. 5 A) as well as z‐projections of immunolabelled rat organs of Corti (Fig. 5 B and C–D) revealed extensive apposition of afferent release sites (CTBP2 immunopuncta) and efferent presynaptic terminals (synapsin immunopuncta). 3‐D reconstructions of the IHC synaptic poles were used to identify CTBP2 and synapsin immunopuncta (Fig. 5 D and E) and calculate the distance between each CTBP2 immunopunctum and its nearest synapsin immunopunctum. The mean distance between a given CTBP2 immunopunctum and its nearest synapsin immunopunctum was 1.96 ± 0.02 μm (n = 1607 values from 3 animals), and 90% of all CTBP2 immunopuncta were within 3 μm of a synapsin immunopunctum (Fig. 5 F). Supporting Information S1 (Video S1) provides an animated version of the 3‐D reconstructions and analysis methodology. In the hippocampus, a combination of empirical findings and predictive modelling indicate that glutamate may diffuse up to 10 μm in 50 ms, even considering diffusional constraints and glutamate uptake (Diamond, 2002). Given similar dynamics in the developing cochlea, glutamate release sites are feasibly close to activate mGluRs present on the efferent presynaptic terminals by glutamate spillover.
Figure 5. Glutamate release sites and efferent presynaptic terminals are closely apposed, consistent with activation of mGluRs on the presynaptic efferent terminals by glutamate released from the IHCs.
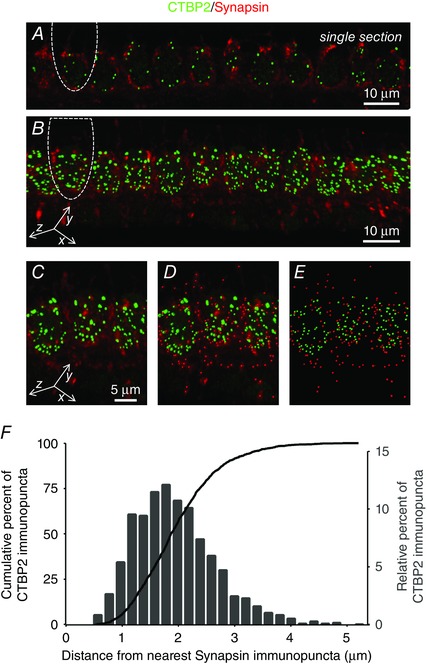
Single optical section (A) and z‐projections (B–D) showing immunolabelled glutamate release sites (CTBP2, green) and efferent presynaptic terminals (synapsin, red). In A and B, the outline of a single inner hair cell has been indicated for reference. D and E, z‐projections additionally (D) or exclusively (E) show immunopuncta detected by Imaris for distance calculations. F, the Euclidean distance between each CTBP2 immunopunctum and its nearest synapsin immunopunctum was calculated from 3‐D reconstructions of the immunolabelled IHC synaptic poles and shown as frequency and cumulative histograms. Supporting Information S1 (Video S1) provides an animated version of the 3‐D reconstructions and analysis methodology.
Discussion
In this work, by examining evoked IPSCs, we show that activation of mGluRs by general and group I mGluR agonists enhances IHC efferent inhibition (Fig. 1). Because this response could be blocked by application of group I mGluR antagonists and specifically by a mGluR1 antagonist, we conclude that enhancement of IHC efferent inhibition is mediated by group I mGluR1s (Fig. 2). Furthermore, by additionally comparing spontaneous and evoked IPSCs, we show that group I mGluR agonists increase quantal content without affecting postsynaptic responsiveness (Fig. 3). Thus, group I mGluR agonists most likely enhance efferent inhibition by increasing the release of ACh from the efferent terminals.
We further show that glutamate released from the IHCs enhances IHC efferent inhibition (also through the activation of group I mGluRs, Fig. 4). In particular, we found that conditions that maximize glutamate release from the IHC and accumulation within the synaptic cleft are required to activate mGluRs, suggesting that mGluRs are present extrasynaptically. Very importantly, during this developmental period, immature IHCs fire APs that can reach instantaneous spike rates of up to 10 events s−1 that persist for minutes (Sendin et al. 2014). Each IHC AP can release hundreds of glutamate‐filled vesicles (Moser & Beutner, 2000; Glowatzki & Fuchs, 2002). Therefore, glutamate spillover is likely to occur in physiological conditions. Because quantal analysis indicates that mGluRs exert their mechanistic effect presynaptically, the simplest model suggests that mGluRs are physically present on the efferent presynaptic terminals. We found that the presynaptic efferent terminals in the developing cochlea are indeed sufficiently close to glutamate release sites to allow activation of mGluRs on the efferent terminals by glutamate spillover (Fig. 5). Because efferent innervation inhibits IHC activity (Glowatzki & Fuchs, 2000), mGluR signalling would establish a negative feedback loop that regulates IHC excitability and potentially is involved in shaping maturation of the afferent auditory system in the developing cochlea (Fig. 6).
Figure 6. mGluR signalling at the IHC efferent synapses establishes a negative feedback loop in the developing cochlea.

Activation of group I mGluR1 receptors (likely on the presynaptic efferent terminal) enhances ACh release (white arrow) and efferent synaptic inhibition of the IHC. IHC inhibition would suppress neurotransmitter release from the IHC (white bar).
Previous examination of the location and function of mGluRs in the cochlea has been limited to studies examining the mature cochlea. A variety of work suggests that group I mGluRs are present in the spiral ganglion neurons and/or hair cells (Niedzielski et al. 1997; Safieddine & Wenthold, 1997; Peng et al. 2004) and serve to increase afferent excitability and/or firing (Kleinlogel et al. 1999; Peng et al. 2004). Furthermore, in the mature cochlea, MOC efferent innervation of the OHCs persists. These efferent synapses are in proximity to afferent synapses that are glutamatergic (Weisz et al. 2009) and also show use‐dependent plasticity (Katz & Elgoyhen, 2014). Although we did not investigate the effects of mGluR activation in the mature cochlea or afferent activity specifically, these previous findings together with our results suggest that mGluRs may modulate signalling at multiple sites and in developmentally regulated ways.
Mechanistically, activation of group I mGluRs generally leads to modulation of ion channels that results in increased neuronal excitability (Niswender & Conn, 2010). Although group I mGluRs are generally localized postsynaptically, they have been found presynaptically (reviewed in Pinheiro & Mulle, 2008). Enhancement of efferent inhibition could arise from mGluR‐mediated potentiation of the Ca2+ currents that support neurotransmitter release from the efferent terminals or inhibition of the K+ currents that repolarize the terminals and inhibit release (Anwyl, 1999). In the developing cochlea, P/Q‐type and N‐type Ca2+ channels support efferent release of neurotransmitter, whereas L‐type Ca2+ channels suppress release by activating Ca2+‐dependent large conductance potassium (BK) channels that repolarize the efferent terminals (Zorrilla de San Martin et al. 2010). Previous work in central neurons has shown that agonists of mGluRs can reduce currents through L‐type Ca2+ channels (Sayer et al. 1992; Sahara & Westbrook, 1993; Pin & Duvoisin, 1995; Schumacher et al. 2000). Thus, group I mGluR activation in the developing cochlea might enhance efferent inhibition by suppressing L‐type Ca2+ channels, preventing activation of BK channels, and prolonging terminal depolarization and neurotransmitter release.
Alternatively, activation of mGluRs present on the postsynaptic IHC might trigger the release of a retrograde neurotransmitter that increases release from the efferent presynaptic terminals. Previous work using a similar preparation and experimental design showed that IHC activity facilitates efferent release via the retrograde signalling molecule nitric oxide (NO; Kong et al. 2013). In the study by Kong and colleagues, NO production and subsequent efferent facilitation was induced by activators of calcium store release in the IHCs. In contrast to our findings, facilitation observed in this earlier work was not dependent on the release of glutamate or the activation of mGluRs. Efferent–IHC synapses are remarkably plastic (Katz & Elgoyhen, 2014), and these previous findings together with our results suggest that various independent mechanisms serve to regulate their strength. Finally, in the CNS (Xu & Chen, 2015), including the medial nucleus of the trapezoid body in the auditory brainstem (Kushmerick et al. 2004), activation of postsynaptic group I mGluRs triggers the release of endocannabinoids that act on presynaptic cannabinoid receptors to modulate, and more specifically inhibit, presynaptic release. Although the possible involvement of cannabinoid signalling in our experiments cannot be excluded, the facilitation of presynaptic release that we observed is not consistent with the inhibition typically observed with cannabinoid receptor activation.
Ultimately, our work indicates that in the developing cochlea, glutamate, released from the IHCs, can activate group I mGluR1s probably present on the efferent presynaptic terminals. Activation of these mGluRs enhances release of ACh from the efferent terminals and subsequent inhibition of the IHCs. Thus, group I mGluR1s establish a local negative feedback loop that integrates afferent and efferent synaptic activity and may be involved in setting IHC activity and shaping auditory development.
Additional information
Competing interests
None declared.
Author contributions
Experiments were performed in the laboratories of J.D.G., S.J.P. and E.G. Z.Y., J.D.G., S.J.P. and E.G. contributed to the conception and design of experiments; collection, assembly, analysis and interpretation of data; and drafting of the article and/or revising it critically for important intellectual content. All authors approved the final version of the manuscript. Experiments with animals were performed at Instituto de Investigaciones en Ingeniería Genética y Biología Molecular (CONICET), Johns Hopkins University School of Medicine, and the University of North Carolina Wilmington. All persons designated as authors qualify for authorship. All those who qualify for authorship are listed.
Funding
We gratefully acknowledge research funding from the US National Institute on Deafness and Other Communication Disorders (NIDCD) R01DC006476 and R01DC012957, from the John Mitchell, Jr. Trust and the David M. Rubenstein Fund for Hearing Research to E.G., NIDCD R03TW009403 to E.G. and J.D.G. and Hearing Health Foundation (formerly Deafness Research Foundation) and the Rosalind Franklin Fellowship from University Medical Center Groningen to S.J.P.
Supporting information
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Video S1: 3D Immunolocliaztion indicates glutamate release sites (anti‐CTBP2, green) and efferent presynaptic terminals (anti‐synpasin, red) are closely apposed.
Supporting information S1: Description of video.
Z. Ye and J. D. Goutman contributed equally to this work.
S. J. Pyott and E. Glowatzki contributed equally to this work.
Linked articles This article is highlighted by a Perspective by Steinert. To read this Perspective, visit https://doi.org/10.1113/JP274196.
References
- Anwyl R (1999). Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev 29, 83–120. [DOI] [PubMed] [Google Scholar]
- Clause A, Kim G, Sonntag M, Weisz CJ, Vetter DE, Rubsamen R & Kandler K (2014). The precise temporal pattern of prehearing spontaneous activity is necessary for tonotopic map refinement. Neuron 82, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J & Katz B (1954). Quantal components of the end‐plate potential. J Physiol 124, 560–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS (2002). A broad view of glutamate spillover. Nat Neurosci 5, 291–292. [DOI] [PubMed] [Google Scholar]
- Doleviczenyi Z, Halmos G, Repassy G, Vizi ES, Zelles T & Lendvai B (2005). Cochlear dopamine release is modulated by group II metabotropic glutamate receptors via GABAergic neurotransmission. Neurosci Lett 385, 93–98. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Velicelebi G & Kuhn R (1999). 2‐Methyl‐6‐(phenylethynyl)‐pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38, 1493–1503. [DOI] [PubMed] [Google Scholar]
- Gerber U (2003). Metabotropic glutamate receptors in vertebrate retina. Doc Ophthalmol 106, 83–87. [DOI] [PubMed] [Google Scholar]
- Glowatzki E, Cheng N, Hiel H, Yi E, Tanaka K, Ellis‐Davies GC, Rothstein JD & Bergles DE (2006). The glutamate‐aspartate transporter GLAST mediates glutamate uptake at inner hair cell afferent synapses in the mammalian cochlea. J Neurosci 26, 7659–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowatzki E & Fuchs PA (2000). Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science 288, 2366–2368. [DOI] [PubMed] [Google Scholar]
- Glowatzki E & Fuchs PA (2002). Transmitter release at the hair cell ribbon synapse. Nat Neurosci 5, 147–154. [DOI] [PubMed] [Google Scholar]
- Goutman JD, Fuchs PA & Glowatzki E (2005). Facilitating efferent inhibition of inner hair cells in the cochlea of the neonatal rat. J Physiol 566, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant L, Yi E & Glowatzki E (2010). Two modes of release shape the postsynaptic response at the inner hair cell ribbon synapse. J Neurosci 30, 4210–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito I, Kohda A, Tanabe S, Hirose E, Hayashi M, Mitsunaga S & Sugiyama H (1992). 3,5‐Dihydroxyphenyl‐glycine: a potent agonist of metabotropic glutamate receptors. Neuroreport 3, 1013–1016. [PubMed] [Google Scholar]
- Katz E & Elgoyhen AB (2014). Short‐term plasticity and modulation of synaptic transmission at mammalian inhibitory cholinergic olivocochlear synapses. Front Syst Neurosci 8, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinlogel S, Oestreicher E, Arnold T, Ehrenberger K & Felix D (1999). Metabotropic glutamate receptors group I are involved in cochlear neurotransmission. Neuroreport 10, 1879–1882. [DOI] [PubMed] [Google Scholar]
- Kong JH, Zachary S, Rohmann KN & Fuchs PA (2013). Retrograde facilitation of efferent synapses on cochlear hair cells. J Assoc Res Otolaryngol 14, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kros CJ, Ruppersberg JP & Rusch A (1998). Expression of a potassium current in inner hair cells during development of hearing in mice. Nature 394, 281–284. [DOI] [PubMed] [Google Scholar]
- Kushmerick C, Price GD, Taschenberger H, Puente N, Renden R, Wadiche JI, Duvoisin RM, Grandes P & von Gersdorff H (2004). Retroinhibition of presynaptic Ca2+ currents by endocannabinoids released via postsynaptic mGluR activation at a calyx synapse. J Neurosci 24, 5955–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litschig S, Gasparini F, Rueegg D, Stoehr N, Flor PJ, Vranesic I, Prezeau L, Pin JP, Thomsen C & Kuhn R (1999). CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol 55, 453–461. [PubMed] [Google Scholar]
- McLachlan EM (1978). The statistics of transmitter release at chemical synapses. Int Rev Physiol 17, 49–117. [PubMed] [Google Scholar]
- McLean WJ, Smith KA, Glowatzki E & Pyott SJ (2009). Distribution of the Na,K‐ATPase α subunit in the rat spiral ganglion and organ of corti. J Assoc Res Otolaryngol 10, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ & Li F (2005). Driving cellular plasticity and survival through the signal transduction pathways of metabotropic glutamate receptors. Curr Neurovasc Res 2, 425–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Holley MC & Kros CJ (2003). Developmental changes in the expression of potassium currents of embryonic, neonatal and mature mouse inner hair cells. J Physiol 548, 383–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL & Kros CJ (2004). A transiently expressed SK current sustains and modulates action potential activity in immature mouse inner hair cells. J Physiol 560, 691–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montana MC & Gereau RW (2011). Metabotropic glutamate receptors as targets for analgesia: antagonism, activation, and allosteric modulation. Curr Pharm Biotechnol 12, 1681–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T & Beutner D (2000). Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc Natl Acad Sci USA 97, 883–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedzielski AS, Safieddine S & Wenthold RJ (1997). Molecular analysis of excitatory amino acid receptor expression in the cochlea. Audiol Neurootol 2, 79–91. [DOI] [PubMed] [Google Scholar]
- Niswender CM & Conn PJ (2010). Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer E, Monaghan DT & Cotman CW (1989). Trans‐ACPD, a selective agonist of the phosphoinositide‐coupled excitatory amino acid receptor. Eur J Pharmacol 166, 585–587. [DOI] [PubMed] [Google Scholar]
- Peng BG, Li QX, Ren TY, Ahmad S, Chen SP, Chen P & Lin X (2004). Group I metabotropic glutamate receptors in spiral ganglion neurons contribute to excitatory neurotransmissions in the cochlea. Neuroscience 123, 221–230. [DOI] [PubMed] [Google Scholar]
- Pin JP & Duvoisin R (1995). The metabotropic glutamate receptors: structure and functions. Neuropharmacology 34, 1–26. [DOI] [PubMed] [Google Scholar]
- Pinheiro PS & Mulle C (2008). Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat Rev Neurosci 9, 423–436. [DOI] [PubMed] [Google Scholar]
- Roper SD (2013). Taste buds as peripheral chemosensory processors. Semin Cell Dev Biol 24, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghi SG, Pyott SJ, Yu Z & Glowatzki E (2014). Glutamatergic signaling at the vestibular hair cell calyx synapse. J Neurosci 34, 14536–14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safieddine S & Wenthold RJ (1997). The glutamate receptor subunit δ1 is highly expressed in hair cells of the auditory and vestibular systems. J Neurosci 17, 7523–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara Y & Westbrook GL (1993). Modulation of calcium currents by a metabotropic glutamate receptor involves fast and slow kinetic components in cultured hippocampal neurons. J Neurosci 13, 3041–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayer RJ, Schwindt PC & Crill WE (1992). Metabotropic glutamate receptor‐mediated suppression of L‐type calcium current in acutely isolated neocortical neurons. J Neurophysiol 68, 833–842. [DOI] [PubMed] [Google Scholar]
- Schumacher TB, Beck H, Steffens R, Blumcke I, Schramm J, Elger CE & Steinhauser C (2000). Modulation of calcium channels by group I and group II metabotropic glutamate receptors in dentate gyrus neurons from patients with temporal lobe epilepsy. Epilepsia 41, 1249–1258. [DOI] [PubMed] [Google Scholar]
- Sendin G, Bourien J, Rassendren F, Puel JL & Nouvian R (2014). Spatiotemporal pattern of action potential firing in developing inner hair cells of the mouse cochlea. Proc Natl Acad Sci USA 111, 1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ & Dingledine R (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HC & Bergles DE (2015). Spontaneous activity in the developing auditory system. Cell Tissue Res 361, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz C, Glowatzki E & Fuchs P (2009). The postsynaptic function of type II cochlear afferents. Nature 461, 1126–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JY & Chen C (2015). Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist 21, 152–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorrilla de San Martin J, Pyott S, Ballestero J & Katz E (2010). Ca2+ and Ca2+‐activated K+ channels that support and modulate transmitter release at the olivocochlear efferent‐inner hair cell synapse. J Neurosci 30, 12157–12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Video S1: 3D Immunolocliaztion indicates glutamate release sites (anti‐CTBP2, green) and efferent presynaptic terminals (anti‐synpasin, red) are closely apposed.
Supporting information S1: Description of video.