

Abstract
ATRX is a chromatin remodelling factor found at a wide range of tandemly repeated sequences including telomeres (TTAGGG)n. ATRX mutations are found in nearly all tumours that maintain their telomeres via the alternative lengthening of telomere (ALT) pathway, and ATRX is known to suppress this pathway. Here, we show that recruitment of ATRX to telomeric repeats depends on repeat number, orientation and, critically, on repeat transcription. Importantly, the transcribed telomeric repeats form RNA–DNA hybrids (R‐loops) whose abundance correlates with the recruitment of ATRX. Here, we show loss of ATRX is also associated with increased R‐loop formation. Our data suggest that the presence of ATRX at telomeres may have a central role in suppressing deleterious DNA secondary structures that form at transcribed telomeric repeats, and this may account for the increased DNA damage, stalling of replication and homology‐directed repair previously observed upon loss of ATRX function.
Keywords: ATRX, G‐quadruplex, R‐loops, telomeres
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Transcription
Introduction
ATRX is a member of the SWI/SNF family of chromatin remodelling factors which is found at a wide range of tandem repeats throughout the genome. ATRX localises to rDNA repeats, telomeric repeats, pericentric repeats, minisatellites and endogenous retroviral sequences and most recently has been found at repeated structures associated with the highly duplicated genes of the zinc finger family 1, 2, 3, 4, 5. Common features of these repeats are that they are heterochromatic, may all form abnormal DNA structures, they are frequently transcribed and most are noted as regions that may be difficult to replicate.
The recruitment of ATRX has only been explored at a few loci. There is strong evidence that ATRX is localised to pericentric heterochromatin via histones modified by H3K9me3. This is thought to be mediated directly via an interaction between its ADD domain and the modified histone tails and indirectly via its interaction with HP1, which also binds H3K9me3‐modified histones 6, 7, 8. By contrast, in the postnatal brain highly abundant MeCP2 is the dominant signal for the localisation of ATRX to pericentric heterochromatin 9. It is not known how ATRX is recruited to other repeats although different mechanisms are clearly involved. Of particular interest is the observation that ATRX localises to G‐rich tandem repeats; these have been shown in vitro to form G‐quadruplex (G4) structures and recombinant ATRX binds to these sequences when folded into a G4 structure but not when denatured 3. However, ATRX binding at one such locus, the ψζ variable number tandem repeat (ψζ VNTR), was noted to vary from one cell type to another indicating that additional factors influence the localisation to these G‐rich sequences.
At present, the functional role of ATRX, when recruited, is also unclear. In particular, the pleiotropic effects seen in cell models, humans and a variety of other organisms when ATRX is inactivated pose the question of whether it has a single or multiple activities. Loss of ATRX function affects diverse nuclear processes including methylation 2, gene expression 3, replication, genome stability 10, 11, 12, 13, 14, mitosis 15 and meiosis 16. The mechanisms underlying these multiple effects remain to be elucidated; however, increasing evidence points to a common role for ATRX as part of a complex with a histone chaperone (DAXX) and the histone variant (H3.3) in remodelling chromatin and replacing canonical histones with H3.3. ATRX provides an ATP‐dependent molecular motor for this complex which has been shown to remodel and replace histones both in vitro and in vivo.
To understand in greater detail how ATRX recognises tandem repeats, we have specifically investigated how it is recruited to telomeres (TTAGGG)n where it plays one of its major functional roles in vivo. Acquired mutations in ATRX have been identified in almost all tumours which maintain their telomere length via the alternative lengthening of telomeres (ALT) pathway 17, 18, 19. In the 15% of cancers that use this pathway, the banks of telomeric repeats are maintained by homology‐directed repair (HDR) rather than by increasing the expression of telomerase 20, 21. The ATRX/DAXX complex suppresses this pathway and thereby acts as an important tumour suppressor 22, 23.
Here, we show that recruitment of ATRX to telomeric repeats, and other repeats, critically depends on their transcription and the presence of RNA–DNA hybrids, known as R‐loops 24. In the absence of ATRX, the formation of R‐loops at transcribed telomeric repeats greatly increases in telomerase‐positive cells. Moreover, re‐introduction of ATRX reduced R‐loop levels in ALT‐positive cells. Increased R‐loops at transcribed telomeric sequences could explain many previously reported phenomena that occur at telomeres in ALT cells in the absence of ATRX. These include the formation of abnormal DNA structures (particularly G4 sequences 25), stalling of replication, induction of a DNA damage response 26 and increased frequency of homologous recombination 27. All of these effects have been linked to the formation of R‐loops which we show here are suppressed by ATRX. In this way, a single activity of ATRX could explain multiple secondary effects seen at telomeres, and other repeats, when ATRX is absent.
Results
Recruitment of ATRX to GC‐rich sequences is associated with their transcription
ATRX binds to many different regions of the genome containing repeats (e.g. telomeres, centromeres, rDNA and tandem repeats). Although the binding of ATRX at AT‐rich pericentric heterochromatin has been shown to depend on H3K9me3, the mechanism of recruitment at other targets is unknown. Using chromatin immunoprecipitation (ChIP), we have previously shown that some ATRX peaks, such as those associated with a minisatellite (ψζ VNTR) in the globin gene cluster, are present only in erythroid cells, when the globin genes are expressed. Other ATRX peaks associated with widely expressed genes appear in all cell types tested 3. This suggests that transcription, or associated aspects of gene expression, may be important for the recruitment of ATRX. To address this, we performed ChIP‐seq in three different mouse tissues [embryonic stem cells (ESCs), mouse embryonic fibroblasts (MEF) and foetal liver cells (FL)] to determine whether recruitment of ATRX is related to gene expression. Strikingly, only a small number of ATRX peaks were shared between cell types; most peaks (genomewide) are specific to the cell type (Fig EV1A). This was also true when we studied ATRX peaks which only lie within genes (Fig EV1B).
Figure EV1. Cell‐type variability in ATRX binding is associated with differential transcription.
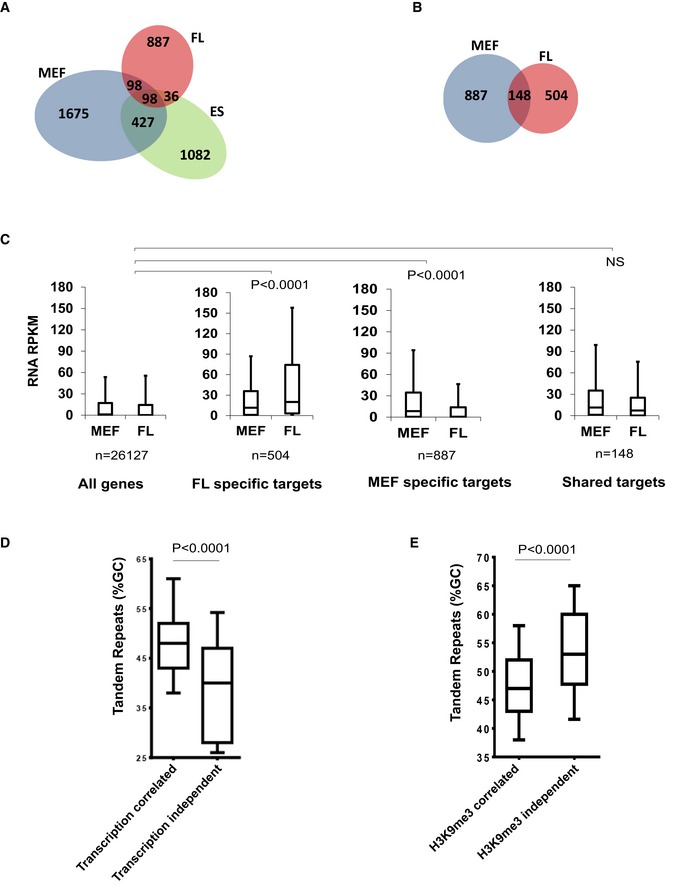
- Overlap between ATRX binding sites in mouse embryonic stem cells (ES), embryonic fibroblasts (MEF) and erythroid foetal liver (FL) cells.
- Overlap between ATRX binding sites at genes in isogenic MEF and FL cells.
- Reads per kilobase of transcript per Million mapped reads (RPKM) of RNA‐seq data in MEF and FL cells at: all genes in the genome; genes which bind ATRX in FL cells but not MEFs; genes which bind ATRX in MEFs but not FL cells; genes which bind ATRX in both MEF and FL cells (shared targets). Boxes represent the 25th, median and 75th percentiles. Whiskers represent the 10th and 90th percentiles. Statistical significance was determined using a Mann–Whitney U‐test on the difference in expression between FL and MEFs (MEF‐FL) in each group compared to (MEF‐FL) values of all genes in the genome.
- %GC content of tandem repeats underlying ATRX peaks which were transcription correlated or transcription independent in MEF and FL cells. Transcription‐independent ATRX TRs (n = 35) had average %GC content lower than transcription‐correlated (n = 169) ATRX TRs. Boxes represent the 25th, median and 75th percentile of %GC content. Statistical significance was assessed using a two‐tailed Mann–Whitney test.
- %GC content of tandem repeats underlying ATRX peaks which were H3K9me3 correlated or H3K9me3 independent in MEF and ES cells. H3K9me3‐independent ATRX TRs (n = 83) had average %GC content higher than H3K9me3‐correlated (n = 622) ATRX TRs. Boxes represent the 25th, median and 75th percentile of %GC content. Statistical significance was assessed using a two‐tailed Mann–Whitney test (see also Table EV1).
We next asked whether the variability of cell‐type‐specific ATRX recruitment is associated with genes specifically expressed in different cell types. To ensure that ATRX binding is not skewed by differences in genomic background, we only analysed ATRX ChIP‐seq peaks in the isogenic MEF and FL generated from a single C57 mouse. The genic sequences that recruit ATRX only in FL are expressed at higher levels (on average) in FL compared to MEF (Fig EV1C). Likewise, MEF‐specific peaks were only seen at genes expressed at higher levels (on average) in MEFs compared to FL. Importantly, we observed no differences between FL and MEF in the expression of genes underlying the shared peaks in both tissues. These observations strongly suggest that recruitment of ATRX occurs predominantly at genic sequences that are being transcribed in the cell types being studied.
Further analysis showed that ATRX recruitment was more likely to be dependent on transcription at tandem repeats with a high GC content (Fig EV1D). Furthermore, tandem repeats at which ATRX recruitment is dependent on H3K9me3 were shown to have a relatively low GC content (Fig EV1E). Unexpectedly, in Suv39h double‐knockout (dn) mouse embryonic fibroblasts in which H3K9me3 was depleted in heterochromatin, ATRX recruitment to telomeric repeats (CCCTAA)n increased (Table EV1). In summary, it appears that there is a mechanism for the recruitment of ATRX to repeated sequences other than via H3K9me3. Recruitment of ATRX to some GC‐rich repeats is clearly associated with their transcription.
ATRX recruitment to telomeric (TTAGGG)n repeats is dependent on their transcription and is correlated to the number of tandem repeats
To investigate further whether ATRX recruitment to G‐rich repeats is dependent on transcription, we created an experimental model in which transcription can be switched on and off to determine its effect on the recruitment of ATRX. We were particularly interested to explore the basis for ATRX binding to G‐rich telomeric repeats since ATRX clearly plays a major function at such repeats in its role as a tumour suppressor.
A vector construct (from M Napierala) was modified. Telomeric sequences were inserted into an artificial intron (from the gene Pem in rat) of a GFP reporter gene, expression of which is controlled by a Tet‐inducible promoter (pCMV/TetO2). To investigate the link between the number of tandem repeats and ATRX recruitment, two different banks of repeats, (TTAGGG)42 and (TTAGGG)71, were engineered into two independent constructs. Each of these constructs was then stably integrated into the genome of 293T‐Rex cells using site‐directed recombination (Fig 1A). Each stable clone was shown to have a single integration at the chosen site (Appendix Fig S1) so that any change in recruitment of ATRX to the newly introduced, ectopic G‐rich sequence could be attributed to the inserted sequence itself and not to different chromatin environments and “position effects” at different integration sites.
Figure 1. ATRX recruitment to telomeric sequences is dependent on transcription and length of repeats.
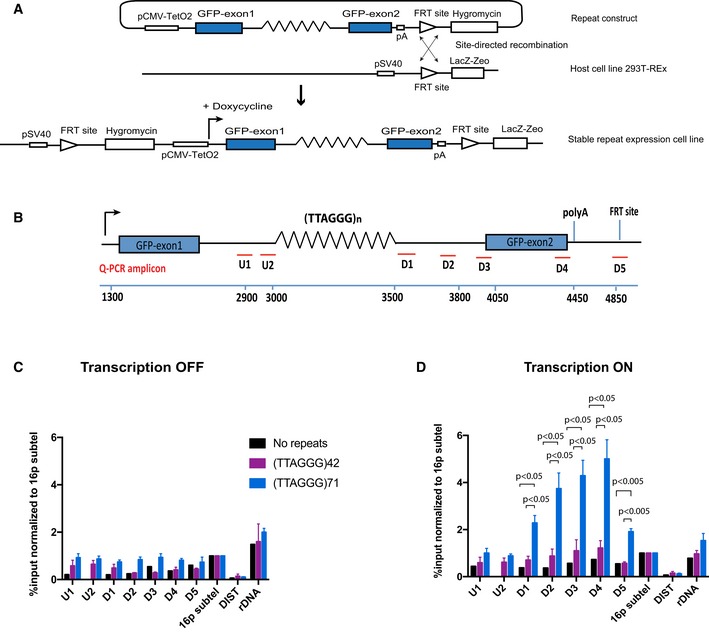
- Diagram of the ectopic reporter gene GFP containing a G‐rich repeat, integrated into the genome of 293T‐Rex at a single FRT site by site‐directed recombination. The integration inactivates LacZ‐Zeo and activates hygromycin resistance, which allows selection of stable clones. The expression of the ectopic gene is regulated by a pCMV‐inducible promoter, which is activated by addition of doxycycline. The blue boxes are GFP exons; lines are introns; and zigzag lines are the G‐rich tandem repeat (see also Appendix Fig S1).
- Diagram shows locations of the qPCR amplicons used to assess ATRX enrichment at the ectopic telomere repeat region. Numbers indicate distance to the start of the ectopic cassette. The diagram is not drawn to scale.
- ATRX ChIP analysis in stable clones containing telomere repeats of indicated sizes and the control without the repeats when the ectopic gene is inactive (transcription off).
- ATRX ChIP analysis in stable clones containing telomere repeats of indicated sizes (clones 5, 6 and 7 for (TTAGGG)71 construct and clones 5 and 6 for (TTAGGG)42 construct) and the control without the repeats (clone EV4 and N2S‐9) when the ectopic gene is activated (transcription on) upon addition of 1 μg/ml doxycycline for 24 h. The enrichment of ATRX is represented as % input normalised to that at 16p telomeric region (16ptel). DIST is a negative control, and ribosomal DNA (rDNA) is a positive control.
First, we looked at ATRX recruitment in the stable cell lines carrying (TTAGGG)42 and (TTAGGG)71, ectopic telomeric repeats. ATRX enrichment was analysed by at least three independent ChIP experiments on at least two different clones. When the reporter gene is silenced in the absence of doxycycline in the media, there was a detectable level of ATRX binding at the ectopic locus as compared to the negative control (DIST) and the positive controls (rDNA and 16p sub‐telomeric region) (Figs 1B and C, and EV2A). Strikingly, when the gene is activated by the addition of doxycycline, ATRX enrichment at the repeat is increased as compared to the no repeat control (Figs 1D and EV2A) and interestingly, the longer telomeric sequence (71 repeats compared to 42 repeats) recruited more ATRX.
Figure EV2. Expression of GFP exon 1 and exon 2 upon doxycycline induction.
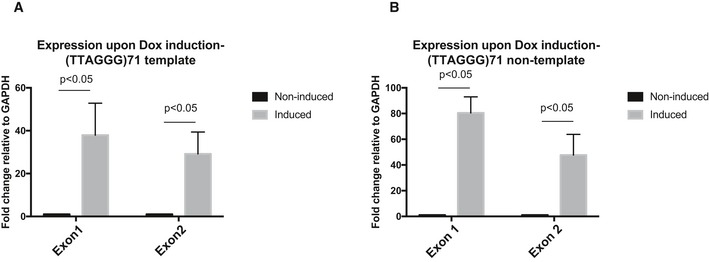
-
A, BReverse transcription quantitative PCR showing that both GFP exon 1 and exon 2 in (A) the (TTAGGG)71 clones with G‐rich strand the template and (B) the reverse orientation clones with G‐rich strand the non‐template are expressed upon transcription induction by doxycycline. Data are presented as fold change of expression relative to GAPDH. “No reverse transcriptase” (no RT) controls were included in each experiment. Data bars represent the mean of at least four biological repeats (± SEM). Statistical significance was determined using Student's t‐test.
Next, we investigated another previously characterised ATRX target, the G‐rich ψζ VNTR, which comprises an imperfect repeat of (GCGGGG)n. This minisatellite lies in the second intron of the ψζ gene within the human alpha‐globin cluster located in the sub‐telomeric region of chromosome 16 (16p13.3). ATRX ChIP was performed on different clones containing the ψζ VNTR minisatellite of different numbers of (GCGGGG) repeats (and sizes of 140, 240, 390 and 490 bp). These originated from four different individuals, and a clone with no repeats (NoVNTR) was included as a control. As for the telomeric constructs, using ATRX ChIP we again observed that ATRX recruitment increased upon induction of transcription of the ectopic gene and that the more the repeats, the more dramatic the increase in ATRX enrichment (Fig EV3A–C).
Figure EV3. ATRX recruitment to the G‐rich ψζ VNTR is dependent on transcription and the length of repeats.
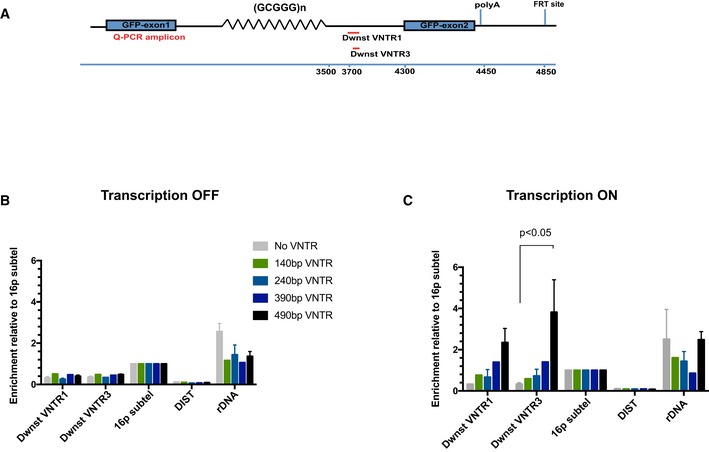
- Diagram shows locations of the qPCR amplicons used to assess ATRX enrichment at the ectopic telomere repeat region. Numbers indicate distance to the start of the ectopic cassette.
- ATRX ChIP analysis in stable clones containing ψζ VNTR of indicated sizes and the control without the VNTR when the ectopic gene is inactive (transcription off). The enrichment of ATRX is represented as % input normalised to that at 16p telomeric region (16ptel).
- ATRX ChIP analysis in stable clones containing ψζ VNTR of indicated sizes when the ectopic gene is switched on (transcription on) by addition of 1 μg/ml doxycycline for 48 h. The enrichment of ATRX is represented as % input normalised to that at 16p telomeric region (16ptel).
These results show that the recruitment of ATRX to G‐rich tandem repeats is dependent on transcription and that the level of ATRX binding upon activation of transcription increases with the number of repeats present. Furthermore, the fact that the same observation regarding ATRX recruitment was made for two completely different GC‐rich sequences indicates that ATRX recruitment is probably determined by shared properties of such repeats.
Of interest, we observed that ATRX recruitment to the arrays of tandem repeats is asymmetrical with more binding downstream of the repeat, as indicated by qPCR at the D1 to D4 amplicons, compared to upstream at the U1 and U2 amplicons (Fig 1D). Moreover, the increase in ATRX enrichment upon transcriptional activation rapidly declined beyond the polyA addition site, as indicated by the amplicon D5, where RNA transcription is terminated (Fig 1D), which further supports the link between transcription and the recruitment of ATRX.
The recruitment of ATRX to telomeric G‐rich repeats depends on their transcription rather than any associated chromatin signatures
To search for the factor or factors that determine the transcription‐dependent recruitment of ATRX to the ectopic repeats, we first tested whether the changes in ATRX binding upon transcriptional activation correlated simply with the presence of RNA Pol II or histone modifications associated with transcription. To examine this, we used the clones containing 71 telomere repeats, (TTAGGG)71, since they showed the highest enrichment of ATRX upon transcription.
Comparing the distribution of RNA Pol II in the uninduced and Tet‐induced states, when the telomeric repeats were transcribed, Pol II was uniformly increased across the entire gene (Appendix Fig S2). RNA Pol II was present both in clones containing the (TTAGGG)71 repeats within the intron of the reporter gene and in clones without the repeats. This clearly shows that ATRX recruitment does not simply depend on the recruitment of Pol II and transcription at this ectopic locus but specifically requires the transcription of the tandem repeats.
We next looked for any evidence for requirement of transcription‐associated histone modifications in the recruitment of ATRX; in particular, we examined trimethylated lysine 4 (H3K4me3) and trimethylated lysine 36 (H3K36me3) on histone H3. Previous studies 6, 8 demonstrated that H3K4me3 inhibits the binding of the ATRX ADD domain to the histone H3 tail and it has further been postulated that H3K4me3 at CpG islands excludes the binding of ADD‐containing DNMT3 enzymes 28, 29. If ATRX recruitment is inhibited by H3K4me3, then we would expect to see a decrease in K4me3 level downstream of the repeats where ATRX is recruited. ChIP analysis shows that upon induction of expression by doxycycline, H3K4me3 modification increases, as expected, with the highest level at the start of the gene and the signal decreasing downstream of the repeats, a pattern which is the inverse of that seen for ATRX (Fig 2A and B). This suggests that although H3K4me3 is not the signal required to recruit ATRX, we cannot rule out the possibility that H3K4me3 levels may modulate the pattern of ATRX recruitment across the repeat.
Figure 2. ATRX binding at the ectopic G‐rich sequence may be modulated by H3K4me3, but is independent of H3K36me3, H3K9me3 and nucleosome occupancy.
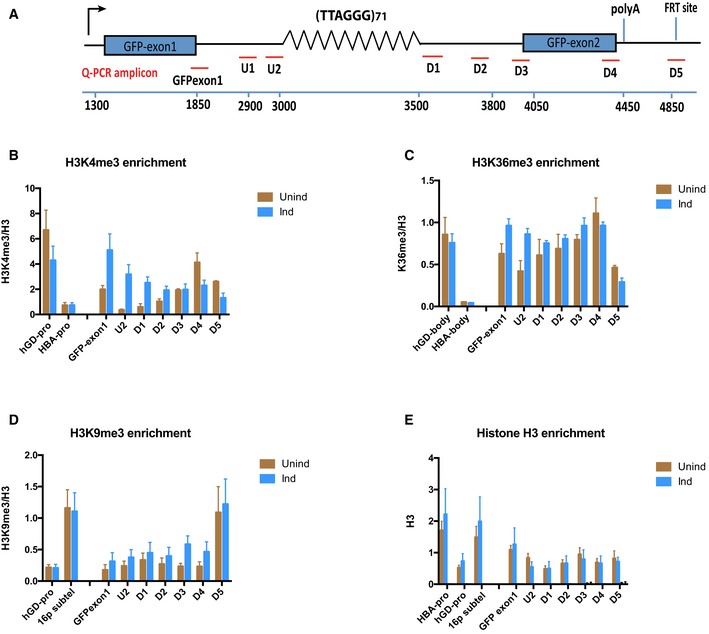
- Diagram shows locations of the qPCR probes used to assess enrichment of relevant histone modifications at the ectopic telomere repeat region. Numbers indicate distance to the start of the ectopic cassette. The diagram is not drawn to scale.
- H3K4me3 ChIP in (TTAGGG)71 clones when transcription is off (Unind) and on (Ind). GAPDH promoter (hGD‐pro) is a positive control, and HBA promoter (HBA‐pro) is a negative control. n = 3.
- H3K36me3 ChIP in (TTAGGG)71 clones when transcription is off (Unind) and on (Ind). hGD‐body (a region in intron 1 of human GAPDH) is a positive control, and HBA body (a region in HBA gene) is a negative control. n = 3.
- H3K9me3 ChIP in (TTAGGG)71 clones when transcription is off (Unind) and on (Ind). hGD‐pro is a negative control, and 16ptel is a positive control. n = 4.
- Histone H3 ChIP in (TTAGGG)71 clones when transcription is off (Unind) and on (Ind) compared with and IgG antibody control. HBA‐pro and 16ptel are positive controls, and hGD‐pro is a negative control. n = 3.
ChIP analysis using antibodies against H3K36me3 also shows little change in this histone mark between induced and non‐induced cells (Fig 2C), suggesting that it is also not required for ATRX recruitment to the G‐rich repeats.
Histones modified by H3K9me3 are required for ATRX recruitment to pericentric heterochromatin. However, ChIP analysis only detected a low level of this histone modification across the ectopic locus with little difference in the enrichment level between induced and non‐induced cells, indicating that, unlike at pericentromeric, heterochromatic repeats, this histone modification is unlikely to be associated with the recruitment of ATRX to telomeric repeats (Fig 2D).
The Drosophila homolog of ATRX, XNP, which lacks the ADD domain, is recruited to nucleosome‐depleted chromatin 30. We tested whether nucleosome depletion was linked to the recruitment of ATRX at the ectopic G‐rich repeat. A low level of H3 can be seen across the gene in non‐induced cells; however, no change in its enrichment was detected upon induction of transcription (Fig 2E). This suggests that nucleosome occupancy per se is not a factor in ATRX targeting to the telomeric repeats in these cells.
We conclude that the recruitment of ATRX to telomeric repeats appears to be independent of histone modifications and nucleosome occupancy and is not simply associated with the presence of RNA Pol II or transcription in general, but specifically with the transcription of G‐rich repeats.
Transcription‐dependent recruitment of ATRX to telomeric repeats correlates with the presence of R‐loops
G‐rich repetitive DNA sequences are prone to form RNA–DNA hybrids (R‐loops) during transcription, and the longer the tandem repeat array, the higher the likelihood of R‐loop formation 31. R‐loops are known to facilitate the formation of G4 structures by stabilising the single‐stranded G‐rich strand to form G‐loops 25. We hypothesised that R‐loops might form at the ectopic tandem repeats, allow the formation of G4 and so lead to the recruitment of ATRX. To test whether ATRX binding is associated with the presence of R‐loops, we used the DNA immunoprecipitation (DIP) technique with the S9.6 monoclonal antibody which specifically recognises the DNA–RNA hybrids in R‐loops. DIP analysis of induced and non‐induced clones carrying the long telomere repeat (TTAGGG)71 shows that R‐loop signals at the ectopic telomere repeats increased upon activation of transcription (Fig 3A and B). To confirm the specificity of the R‐loop signal, E. coli recombinant RNase H, an enzyme that specifically degrades the RNA strand in the RNA–DNA hybrids was added to the samples prior to immunoprecipitation. Following the treatment, the DIP signal for the ectopic repeat was substantially reduced, suggesting that the R‐loops detected were genuine (Fig 3B). Interestingly, ATRX recruitment upon activation of transcription does not directly mirror the distribution of R‐loops across the repeat but, as previously noted, accumulates downstream of the R‐loops.
Figure 3. R‐loop formation increases upon transcriptional induction of the ectopic telomere repeat.
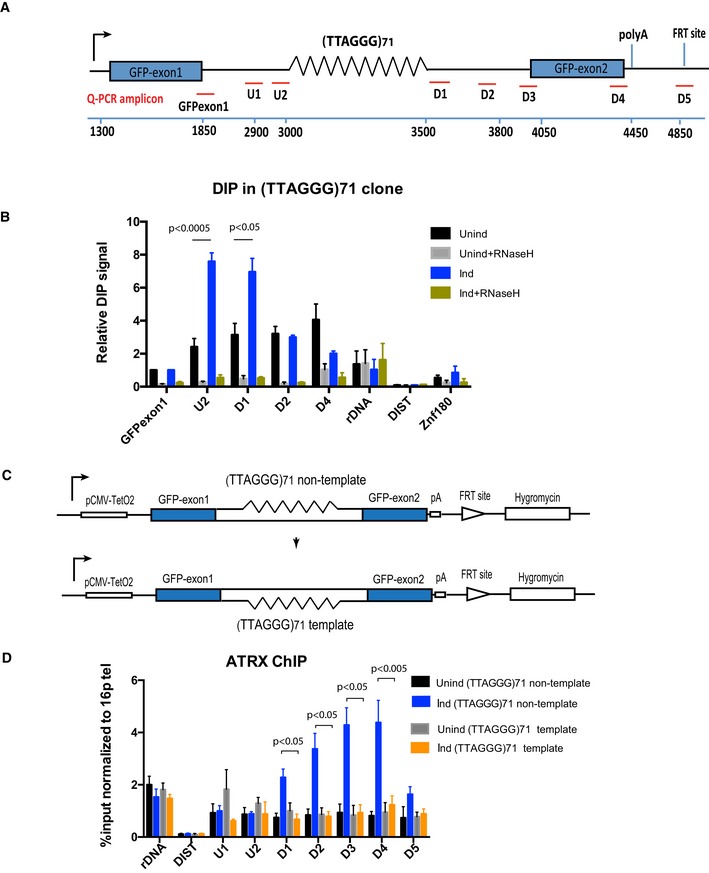
- Diagram shows the ectopic locus and locations of the qPCR probes used to assess R‐loop signal and ATRX enrichment at the ectopic telomere repeat region.
- DIP analysis in cells containing the (TTAGGG)71 ectopic repeat when transcription is off (Unind) and on (Ind). As a control, DIP samples were also treated with E. coli recombinant RNase H for 5 h at 37°C prior to immunoprecipitation with S9.6 antibody. Ribosomal DNA is a positive control, and DIST and Znf180 are negative controls. DIP signal is expressed as % input normalised to the non‐repetitive region of GFP exon 1. Data bars represent the average value from four independent experiments (± SEM) from three independent clones (5, 6 and 7) for the ectopic repeat (TTAGGG)71. Statistical significance was determined using Student's paired t‐test.
- Diagram shows the inversion of the repeat region. The G‐rich strand in the original orientation construct is the non‐template strand, whereas it is the template strand in the reverse orientation construct.
- ATRX ChIP analysis in the reverse orientation clone when transcription is inactive (Unind) and active (Ind) upon addition of 1 μg/ml doxycycline for 24 h. ATRX enrichment is presented as % input normalised to that of 16ptel region. Data bars are the average values of at least three independent experiments (± SEM) from two clones (3 and 4) of the reverse orientation and three clones (5, 6 and 7) of the original orientation. rDNA and DIST are a positive and negative control, respectively. Statistical significance was determined using Student's paired t‐test.
G‐loops have been shown to form only when the G‐rich stand is the non‐template strand for transcription 25. To test the hypothesis that ATRX recruitment correlates with G4 and R‐loop formation, we reversed the orientation of the repeat such that the G‐rich strand is on the template strand (Fig 3C). In this orientation, the non‐template strand is C‐rich and unable to form G4 and the R‐loops which comprise hybrids of C‐rich RNA and G‐rich DNA are less stable than when formed from G‐rich RNA and C‐rich DNA 31, 32. As expected, there was a reduction in R‐loops in the reverse orientation, reflecting their lower stability (Fig EV4) but importantly, reversing the orientation also led to the loss of ATRX enrichment (Figs 3D and EV2B) downstream of the repeat when transcription is induced. In summary, these data suggest that the transcription‐dependent recruitment of ATRX depends on the orientation of the repeat and strongly suggest that it is associated with the presence of DNA secondary structure.
Figure EV4. R‐loop formation preferentially occurs when the G‐rich strand is the non‐template strand.
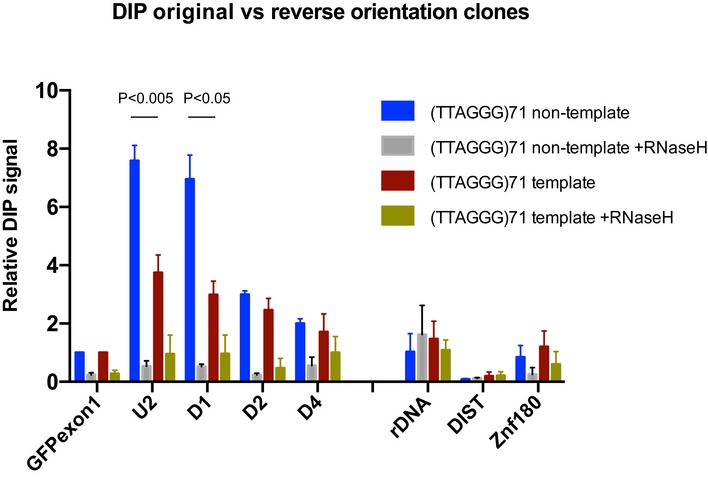
DIP analysis in clones containing the ectopic telomeric sequence with the G‐rich strand being the non‐template strand and in clones containing the reverse oriented telomeric sequence with the G‐rich strand being the template strand after activation of transcription by doxycycline. DIP samples were also treated with E. coli RNase H prior to immunoprecipitation with S9.6 antibody as a control. DIP signal is expressed as % input normalised to the non‐repetitive region of GFP exon 1. Data bars represent the average value from four independent experiments ± SEM. Statistical significance was determined by unpaired Student's t‐test.
Changes in the level of R‐loops are associated with changes in the recruitment of ATRX
If it were the R‐loops per se which trigger the recruitment of ATRX to telomeric repeats, then modulating R‐loops, independently of transcription, should lead to changes in ATRX binding. RNase H1 over‐expression or depletion has been previously used as a tool to decrease or increase R‐loops, respectively, but this approach is not always successful; Arora et al 27 found that RNase H1 over‐expression only affected telomeric R‐loops in the context of ALT cells, while telomerase‐positive cells were insensitive. Consistent with this, we found that, in our cells, over‐expression or deletion of RNase H1 did not lead to changes in R‐loops.
Therefore, to explore further the role of R‐loops in recruiting ATRX, we took an alternative approach of using camptothecin (CPT), an inhibitor of topoisomerase I. This enzyme relieves transcription‐induced DNA supercoiling which is known to affect the level of R‐loops 33. Treatment with CPT has been shown to lead to a transient increase in R‐loops followed, over longer time periods, by their abolition. The kinetics of this process appear to vary at different genes which is possibly related to the different abundance of R‐loops at different loci 34, 35. Transcription of the (TTAGGG)71 construct was induced in these cells for 24 h, and then, they were treated with 10 μM CPT for 30 min. Following CPT treatment, DIP and ATRX ChIP were performed. Consistent with previous studies 36, R‐loop enrichment is increased and persists at ribosomal DNA (Fig 4A). Strikingly, following treatment with CPT, R‐loops are decreased at both the ectopic telomere repeat and endogenous telomeres as compared to the control (Fig 4B–D). Of interest, these changes in R‐loops coincide with a reduction of ATRX recruitment at the ectopic telomere repeat (Fig 4E) and the endogenous telomeres (Fig 4F and G), as well as increased recruitment of ATRX at ribosomal DNA (Fig 4H). The changes in ATRX binding at the ectopic repeat and at rDNA are not simply correlated with transcription as this is decreased at both loci (Appendix Fig S3). It therefore seems likely the link between gene expression, R‐loops and ATRX recruitment does not always depend on active transcription, but rather on the existence of R‐loops as a result of prior transcription. These results strongly suggest that ATRX recruitment depends on the presence of R‐loops at telomeric tandem repeats.
Figure 4. Modulating R‐loop levels leads to changes in ATRX binding.
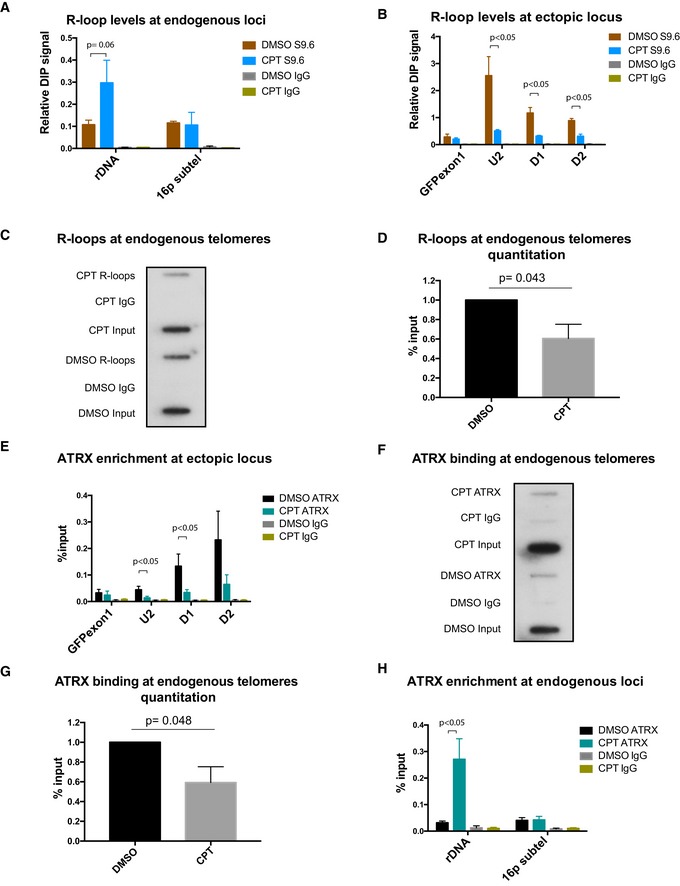
-
ADIP analysis at endogenous regions of ribosomal DNA and 16p subtel. n = 3.
-
BDIP analysis at the ectopic telomeric sequence (TTAGGG)71, treated with 10 μM camptothecin for 30 min or with solvent DMSO, following induction of transcription by 1 μg/ml doxycycline for 24 h. n = 3.
-
C, DRepresentative blot (C) showing a decrease in R‐loops at endogenous telomeres in the camptothecin (CPT)‐treated cells compared to the control DMSO‐treated cells. Immunoprecipitated DNA, using S9.6 antibody, was slot blotted and probed with telomeric probes. Quantification is shown in (D) for three biological replicates. R‐loop levels are expressed as % input.
-
EATRX enrichment at the ectopic telomeric sequence (TTAGGG)71, in cells treated with 10 μM camptothecin for 30 min or with solvent DMSO control, following induction of transcription by 1 μg/ml doxycycline for 24 h. n = 3.
-
F, GRepresentative blot (F) showing a reduction in ATRX binding at endogenous telomeres in the CPT‐treated cells compared to the DMSO control. Immunoprecipitated DNA, using H300 antibody, was slot blotted and probed with telomeric probes. Quantitation is shown in (G) for three biological replicates. ATRX enrichment is normalised to input.
-
HATRX ChIP analysis at endogenous regions of ribosomal DNA and 16p subtel. n = 3. Enrichment of R‐loops or ATRX is presented as % input.
Loss of ATRX results in an increase in R‐loops
To investigate the role of ATRX at R‐loop forming sequences, we knocked down ATRX in clones containing long arrays of telomere repeats (TTAGGG)71 using several lentivirus expressing short‐hairpin RNAs (shATRX2, shATRX90 and shATRX91) which almost completely abolish the expression of both full‐length and truncated isoforms of ATRX (Figs 5A and EV5A). As a consequence, RNA–DNA hybrids (detected by DIP) at the transcribed ectopic telomere repeats increased ~twofold in the ATRX‐depleted cells (shATRX2) to ~fivefold (shATRX90 and shATRX91) compared to the control (Figs 5B and EV5B), and we also observed an increase in the abundance of R‐loops at ribosomal DNA in the absence of ATRX (Figs 5C and EV5C). This change is unlikely to be due to the increase in transcription through the repeats as our FACS analysis shows that GFP expression goes down when ATRX is knocked down (Appendix Fig S4).
Figure 5. ATRX modulates R‐loop formation.
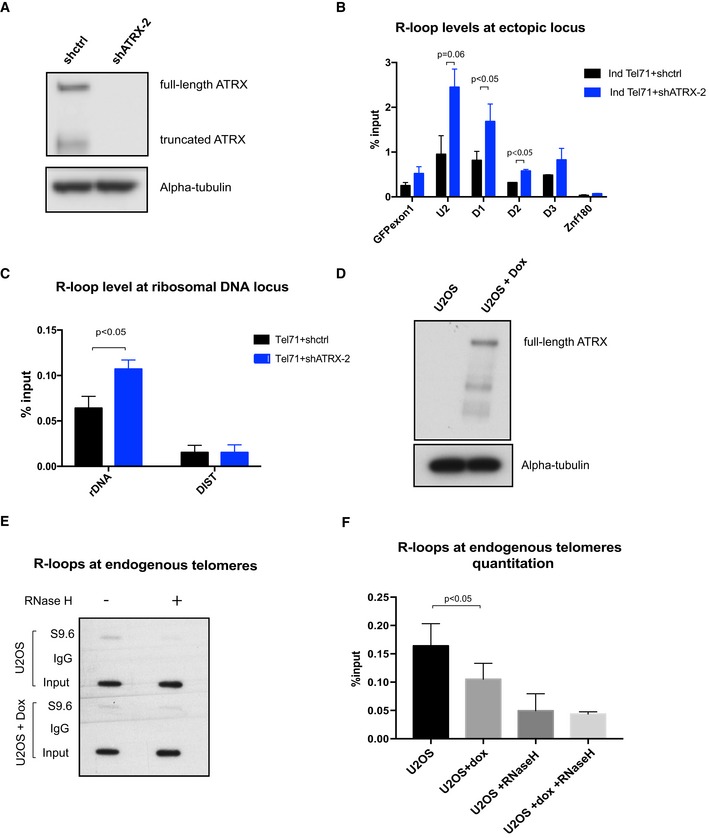
- Western blot analysis of whole‐cell extract of cells containing the (TTAGGG)71 ectopic repeat treated with lentiviral shRNA against ATRX (shATRX‐2) or with shRNA control (shctrl). Western blot membrane was probed with anti‐ATRX and anti‐alpha‐tubulin antibody.
- DIP analysis in cells containing the (TTAGGG)71 ectopic repeat treated with shRNA against ATRX or shRNA control, followed by transcription induction by addition of 1 μg/ml doxycycline for 24 h. The enrichment of R‐loops at the ectopic repeats was measured by qPCR. n = 3. Znf180 is a negative control.
- DIP analysis showing increase in R‐loops at ribosomal DNA region in the ATRX knockdown cells. DIST is a negative control. n = 3 (see also Fig EV5).
- Western blot analysis showing re‐expression of ATRX in U‐2 OS 22/3 cell line upon induction by doxycycline for 2 days.
- R‐loops at endogenous telomeres in U‐2 OS 22/3 before and after 2 days of doxycycline‐induced ATRX re‐expression. DIP was performed with genomic DNA from these cell lines. As a control, the samples were treated with recombinant RNase H prior to the immunoprecipitation. Recovered DNA was slot blotted on a Zeta‐probe blotting membrane and then hybridised with 32P‐labelled telomeric oligos.
- Quantitation of the S9.6 signal relative to the inputs in (E). n = 3.
Figure EV5. ATRX modulates R‐loops.
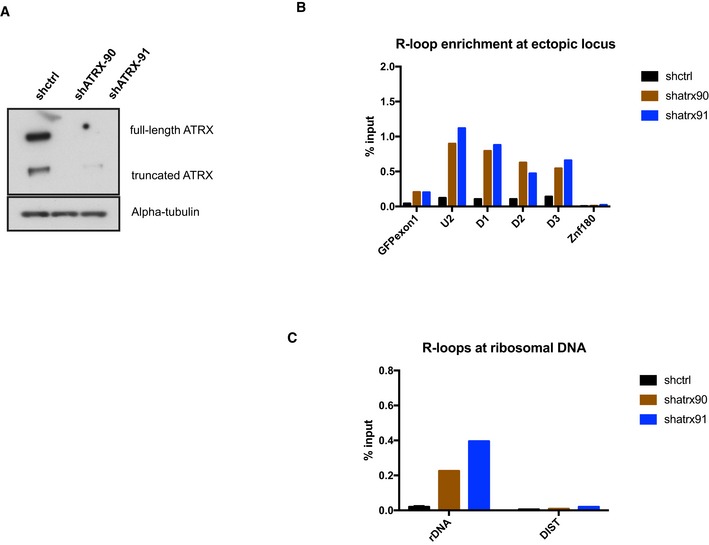
- Western blot analysis of whole‐cell extract of cells with the ectopic telomeric sequence (TTAGGG)71 treated with lentiviral shRNAs against ATRX (shATRX‐90 and shATRX‐91) or with shRNA control. The Western blot membrane was probed with anti‐ATRX and anti‐alpha‐tubulin antibody.
- DIP analysis in clones treated with shRNA against ATRX or shRNA control, followed by transcription induction by addition of 1 μg/ml doxycycline for 24 h.
- DIP analysis showing an increase in R‐loops at ribosomal DNA region in the ATRX knockdown cells. DIST is a negative control.
It has previously been reported that cancer cells harbouring an active ALT pathway characteristically exhibit a higher level of R‐loops at telomeres. We reasoned that if ATRX suppresses R‐loop formation, then re‐introduction of ATRX in ATRX‐deficient cells should decrease the abundance of these structures. As expected, re‐expressing ATRX in U‐2 OS cells 22, which are normally deficient of ATRX, led to a ~40% reduction in RNA–DNA hybrids (Fig 5D–F). This strongly suggests that ATRX normally plays a role in resolving R‐loops or suppressing their formation at G‐rich sequences.
Discussion
The localisation of ATRX at GC‐rich tandem repeats, including telomeres, is well documented; however, the primary sequences of such repeats are not sufficient to recruit ATRX. The pattern of ATRX localisation varies from one cell type to another (3 and this study) and may change during differentiation 10. Here, we have shown that the different patterns of ATRX recruitment reflect the transcriptional status of its targets and this is most notable for tandem repeats with a high GC content including telomeric repeats.
It has been previously shown that recruitment of ATRX to A/T‐rich pericentric heterochromatin occurs via its interaction with H3K9me3; however, here we have shown that this chromatin modification is not required to recruit ATRX to GC‐rich tandem repeats. In fact, when H3K9me3 at telomeres was virtually abolished by inactivating the histone methyltransferases SUV39H1 and SUV39H2, ATRX binding increased at telomeres, indicating that this mark does not play a significant role in recruiting ATRX to telomeric repeats. These observations suggest that different mechanisms are used to recruit ATRX to different types of targets and for GC‐rich tandem repeats in particular, transcription appears to play an important role. Transcription of telomeres producing telomere repeat containing RNA (TERRA) 37 may play a role in recruitment of ATRX. Indeed, the modest increase in binding of ATRX at telomeres in the SUV39H1/2 double knockout may reflect an increase in TERRA transcription which is usually repressed by H3K9me3 38.
To explore the factors involved in the recruitment of ATRX to telomeres, we devised an experimental system incorporating a telomeric repeat within an intron of a GFP reporter under the control of a doxycycline‐inducible promoter. Using this system, we showed that ATRX recruitment was substantially increased on transcription and the amount of ATRX was also directly related to the presence, number and orientation of repeats in the tandem array. Since RNA Pol II was elevated across the reporter gene whether it included the repeats (ATRX recruited) or contained no repeats (ATRX not recruited), we concluded that RNA Pol II alone was insufficient to recruit ATRX. Similarly, we showed that none of the chromatin modifications associated with transcription correlated with ATRX recruitment. Furthermore, in contrast to a previous report 30, we observed no anti‐correlation between histone H3 enrichment and ATRX.
Most importantly, this study provides the first evidence that ATRX targeting to G‐rich tandem repeats is associated with the presence of R‐loops at arrays of repeats that are being, or have been transcribed. During transcription, nascent RNA transcripts with high G content thread back and invade the open DNA duplex, giving rise to R‐loops 24. Moreover, we found that ATRX recruitment was dependent on the orientation of the telomeric repeat and was observed only when the transcriptional non‐template strand was G‐rich. It has been shown that the RNA–DNA hybrids in R‐loops extend the lifetime of the unpaired guanines and facilitate the formation of G4 DNA structures which, in turn, stabilise R‐loops 25. We and others have previously shown that GC‐rich tandem repeats (including telomeric repeats), that are bound by ATRX, can form G4 DNA and that recombinant full‐length ATRX can directly bind G4 structures in vitro 3. The dependence of ATRX recruitment on the orientation of the tandem repeat is therefore consistent with the formation of G4 when the G‐rich sequence is on the non‐template strand 25.
The increased ATRX binding seen with larger arrays containing more repeats is consistent with their greater propensity to form R‐loops and G4 31. Importantly, modulation of R‐loop levels by treating cells with camptothecin led to changes in the ATRX binding level which at different loci faithfully reflected the levels of R‐loops observed. These data strongly suggest that the presence of R‐loops plays a critical role in the recruitment of ATRX to G‐rich telomeric tandem repeats. Furthermore, the increased recruitment of ATRX observed at rDNA following treatment with camptothecin occurred despite a reduction in transcription, suggesting that the recruitment of ATRX is associated with the presence of R‐loops rather than the process of transcription. It remains a possibility that transcription leads to the formation of G4 RNA and that this is responsible for the recruiting ATRX, but since this would be obstructive to the formation of R‐loops, our observations make this model less likely.
This study shows that ATRX may not only be recruited in the presence of R‐loops but it may also play a role in processing them. When ATRX is depleted, the enrichment of R‐loops increases at the ectopic telomeric repeats. In this context, it was of interest that the distribution of ATRX was asymmetrical in relationship to the repeat. It is conceivable that ATRX binds upstream of the secondary structure, translocates in a 5′–3′ direction and accumulates downstream of it as it processes these structures. Future studies will aim to determine how the R‐loops are processed.
These new findings add significantly to previous observations on the potential role of ATRX in transcription, replication and DNA repair and suggest a model in which the formation of R‐loops plays a central role (Fig 6). The presence of R‐loops and G4 DNA has both been implicated in stalling of replication, causing a subsequent DNA damage response 26, 39, 40. Consistent with a role for ATRX in resolving such structures, directly or indirectly, when ATRX is absent, there is an increased frequency of replication stalling and DNA damage 13, 14. Given the proven role of ATRX/DAXX/H3.3 as a chromatin remodelling complex, the simplest explanation is that recruitment of ATRX by R‐loops and their associated G4 structures may enable this complex to re‐establish a normal chromatin structure during transcription and/or replication via the insertion of histone H3.3, as seen at telomeres when ATRX is re‐introduced to ATRX‐negative ALT cells 22. Finally, in ALT tumours, the defect in DNA replication at telomeres is thought to lead to collapse of replication forks, double‐strand break formation and the triggering of homology‐directed repair. Re‐expression of ATRX suppresses the ALT phenotype 22. Here, we show that re‐expressed ATRX also decreases telomeric R‐loops in ALT cells. Given that telomeric RNA–DNA hybrids are one of the substrates for homologous recombination in ALT 27, our data suggest that ATRX may in part prevent the triggering of ALT by suppressing these recombinogenic R‐loops. The findings in this report demonstrate how transcription may generate the endogenous lesions that are probably critical to this process and help determine the role of ATRX in their resolution.
Figure 6. Model explaining the role of ATRX at R‐loop sites.
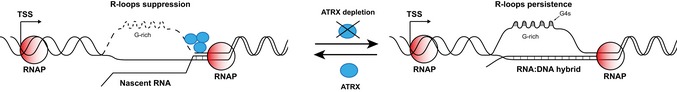
G‐rich regions are prone to form R‐loops during transcription, and these promote G4 formation. In the wild type, ATRX is recruited to these structures where it resolves R‐loops or recruits other enzymes that degrade R‐loops leading to dissolution of G4. In the absence of ATRX, R‐loops and G4s persist, which may cause impaired gene expression, stalling of replication and DNA damage.
Materials and Methods
PCR amplification of G‐rich tandem repeats
The human telomere repeats were amplified from the genomic DNA of patients that have a truncated chromosome repaired with telomere sequences using the primer pair TEL‐REPT ORI1F/R (Table EV2). The PCR was performed with Faststart Taq polymerase (Sigma‐Aldrich, St Louis, MO, USA) and the thermal cycles as follows: 95°C for 5 min, 35 cycles of 95°C for 30 s, 57.7°C for 30 s, 72°C for 1 min 30 s and 72°C for 10 min.
The ψζ VNTR from human genomic DNA of anonymous donors was amplified using primers ψζ VNTR F and R (Table EV2). The reaction was carried out with the addition of DMSO buffer (16.6 mM (NH4)2SO4, 67 mM Tris–HCl, 10% DMSO and 10 mM 2‐mercaptoethanol) and 0.7 M betaine to overcome the high GC content and the highly repetitive nature of the template. Thermal cycles were as follows: 94°C for 2 min, 35 cycles of 94°C for 1 min, 55°C for 30 s, 72°C for 1 min 30 s and 72°C for 10 min.
Plasmid construction
The pcDNA5/FRT/GFP GAA plasmid (a kind gift from Dr. Marek Napierala, University of Alabama at Birmingham) has the backbone of pcDNA5/FRT/TO (Invitrogen, ThermoFisher Scientific, Waltham, MA, USA) with the GFP gene in the multi‐cloning site. This GFP gene has two exons and an exogenous intron (intron of rat Pem gene) with a stretch of GAA repeats flanked by Bsu36I and BssHII recognition sequences. Its expression is driven by a CMV promoter, which is under the regulation of two consecutive tetracycline operators. The vector also has a homologous recombination site, FRT, which can be used to integrate the vector into the genome. The PCR products of telomeric sequence or ψζ VNTR were double‐digested with Bsu36I and BssHII and then used to replace the GAA repeat on the pcDNA5/FRT/GFP GAA vector to make the pcDNA/FRT/GFP TEL or pcDNA/FRT/GFP ψζ VNTR constructs, respectively.
To construct the No repeats control vector, the telomeric repeat stretch was removed from the pcDNA/FRT/GFP TEL.
Cell culture
The 293TRex (Flp‐In T‐REx‐293, R780‐07, Invitrogen) cell line contains a single FRT site in the genome and stably expresses the tetracycline repressor. 293TREx cells were cultured in DMEM (Invitrogen) supplemented with 10% foetal calf serum, 1% l‐glutamine and 1% peni‐streptomycin. Blasticidin and zeocin were added in the medium at concentrations of 15 and 100 μg/ml, respectively, in order to maintain the FRT site in the genome and the expression of the tetracycline repressor.
Generation of stable cell lines
293TRex cells were cultured in 6‐well plates until a confluence of more than 90% was achieved. The cells were then co‐transfected with 1 μg of telomeric constructs (or ψζ VNTR constructs) and 9 μg of plasmid pOG44, which produces the Flp recombinase enzyme to generate stable expression cell lines. Forty‐eight hours after transfection, the cells were cultured in a selective medium (DMEM 10% FCS 1% l‐glutamine 1% pen‐strep) containing 200 μg/ml hygromycin B and 15 μg/ml blasticidin until foci were identified. Clones were selected and screened by Southern blot and sequencing.
To induce transcription of the ectopic GFP locus, 1 μg/ml doxycycline (which has the same mechanism of action as tetracycline) was added into the medium for 24 or 48 h.
To generate stable ATRX knockdown cell lines, lentiviral particles carrying the pLKO‐shATRX expression vectors (41 and Vera Lukashchuk personal communication) which produce short‐hairpin RNAs targeting both full‐length and truncated forms of ATRX were used to infect the cells. Clonal selection and maintenance were performed using 1 μg/ml puromycin. A control vector carrying a scrambled short hairpin was also created to make a stable clone that express wild‐type ATRX.
Southern blot
15 μg of genomic DNA from each clone was digested with KpnI or AflII restriction enzyme at 37°C for 5 h. The digestion products were run on 1% agarose gel at 50 V overnight. After that, the gel was soaked in 1 M NaOH solution for 1 h and then was neutralised for 2 h in neutralisation buffer (1 M Tris, 3 M NaCl, 80 ml/l concentrated HCl pH 7.4). DNA was blotted onto Zeta‐probe nylon membrane (Bio‐Rad, Hercules, CA, USA). The blot was then hybridised with GFP exon 2 probe, which was labelled with 32P at 42°C overnight. After probing, the membrane was washed with 2× SSC (0.03 M sodium citrate and 0.3 M NaCl) and then with 0.1% SSC before developing onto a film.
Western blot
1 × 107 cells were lysed using high salt RIPA buffer (50 mM Tris 7.4, 1 M NaCl, 0.2% SDS, 1% deoxycholic acid, 1% NP‐40 and protease inhibitor cocktail). The lysate was incubated with 50 μl of DNase I (Roche, Welwyn Garden City, UK) for 20 min at room temperature. After that, cellular protein was collected by centrifugation at 16,000 g for 10 min at 4°C. The concentration of whole‐cell protein was measured using a DC protein assay kit (Bio‐Rad). For Western blot, 50 μg of protein was denatured and then run on NuPAGE 4–12% Bis–Tris gel at 150V. Then, the protein on the gel was transferred onto a PVDF Immobilon‐P membrane (Millipore, Watford, Herts, UK). The membrane was then incubated with monoclonal anti‐ATRX antibody 39f at 1:10 dilution, washed and developed onto a film.
Chromatin immunoprecipitation (ChIP)
ChIP for ATRX
ATRX chromatin immunoprecipitation was described previously 3. Approximately 3 × 107–5 × 107 cells were fixed with 2 mM EGS at room temperature for 45 min and then with 1% formaldehyde for 20 min. The cells were then lysed in a stepwise manner, first, by lysis buffer 1 (100 mM HEPES, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP‐40 and 0.25% Triton X‐100), by lysis buffer 2 (200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA and 10 mM Tris pH 8) and then lysis buffer 3 (1 mM EDTA, 0.5 mM EGTA, 1 mM Tris–HCl pH 8, 100 mM NaCl, 0.1% sodium deoxycholate and 0.5% N‐lauroyl sarcosine). Chromatin was subjected to sonication for 30 min (30 s on, 30 s off; high power, Bioruptor; Diagenode s.a., Seraing, Belgium) to yield DNA fragments of under 500 bp. The chromatin was pre‐cleared with Protein A Dynabeads (10002D, Life Technologies, Carlsbad, CA, USA), which were pre‐blocked with 0.5% BSA, for 1 h. 50 μl of the sonicated chromatin was taken as an input. The remaining chromatin was immunoprecipitated with Dynabeads pre‐coated with 10 μg of ATRX antibody H300 (sc‐15408, Santa Cruz Biotechnology, Dallas, TX, USA) or with 10 μg of rabbit IgG control (X0903, Dako UK Ltd, Ely, Cambs, UK) overnight at 4°C. The chromatin–antibody–bead complexes were washed five times with wash buffer (50 mM HEPES, 1 mM EDTA, 0.7% sodium deoxycholate, 500 mM LiCl and protease inhibitor cocktail) and once with ice‐cold PBS. All the washes were carried out for 5 min at 4°C. The captured chromatin was eluted with 0.5% SDS and 0.1 M sodium bicarbonate at room temperature. The cross‐link was reversed with 0.2 M sodium chloride at 65°C for 4 h. DNA was purified and precipitated with ethanol, sodium acetate and carrier glycogen (Life Technologies).
ChIP for other factors
This technique was performed using reagents from the ChIP Assay kit (17‐295, Millipore). Approximately 1 × 107 cells were cross‐linked with 1% formaldehyde for 10 min at room temperature. The cells were lysed in SDS lysis buffer for 10 min on ice. Chromatin was subjected to sonication for 12 min (30 s on, 30 s off, high power, Bioruptor, Diagenode). The sonicated chromatin was then diluted 1 in 10 in ChIP dilution buffer. 10 μl of the diluted chromatin was taken as an input per antibody. 60 μl of protein A/Salmon sperm DNA Agarose beads (from the kit) was added to pre‐clear every 2 ml of diluted chromatin for 1 h at 4°C. The diluted chromatin was then incubated with 10 μg of RNA Pol II antibody (sc‐899, Santa Cruz Biotechnology), 2 μg of H3K4me3 antibody (Ab8580, Abcam, Milton, Cambs, UK), 10 μg of H3K36me3 antibody (61101, Active Motif, Carlsbad, CA, USA), 10 μg of H3K9me3 antibody (Ab8898, Abcam) or H3 antibody (Ab1791, Abcam) overnight at 4°C. After that, 60 μl of agarose beads was added to the antibody–chromatin IP mix and was incubated for 1 h. The antibody–chromatin–bead capturing complexes were washed once in low salt buffer, high salt buffer, LiCl buffer and twice in TE buffer at 4°C. The captured chromatin was eluted in 1% SDS and 0.1 M NaHCO3.
Reverse transcription
RNA was extracted from 105–106 cells using the standard TRI Reagent® Protocol (Sigma). cDNA was produced from 1 μg of RNA in a 20 μl reaction volume using the following commercial kits for first‐strand cDNA synthesis: RETROscript (Ambion), SuperScript III (Invitrogen) or high capacity RNA‐to‐cDNA Kit (Thermo Fisher Scientific).
Quantitative PCR
Quantitative PCR was performed using SYBR green master mix (4309155, Applied Biosystem, Foster City, CA, USA). SYBR primers (Table EV2) were designed manually or using MacVector software (MacVector Inc., Apex, NC, USA). All primers were tested by a five‐point series dilution using genomic DNA. All qPCRs were performed at least three times. ChIP or DIP enrichment was determined relative to input.
ChIP‐seq analysis and H3K9me3 dependency of ATRX targets
ChIP‐seq data of ATRX in mouse ES cells (GSE22162) and mouse immortalised MEFs (GSE96768) were imported into Seqmonk (v0.24.1) (http://www.bioinformatics.babraham.ac.uk/projects/seqmonk/). ATRX binding sites were identified in Seqmonk using the contig probe generator. ATRX and H3K9me3 ChIP‐seq (GSE57092) reads were quantitated under ATRX peaks and normalised for probe length and total mapped reads. An ATRX peak was designated as H3K9me3 correlated if the peak was unique to a single cell type, and both ATRX and H3K9me3 reads were greater than twofold enriched in a single cell type relative to the other. An ATRX peak was designated as H3K9me3 independent if the peak was unique to a single cell type, ATRX reads were greater than twofold enriched relative to the other, and H3K9me3 reads were greater than twofold enriched at the corresponding site in the non‐ATRX bound cell type. ATRX peaks which overlapped a tandem repeat over 100 bp in length were isolated. The %GC content of tandem repeats was obtained from the UCSC genome browser.
RNA‐seq analysis and transcription dependency analysis of ATRX targets
Fragments per kilobase of transcript per million mapped read (FPKM) values of RNA‐seq for every gene were calculated in MEF and FL cells (GSE27843 and GSE26086, respectively). Average FPKM values were calculated in each cell type after excluding genes with zero values. A gene was designated as “expressed” in a particular cell type if FPKM values were greater than the 25th percentile FPKM in the cell type analysed. A gene was designated as “not expressed” if FPKM values were under the 25th percentile or zero in the cell type analysed. ATRX ChIP‐seq was performed in C57 MEFs and FLs (GSE96768). An ATRX peak was designated as transcription correlated if the peak was unique to a single cell type and overlapped a gene which was expressed in that cell type and not expressed in the non‐ATRX bound cell type. An ATRX peak was designated as transcription independent if the peak was unique to a single cell type and overlapped a gene which was not expressed in that cell type and expressed in the non‐ATRX bound cell type. ATRX peaks which overlapped a tandem repeat over 100 bp in length were isolated. The %GC content of tandem repeats was obtained from the UCSC genome browser.
DNA immunoprecipitation
Cells, cultured in a 10‐cm plate to 70–80% confluency, untreated or treated with doxycycline as described above, were harvested by scraping in ice‐cold PBS in a cold room (4°C). They were collected by centrifugation at 500 g for 5 min at 4°C. The cells were then lysed by incubating with 600 μl of cell lysis buffer (0.5% NP‐40, 85 mM KCl and 5 mM PIPES) on ice for 10 min. Nuclei were collected by centrifugation at 500 g for 5 min at 4°C. Then, 800 μl of nuclear lysis buffer (1% SDS, 25 mM Tris–HCl pH 8.0 and 5 mM EDTA) was added and incubated on ice for another 10 min, followed by an incubation with 20 μl of proteinase K (Tritirachium album, ≥ 800 units/ml, Sigma‐Aldrich) for 5 h to overnight. Genomic DNA was isolated from the cell lysate by isopropanol and washed with 70% ethanol.
For RNase H control, 50 μg DNA was treated with 5 μl recombinant RNase H (5 U/μl, New England Biolabs, Ipswich, MA, USA) in a total volume of 100 μl for 5 h or overnight. After the treatment, DNA was diluted with 300 μl of IP dilution buffer (0.001% SDS, 1.1% Triton X‐100, 0.12 mM EDTA, 16 mM Tris–HCl pH 8.0, 160 mM NaCl and protease inhibitor cocktail). DNA was sonicated for 15 min (30 s on, 30 s off) with high power setting by the Diagenode Bioruptor. Sonicated DNA was incubated with either 10 μg of S9.6 antibody or of normal mouse IgG (sc‐2025, Santa Cruz) overnight at 4°C. The antibody‐R‐loops complexes were pulled down by incubating with 60 μl Protein A Dynabeads pre‐blocked with 1% BSA for 2 h. The beads were then collected and washed once with buffer A (20 mM Tris–HCl pH 8.0, 2 mM EDTA, 0.1% SDS, 1% Triton X‐100, 165 mM NaCl and protease inhibitor cocktail), buffer B (20 mM Tris–HCl pH 8.0, 2 mM EDTA, 0.1% Triton X‐100, 0.55 M NaCl and protease inhibitor cocktail) and buffer C (100 mM Tris–HCl pH 8.0, 1 mM EDTA, 1% NP‐40, 0.01% sodium deoxycholate, 250 mM LiCl and protease inhibitor cocktail). Each wash was done for 5 min at 4°C. After that, the beads were washed twice with buffer D (100 mM Tris–HCl pH 8.0, 1 mM EDTA and protease inhibitor cocktail) at 4°C and once at room temperature. Captured DNA was eluted twice in 250 μl elution buffer (1% SDS and 0.1 M NaHCO3). Eluted DNA, together with the inputs, was purified using a QIAGEN PCR purification kit.
Flow cytometry
Cells were harvested and washed once in PBS. They were then resuspended in 1× Annexin V binding buffer containing Alexa‐Fluor 647‐conjugated Annexin V (1:100) (A23204) and Hoechst 33342 (1:5,000) (H3570, Invitrogen) analysed on the CyAn Advanced Digital Processing (ADP) High‐Performance Flow Cytometer (Beckman Coulter, High Wycombe, UK). Cells that are double‐negative for Annexin V and Hoechst were analysed for GFP expression.
Statistical analysis
Student's t‐test was used to test for significance of the difference in the graphs, except where stated otherwise. A two‐sample equal variance with normal distribution was used. Except where stated, paired, two‐tailed t‐test was used. The difference is considered significant if the P‐value is < 0.05. Graphs and error bars represent the mean values ± SEM, unless otherwise stated. The number of independent experiments for each data bar is indicated in the figure legends.
Data availability
Gene Expression Omnibus GSE22162 3.
Gene Expression Omnibus GSE27843 42.
Author contributions
DTTN helped design the experiments and generated most of the data and co‐wrote the manuscript; HPJV carried out the ATRX Chip in mouse tissues and analysed the ChIP and RNA‐seq data; BX carried out the probing for the slot blot experiments; CS prepared the shATRX lentivirus; CB isolated the telomeric DNA; HA and CAF prepared experimental material; JK helped with validating the ATRX ChIP‐seq data; JAS and JAS‐S carried the animal work; SB assisted with cell culture; DC advised on the conduct of the experiments; NEG assisted with the bioinformatic analysis; TJ provided genomewide H3K9me3 profiles across repeat sequences in wt and Suv39 h dn MEFs; DRH advised on the design of the experiments and the interpretation of the data and co‐wrote the manuscript; RJG devised the project, helped in the design of the experiments and the interpretation of the data and co‐wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Acknowledgements
The pcDNA5/FRT/GFP GAA plasmid was a kind gift from Dr. Marek Napierala, University of Alabama at Birmingham. Konstantina Skourti‐Stathaki, Nick Proudfoot and Natalia Gromak advised over the R‐loop assays. The Computational Biology Research Group, WIMM helped with the bioinformatics. The H3K9me3 repeat profiles were provided by Inti de la Rosa‐Velazquez with the help of Megumi Onishi‐Seebacher. The cell sorting and analysis were performed in the WIMM FACS facility with help from Ghadeer Almuhaini. The facility is supported by the MRC HIU; MRC MHU (MC_UU_12009); NIHR Oxford BRC and John Fell Fund (131/030 and 101/517), the EPA fund (CF182 and CF170) and by the WIMM Strategic Alliance awards G0902418 and MC_UU_12025. DN was funded by the Marie Curie FP7 ITN “DISCHROM” fellowship. This work was supported by the Medical Research Council [grant number MC_UU_12025/unit programme MC_UU_12009/3].
EMBO Reports (2017) 18: 914–928
References
- 1. McDowell TL, Gibbons RJ, Sutherland H, O'Rourke DM, Bickmore WA, Pombo A, Turley H, Gatter K, Picketts DJ, Buckle VJ et al (1999) Localization of a putative transcriptional regulator (ATRX) at pericentromeric heterochromatin and the short arms of acrocentric chromosomes. Proc Natl Acad Sci USA 96: 13983–13988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gibbons RJ, McDowell TL, Raman S, O'Rourke DM, Garrick D, Ayyub H, Higgs DR (2000) Mutations in ATRX, encoding a SWI/SNF‐like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet 24: 368–371 [DOI] [PubMed] [Google Scholar]
- 3. Law MJ, Lower KM, Voon HP, Hughes JR, Garrick D, Viprakasit V, Mitson M, De Gobbi M, Marra M, Morris A et al (2010) ATR‐X syndrome protein targets tandem repeats and influences allele‐specific expression in a size‐dependent manner. Cell 143: 367–378 [DOI] [PubMed] [Google Scholar]
- 4. Elsasser SJ, Noh KM, Diaz N, Allis CD, Banaszynski LA (2015) Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522: 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Valle‐Garcia D, Qadeer ZA, McHugh DS, Ghiraldini FG, Chowdhury AH, Hasson D, Dyer MA, Recillas‐Targa F, Bernstein E (2016) ATRX binds to atypical chromatin domains at the 3’ exons of zinc finger genes to preserve H3K9me3 enrichment. Epigenetics 11: 398–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dhayalan A, Tamas R, Bock I, Tattermusch A, Dimitrova E, Kudithipudi S, Ragozin S, Jeltsch A (2011) The ATRX‐ADD domain binds to H3 tail peptides and reads the combined methylation state of K4 and K9. Hum Mol Genet 20: 2195–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eustermann S, Yang JC, Law MJ, Amos R, Chapman LM, Jelinska C, Garrick D, Clynes D, Gibbons RJ, Rhodes D et al (2011) Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat Struct Mol Biol 18: 777–782 [DOI] [PubMed] [Google Scholar]
- 8. Iwase S, Xiang B, Ghosh S, Ren T, Lewis PW, Cochrane JC, Allis CD, Picketts DJ, Patel DJ, Li H et al (2011) ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental‐retardation syndrome. Nat Struct Mol Biol 18: 769–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nan X, Hou J, Maclean A, Nasir J, Lafuente MJ, Shu X, Kriaucionis S, Bird A (2007) Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc Natl Acad Sci USA 104: 2709–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wong LH, McGhie JD, Sim M, Anderson MA, Ahn S, Hannan RD, George AJ, Morgan KA, Mann JR, Choo KH (2010) ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res 20: 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huh MS, Price O'Dea T, Ouazia D, McKay BC, Parise G, Parks RJ, Rudnicki MA, Picketts DJ (2012) Compromised genomic integrity impedes muscle growth after Atrx inactivation. J Clin Invest 122: 4412–4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Watson LA, Solomon LA, Li JR, Jiang Y, Edwards M, Shin‐ya K, Beier F, Berube NG (2013) Atrx deficiency induces telomere dysfunction, endocrine defects, and reduced life span. J Clin Invest 123: 2049–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leung JW, Ghosal G, Wang W, Shen X, Wang J, Li L, Chen J (2013) Alpha thalassemia/mental retardation syndrome X‐linked gene product ATRX is required for proper replication restart and cellular resistance to replication stress. J Biol Chem 288: 6342–6350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clynes D, Jelinska C, Xella B, Ayyub H, Taylor S, Mitson M, Bachrati CZ, Higgs DR, Gibbons RJ (2014) ATRX dysfunction induces replication defects in primary mouse cells. PLoS One 9: e92915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ritchie K, Seah C, Moulin J, Isaac C, Dick F, Berube NG (2008) Loss of ATRX leads to chromosome cohesion and congression defects. J Cell Biol 180: 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De La Fuente R, Viveiros MM, Wigglesworth K, Eppig JJ (2004) ATRX, a member of the SNF2 family of helicase/ATPases, is required for chromosome alignment and meiotic spindle organization in metaphase II stage mouse oocytes. Dev Biol 272: 1–14 [DOI] [PubMed] [Google Scholar]
- 17. Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S et al (2011) Altered telomeres in tumors with ATRX and DAXX mutations. Science 333: 425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, De S, Petrini JH, Sung PA, Jasin M et al (2012) Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet 8: e1002772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482: 226–231 [DOI] [PubMed] [Google Scholar]
- 20. Bryan TM, Englezou A, Dalla‐Pozza L, Dunham MA, Reddel RR (1997) Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor‐derived cell lines. Nat Med 3: 1271–1274 [DOI] [PubMed] [Google Scholar]
- 21. Dunham MA, Neumann AA, Fasching CL, Reddel RR (2000) Telomere maintenance by recombination in human cells. Nat Genet 26: 447–450 [DOI] [PubMed] [Google Scholar]
- 22. Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, Taylor S, Higgs DR, Gibbons RJ (2015) Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat Commun 6: 7538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Napier CE, Huschtscha LI, Harvey A, Bower K, Noble JR, Hendrickson EA, Reddel RR (2015) ATRX represses alternative lengthening of telomeres. Oncotarget 6: 16543–16558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aguilera A, Garcia‐Muse T (2012) R loops: from transcription byproducts to threats to genome stability. Mol Cell 46: 115–124 [DOI] [PubMed] [Google Scholar]
- 25. Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N (2004) Intracellular transcription of G‐rich DNAs induces formation of G‐loops, novel structures containing G4 DNA. Genes Dev 18: 1618–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, Niedzwiedz W (2015) The fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol Cell 60: 351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arora R, Lee Y, Wischnewski H, Brun CM, Schwarz T, Azzalin CM (2014) RNaseH1 regulates TERRA‐telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat Commun 5: 5220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aalfs JD, Narlikar GJ, Kingston RE (2001) Functional differences between the human ATP‐dependent nucleosome remodeling proteins BRG1 and SNF2H. J Biol Chem 276: 34270–34278 [DOI] [PubMed] [Google Scholar]
- 29. Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument‐Bromage H, Tempst P, Lin SP, Allis CD et al (2007) DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448: 714–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schneiderman JI, Orsi GA, Hughes KT, Loppin B, Ahmad K (2012) Nucleosome‐depleted chromatin gaps recruit assembly factors for the H3.3 histone variant. Proc Natl Acad Sci USA 109: 19721–19726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reddy K, Tam M, Bowater RP, Barber M, Tomlinson M, Nichol Edamura K, Wang YH, Pearson CE (2011) Determinants of R‐loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res 39: 1749–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ratmeyer L, Vinayak R, Zhong YY, Zon G, Wilson WD (1994) Sequence specific thermodynamic and structural properties for DNA.RNA duplexes. Biochemistry 33: 5298–5304 [DOI] [PubMed] [Google Scholar]
- 33. El Hage A, French SL, Beyer AL, Tollervey D (2010) Loss of Topoisomerase I leads to R‐loop‐mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev 24: 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marinello J, Chillemi G, Bueno S, Manzo SG, Capranico G (2013) Antisense transcripts enhanced by camptothecin at divergent CpG‐island promoters associated with bursts of topoisomerase I‐DNA cleavage complex and R‐loop formation. Nucleic Acids Res 41: 10110–10123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marinello J, Bertoncini S, Aloisi I, Cristini A, Malagoli Tagliazucchi G, Forcato M, Sordet O, Capranico G (2016) Dynamic effects of topoisomerase I inhibition on R‐loops and short transcripts at active promoters. PLoS One 11: e0147053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yeo AJ, Becherel OJ, Luff JE, Cullen JK, Wongsurawat T, Jenjaroenpun P, Kuznetsov VA, McKinnon PJ, Lavin MF (2014) R‐loops in proliferating cells but not in the brain: implications for AOA2 and other autosomal recessive ataxias. PLoS One 9: e90219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J (2007) Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318: 798–801 [DOI] [PubMed] [Google Scholar]
- 38. Arnoult N, Van Beneden A, Decottignies A (2012) Telomere length regulates TERRA levels through increased trimethylation of telomeric H3K9 and HP1alpha. Nat Struct Mol Biol 19: 948–956 [DOI] [PubMed] [Google Scholar]
- 39. Sarkies P, Reams C, Simpson LJ, Sale JE (2010) Epigenetic instability due to defective replication of structured DNA. Mol Cell 40: 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sarkies P, Murat P, Phillips LG, Patel KJ, Balasubramanian S, Sale JE (2012) FANCJ coordinates two pathways that maintain epigenetic stability at G‐quadruplex DNA. Nucleic Acids Res 40: 1485–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lukashchuk V, McFarlane S, Everett RD, Preston CM (2008) Human cytomegalovirus protein pp71 displaces the chromatin‐associated factor ATRX from nuclear domain 10 at early stages of infection. J Virol 82: 12543–12554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lienert F, Mohn F, Tiwari VK, Baubec T, Roloff TC, Gaidatzis D, Stadler MB, Schübeler D (2011) Genomic prevalence of heterochromatic H3K9me2 and transcription do not discriminate pluripotent from terminally differentiated cells. PLoS Genet 6: e1002090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Flygare J, Rayon Estrada V, Shin C, Gupta S, Lodish HF (2011) HIF1alpha synergizes with glucocorticoids to promote BFU‐E progenitor self‐renewal. Blood 117: 3435–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bulut‐Karslioglu A, De La Rosa‐Velázquez IA, Ramirez F, Barenboim M, Onishi‐Seebacher M, Arand J, Galán C, Winter GE, Engist B, Gerle B et al (2014) Suv39 h‐dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol Cell 55: 277–290 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Data Availability Statement
Gene Expression Omnibus GSE22162 3.
Gene Expression Omnibus GSE27843 42.