

Abstract
Base excision repair (BER) is one of the most frequently used cellular DNA repair mechanisms and modulates many human pathophysiological conditions related to DNA damage. Through live cell and in vitro reconstitution experiments, we have discovered a major sub‐pathway of conventional long‐patch BER that involves formation of a 9‐nucleotide gap 5′ to the lesion. This new sub‐pathway is mediated by RECQ1 DNA helicase and ERCC1‐XPF endonuclease in cooperation with PARP1 poly(ADP‐ribose) polymerase and RPA. The novel gap formation step is employed during repair of a variety of DNA lesions, including oxidative and alkylation damage. Moreover, RECQ1 regulates PARP1 auto‐(ADP‐ribosyl)ation and the choice between long‐patch and single‐nucleotide BER, thereby modulating cellular sensitivity to DNA damage. Based on these results, we propose a revised model of long‐patch BER and a new key regulation point for pathway choice in BER.
Keywords: BER pathway switch, oxidative damage, PARP inhibition, RECQ1, XPF‐ERCC1
Subject Categories: DNA Replication, Repair & Recombination
Introduction
The base excision repair (BER) pathway is likely the most frequently used DNA repair mechanism in the cell (Zharkov 2008; Wallace, 2014). Many spontaneous chemical reactions within the cell, including base hydrolysis, oxidation, and alkylation, can result in base alterations that require repair by the BER pathway in order to ensure faithful copying of the genome (Lindahl, 1993). As the cell's major defense against damage, BER modulates a variety of human conditions related to DNA damage, including carcinogenesis, resistance to chemotherapy, neurodegeneration, and aging (Jackson & Bartek, 2009).
BER involves many enzymes and can occur via different sub‐pathways that differ in repair patch size, transaction mechanism, and requisite proteins (Hazra et al, 2007; Wallace, 2014). BER is initiated by DNA adduct‐specific monofunctional and bifunctional DNA glycosylases that excise the damaged base by hydrolytic cleavage of the N‐glycosidic bond between the base and the sugar (Zharkov & Grollman, 2005). Monofunctional DNA glycosylase activity leads to the formation of an abasic (AP) site, which is subsequently cleaved on the 5′ side by apurinic/apyrimidinic endonuclease 1 (APE1), leaving a 3′‐OH and a 5′‐deoxyribose phosphate (dRP; Wilson & Barsky, 2001). Bifunctional DNA glycosylases have a glycosylase‐associated AP lyase activity which cleaves the resulting AP site on the 3′ side, leaving a 3′‐α,β unsaturated aldehyde (ring‐opened sugar) or a 3′‐phosphate (Wallace, 2013). The ring‐opened sugar is cleaved by APE1 while the phosphate is cleaved by the phosphatase activity of polynucleotide kinase (PNK), both yielding a 3′‐OH (Karimi‐Busheri et al, 1998). After base excision and AP‐site incision, poly(ADP‐ribose) polymerase 1 (PARP1) may play an enzymatic role at the resulting 1‐nucleotide (nt) gap, possibly by inducing a relaxed chromatin structure and allowing access of repair enzymes to the DNA and/or by coordinating the actions of downstream BER events (Dantzer et al, 1999; Prasad et al, 2001; Le Page et al, 2003). While intuitively it seems that PARP1, as a nick sensor, should be important for the repair of single‐strand breaks (SSBs), it is unclear why these intermediates generated during BER require a sensor as it has been shown that BER enzymes interact with each other to coordinate the reaction cascade and avoid the exposure of unstable intermediates (Wilson & Kunkel, 2000). Furthermore, some studies have demonstrated that BER can occur in the absence of PARP1 (Allinson et al, 2003). Therefore, the precise mechanistic role of PARP1 in BER in cells is not fully understood (De Vos et al, 2012).
The remaining steps of BER are described as either the single‐nucleotide BER (SN‐BER) or long‐patch BER (LP‐BER) sub‐pathways. In the SN‐BER pathway, DNA polymerase β (POLβ) exerts its dRP lyase activity to cleave the 5′‐dRP created after AP‐site incision by APE1 (Matsumoto & Kim, 1995). POLβ subsequently incorporates 1 nt, and the nick is sealed by DNA ligase III (LIGIII). In the LP‐BER pathway, the replicative DNA polymerases δ and ε (POLδ, POLε), in combination with the other replication accessory factors, proliferating cell nuclear antigen (PCNA), and replication factor C (RFC), incorporate nucleotides at the single‐nucleotide gap and continue to incorporate 2 to 10 nts by strand displacement on the 3′ side of the original lesion site (Klungland & Lindahl, 1997; Pascucci et al, 1999). POLβ has also been implicated in LP‐BER as it has been shown to initiate DNA synthesis with 1‐nt incorporation after which POLδ and POLε proceed with longer patch formation (Dianov et al, 1999). The flap created by strand displacement by these polymerases is cleaved by flap endonuclease 1 (FEN1), and the resulting nick is sealed by DNA ligase I (LIGI). To date, the mechanisms of SN‐BER and LP‐BER have largely been elucidated through in vitro experiments and cell‐free extract‐based assays, and very few studies have detailed BER sub‐pathway mechanisms in live cells.
We report here the discovery of a novel sub‐pathway of LP‐BER, which we have elucidated using plasmid‐based and genomic DNA‐based in‐cell repair assays and in vitro reconstitution experiments. In this sub‐pathway, a 9‐nt gap is formed 5′ to the lesion site by the combined action of the RecQ family DNA helicase RECQ1 (also known as RECQL or RECQL1) and the endonuclease ERCC1‐XPF in cooperation with PARP1 and replication protein A (RPA). Formation of this gap is critical for rapid repair of a variety of BER substrates, including 8‐oxoguanine (8‐oxoG), 5‐hydroxyuracil (5‐OHU), 1, N6‐ethenoadenine (εA), and methylated bases, whose repair is initiated by a wide array of monofunctional and bifunctional DNA glycosylases. We found that RECQ1 regulates the autoribosylation status of PARP1, which regulates BER sub‐pathway preference and cellular toxicity to DNA damage. Our studies thus define a new LP‐BER sub‐pathway and a crucial regulation point for pathway choice in BER.
Results
A new sub‐pathway of LP‐BER in live cells involves 5′ patch formation
We utilized a modified version of a plasmid‐based assay developed previously in our laboratory for in‐cell repair of a representative BER substrate DNA adduct, εA (Choudhury et al, 2008; Fig 1A). After transfection of a plasmid (M13mp18) containing a single εA within a restriction site into human cells, restriction digestions detect the presence or absence (complete repair) of the adduct by transformation of the digestion products into E. coli and counting the transformants. Completely repaired DNA will be linearized by restriction digestion and will not form plaques, while unrepaired DNA or products of incomplete BER will be resistant to digestion, remain circular, and form plaques. To monitor the patch size, different plasmids were generated containing C:A mismatches within different restriction sites (inhibiting digestion) at various distances and on both sides (5′ or 3′) of the εA‐containing restriction sites (oligonucleotides used to generate various plasmids shown in Appendix Table S1). Thus, combinations of appropriate restriction enzymes were used for the assay. One enzyme was utilized to probe for repair at the damage site, and one enzyme was utilized to probe for patch formation. Using this assay, the percentage of repair events that occur via 5′ and 3′ patch formation can be quantified. Resolution of the parent lesion (εA) is denoted “εA repair” while resolution of a neighboring mismatch is used to detect patch formation and is denoted “patch formation”. Mismatch resolution only occurs during patch formation in BER (instigated by the parent lesion) and not via the mismatch repair pathway (Appendix Fig S1D). After extensive optimization of the plaque‐based repair patch assay (Appendix Fig S1A–D is representative), a survey experiment in HCT116 cells with various εA/mismatch constructs was performed with mismatches spanning as far as 12 bases 5′ to εA and 13 bases 3′ to εA to determine the extent of repair patch formation. Notably, in‐cell repair of εA over 24 h generated a patch size of 12 bases 3′ to the lesion as well as 7–9 bases 5′ to the lesion (Fig 1B and Appendix Fig S2). These conventional patch size definitions include the original lesion site in the nucleotide count for both sides. Therefore, the total patch size in cells is 18–20 nts.
Figure 1. A new 5′ patch formation‐mediated BER sub‐pathway in living cells.
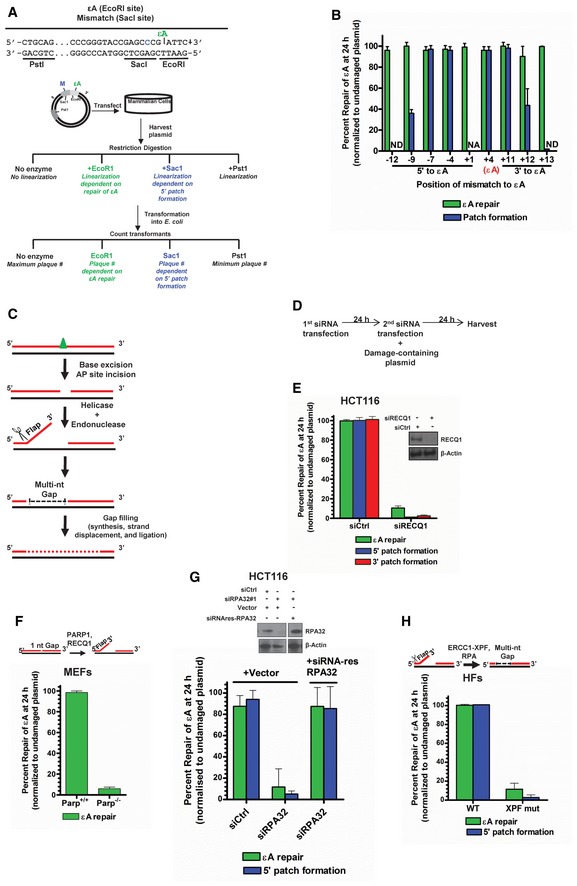
-
AExperimental strategy of the plaque‐based assay for detecting patch formation during BER. Plasmid #4 (see Appendix Table S1) is used as a representative example. Sequence of relevant restriction sites is shown in which green “εA” indicates the position of εA (at EcoRI site), which inhibits EcoRI digestion, and the blue “C” indicates the position of the C:A mismatch (at SacI site), which inhibits SacI digestion. The PstI site is used as a control and is unmodified. The schematic of the plasmid shows the green εA within the EcoRI site and a blue “M” indicating the mismatch within the SacI site. After transfection of the plasmid into cells, the plasmid is harvested for digestion with indicated enzymes and transformation into E. coli. Repair at the modified restriction sites will yield linearized plasmid and no transformants (plaques) while unrepaired restriction sites will be resistant to digestion and will produce transformants.
-
BεA repair patch mapping. Plasmids used are listed in Appendix Table S1. NA indicates “not applicable”, and ND indicates “repair and/or 5′ patch formation not detectable”.
-
CSchematic representation of BER 5′ patch formation hypothesis.
-
DExperimental timeline for plaque‐based repair assay in the presence of gene‐specific siRNA.
-
EεA repair, 5′ patch formation, and 3′ patch formation were monitored by plaque assay in control and RECQ1 KD HCT116 cells using plasmids #4 (to monitor 5′ patch formation) and #2 (to monitor 3′ patch formation). Inset shows Western blot analysis of control and RECQ1 KD. Inset shows Western blot analysis of control and RECQ1 KD cells.
-
F–HεA repair was monitored by plaque assay using plasmid #4 in (F) Parp +/+ and Parp −/− MEFs, (G) control and RPA KD HCT116 cells complemented with empty vector or siRNA‐resistant RPA32 (siRNA res‐RPA32), and (H) wild‐type (WT) and XPF mutant (XPF mut) HFs. Schematics above (F) and (H) depict the proposed action of indicated proteins in BER. Inset in (G) shows Western blot analysis of control and RPA KD cells complemented with empty vector or siRNA res‐RPA32. Space between samples indicates where samples were separated by gel lanes.
While a 3′ patch of 12 nucleotides could indicate LP‐BER as the mechanism for εA repair and is consistent with the patch size reported for traditional LP‐BER by others (Sattler et al, 2003), patch formation on the 5′ side of the lesion site is newly revealed in this study and may indicate NER activity. However, our previous studies demonstrated that εA repair did not occur via the NER pathway (Choudhury et al, 2008). To dissect the repair machineries involved in the repair process, we probed for the roles of bona fide BER proteins, including MPG, APE1, POLβ, replicative polymerases (POLδ/ε), and FEN1 in repair of εA. Additionally, using XPA‐deficient human fibroblasts, we probed for the role of XPA, a critical NER protein, in εA repair to confirm our previously published result. MPG and POLβ were tested using knockout mouse embryonic fibroblasts (MEFs), and APE1 and FEN1 were tested following knockdown (KD) using gene‐specific siRNA. The role of replicative polymerases was determined by treating the cells with aphidicolin, a replicative polymerase inhibitor (Weiser et al, 1991). Repair of εA and 5′ patch formation were dependent on MPG (Fig EV1A), APE1 (Fig EV1B), FEN1 (Fig EV1B), and replicative polymerases (Fig EV1C), but were independent of POLβ (Fig EV1A) and XPA (Fig EV1D), indicating that the repair mechanism we observed was LP‐BER (Sobol et al, 1996; Klungland & Lindahl, 1997; Matsumoto et al, 1999). This finding was consistent with BER studies for other adducts in live cells, which generally report LP‐BER preference for repair (Sattler et al, 2003; Masaoka et al, 2009). Moreover, our finding that POLβ is not the preferred polymerase for repair under basal conditions is also consistent with another BER study using similar assay conditions (Masaoka et al, 2009). However, the discovery of patch formation 5′ to the lesion and a larger total patch size (~20 nucleotides) compared to patch size of 2–12 nucleotides reported for traditional LP‐BER suggested a new LP‐BER sub‐pathway mediated by 5′ patch formation. Therefore, we sought to further elucidate this new LP‐BER mechanism.
Figure EV1. Newly identified 5′ patch formation step occurs in the LP‐BER sub‐pathway.
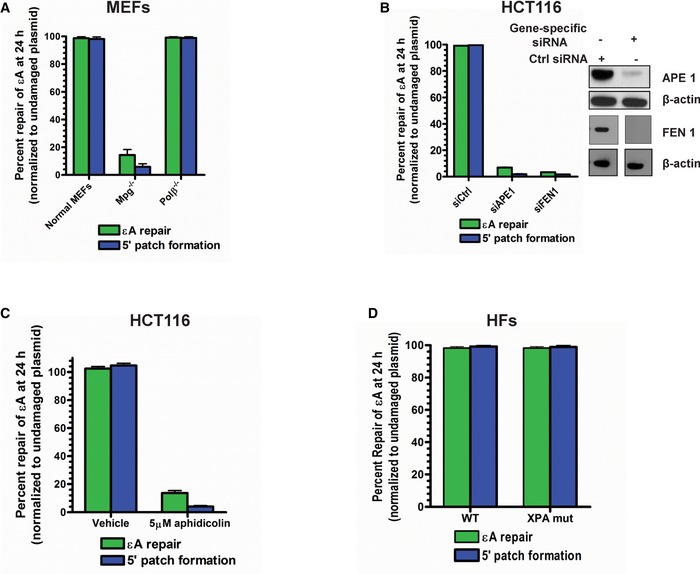
-
A–DεA repair and 5′ patch formation in plasmid #4 (Appendix Table S1) were monitored 24 h post‐plasmid transfection under various conditions: (A) wild‐type, Mpg −/−, and Polb −/− MEFs, (B) control, APE1 KD, and FEN1 KD HCT116 cells, (C) under vehicle or aphidicolin treatment in HCT116 cells, and (D) wild‐type and XPA mutant HFs. For KD experiments, control or gene‐specific (APE1 or FEN1) siRNAs were transfected into HCT116 cells and repair was analyzed in the same manner as described in Fig 1D. APE1 and FEN1 expression was analyzed by Western blot, which is shown in the inset in (B). Space between samples indicates where samples were separated by gel lanes.
RECQ1, PARP1, RPA, and ERCC1‐XPF are necessary for 5′ patch formation and BER
As 5′ patch formation has not previously been associated with BER and is unlikely to be accomplished by currently known BER proteins, we hypothesized the involvement of a 3′–5′ helicase at the incised AP site and an associated 3′ flap endonuclease (Fig 1C). Among the candidate 3′–5′ DNA helicases, the RecQ family helicases are known to be involved in many ways in DNA replication and genome maintenance (Hickson, 2003; Croteau et al, 2014). To date, little information is known about the functions of the most abundant RecQ family helicase, RECQ1, but it has been shown to be involved in non‐homologous end‐joining (NHEJ; Parvathaneni et al, 2013) and restoring regressed replication forks (Berti et al, 2013) and has been implicated in homologous recombination (HR; Sharma & Brosh, 2008). Additionally, we had demonstrated the involvement of RECQ1 in DNA damage response to oxidative DNA damage in cells (Sharma & Brosh, 2007; Sharma et al, 2012), and others had shown RECQ1 to have a role in resistance to alkylating agents (Mendoza‐Maldonado et al, 2011; Agnihotri et al, 2014). We reasoned that RECQ1 as a 3′–5′ DNA helicase could play a role in the proposed model of 5′ gap formation during BER (Fig 1C). To probe for the role of RECQ1 in BER, we used RECQ1‐specific siRNA in the plaque‐based repair assay (Fig 1D). Indeed, in RECQ1 KD HCT116 cells, only ≈10% of εA was repaired in 24 h, and both 5′ and 3′ patch formations were abolished (Fig 1E), indicating that RECQ1 is required for total repair and patch synthesis during 5′ gap‐mediated LP‐BER. We confirmed the critical role of RECQ1 in BER by testing repair in RECQ1 KD HeLa cells (Fig EV2A). Importantly, RECQ1 siRNA did not affect the expression of other RecQ family helicases, indicating that RECQ1 is specifically needed for 5′ patch formation since other available RecQ helicases could not rescue the 5′ patch formation in the absence of RECQ1 (Fig EV2B).
Figure EV2. RECQ1 is required for 5′ and 3′ patch formation during BER in human cells.
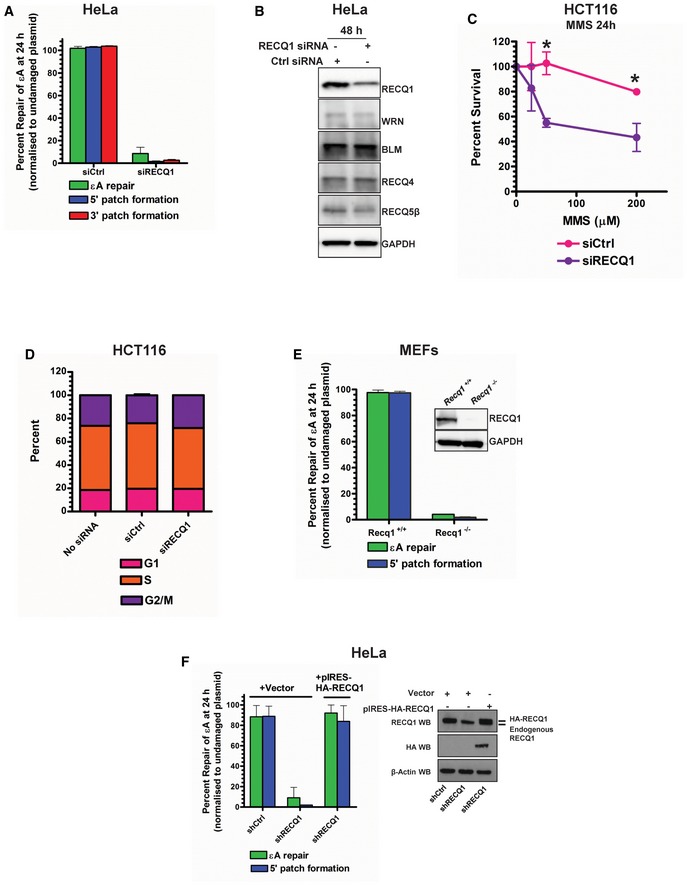
-
AεA repair, 5′ patch formation, and 3′ patch formation were monitored by plaque assay 24 h post‐plasmid transfection (plasmids #2 and #4 for monitoring the 3′ and 5′ patch formation, respectively) in control and RECQ1 KD HeLa cells in the same manner as described in Fig 1D.
-
BWestern blot analysis of RECQ family helicases in HeLa cells 48 h post‐RECQ1 siRNA transfection.
-
CSurvival of control and RECQ1 KD HCT116 cells treated for 24 h with indicated doses of MMS was measured by cell counting. Asterisk (*) denotes P‐value of Student's t‐test < 0.05.
-
DCell cycle distribution was monitored in control and RECQ1 KD HCT116 cells 24 h post‐siRNA transfection by DNA content analysis (propidium iodide staining) using flow cytometry.
-
E, FεA repair and 5′ patch formation were monitored 24 h post‐plasmid transfection (plasmid #4) by plaque assay in (E) Recq1 +/+ and Recq1 −/− MEFs and (F) stable control and RECQ1 KD cells complemented with empty vector or shRNA‐resistant RECQ1. Insets show Western blot for RECQ1 in MEFs (E) and RECQ1 stable KD HeLa cells (F).
Consistent with our findings and other reports, RECQ1 KD HCT116 cells were sensitized to methylmethane sulfonate (MMS), an alkylating agent that produces adducts such as 3‐methyladenine and 7‐methylguanine, which are substrates of MPG‐mediated BER (Beranek, 1990; Fig EV2C). Since some evidence has shown that cell cycle phase may affect BER sub‐pathway selection (Mjelle et al, 2015), and stable RECQ1 KD has been shown to affect cell cycle progression in some cell lines (Thangavel et al, 2010; Popuri et al, 2012), we also monitored cell cycle phase distribution and found that transient knockdown of RECQ1 did not significantly affect the cell cycle after 24 h (Fig EV2D). The role of RECQ1 in BER 5′ patch formation was further confirmed in RECQ1‐null MEFs (Fig EV2E). We finally confirmed a specific role for RECQ1 in BER by utilizing a previously published complementing system in which HeLa cells stably expressing RECQ1 shRNA were complemented with a shRNA‐resistant HA‐tagged RECQ1 expression plasmid (pIRES‐HA‐RECQ1; Berti et al, 2013). We tested εA repair and 5′ patch formation in this experimental system and found that complementation with shRNA‐resistant RECQ1 completely restored εA repair and the associated 5′ patch formation (Fig EV2F), confirming that RECQ1 had a specific and critical role in 5′ patch‐mediated LP‐BER.
While it is true that our results and the reports of others showed that LP‐BER is the predominant BER sub‐pathway in cells, it has been suggested that LP‐BER may be the preferred pathway during S‐phase (when replicative enzymes are most abundant; Akbari et al, 2009) or in proliferating cells in general (Mjelle et al, 2015). It was not clear whether or not the RECQ1‐mediated LP‐BER sub‐pathway we had identified occurred only in proliferating cells, since our experiments had utilized proliferating cells exclusively. Therefore, we investigated the mechanism of repair in stable RECQ1 KD HeLa cells that were arrested in G1 phase after 33 h of 20 μM lovastatin treatment (Appendix Fig S3A). Lovastatin is a chemical inhibitor of HMG‐CoA reductase, which is required for mevalonate synthesis, and inhibition of this enzyme leads to G1 arrest in multiple cell types (Keyomarsi et al, 1991). Interestingly, RECQ1‐mediated LP‐BER with 5′ patch formation occurred in both vehicle‐treated and lovastatin‐treated cells, indicating that the LP‐BER sub‐pathway we discovered was occurring in both proliferating and non‐dividing or G1‐arrested cells (Appendix Fig S3B).
PARP1 is a multifunctional enzyme that interacts with RECQ1 (Sharma et al, 2012; Berti et al, 2013) and plays an important but not well‐understood role in BER (De Vos et al, 2012). Using PARP1 null MEFs, we tested for PARP1's involvement in BER of εA and found that PARP1 was critical for repair (Fig 1F). Another well‐known interaction partner and stimulator of RECQ1 is RPA (Cui et al, 2003), which has also been shown to stimulate LP‐BER (DeMott et al, 1998). Moreover, RFA1 (yeast RPA homolog)‐deficient yeast demonstrated increased sensitivity to MMS treatment (Umezu et al, 1998). Therefore, we reasoned that RPA may be involved in the 5′ patch formation during LP‐BER. Indeed, by siRNA KD of RPA32 (siRPA32 #1), which results in destruction of the entire RPA complex (Grudic et al, 2007), we found that RPA was essential for BER of εA, including 5′ patch formation in HCT116 (Fig 1G) and HeLa cells (Fig EV3A). We further confirmed this result by complementing RPA KD HCT116 cells with a siRNA‐resistant RPA expression plasmid (Fig 1G), which restored εA repair and 5′ patch formation. Moreover, similar to the results of the RPA KD with siRPA32 #1, a second siRNA against RPA32 (siRPA32 #2) used in HCT116 cells eliminated both repair and 5′ patch formation (Fig EV3B).
Figure EV3. RPA and XPF are required for 5′ patch formation during BER in human cells.
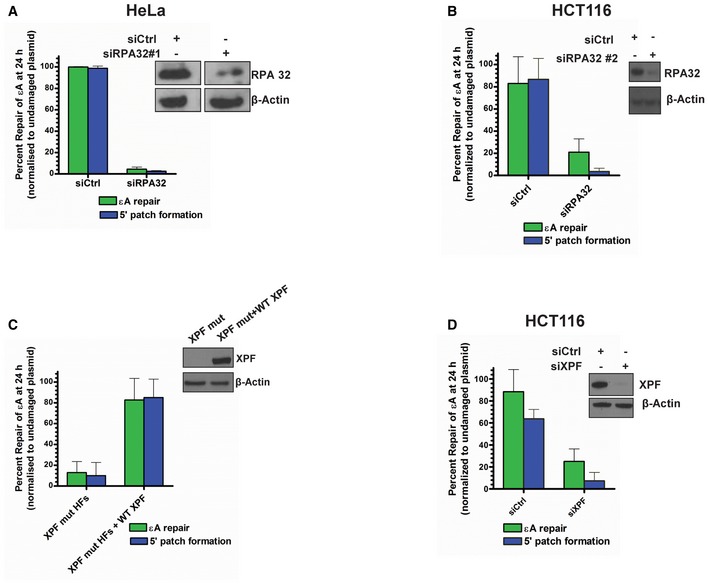
-
A–DεA repair and 5′ patch formation were monitored by plaque assay 24 h post‐plasmid transfection (plasmid #4) in the same manner as described in Fig 1D in (A) control and RPA KD HeLa cells (using RPA siRNA #1), (B) control and RPA KD HCT116 cells (using RPA siRNA #2), (C) parental XPF mutant HFs and XPF mutant HFs lentivirally transduced with WT XPF, and (D) control and XPF KD HCT116 cells. Insets in panels show Western blots for RPA and XPF, as indicated. Space between samples indicates where samples were separated by gel lanes, which is shown in the inset in (A).
Next, we reasoned that the ERCC1‐XPF heterodimer, first identified as the endonuclease catalyzing the 5′ incision during NER (Sijbers et al, 1996), could incise the DNA on the 5′ side of the lesion. We hypothesized that ERCC1‐XPF might function as a 3′ flap endonuclease in LP‐BER once RECQ1 unwinds the helix post‐AP‐site incision (Fig 1C). Indeed, ERCC1‐XPF has roles in interstrand cross‐link (ICL) repair and double‐strand break (DSB) repair (Niedernhofer et al, 2004; Ahmad et al, 2008; Bhagwat et al, 2009) and, importantly, has been implicated in the repair of oxidative damage, particularly when 3′ blocked ends are involved (Fisher et al, 2011). In fact, XPF‐deficient Chinese hamster ovary (CHO) cells demonstrated dramatic sensitivity to H2O2 compared to wild‐type CHO cells (Fisher et al, 2011). Furthermore, during NER ERCC1‐XPF is positioned by RPA (de Laat et al, 1998), which we had already shown to be necessary for repair (Fig 1G), and it seemed reasonable that RPA may be able to position ERCC1‐XPF at a similar DNA structure during BER. By testing εA repair and 5′ patch formation in ERCC1‐XPF‐deficient (XPF mutant) human fibroblasts (HFs), we found ERCC1‐XPF was essential for BER in our assay, including 5′ patch formation (Fig 1H). Repair of εA and 5′ patch formation were abrogated in XPF mutant HFs but were restored in XPF mutant HFs lentivirally transduced with a wild‐type XPF‐expressing viral expression construct (Staresincic et al, 2009; Fig EV3C). We further confirmed that XPF was playing a role in repair of εA using siRNA against XPF in HCT116 cells. Again, KD of XPF resulted in abrogation of both repair of εA and 5′ patch formation, confirming that XPF was indeed playing a role in 5′ patch‐mediated LP‐BER (Fig EV3D). Since it was possible that another endonuclease, Mus81, with similar substrate specificity could conceivably fulfill the role of ERCC1‐XPF in BER 5′ patch formation, we measured Mus81 mRNA expression in cell lines tested for repair, including wild‐type and XPF mutant fibroblasts and HCT116 cells (Appendix Fig S4). We found that the Mus81 expression levels in XPF mutant cells, which were BER‐deficient (Fig 1H), are similar to HCT116 cells, which are BER‐proficient (Fig 1B). Thus, Mus81 is likely not needed for 5′ patch formation since expression of Mus81 could not rescue the 5′ patch formation in the XPF mutant HFs.
5′ repair patch in BER is created by the formation of a 5′ gap
The mechanism of the 5′ patch formation was subsequently studied through a series of biochemical assays using cell extracts and purified proteins. Our proposed model (Fig 1C) and the newly identified members of BER, including RECQ1, ERCC1‐XPF, and RPA, suggested the formation of a 5′ gap. Utilizing a strategy previously used for mismatch repair (MMR) studies (Zhang et al, 2005), gap formation on the 5′ side of the original lesion site should therefore result in loss of dsDNA structure at restriction sites 5′ to the lesion, rendering them resistant to restriction enzyme digestion, which can be subsequently detected by resolution on an agarose gel. To investigate the product of BER enzymes prior to polymerase action, either (a) aphidicolin‐treated (replicative polymerase‐inhibited) HCT116 nuclear cell extracts or (b) purified proteins were incubated with the εA‐containing plasmid (#3, Appendix Table S1), in which εA was placed at the EcoRI site, and reaction products were deproteinized then analyzed. The linear M13mp18 plasmid DNA and the expected sizes and results for digestion are depicted in Fig 2A with the sequence shown for the MCS region and BspHI site, which is a distant site used to linearize the plasmid so that sensitivity to MCS region sites could be readily ascertained by gel electrophoresis. Gap formation on the 5′ side of the lesion was observed in reaction products from εA‐containing plasmid DNA but not undamaged plasmid with HCT116 nuclear extracts (Fig 2B, lanes 1–6). Interestingly, 5′ gap formation did not occur in reaction products from εA‐containing plasmid incubated with RECQ1 KD HCT116 nuclear extracts (Fig 2B, compare lanes 4 and 7). These results confirmed our prediction that BER employs a mechanism that includes RECQ1‐dependent formation of a gap on the 5′ side of the lesion. Additionally, we confirmed that RECQ1 was not involved in 3′ patch formation during BER using a similar strategy with εA at the PstI site (plasmid #1, Appendix Table S1; Fig 2B, lanes 10 and 12). This observation is consistent with the proposed mechanism of LP‐BER in which the 3′ patch formation occurs via strand displacement by replicative polymerases (Klungland & Lindahl, 1997).
Figure 2. 5′ gap formation during BER in vitro .
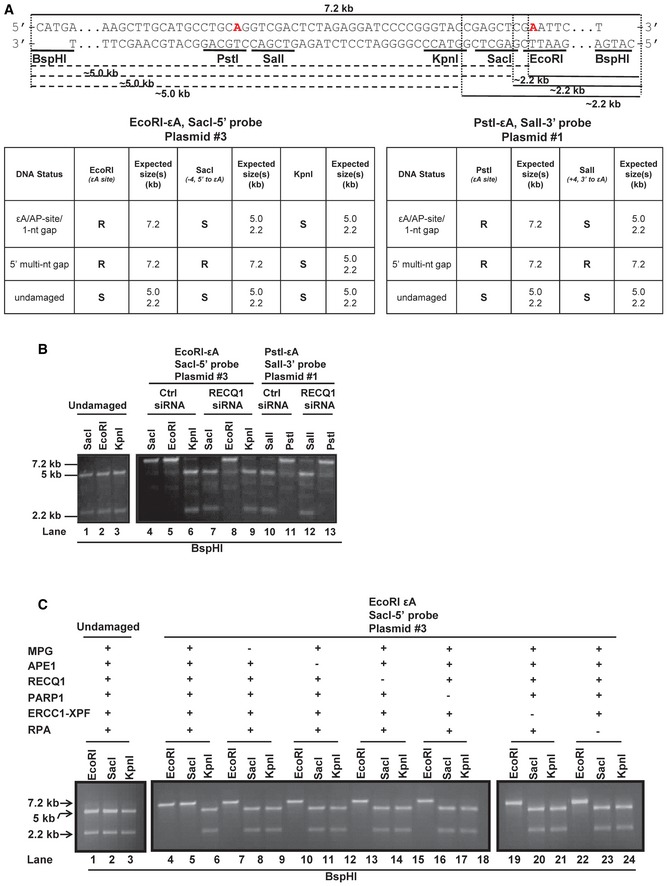
- The MCS region of M13mp18 is shown with the EcoRI (εA), SacI (−4, 5′ to εA), KpnI (−12, 5′ to εA), and BspHI restriction sites noted. The red “A” within the EcoRI site indicates the adenine that is modified to εA for assays utilizing plasmid #3 (see Appendix Table S1). The red “A” within the PstI site indicates the adenine that is modified to εA for assays utilizing plasmid #1 (see Appendix Table S1). After incubation with HCT116 nuclear extracts, aliquots of the reaction products were double digested with EcoRI/BspHI, SacI/BspHI, or KpnI/BspH1. The dotted lines indicate digestion of each enzyme (+ BspHI) and the resulting product sizes, 5 kb and 2.2 kb. Tables indicate the expected digestion results of the assay according to the DNA structure of reaction products. R = resistant, S = sensitive. The details of the plasmids used are found in Appendix Table S1.
- Lanes 1–3, undamaged M13mp18 DNA was incubated with aphidicolin‐treated HCT116 nuclear extracts for 1 h at 37°C. Samples were subsequently aliquoted and digested with EcoRI, SacI, and KpnI in combination with BspHI, and reaction products were resolved on a 1% agarose gel. Lanes 4‐13, M13mp18‐εA DNA was incubated with aphidicolin‐treated control and RECQ1 KD HCT116 nuclear extracts for 1 h at 37°C. Samples were subsequently aliquoted and digested with EcoRI, SacI, and KpnI (lanes 4–9) or PstI and SalI (lanes 10–13) in combination with BspHI, and reaction products were resolved on a 1% agarose gel.
- Undamaged (lanes 1–3) and M13mp18‐εA (lanes 4–24) DNA were incubated with purified proteins MPG, APE1, RECQ1, PARP1, ERCC1‐XPF, and RPA and processed as described in (B). Spaces between lanes indicate where samples were separated by lanes or where two gels were required to accommodate the number of samples.
Source data are available online for this figure.
To identify the enzymes essential to the formation of the 5′ gap, we reconstituted the in vitro gap formation assay with purified proteins, including MPG, APE1, PARP1, RECQ1, ERCC1‐XPF, and RPA, which were identified in the in‐cell repair studies, and the εA plasmid #3 as above (Appendix Table S1). When all six proteins were present, 5′ gap formation was observed (Fig 2C, lanes 4–6), but elimination of any one of the six proteins resulted in abolition of the gap formation, indicating that all six of these proteins are required for 5′ gap formation during repair (Fig 2C, compare lanes 7–24 to 4–6).
The BER 5′ gap is 9 nts in length
To measure the exact size of the 5′ gap, plasmid DNA containing εA at the EcoRI site (plasmid #3) was incubated with purified proteins, and reaction products were subsequently digested with HindIII and NarI, radiolabeled and resolved on a denaturing polyacrylamide gel where the size of the labeled fragments indicated the location of the incision site and thus the size of the 5′ gap that was formed (Fig 3A). Interestingly, reactions containing all six proteins resulted in a 5′ gap that was precisely 9 nt in length (including the original lesion site) (Fig 3B, lane 2, 43‐mer product). Similar to the results of the in vitro gap formation assay (Fig 2C), the 9‐nt gap was not observed when any one of the six proteins was omitted (Fig 3B, lanes 3–7). However, individual omission of RECQ1, PARP1, RPA, or ERCC1‐XPF did not inhibit 1‐nt gap formation, indicating that the activities of MPG and APE1 are independent and precede those of the other four proteins (Fig 3B, lanes 3–7, 51‐mer product). The 8 nts 5′ to the lesion (Fig 3B) combined with the 12 nts 3′ to the lesion (Fig 1B) results in a total patch size of precisely 20 nts in the newly discovered 5′ gap‐mediated LP‐BER.
Figure 3. RECQ1 and PARP1 at the BER 1‐nt gap intermediate.
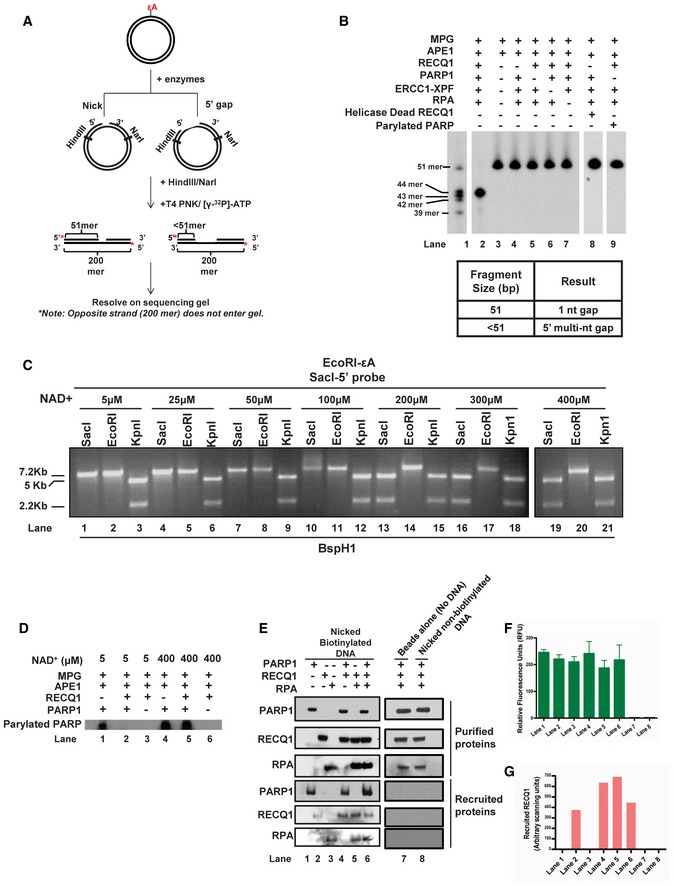
-
ASchematic depiction of gap mapping assay. M13mp18‐εA plasmid DNA (plasmid #3) was treated with the indicated purified proteins for 1 h at 37°C. Samples were subsequently digested with HindIII and NarI, and the 5′ termini were radiolabeled by polynucleotide kinase‐mediated exchange reaction with [γ‐32P]‐ATP (red stars). The radiolabeled reaction products were resolved on a 10% denaturing polyacrylamide gel.
-
BResults of gap mapping assay. Table lists the expected results interpreted by labeled fragment sizes.
-
CIncreasing concentrations of NAD+ were incubated for 1 h at 37°C with M13mp18‐εA plasmid DNA (plasmid #3) and indicated proteins and processed as described in Fig 2C.
-
DPARP PARylation with [γ‐32P]‐NAD+. M13mp18‐εA DNA was incubated with the indicated proteins and [γ‐32P]‐NAD+ for 1 h at 37°C. Reactions were subsequently analyzed by SDS–PAGE followed by autoradiography.
-
E–GBiotinylated or non‐biotinylated M13mp18 plasmid DNA (plasmid #3) with an incised AP site were incubated with the indicated proteins at room temperature for 20 min. DNA was collected using streptavidin‐coated magnetic beads and a magnetic separator. Samples collected on the magnet were immunoblotted for respective proteins and (F) tested for DNA content by the PicoGreen dsDNA assay. Graph depicts the RFU for corresponding samples from (E). Spaces between lanes indicate two separate gels. (G) Graph depicts the quantitation of RECQ1 band from RECQ1 immunoblot in (E). In (F & G) data are presented as mean ± SD from three independent experiments.
Source data are available online for this figure.
5′ gap formation during BER is modulated by PARP1–RECQ1 relationship
The results of the gap mapping experiment (Fig 3B) indicate that the formation of the 5′ gap occurs post‐base excision and AP‐site incision, and the single‐nucleotide gap (1‐nt gap) is the critical DNA substrate for the other four proteins (PARP1, RECQ1, RPA, and ERCC1‐XPF) to form the 5′ gap. PARP1 undergoes auto‐poly(ADP)ribosylation (PARylation) upon binding to the 1‐nt gap intermediate formed during BER, a mechanism by which PARP1 regulates DNA repair (Dantzer et al, 1999). However, when PARP1 was PARylated before addition to the repair reaction, gap formation was not observed (Fig 3B, lane 9). We also found that gap formation was inhibited by addition of higher concentrations of PARP1's substrate for PARylation, NAD+ (≥ 200 μM; Fig 3C). Interestingly, gap formation did occur in the presence of physiologically relevant concentrations of NAD+, which is found at a concentration of ~100 μM in the nucleus and cytoplasm (Fig 3C; Koch‐Nolte et al, 2011). We had previously reported that RECQ1‐deficient cells demonstrate hyperactivated PARP1 in response to treatment with a damaging agent, indicating that RECQ1 may have a regulatory role in PARP1 activation (Sharma et al, 2012). Using 32P‐labeled NAD+, we monitored PARP1 autoPARylation in the presence and absence of RECQ1. PARP1 autoPARylation induced by 1‐nt‐gapped DNA was significantly inhibited by the presence of RECQ1 with 5 μM NAD+ (Fig 3D, lanes 1 and 2), a concentration of NAD+ at which gap formation was also observed (Fig 3C, lanes 1–3). However, PARP autoPARylation was unaffected by the presence of RECQ1 under excess NAD+ (400 μM; Fig 3D, lanes 4 and 5), a concentration of NAD+ at which gap formation was not observed (Fig 3C, lanes 19–21). These results indicate that RECQ1 may be acting as a biological PARP1 inhibitor, and this inhibition may be critical for the gap formation initiated by RECQ1 in BER.
Gap formation 5′ to the lesion requires RECQ1 helicase activity since no gap formation was observed with a catalytically inactive RECQ1 mutant (Sharma et al, 2005) (Fig 3B, lane 8). However, it was unclear whether RECQ1 could be recruited to the BER intermediate alone or through interaction with other proteins. We reasoned that both PARP1 and RPA may be able to recruit RECQ1 to the 1‐nt‐gapped DNA since both proteins have a high affinity for nicks and 1‐nt gaps (de Murcia & Menissier de Murcia, 1994; Wold, 1997) and both interact with RECQ1 (Cui et al, 2003; Sharma et al, 2012). Therefore, we utilized recombinant PARP1, RECQ1, and RPA proteins and a 1‐nt‐gapped DNA substrate in order to determine which protein, if any, recruited RECQ1 to the 1‐nt gap site. Notably, PARP1, RPA, and RECQ1 were recruited to the 1‐nt‐gapped substrate independently (Fig 3E, lanes 1–3), but RECQ1 recruitment to the substrate was significantly increased in the presence of PARP1 or RPA (Fig 3E, lanes 2, 4 and 5, and Fig 3G). Slightly less RECQ1 was recruited in the presence of both proteins (Fig 3E, lane 6 and Fig 3G), which may be due to crowding at the nick site. Importantly, variation in recruitment of proteins was not due to unequal recovery of DNA samples since quantification of the DNA in recruitment samples indicated that there was no significant difference in the amount of DNA collected for each reaction (Fig 3F). Moreover, we confirmed that differential recruitment of proteins was not due to non‐specific interaction with the beads alone (Fig 3E, lane 7). We also verified that proteins were not recruited via non‐specific interaction between the DNA and streptavidin‐coated magnetic beads by using non‐biotinylated nicked DNA (Fig 3E, lane 8). Interestingly, our elucidation of the 5′ gap formation step in LP‐BER revealed the potential importance of the relationship between RECQ1, PARP1, and RPA in regulating BER.
Interestingly, we found that the involvement of other RecQ family helicases in BER supported our conclusions about the importance of PARP1 for 5′ patch formation. It is true that none of the RecQ family helicases have been shown to play a role in BER specifically as helicases although they have been shown to play some indirect or non‐helicase roles in BER by stimulating other proteins or affecting protein expression (Croteau et al, 2014). In fact, only RECQ5 has been shown to modulate expression of PARP1, as one study reported that KD of RECQ5 resulted in a significant decrease in PARP1 expression (Tadokoro et al, 2012). Indeed, we found that while repair of εA was independent of BLM, WRN, and RECQ4, it was somewhat dependent on RECQ5 (Appendix Fig S6). Moreover, 5′ patch formation was completely abrogated by RECQ5 KD (Appendix Fig S6), supporting our finding that PARP1 is required for 5′ patch formation. In future, it will certainly be interesting to investigate the apparently complex relationships among RECQ5, RECQ1, and PARP1 in LP‐BER.
PARP activity‐mediated SN‐BER is the mechanism of repair in the absence of RECQ1
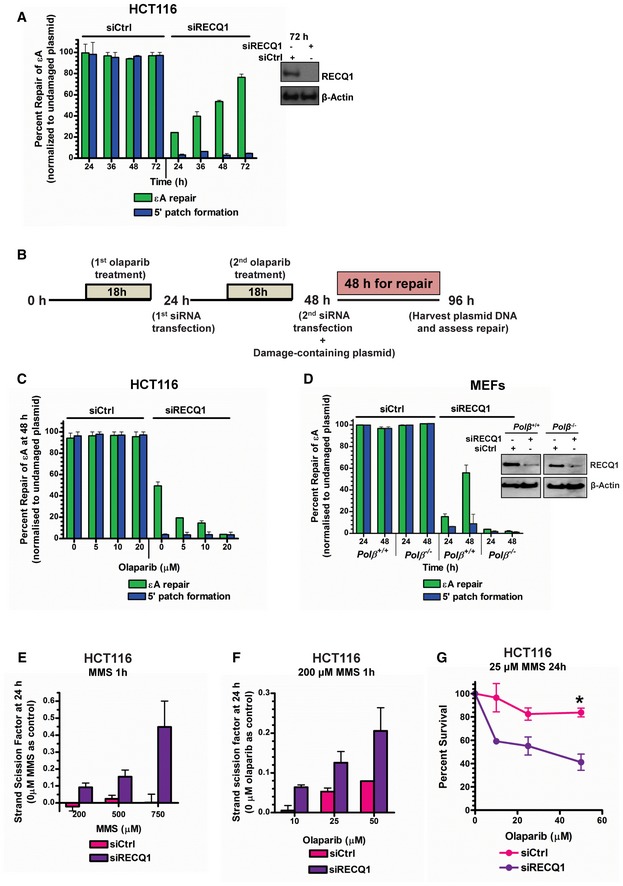
We further explored the inhibition of PARP1 by RECQ1 and consequent control of BER mechanism using the in‐cell repair assay. While repair was nearly complete in control cells, only ~20% of εA was repaired in 24 h in the absence of RECQ1 (Fig 4A). We reasoned that RECQ1 KD should allow PARP1 PARylation and activation of the SN‐BER pathway since it is known that PARP1 PARylation coordinates SN‐BER (Caldecott et al, 1996; Dantzer et al, 1999). Under RECQ1 KD conditions in HCT116 cells, repair of εA and 5′ patch formation were monitored for an extended period of time (72 h) post‐transfection of the εA‐containing plasmid #4. Interestingly, repair of εA increased linearly with time and by 72 h post‐transfection had reached ~70% complete repair (Fig 4A). However, 5′ patch formation did not occur concurrently with εA repair, indicating the 5′ gap‐mediated LP‐BER mode of repair was not functioning (Fig 4A), which was expected in the absence of RECQ1. To verify that the RECQ1‐independent BER mechanism was not slower due to the presence of the mismatch in close proximity to εA, we performed the same long‐term repair assay in RECQ1 KD HCT116 cells using a plasmid containing only εA (plasmid #3). Even without a nearby mismatch, εA was repaired at a slow rate in the absence of RECQ1, reaching ~70% repair by 72 h post‐transfection (Appendix Fig S5A), which was consistent with the result observed in the plasmid containing the 5′ mismatch. We probed for the role of PARP activity in the RECQ1‐independent repair pathway with the addition of olaparib, a small‐molecule PARP inhibitor (PARPi), to the plaque‐based repair assay (Fig 4B). Interestingly, the RECQ1‐independent, slower mechanism of BER was abrogated in the presence of olaparib in HCT116 (Fig 4C) and HeLa cells (Appendix Fig S5B). The RECQ1‐independent BER mechanism was also abrogated in POLβ‐deficient cells (Fig 4D). Since the RECQ1‐independent BER mechanism repaired DNA only at the site of the lesion and was PARP1‐activity and POLβ‐dependent, we concluded that this RECQ1‐independent mechanism of repair was SN‐BER. Taken together, our results (Figs 3 and 4A–D) suggest that the physical and functional interaction between RECQ1 and PARP1 leads to LP‐BER sub‐pathway preference in cells.
Figure 4. RECQ1‐PARP1 regulation of BER sub‐pathway preference.
-
AεA repair and 5′ patch formation were monitored by plaque assay post‐plasmid transfection (plasmid #4) in control and RECQ1 KD HCT116 cells at 24–72 h. Inset shows Western blot analysis of RECQ1 KD in HCT116 cells.
-
BExperimental timeline for plaque‐based repair assay in the presence of olaparib and RECQ1 siRNA.
-
C, DεA repair and 5′ patch formation were monitored by plaque assay post‐plasmid transfection (plasmid #4) in (C) olaparib‐treated control and RECQ1 KD HCT116 cells at 48 h, and (D) control and RECQ1 KD Polb +/+ and Polb −/− MEFs at 24–48 h. Inset in (D) shows Western blot analysis of RECQ1 KD in MEFs.
-
ESSBs were quantified in control and RECQ1 KD HCT116 cells 24 h after treatment for 1 h with indicated doses of MMS.
-
FSSBs were quantified in control and RECQ1 KD HCT116 cells 24 h after treatment for 1 h with 200 μM MMS (sub‐lethal 1‐h treatment) in the presence of indicated doses of olaparib.
-
GSurvival of control and RECQ1 KD HCT116 treated for 24 h with 25 μM MMS (sub‐lethal 24‐h treatment) and indicated doses of olaparib was measured by cell counting. Asterisk (*) denotes P‐value of Student's t‐test < 0.05. Insets in (A) and (D) show Western blot analysis of RECQ1 KD in HCT116 cells and MEFs, respectively.
RECQ1‐PARP1 regulation of BER in genomic DNA
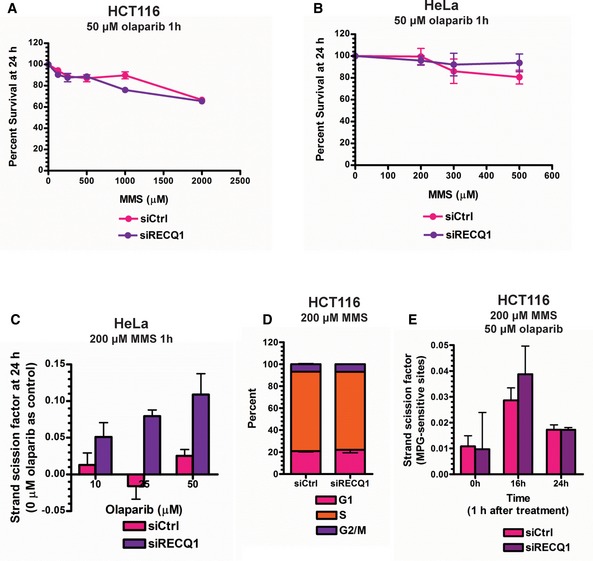
Since RECQ1‐PARP1 regulation of BER was occurring at the 1‐nt gap intermediate in the plasmid DNA, it was possible that the slower εA repair observed in cells under RECQ1 KD conditions (Fig 4A) resulted in the significant accumulation of 1‐nt‐gapped DNA, which could be measured as single‐strand breaks (SSBs). Therefore, to further demonstrate our newly discovered BER regulation mechanism in genomic DNA, especially in the context of chromatin, we treated RECQ1‐proficient or RECQ1‐deficient HCT116 for 1 h with increasing doses of alkylating agent MMS (sub‐lethal, determined by survival assay in Fig EV4A). SSBs were measured by the fast micromethod DNA SSB assay 24 h later (Elmendorff‐Dreikorn et al, 1999). Of note, sub‐lethal doses were selected in order to more appropriately mimic the conditions used in the plasmid‐based in‐cell repair assays and to observe repair in the absence of any off‐target effects that may be produced by lethal doses of DNA‐damaging agents. SSBs increased linearly with MMS dose in RECQ1‐deficient cells while RECQ1‐proficient cells under the same conditions exhibited little or no increase in SSBs (Fig 4E), demonstrating that RECQ1‐mediated BER is indeed the major mechanism of BER and confirmed the results shown in our plasmid‐based assays. To probe for PARP activity‐dependent SN‐BER, we performed the same experiment with a single sub‐lethal dose of MMS (200 μM for 1 h) in combination with increasing doses of olaparib (sub‐lethal doses determined by survival assay in Fig EV4A and B). Again, accumulation of SSBs was observed in RECQ1 KD HCT116 cells (Fig 4F) and HeLa cells (Fig EV4C) with increasing concentrations of olaparib, indicating that PARP activity‐mediated repair was occurring primarily in the absence of RECQ1, which confirmed the results of plasmid‐based assays. This increase was not due to cell cycle changes and dilution of damage by replication in the control cells as there was no difference in cell cycle phase between treated control and RECQ1 KD cells (Fig EV4D). We also did not observe a difference in parent damage formation (MPG‐sensitive sites) between control and RECQ1 KD cells over time after treatment with MMS (Fig EV4E). MPG‐sensitive sites peaked at 12 h post‐treatment, and at 24 h post‐treatment, MPG‐sensitive sites were detected at low levels in both control and RECQ1 KD cells, indicating that alkylated bases had been removed by MPG as expected (Fig EV4E). Therefore, we concluded that the SSB accumulation 24 h after PARP inhibitor treatment in RECQ1 KD cells treated with MMS was, in fact, the accumulation of BER intermediates due to inhibition of both LP‐BER and SN‐BER mechanisms. We further confirmed this result by analyzing cell survival in control and RECQ1 KD HCT116 cells treated with MMS for 24 h at 25 μM, which is a sub‐lethal 24‐h treatment (Fig EV2C), and increasing concentrations of PARPi. Indeed, the RECQ1 KD cells were more sensitive than the control cells to alkylating agent in combination with PARPi (Fig 4G), which is consistent with the absence of both BER sub‐pathways.
Figure EV4. Optimization of alkylation damage repair assays in genomic DNA.
-
A, BMTT assays were performed in control and RECQ1 KD (A) HCT116 cells and (B) HeLa cells 24 h after treatment with increasing doses of MMS for 1 h in combination with 50 μM olaparib.
-
CSSBs were quantified in control and RECQ1 KD HeLa cells 24 h after treatment for 1 h with 200 μM MMS (sub‐lethal 1‐h treatment) in the presence of indicated doses of olaparib.
-
DCell cycle distribution measured by DNA content analysis (propidium iodide staining) using flow cytometry was monitored in control and RECQ1 KD HCT116 cells 24 h after treatment with 200 μM MMS in combination with 50 μM of olaparib for 1 h.
-
EMPG‐sensitive sites were quantified in genomic DNA isolated from control and RECQ1 KD HCT116 cells 0, 16, and 24 h after 1‐h treatment with 200 μM MMS in combination with 50 μM of olaparib.
5′ gap formation‐mediated LP‐BER is relevant for repair of oxidative DNA damage
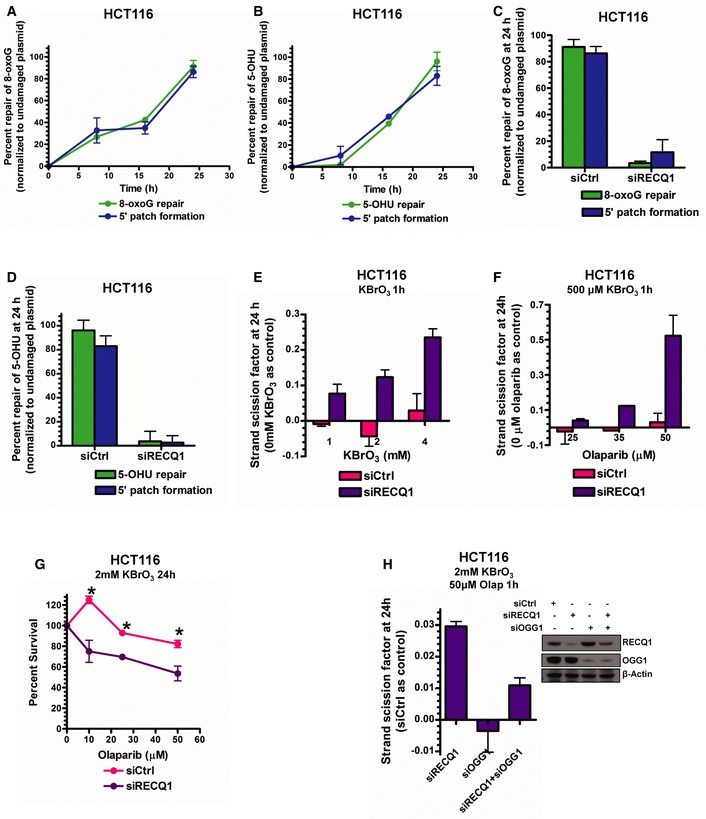
Our next goal was to determine the relevance of the 5′ gap formation step in the repair of other adducts since we had only tested εA. Ethenoadenine, as mentioned previously, is recognized and excised by the monofunctional DNA glycosylase MPG, but it was unclear whether or not 5′ gap formation occurred during repair initiated by bifunctional DNA glycosylases. Therefore, we tested for 5′ gap formation during the repair of two other well‐known and representative BER substrate DNA adducts, 8‐oxoguanine (8‐oxoG) and 5‐hydroxyuracil (5‐OHU), which are repaired by bifunctional DNA glycosylases. 8‐oxoG is recognized and excised by 8‐oxoguanine DNA glycosylase (OGG1) while 5‐OHU is recognized by nth‐like DNA glycosylase 1 (NTH1) and Nei family DNA glycosylases (NEIL1, NEIL2; Hazra et al, 2007). For measuring the repair of these adducts, we used a modified version of our in‐cell repair assay which uses a real‐time PCR‐based readout instead of the plaque‐based readout and was recently used by our laboratory in a separate study (Woodrick et al, 2015; see Materials and Methods section for details). The repair of both adducts occurred via 5′ gap formation (Fig 5A and B) and was dependent on RECQ1 (Fig 5C and D) in HCT116 cells. Additionally, we observed RECQ1‐dependent repair of 8‐oxoG and 5‐OHU in HeLa cells (Appendix Fig S7A and B). Similar to our previous report with H2O2 (Sharma et al, 2012), RECQ1 KD HCT116 cells were significantly sensitized to KBrO3, an oxidizer that predominantly induces the formation of 8‐oxoG in genomic DNA (Ballmaier & Epe, 1995), which was consistent with the results of our repair assays indicating a role for RECQ1 in 8‐oxoG repair (Appendix Fig S7C).
Figure 5. 5′ patch formation in BER of oxidative DNA damage.
-
A, B8‐oxoG repair and 5′ patch formation (A) and 5‐OHU repair and 5′ patch formation (B) were monitored by real‐time PCR‐based repair assay in HCT116 cells for 24 h post‐plasmid transfection (plasmids #13 and 14, respectively).
-
C, D8‐oxoG repair and 5′ patch formation (C) and 5‐OHU repair and 5′ patch formation (D) were monitored by real‐time PCR‐based repair assay in control and RECQ1 KD HCT116 cells 24 h post‐plasmid transfection in the same manner as described in Fig 1D.
-
ESSBs were quantified in control and RECQ1 KD HCT116 cells 24 h after treatment for 1 h with indicated doses of KBrO3.
-
FSSBs were quantified in control and RECQ1 KD HCT116 cells 24 h after treatment for 1 h with 500 μM KBrO3 (sub‐lethal 1‐h treatment) in the presence of indicated doses of olaparib.
-
GSurvival of control and RECQ1 KD HCT116 treated for 24 h with 2 mM KBrO3 (sub‐lethal 24‐h treatment) and increasing doses of olaparib was measured by cell counting. Asterisk (*) denotes P‐value of Student's t‐test < 0.05.
-
HSSBs were quantified in control, RECQ1 KD, OGG1 KD, and RECQ1/OGG1 double KD HCT116 cells 24 h after treatment for 1 h with 2 mM KBrO3 (sub‐lethal 1‐h treatment) in the presence of 50 μM olaparib. Inset shows Western blot analysis of RECQ1 KD and OGG1 KD in HCT116 cells.
Finally, to validate the RECQ1‐PARP1 BER regulation mechanism in genomic DNA for oxidative DNA adducts, RECQ1‐proficient or RECQ1‐deficient HCT116 cells were treated for 1 h with increasing sub‐lethal doses of KBrO3 (determined by survival assay in Fig EV5A and B). SSBs increased linearly with KBrO3 dose in RECQ1‐deficient HCT116 cells while RECQ1‐proficient HCT116 cells under the same conditions exhibited little or no increase in SSBs (Fig 5E), demonstrating that RECQ1‐mediated repair is indeed the major mechanism of BER of oxidative DNA damage. We then probed for PARP activity‐dependent SN‐BER by performing the same experiment with a single sub‐lethal dose of KBrO3 (500 μM for 1 h) in combination with increasing doses of olaparib (doses determined by survival assay in Fig EV5A and B). Again, accumulation of SSBs was observed in RECQ1 KD HCT116 cells (Fig 5F) and HeLa cells (Fig EV5C) with increasing concentration of olaparib, confirming that PARP activity‐mediated repair of oxidative DNA damage was occurring in the absence of RECQ1. As previously demonstrated in MMS‐treated cells, we did not observe a difference in cell cycle phase between KBrO3‐treated control and RECQ1 KD cells (Fig EV5D). Furthermore, Fpg (E. coli functional homolog of human OGG1)‐sensitive sites peaked at 16 h post‐treatment, and at 24 h post‐treatment, Fpg‐sensitive sites were low to undetectable in both control and RECQ1 KD cells, indicating that 8‐oxoG had been removed by OGG1 as expected (Fig EV5E). The accumulation of SSBs 24 h after KBrO3 and PARP inhibitor treatment in RECQ1 KD cells was then concluded to be the result of BER intermediate accumulation due to inhibition of both LP‐BER and SN‐BER of oxidative DNA damage. We further confirmed this result by analyzing cell survival in control and RECQ1 KD HCT116 cells treated with 2 mM KBrO3 for 24 h, which is a sub‐lethal 24‐h treatment (see Appendix Fig S7C), and increasing concentrations of PARPi. Indeed, the RECQ1 KD cells were slightly more sensitive than the control cells to oxidizing agent in combination with PARPi (Fig 5G), but not as sensitive as the MMS‐treated RECQ1 KD cells (Fig 4G). We reasoned that the resistance to KBrO3/olaparib‐induced cell killing could be due to the G1‐arrest induced by the KBrO3/olaparib treatment (Fig EV5D), which would bar conversion of SSB BER intermediates to lethal double‐strand breaks. The MMS/olaparib treatment did not induce a G1 arrest (Fig EV4D), which then probably led to greater sensitivity. Therefore, to confirm that we were, in fact, probing for BER in RECQ1 KD cells after treatment with oxidative DNA‐damaging agent, we quantified SSBs in control, RECQ1 KD, OGG1 KD, and RECQ1/OGG1 double KD cells in the presence of 2 mM KBrO3 and 50 μM olaparib. We reasoned that KD of OGG1 would lead to inhibition of 8‐oxoG‐induced BER initiation, and thus, no SSBs would accumulate with or without RECQ1. Indeed, we found that, compared to the control cells, SSBs accumulated in the RECQ1 KD only cells while no SSBs were detected in OGG1 KD only cells (Fig 5H). When both RECQ1 and OGG1 were knocked down, SSB accumulation noticeably decreased compared to RECQ1 KD alone, confirming that the SSBs we observed in RECQ1 KD cells were, in fact, BER intermediates (Fig 5H). Importantly, complete abrogation of SSB accumulation under RECQ1/OGG1 double KD was not achieved, possibly due to other oxidative DNA adducts induced by KBrO3 treatment (Ballmaier & Epe, 1995), which can be removed by various DNA glycosylases during BER initiation, resulting in the formation of SSBs in the absence of RECQ1.
Figure EV5. Optimization of oxidative damage repair assays in genomic DNA.
-
A, BMTT assays were performed in control and RECQ1 KD (A) HCT116 cells and (B) HeLa cells 24 h after treatment with increasing doses of KBrO3 for 1 h in combination with 50 μM olaparib.
-
CSSBs were quantified in control and RECQ1 KD HeLa cells 24 h after treatment for 1 h with 500 μM KBrO3 (sub‐lethal 1‐h treatment) in the presence of indicated doses of olaparib.
-
DCell cycle distribution measured by DNA content analysis (propidium iodide staining) using flow cytometry was monitored in control and RECQ1 KD HCT116 cells 24 h after treatment with 500 μM KBrO3 in combination with 50 μM olaparib for 1 h.
-
EE. coli Fpg‐sensitive sites were quantified in genomic DNA isolated from control and RECQ1 KD HCT116 cells 0, 16, and 24 h after 1‐h treatment with 500 μM KBrO3 in combination with 50 μM olaparib.
Discussion
Collectively, the results of this study elucidate a previously undiscovered LP‐BER sub‐pathway. We describe a pathway in which an adduct is recognized and excised by a DNA glycosylase, and the resulting AP site is incised by APE1, leaving a single‐nucleotide gap (Fig 6A). The single‐nucleotide gap recruits PARP1, RECQ1, and RPA, and the DNA duplex is subsequently unwound 3′–5′ by RECQ1 while RPA, as a single‐stranded DNA binding protein, presumably coats the opposite strand and stabilizes the flap structure (Fig 6B). The 3′ flap is strictly 8 nt in length and is incised by ERCC1‐XPF, creating a final gap size of 9 nts (Fig 6C), after which repair synthesis occurs with the activity of replicative polymerases and FEN1 proceeding 5′–3′ and incorporating multiple nucleotides on both sides of the original lesion site, up to 20 nts total (Fig 6D). Interestingly, our work shows that this pathway occurs as described when PARP autoPARylation is inhibited by RECQ1 (Fig 6B). However, in the absence of RECQ1, PARP autoPARylation mediates the POLβ‐dependent SN‐BER pathway, resulting in 1‐nt incorporation at the damage site (Fig 6E).
Figure 6. New 5′ gap‐mediated LP‐BER model.
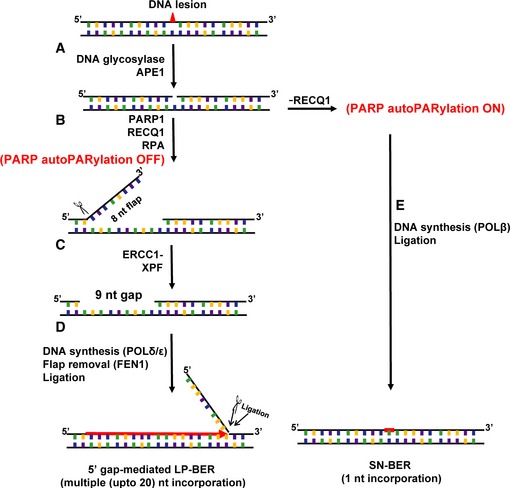
Schematic representation of the results of the present study.
Relevance of proposed model to conventional LP‐BER
Our studies suggest that RECQ1‐mediated 5′ gap formation is a step in a new sub‐pathway of the conventional LP‐BER pathway. In an in vitro reconstitution study of the LP‐BER pathway with replicative polymerases, an important question was raised regarding the use of these polymerases during LP‐BER since the nicked AP site (1‐nt gap) was not the usual 3′ recessed end utilized by replicative polymerases (Matsumoto et al, 1999). However, our results suggest that, if the 5′ gap‐mediated LP‐BER sub‐pathway is in effect, these replicative polymerases have a gapped template available, which is created by the activities of RECQ1 and ERCC1‐XPF and does indeed contain a 3′ recessed end, to initiate strand synthesis. Interestingly, it was demonstrated previously that POLδ preferentially binds larger gapped DNA (9‐13 nts) over 1‐nt gaps (Sharova et al, 2000). It is possible that the 5′ gap‐mediated LP‐BER sub‐pathway leads to increased patch sizes due to the gapped template produced. For example, in in vitro reconstitution experiments for conventional LP‐BER, which did not include RECQ1 and ERCC1‐XPF, only a 1‐nt gap was available for the polymerase, and LP‐BER patch sizes were only ~2 nt in length (Matsumoto et al, 1999). In extracts, presumably expressing RECQ1 and XPF‐ERCC1, LP‐BER experiments demonstrated patch sizes up to 6 or 7 nts, indicating more robust polymerase activity (Frosina et al, 1996; Klungland & Lindahl, 1997). Overall, these observations suggest that polymerase activity during LP‐BER in vivo may be more efficient than what has been observed in in vitro reconstitution experiments since those experiments lacked the critical enzymes necessary to create the preferred larger gap for replicative polymerase activity. It should be noted that we cannot eliminate the possibility that the conventional LP‐BER sub‐pathway may still be operating in live cells. However, it requires further investigation in the future to determine the contribution of the conventional sub‐pathway (with exclusive 3′ patch formation) to overall BER events in live cells. Moreover, it is true that limitations in the size of the MCS on the M13mp18 plasmid required that we place the adduct and/or the mismatch at different restriction sites on the MCS in order to more finely map the patch size for BER (Fig 1B). One might reason that different sequences could affect BER sub‐pathway selection and could possibly explain discrepancies in the reported cell‐free extract results described by others and the results of our experiments performed in live cells. However, our results do suggest that the majority of BER events under basal conditions in cells occur via the newly discovered 5′ gap‐mediated LP‐BER.
Regarding the initiation and regulation of the 5′ gap formation step, Fig 3 is instructive. PARP1 and RPA are nick sensor proteins that transport RECQ1 to the 1‐nt gap site (Fig 3E) via the well‐established interaction among the proteins. RECQ1 exerts its catalytic function, unwinding the duplex 3′–5′, and the resulting flap is resolved by ERCC1‐XPF, creating the gap (9 nts) utilized by the replicative polymerases. RPA is also present at the 1‐nt gap site (Fig 3E) and is absolutely necessary for 5′ gap formation during BER. The scope of the present study does not include an investigation of how the size of the 5′ gap is regulated, but it might be interesting in the future to explore how positioning of ERCC1‐XPF by RPA may play a key role in regulation of the gap size (de Laat et al, 1998). Another feasible mechanism of gap size regulation may be the strong affinity of PARP1 for nicked DNA, which may prevent RECQ1 from unwinding the DNA further.
Similarities between LP‐BER and NER
It is interesting to consider how the new 5′ gap‐mediated LP‐BER sub‐pathway is similar to NER. It had been proposed that LP‐BER may be similar to NER since replicative polymerases and DNA ligase I (LIG1) are required for both pathways (Cappelli et al, 1999). Interestingly, Cappelli et al even tested NER proteins (XPD, CSB, and XPB) for their involvement in BER but did not find involvement of those proteins (Cappelli et al, 1999). Our study has now identified ERCC1‐XPF as a critical LP‐BER enzyme, and its enzymatic function during 5′ gap‐mediated LP‐BER is similar to its function in NER. In the case of NER, after damage recognition the DNA helix is unwound and opened up on both sides of the lesion by the DNA opening activity of XPB and the 5′–3′ helicase activity of XPD (Coin et al, 2007). The resulting bubble structure is stabilized by RPA and XPA, and dual incision occurs on the 5′ side of the bubble structure by ERCC1‐XPF and the 3′ side by XPG (O'Donovan et al, 1994; Sijbers et al, 1996). In our present characterization of the new BER sub‐pathway, RECQ1 functions to unwind the helix in the 3′–5′ direction, generating a similar ss/dsDNA junction for ERCC1‐XPF to incise. It will be interesting to determine whether there is an auxiliary factor in addition to RPA that helps to position ERCC1‐XPF at the junction, similar to XPA in NER.
Consistency with other reports of BER in live cells
Studies in living cells have reported that 55–80% of 8‐oxoG repair events occur via LP‐BER (Sattler et al, 2003), which is consistent with our present study, demonstrating that 8‐oxoG repair occurred via LP‐BER. Furthermore, Masaoka et al demonstrated in live cells that 80% of repair of uracil, a BER substrate, occurred by a POLβ‐ and PARP1 activity‐independent mechanism (Masaoka et al, 2009), which could be our newly identified RECQ1‐dependent LP‐BER sub‐pathway since we found this sub‐pathway is POLβ‐ and PARP1 activity‐independent (Fig 4). It is interesting to note, that when many DNA adducts are analyzed for repair in cell‐free extract‐based assays, the LP‐BER mechanism is somewhat obscured, and SN‐BER is identified as the primary mechanism for repair. For example, studies using cell extracts showed that 8‐oxoG is primarily repaired by the SN‐BER pathway (Dianov et al, 1998; Fortini et al, 1999), but live cell experiments showed LP‐BER to be the primary mechanism for 8‐oxoG repair (Sattler et al, 2003). Interestingly, the same cell extract study by Fortini et al examined repair of εA and found both LP‐BER and SN‐BER to be relevant for repair of the adduct (Fortini et al, 1999), but our study demonstrates exclusive LP‐BER for repair of εA in live cells (Fig EV1). Additionally, uracil repair in bovine testis extracts was demonstrated to occur primarily via POLβ‐mediated SN‐BER (Singhal et al, 1995), but live cell studies demonstrated a POLβ‐independent mechanism for uracil repair (Masaoka et al, 2009). It is possible that these discrepancies between cell‐free extract‐based assays and those performed in live cells may be due to assay conditions that are inadvertently unsuitable for our newly identified LP‐BER enzymes, including RECQ1 and XPF‐ERCC1.
While it had been previously concluded that POLβ was the major BER DNA polymerase in the repair of alkylation damage (Ochs et al, 1999), our results indicate that under basal conditions, it is not the preferred polymerase. The apparent discrepancy may simply be due to differences in assay design and purpose. In our study, we aimed to elucidate the mechanism of DNA repair under basal conditions, while studies showing sensitivity of POLβ‐null MEFs necessarily involved treatment of cells with increasing doses of alkylating agents which may have effects on DNA polymerase and/or sub‐pathway selection (Ochs et al, 1999). It is not clear yet how DNA damage load or off‐target effects coming from damaging agent treatment may affect BER sub‐pathway selection and would be an interesting future research focus.
RECQ1 and PARP1 regulation of LP‐BER and SN‐BER
Remarkably, our elucidation of the novel 5′ gap formation step in LP‐BER reveals an intriguing relationship between RECQ1 and PARP1 in regulation of BER sub‐pathways. Specifically, RECQ1 inhibits PARP1 activity (Fig 3D), which inhibits SN‐BER and promotes RECQ1‐dependent LP‐BER as the preferred mechanism for repair. In the absence of RECQ1, repair proceeds by the SN‐BER mechanism via the activity of PARP (Fig 4C). Interestingly, the plasmid‐based assay indicated that SN‐BER was a slower mechanism of repair than LP‐BER (Fig 4A), and indeed, RECQ1 KD cells, which rely exclusively on SN‐BER, were sensitized to DNA‐damaging agent treatment (Fig EV2C and Appendix Fig S7C). The addition of a PARPi to RECQ1 KD cells inhibited the SN‐BER mechanism of repair and further sensitized cells to the DNA‐damaging agent compared to the control cells (Figs 4G and 5G). While our results indicate that the primary regulator of the BER pathway preference is the physical and functional interaction between RECQ1 and PARP1, it should be noted that others have reported BER in the absence of PARP1 in mouse extracts (Allinson et al, 2003), indicating that BER mechanisms that are independent of the PARP1‐RECQ1 regulation shown in this study may exist and can be explored in the future.
It is interesting to consider the possible physiological conditions under which the relationship between RECQ1 and PARP1 may be disrupted, which would conceivably alter BER sub‐pathway selection in the cell. To date, no physiologically relevant conditions have been discovered or characterized that would render RECQ1 unable to interact with PARP1. However, recently identified mutations in RECQ1 in the human population were found to confer breast cancer susceptibility (Cybulski et al, 2015; Sun et al, 2015). It is intriguing to consider how proficiently these RECQ1 mutants may function in BER. It is possible that some of these mutants may have disrupted interaction with PARP1 and thus confer an “SN‐BER preference” to segments of the population harboring these RECQ1 mutations. Overall, our results could lead to intriguing future studies comparing the preference of LP‐BER or SN‐BER in cells and the consequences of that preference. SN‐BER has lower fidelity than LP‐BER (Akbari et al, 2009), so it is possible that the use of SN‐BER by cells with mutant RECQ1 promotes genomic instability. If so, the RECQ1‐mediated inhibition of the SN‐BER sub‐pathway of repair may be functioning as a genome maintenance mechanism.
The present study demonstrates a new 5′ gap‐mediated sub‐pathway of LP‐BER pathway mediated by RECQ1, PARP1, ERCC1‐XPF, and RPA and has described a novel DNA transaction mechanism for BER of a variety of adducts. The distinct nature of the 9‐nt 5′ gap formed by these proteins during repair is itself a platform for further study to determine how the formation of the gap is regulated. Additionally, the presumably intricate connection between gap formation and repair synthesis is now a novel mechanism to investigate and its elucidation would be highly significant in progressing the present understanding of BER. Intriguingly, our findings indicate that this step is a novel component in BER sub‐pathway selection and identifies RECQ1 and PARP1 as major components that govern LP‐/SN‐BER regulation. Overall, our characterization of a new sub‐pathway of LP‐BER provides numerous new opportunities for advancement in the fields in which BER is a critical player, including aging, neurodegeneration, and cancer.
Materials and Methods
A full description of all experimental methods can be found in the Appendix Supplementary Methods section.
εA/8‐oxoG/5‐OHU‐M13mp18 in vitro construct preparation
The εA/8‐oxoG/5‐OHU‐M13mp18 in vitro construct preparation included three main steps: phosphorylation of the adduct‐containing primer oligonucleotide, annealing of the oligonucleotide to the ssDNA, and the primer extension reaction. Each of these steps is described in detail in the Appendix Supplementary Methods.
In‐cell repair and patch formation assay
For monitoring in‐cell repair of adducts and BER patch formation, cells were seeded in a 6‐well plate overnight at 37°C. Transfection of plasmid DNA was performed, and cells were harvested and plasmid DNA recovered using Mini Prep Kit (Qiagen) at different time points post‐plasmid transfection (24, 36, 48, 72 h) as indicated in the text (Choudhury et al, 2008). After retrieving the plasmid DNA, digestions were performed with appropriate enzymes corresponding to the restriction site placement of adduct and/or mismatches. The digested plasmid DNA was subsequently analyzed as previously described using the plaque assay (Choudhury et al, 2008) or by real‐time PCR (Woodrick et al, 2015).
In vitro gap formation and mapping assays
In vitro gap formation and mapping assays were performed with εA‐ or control‐M13mp18 plasmid DNA and aphidicolin‐treated HCT116 nuclear extract (or purified proteins as indicated). Reaction products were recovered following phenol–chloroform extraction and ethanol precipitation. A previously described method was slightly modified (Zhang et al, 2005) to monitor gap formation in the recovered products. Recovered plasmid was aliquoted and digested with restriction enzymes as indicated and resolved on a 1% agarose gel. For the mapping assay (Fig 3A), recovered plasmid was digested with HindIII and NarI and radiolabeled at 5′ termini by T4 PNK (Thermo Scientific)‐mediated exchange reaction with [γ‐32P]‐ATP according to the manufacturer's protocol. Radiolabeled products were then resolved on 10% polyacrylamide sequencing gel containing 8 M urea.
Cell survival and fast micromethod DNA SSB assays
Cells were seeded in a 6‐well plate (SSB assay) or 24‐well plate (survival assay). The following day cells were treated for 1 h (SSB assay) or 24 h (survival assay) with the indicated doses of DNA‐damaging agent (MMS or KBrO3) with or without olaparib. For 1‐h treatment experiments (SSB assay), treatment media were removed and replaced with growth media with or without olaparib for the remaining 24 h. Cell survival was assessed by cell counting or tetrazolium salt, 3‐4,5 dimethylthiazol‐2,4 diphenyl tetrazolium bromide (MTT) assay.
To detect single‐strand breaks, cells were treated with olaparib and non‐lethal dose of damaging agent as stated above, and fast micromethod DNA SSB assay was performed 24 h later following a published method (Elmendorff‐Dreikorn et al, 1999) using PicoGreen dsDNA reagent (Life Technologies), a fluorescent nucleic acid stain for dsDNA in solution. Fluorescence by PicoGreen dsDNA reagent in solution with cellular DNA was monitored every 2 min for 30 min in the presence of 0.1 M NaOH (pH 12.4). The rate of unwinding was calculated using the amount of fluorescence at 0 min as reference, and the results are calculated as the log10 of the ratio of the percentage of dsDNA from treated and control samples, respectively (Schroder et al, 2006).
Author contributions
JW and SG wrote the manuscript, designed, and performed experiments. SCa, SP, SCh, YB, PK, FS, and YS performed experiments. RR, SS, ODS, HVE, and AC edited the manuscript and designed experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Drs. Mitchell Jung of Georgetown University and Altaf Sarkar of Lawrence Berkeley National Laboratory for Parp +/+ and Parp −/− MEFs and RPA protein, respectively. We thank Dr. Robert Brosh of National Institute on Aging for Recql +/+ and Recql −/− MEFs and Dr. Alessandro Vindigni of St. Louis University for shRNA‐resistant RECQ1 expression constructs. We also thank Dr. Soumendra K. Karmahapatra for optimization of APE1 knockdown experiments. We are grateful to Dr. Tapan Biswas of University of California at San Diego and Dr. Sankar Mitra of Houston Methodist Research Institute of Texas for their reading and valuable discussion of the manuscript. This work is supported by National Institutes of Health Grants [RO1 CA92306 to R.R.], [PO1 CA092584 to O.S], [SC1GM093999 to S.S.], and Cancer Center Support grant [P30 CA051008] for use of Shared Resources.
The EMBO Journal (2017) 36: 1605–1622
Contributor Information
Sudha Sharma, Email: sudha.sharma@howard.edu.
Rabindra Roy, Email: rr228@georgetown.edu.
References
- Agnihotri S, Burrell K, Buczkowicz P, Remke M, Golbourn B, Chornenkyy Y, Gajadhar A, Fernandez NA, Clarke ID, Barszczyk MS, Pajovic S, Ternamian C, Head R, Sabha N, Sobol RW, Taylor MD, Rutka JT, Jones C, Dirks PB, Zadeh G et al (2014) ATM regulates 3‐methylpurine‐DNA glycosylase and promotes therapeutic resistance to alkylating agents. Cancer Discov 4: 1198–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad A, Robinson AR, Duensing A, van Drunen E, Beverloo HB, Weisberg DB, Hasty P, Hoeijmakers JH, Niedernhofer LJ (2008) ERCC1‐XPF endonuclease facilitates DNA double‐strand break repair. Mol Cell Biol 28: 5082–5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbari M, Pena‐Diaz J, Andersen S, Liabakk NB, Otterlei M, Krokan HE (2009) Extracts of proliferating and non‐proliferating human cells display different base excision pathways and repair fidelity. DNA Repair 8: 834–843 [DOI] [PubMed] [Google Scholar]
- Allinson SL, Dianova II, Dianov GL (2003) Poly(ADP‐ribose) polymerase in base excision repair: always engaged, but not essential for DNA damage processing. Acta Biochim Pol 50: 169–179 [PubMed] [Google Scholar]
- Ballmaier D, Epe B (1995) Oxidative DNA damage induced by potassium bromate under cell‐free conditions and in mammalian cells. Carcinogenesis 16: 335–342 [DOI] [PubMed] [Google Scholar]
- Beranek DT (1990) Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res 231: 11–30 [DOI] [PubMed] [Google Scholar]
- Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza‐Maldonado R, Marino F, Lucic B, Biasin V, Gstaiger M, Aebersold R, Sidorova JM, Monnat RJ Jr, Lopes M, Vindigni A (2013) Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20: 347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwat N, Olsen AL, Wang AT, Hanada K, Stuckert P, Kanaar R, D'Andrea A, Niedernhofer LJ, McHugh PJ (2009) XPF‐ERCC1 participates in the Fanconi anemia pathway of cross‐link repair. Mol Cell Biol 29: 6427–6437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott KW, Aoufouchi S, Johnson P, Shall S (1996) XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP‐ribose) polymerase, and DNA ligase III is a novel molecular ‘nick‐sensor’ in vitro . Nucleic Acids Res 24: 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappelli E, Carrozzino F, Abbondandolo A, Frosina G (1999) The DNA helicases acting in nucleotide excision repair, XPD, CSB and XPB, are not required for PCNA‐dependent repair of abasic sites. Eur J Biochem 259: 325–330 [DOI] [PubMed] [Google Scholar]
- Choudhury S, Adhikari S, Cheema A, Roy R (2008) Evidence of complete cellular repair of 1, N6‐ethenoadenine, a mutagenic and potential damage for human cancer, revealed by a novel method. Mol Cell Biochem 313: 19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin F, Oksenych V, Egly JM (2007) Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol Cell 26: 245–256 [DOI] [PubMed] [Google Scholar]
- Croteau DL, Popuri V, Opresko PL, Bohr VA (2014) Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem 83: 519–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Klima R, Ochem A, Arosio D, Falaschi A, Vindigni A (2003) Characterization of the DNA‐unwinding activity of human RECQ1, a helicase specifically stimulated by human replication protein A. J Biol Chem 278: 1424–1432 [DOI] [PubMed] [Google Scholar]
- Cybulski C, Carrot‐Zhang J, Kluzniak W, Rivera B, Kashyap A, Wokolorczyk D, Giroux S, Nadaf J, Hamel N, Zhang S, Huzarski T, Gronwald J, Byrski T , Szwiec M, Jakubowska A, Rudnicka H, Lener M, Masojc B, Tonin PN, Rousseau F et al (2015) Germline RECQL mutations are associated with breast cancer susceptibility. Nat Genet 47: 643–646 [DOI] [PubMed] [Google Scholar]
- Dantzer F, Schreiber V, Niedergang C, Trucco C, Flatter E, De La Rubia G, Oliver J, Rolli V, Menissier‐de Murcia J, de Murcia G (1999) Involvement of poly(ADP‐ribose) polymerase in base excision repair. Biochimie 81: 69–75 [DOI] [PubMed] [Google Scholar]
- De Vos M, Schreiber V, Dantzer F (2012) The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol 84: 137–146 [DOI] [PubMed] [Google Scholar]
- DeMott MS, Zigman S, Bambara RA (1998) Replication protein A stimulates long patch DNA base excision repair. J Biol Chem 273: 27492–27498 [DOI] [PubMed] [Google Scholar]
- Dianov G, Bischoff C, Piotrowski J, Bohr VA (1998) Repair pathways for processing of 8‐oxoguanine in DNA by mammalian cell extracts. J Biol Chem 273: 33811–33816 [DOI] [PubMed] [Google Scholar]
- Dianov GL, Prasad R, Wilson SH, Bohr VA (1999) Role of DNA polymerase beta in the excision step of long patch mammalian base excision repair. J Biol Chem 274: 13741–13743 [DOI] [PubMed] [Google Scholar]
- Elmendorff‐Dreikorn K, Chauvin C, Slor H, Kutzner J, Batel R, Muller WE, Schroder HC (1999) Assessment of DNA damage and repair in human peripheral blood mononuclear cells using a novel DNA unwinding technique. Cell Mol Biol 45: 211–218 [PubMed] [Google Scholar]
- Fisher LA, Samson L, Bessho T (2011) Removal of reactive oxygen species‐induced 3′‐blocked ends by XPF‐ERCC1. Chem Res Toxicol 24: 1876–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortini P, Parlanti E, Sidorkina OM, Laval J, Dogliotti E (1999) The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J Biol Chem 274: 15230–15236 [DOI] [PubMed] [Google Scholar]
- Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E (1996) Two pathways for base excision repair in mammalian cells. J Biol Chem 271: 9573–9578 [DOI] [PubMed] [Google Scholar]
- Grudic A, Jul‐Larsen A, Haring SJ, Wold MS, Lonning PE, Bjerkvig R, Boe SO (2007) Replication protein A prevents accumulation of single‐stranded telomeric DNA in cells that use alternative lengthening of telomeres. Nucleic Acids Res 35: 7267–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra TK, Das A, Das S, Choudhury S, Kow YW, Roy R (2007) Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair 6: 470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson ID (2003) RecQ helicases: caretakers of the genome. Nat Rev Cancer 3: 169–178 [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J (2009) The DNA‐damage response in human biology and disease. Nature 461: 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi‐Busheri F, Lee J, Tomkinson AE, Weinfeld M (1998) Repair of DNA strand gaps and nicks containing 3′‐phosphate and 5′‐hydroxyl termini by purified mammalian enzymes. Nucleic Acids Res 26: 4395–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyomarsi K, Sandoval L, Band V, Pardee AB (1991) Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res 51: 3602–3609 [PubMed] [Google Scholar]
- Klungland A, Lindahl T (1997) Second pathway for completion of human DNA base excision‐repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J 16: 3341–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch‐Nolte F, Fischer S, Haag F, Ziegler M (2011) Compartmentation of NAD+‐dependent signalling. FEBS Lett 585: 1651–1656 [DOI] [PubMed] [Google Scholar]
- de Laat WL, Appeldoorn E, Sugasawa K, Weterings E, Jaspers NG, Hoeijmakers JH (1998) DNA‐binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev 12: 2598–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Page F, Schreiber V, Dherin C, De Murcia G, Boiteux S (2003) Poly(ADP‐ribose) polymerase‐1 (PARP‐1) is required in murine cell lines for base excision repair of oxidative DNA damage in the absence of DNA polymerase beta. J Biol Chem 278: 18471–18477 [DOI] [PubMed] [Google Scholar]
- Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362: 709–715 [DOI] [PubMed] [Google Scholar]
- Masaoka A, Horton JK, Beard WA, Wilson SH (2009) DNA polymerase beta and PARP activities in base excision repair in living cells. DNA Repair 8: 1290–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y, Kim K (1995) Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 269: 699–702 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Kim K, Hurwitz J, Gary R, Levin DS, Tomkinson AE, Park MS (1999) Reconstitution of proliferating cell nuclear antigen‐dependent repair of apurinic/apyrimidinic sites with purified human proteins. J Biol Chem 274: 33703–33708 [DOI] [PubMed] [Google Scholar]
- Mendoza‐Maldonado R, Faoro V, Bajpai S, Berti M, Odreman F, Vindigni M, Ius T, Ghasemian A, Bonin S, Skrap M, Stanta G, Vindigni A (2011) The human RECQ1 helicase is highly expressed in glioblastoma and plays an important role in tumor cell proliferation. Mol Cancer 10: 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mjelle R, Hegre SA, Aas PA, Slupphaug G, Drablos F, Saetrom P, Krokan HE (2015) Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair 30: 53–67 [DOI] [PubMed] [Google Scholar]
- de Murcia G, Menissier de Murcia J (1994) Poly(ADP‐ribose) polymerase: a molecular nick‐sensor. Trends Biochem Sci 19: 172–176 [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, Kanaar R (2004) The structure‐specific endonuclease Ercc1‐Xpf is required to resolve DNA interstrand cross‐link‐induced double‐strand breaks. Mol Cell Biol 24: 5776–5787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs K, Sobol RW, Wilson SH, Kaina B (1999) Cells deficient in DNA polymerase beta are hypersensitive to alkylating agent‐induced apoptosis and chromosomal breakage. Cancer Res 59: 1544–1551 [PubMed] [Google Scholar]
- O'Donovan A, Davies AA, Moggs JG, West SC, Wood RD (1994) XPG endonuclease makes the 3′ incision in human DNA nucleotide excision repair. Nature 371: 432–435 [DOI] [PubMed] [Google Scholar]
- Parvathaneni S, Stortchevoi A, Sommers JA, Brosh RM Jr, Sharma S (2013) Human RECQ1 interacts with Ku70/80 and modulates DNA end‐joining of double‐strand breaks. PLoS ONE 8: e62481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascucci B, Stucki M, Jonsson ZO, Dogliotti E, Hubscher U (1999) Long patch base excision repair with purified human proteins. DNA ligase I as patch size mediator for DNA polymerases delta and epsilon. J Biol Chem 274: 33696–33702 [DOI] [PubMed] [Google Scholar]
- Popuri V, Croteau DL, Brosh RM Jr, Bohr VA (2012) RECQ1 is required for cellular resistance to replication stress and catalyzes strand exchange on stalled replication fork structures. Cell Cycle 11: 4252–4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R, Lavrik OI, Kim SJ, Kedar P, Yang XP, Vande Berg BJ, Wilson SH (2001) DNA polymerase beta ‐mediated long patch base excision repair. Poly(ADP‐ribose)polymerase‐1 stimulates strand displacement DNA synthesis. J Biol Chem 276: 32411–32414 [DOI] [PubMed] [Google Scholar]
- Sattler U, Frit P, Salles B, Calsou P (2003) Long‐patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Rep 4: 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder HC, Batel R, Schwertner H, Boreiko O, Muller WE (2006) Fast micromethod DNA single‐strand‐break assay. Methods Mol Biol 314: 287–305 [DOI] [PubMed] [Google Scholar]
- Sharma S, Brosh RM Jr (2007) Human RECQ1 is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PLoS ONE 2: e1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Brosh RM Jr (2008) Unique and important consequences of RECQ1 deficiency in mammalian cells. Cell Cycle 7: 989–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Sommers JA, Choudhary S, Faulkner JK, Cui S, Andreoli L, Muzzolini L, Vindigni A, Brosh RM Jr (2005) Biochemical analysis of the DNA unwinding and strand annealing activities catalyzed by human RECQ1. J Biol Chem 280: 28072–28084 [DOI] [PubMed] [Google Scholar]
- Sharma S, Phatak P, Stortchevoi A, Jasin M, Larocque JR (2012) RECQ1 plays a distinct role in cellular response to oxidative DNA damage. DNA Repair 11: 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharova NP, Abramova EB, Dmitrieva SB, Dimitrova DD, Mikhailov VS (2000) Preferential interaction of loach DNA polymerase delta with DNA duplexes containing single‐stranded gaps. FEBS Lett 486: 14–18 [DOI] [PubMed] [Google Scholar]
- Sijbers AM, de Laat WL, Ariza RR, Biggerstaff M, Wei YF, Moggs JG, Carter KC, Shell BK, Evans E, de Jong MC, Rademakers S, de Rooij J, Jaspers NG, Hoeijmakers JH, Wood RD (1996) Xeroderma pigmentosum group F caused by a defect in a structure‐specific DNA repair endonuclease. Cell 86: 811–822 [DOI] [PubMed] [Google Scholar]
- Singhal RK, Prasad R, Wilson SH (1995) DNA polymerase beta conducts the gap‐filling step in uracil‐initiated base excision repair in a bovine testis nuclear extract. J Biol Chem 270: 949–957 [DOI] [PubMed] [Google Scholar]
- Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH (1996) Requirement of mammalian DNA polymerase‐beta in base‐excision repair. Nature 379: 183–186 [DOI] [PubMed] [Google Scholar]
- Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand‐Sauthier I, Giglia‐Mari G, Clarkson SG, Vermeulen W, Scharer OD (2009) Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J 28: 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Wang Y, Xia Y, Xu Y, Ouyang T, Li J, Wang T, Fan Z, Fan T, Lin B, Lou H, Xie Y (2015) Mutations in RECQL gene are associated with predisposition to breast cancer. PLoS Genet 11: e1005228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro T, Ramamoorthy M, Popuri V, May A, Tian J, Sykora P, Rybanska I, Wilson DM III, Croteau DL, Bohr VA (2012) Human RECQL5 participates in the removal of endogenous DNA damage. Mol Biol Cell 23: 4273–4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Mendoza‐Maldonado R, Tissino E, Sidorova JM, Yin J, Wang W, Monnat RJ Jr, Falaschi A, Vindigni A (2010) Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol Cell Biol 30: 1382–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umezu K, Sugawara N, Chen C, Haber JE, Kolodner RD (1998) Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics 148: 989–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SS (2013) DNA glycosylases search for and remove oxidized DNA bases. Environ Mol Mutagen 54: 691–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SS (2014) Base excision repair: a critical player in many games. DNA Repair 19: 14–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser T, Gassmann M, Thommes P, Ferrari E, Hafkemeyer P, Hubscher U (1991) Biochemical and functional comparison of DNA polymerases alpha, delta, and epsilon from calf thymus. J Biol Chem 266: 10420–10428 [PubMed] [Google Scholar]
- Wilson SH, Kunkel TA (2000) Passing the baton in base excision repair. Nat Struct Biol 7: 176–178 [DOI] [PubMed] [Google Scholar]
- Wilson DM III, Barsky D (2001) The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat Res 485: 283–307 [DOI] [PubMed] [Google Scholar]
- Wold MS (1997) Replication protein A: a heterotrimeric, single‐stranded DNA‐binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem 66: 61–92 [DOI] [PubMed] [Google Scholar]
- Woodrick J, Gupta S, Khatkar P, Dave K, Levashova D, Choudhury S, Elias H, Saha T, Mueller S, Roy R (2015) A novel method for monitoring functional lesion‐specific recruitment of repair proteins in live cells. Mutat Res 775: 48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, Li GM (2005) Reconstitution of 5′‐directed human mismatch repair in a purified system. Cell 122: 693–705 [DOI] [PubMed] [Google Scholar]
- Zharkov DO, Grollman AP (2005) The DNA trackwalkers: principles of lesion search and recognition by DNA glycosylases. Mutat Res 577: 24–54 [DOI] [PubMed] [Google Scholar]
- Zharkov DO (2008) Base excision DNA repair. Cell Mol Life Sci 65: 1544–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5