

Abstract
The transcription and replication machinery of negative‐stranded RNA viruses presents a possible target for interference in the viral life cycle. We demonstrate the validity of this concept through the use of cytosolically expressed single‐domain antibody fragments (VHHs) that protect cells from a lytic infection with vesicular stomatitis virus (VSV) by targeting the viral nucleoprotein N. We define the binding sites for two such VHHs, 1004 and 1307, by X‐ray crystallography to better understand their inhibitory properties. We found that VHH 1307 competes with the polymerase cofactor P for binding and thus inhibits replication and mRNA transcription, while binding of VHH 1004 likely only affects genome replication. The functional relevance of these epitopes is confirmed by the isolation of escape mutants able to replicate in the presence of the inhibitory VHHs. The escape mutations allow identification of the binding site of a third VHH that presumably competes with P for binding at another site than 1307. Collectively, these binding sites uncover different features on the N protein surface that may be suitable for antiviral intervention.
Keywords: antiviral, nanobody, negative‐strand RNA viruses, vesicular stomatitis virus, VHH
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Structural Biology
Introduction
Vesicular stomatitis virus (VSV) is a member of the Rhabdoviridae family, which includes the human pathogen rabies virus. The VSV single‐stranded RNA genome is negative‐sense, non‐segmented, and encodes five viral proteins: the nucleoprotein N, the phosphoprotein P, matrix protein M, glycoprotein G, and the RNA‐dependent RNA polymerase L. Expression levels of the viral proteins correlate with their position within the single‐stranded RNA genome, with N being the most abundant and L the least abundant. The VSV genome is tightly encapsidated by N to form a nucleocapsid (N‐RNA), which serves as the template for RNA synthesis. In the absence of N, transcription can be initiated, but no full‐length RNAs are produced 1. As it encapsidates the 11,161 nucleotide genome, the nucleocapsid adopts a bullet shape in the virion, whereas it shows an elongated, more flexible representation in the cytosol of infected cells 2. Coexpression of N and the RNA polymerase cofactor P in E. coli results in ring‐shaped, decameric nucleocapsid‐like particles that encapsidate bacterial RNA non‐specifically 3.
Crystallographic analysis has provided molecular details of RNA encapsidation and N oligomerization 3. The N protein consists of an N‐ and a C‐terminal lobe in between which the RNA is packed. Each N protomer makes cross‐molecular contacts with three neighboring N protomers, for which an extension of the N‐terminal lobe (N‐arm) and a large loop in the C‐terminal lobe (C‐loop) are critical. Removal of the N‐arm reduces incorporation of RNA 4, while mutations in the C‐loop affect VSV RNA replication and transcription differentially 5. In the nucleocapsid, the RNA is largely protected against digestion with RNAse; only harsh treatment with RNAse leads to RNA degradation 6. How the polymerase L gains access to the tightly encapsidated RNA in the course of transcription remains elusive. The L protein is unable to bind to the nucleocapsid directly. Instead, P, a non‐enzymatic polymerase cofactor that interacts with both L and N, mediates this interaction. P can interact with N in two different ways. First, the extreme N‐terminus of P can chaperone the free N (N0) to prevent it from premature oligomerization and association with random cellular RNA. Instead, it directs N to encapsidate the viral RNA 7. The second interaction is mediated by the C‐terminal domain of P (PCTD), which binds to the C‐lobes of two adjoining N protomers and thus a nucleocapsid‐specific interface 8. This interaction properly positions the L protein, which in turn may impose a conformational change on N that permits access to the RNA. As the complex moves along the template, N folds back and encapsidates the RNA, while N0 molecules encapsidate the newly synthesized strand of RNA.
The three proteins essential for VSV transcription and replication, N, P, and L, provide attractive possible targets for intervention in the virus life cycle. We have explored the use of single‐domain antibody fragments as antiviral agents that can be expressed intracellularly. We used protein domains derived from the variable region of the heavy chain of camelid heavy‐chain‐only antibodies (VHHs), which retain their antigen‐binding properties in the cytosol. We immunized an alpaca with inactivated VSV and selected VHHs using a lentiviral screening approach that relies on inducible expression of cytosolic VHHs and selection of cells that survive a lethal dose of VSV 9. All four identified VHHs are specific for VSV (VHH 1001, 1004, 1014, and 1307). When expressed cytosolically, they target N and impede VSV replication by blocking viral mRNA transcription. In an in vitro transcription assay with purified L, P, and N‐RNA template, only two VHHs (1001 and 1307) blocked mRNA transcription, indicating that the identified VHHs inhibit the virus in different ways. Competition analysis showed that all four N‐specific VHHs recognize distinct epitopes 9.
To provide a molecular explanation for the inhibitory properties of N‐specific VHHs, we defined the VHH binding sites on N for three of the identified VHHs. We obtained crystal structures of N in complex with two VHHs and could map the binding site of a third VHH by identifying VSV escape mutants that emerged in the presence of the inhibitory VHH. The defined binding sites provide a rationale for the inhibitory properties of the VHHs and explain relevance and function of the bound subdomains during viral replication.
Results
Inhibitory characteristics of VSV N‐specific VHHs
We reported the identification of several VSV N‐specific VHHs that protect cells from VSV infection when expressed cytosolically 9. To uncover the underlying vulnerabilities of the virus, we characterized the inhibitory properties of three N‐specific VHHs in more detail. First, we tested the effect of the VHHs on VSV infection in A549 cell derivatives in which cytosolic VHH expression is doxycycline‐inducible. We induced VHH expression and infected cells 24 hours (h) later with VSV‐GFP, a VSV Indiana strain that—in addition to the structural proteins of VSV—expresses GFP as a measure of a successful infection. We infected cells with different doses (multiplicities of infection, MOIs) of virus to test whether increasing amounts of virus could overcome the inhibitory potential of the VHHs (Fig 1A). Cells were harvested 4 h post‐infection, and the percentage of GFP‐positive cells was assessed by flow cytometry. All cell lines that expressed any of the N‐specific VHHs blocked VSV when infected at an MOI of 10, while the cell line that expressed the control VHH was readily infected (control VHH: αNP‐VHH1, influenza A nucleoprotein specific 10). An up to 32‐fold increase in the amount of virus barely allowed infection of cells expressing VHH 1001. For cells that expressed VHH 1004, the fraction of GFP‐positive cells increased with increasing MOI, reaching 37% GFP‐positive cells when infected with the highest dose (MOI 320). Inhibition of infection in cells that expressed VHH 1307 could be more easily overcome by increasing the virus dose, yielding more than 80% of GFP‐positive cells at a fourfold increase of virus (MOI 40). Two VHH characteristics might contribute to the differential response to increasing doses of virus: (I) the expression level of the VHH and (II) the mechanism by which the VHH inhibits the virus. To assess VHH expression levels, we stained for the HA‐tagged VHHs with anti‐HA‐DL650 and quantified fluorescence intensity by flow cytometry (Fig 1B). Expression levels were highest for VHH 1001 and low for VHH 1307, explaining their robustness and vulnerably to increasing doses of virus, respectively. Albeit mediating a clear antiviral effect after doxycycline addition and only mild susceptibility to increasing amounts of virus, levels of VHH 1004 were too low to be detected in this assay. Instead, we verified doxycycline‐induced expression of VHH 1004 by immunoblot (Fig 1C). The comparably low expression levels suggest a potent mechanism of inhibition for VHH 1004.
Figure 1. Inhibitory properties of VSV N‐specific VHHs.
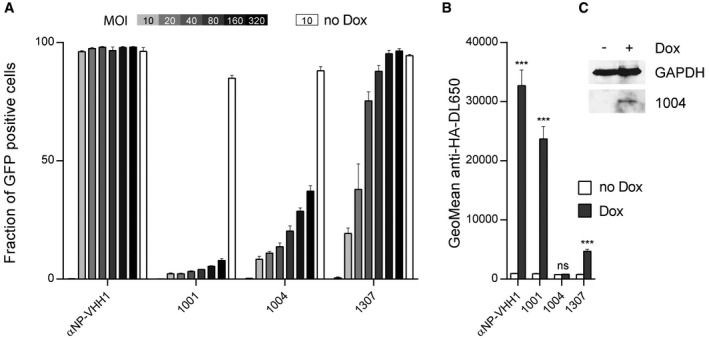
- VSV N‐specific VHHs differentially cope with increasing doses of VSV. A549 cells expressing VHHs in a doxycycline (Dox)‐inducible manner were seeded 24 h before VSV infection. VHH expression was induced (gray to black bars) with Dox, or cells were left untreated (white bars). Each cell line was infected with increasing amounts of VSV‐GFP (MOI = 0–320). Cells were harvested 4 h post‐infection, and the percentage of infected cells (GFP positive) was quantified by flow cytometry. Average data from three independent experiments (± s.e.m.; n = 3) are shown.
- Quantification of VHH expression levels in A549 cells. Cells were seeded, and HA‐tagged VHH expression was induced with Dox. After 24 h, cells were harvested and stained for VHH‐HA using anti‐HA‐DL650 antibodies. The geometric mean of anti‐HA‐DL650 fluorescence was quantified by flow cytometry and compared to uninduced cells lines. Average data from three independent experiments (± s.e.m.) are shown (n = 3; t‐test; ***P < 0.001; ns = non‐significant). Exact P‐values are shown in Table EV1.
- Lysates of A549 cell lines inducibly expressing VHH 1004 were subjected to immunoblot analysis using GAPDH and HA‐tag (VHH‐HA) antibodies.
VHH 1004 in complex with N‐RNA
To uncover the inhibitory mechanism and epitope of the identified VHHs, we defined the binding site on N by X‐ray crystallography. We produced recombinant VHHs and N‐RNA individually in E. coli and purified the proteins. We then combined VHH and N‐RNA in a 3:1 molar ratio and purified the complex by size‐exclusion chromatography. For all VHH:N‐RNA complexes, we detected a clear shift in the elution profile, indicative of VHH binding to the N‐RNA 10‐mer ring (Fig 2A). The collected peak fractions were concentrated to 2.5–5 mg/ml and used to set up crystal screens of the VHH:N‐RNA complexes. We were able to obtain diffraction quality crystals for N‐RNA in complex with VHH 1004 and VHH 1307, respectively.
Figure 2. Crystal structures of VHH 1004 and VHH 1307 in complex with VSV N‐RNA.
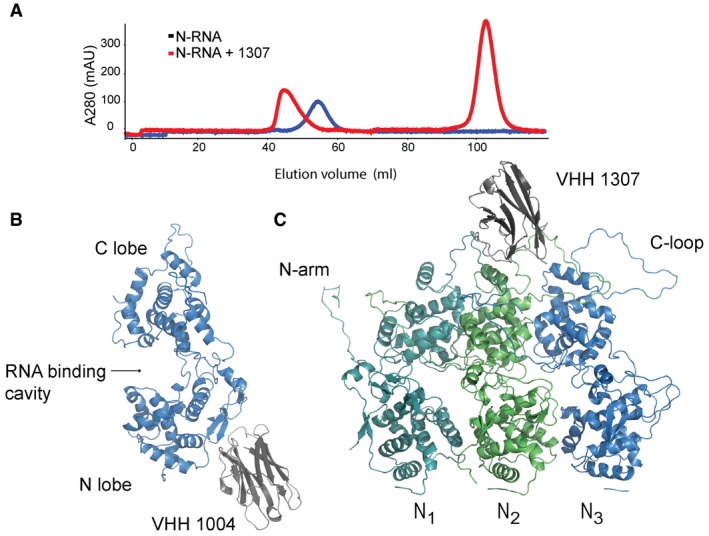
-
APurified N‐RNA alone (blue), or preincubated with an excess of VHH (red), was subjected to size‐exclusion chromatography on a Superdex 200 column. Absorbance at 280 nm of the elution profile is displayed. Data exemplarily shown for N‐RNA and N‐RNA in complex with VHH 1307.
-
B, CRibbon representation of VHH 1004 and 1307 in complex with VSV N. (B) Side view of VHH 1004 (gray) bound to the N‐lobe of a single N protomer (blue). (C) VHH 1307 (gray) and three N protomers are displayed (dark cyan, green, and blue); view from the outside of the 10‐mer ring. The VHH binds to the C‐lobes of two adjacent N protomers. In the depicted structure, the VHH interacts with the left N protomer (N1, dark cyan) and the N protomer in the center (N2, green).
Crystals of the N‐RNA:1004 complex diffracted to 5.45 Å (Table 1). We solved the structure by molecular replacement (MR) (details in the Materials and Methods section) and refined to a final R work of 33.9% and R free of 34.0%. Besides the bound VHH, the structure resembles previously characterized N‐RNA structures, in which N forms a 10‐mer ring that encapsidates RNA (Cα RMSD 0.89–0.99 Å). VHH 1004 binds the N‐terminal lobe of N and decorates the circumference of the 10‐mer N‐RNA ring (Figs 2B and 4, see Fig EV1 for omit and composite omit map). The VHH buries an area of ~550 Å2 on the surface of N. The VHH contacts residues of a single N protomer and may thus also bind to monomeric N0. VHH 1004, when expressed in the cytosol, strongly inhibits VSV replication, whereas in vitro transcription catalyzed by purified components (L, P, and N‐RNA) is barely affected by its presence 9. To replicate or transcribe the RNA, the VSV polymerase L must access the encapsidated RNA. Because VHH 1004 does not cover the known P binding sites on N 7, 8, it either prevents replication of viral genomes or manipulates N in such a way that it fails to correctly encapsidate newly synthesized RNA. Both scenarios would substantially reduce the amount of genomes that serve as templates for mRNA transcription in infected cells, while leaving mRNA transcription per se unaffected 9.
Table 1.
Data collection and refinement statistics
| Protein | VSV N:VHH 1004 native | VSV N:VHH 1307 native |
|---|---|---|
| Organism | Vesicular stomatitis virus, Vicugna pacos | Vesicular stomatitis virus, Vicugna pacos |
| PDB ID | 5UKB | 5UK4 |
| Data collection | ||
| Space group | P21212 | P1 |
| a, b, c (Å) | 240.117, 335.497, 75.899 | 147.555, 156.008, 217.450 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 79.24, 75.66, 62.27 |
| Wavelength (Å) | 0.9791 | 0.9791 |
| Resolution range (Å) | 195.26–5.50 (5.70–5.50) a | 128.45–3.20 (3.31–3.20) |
| Total reflections | 227,207 | 614,436 |
| Unique reflections | 20,655 | 250,547 |
| Completeness (%) | 99.7 (98.3) | 92.4 (92.5) |
| Redundancy | 11.0 (5.5) | 2.5 (2.4) |
| R sym (%) | 16.6 (100.0) | 20.4 (67.5) |
| R p.i.m. (%) | 5.2 (46.0) | 15.2 (50.2) |
| I/σ | 16.2 (0.86) | 5.1 (1.4) |
| CC1/2 (%) | 99.7 (55.6) | 99.3 (61.9) |
| Refinement | ||
| Resolution range (Å) | 195.26–5.50 | 128.45–3.20 |
| R work (%) | 33.9 | 23.6 |
| R free (%) | 34.0 | 28.7 |
| Coordinate error (Å) | 1.01 | 0.47 |
| Number of reflections | ||
| Total | 20,603 | 250,544 |
| R free reflections | 2,000 | 1,990 |
| Number of non‐hydrogen atoms | 21,875 | 88,846 |
| Protein atoms | 21,875 | 88,846 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.004 | 0.002 |
| Bond angles (°) | 0.85 | 0.525 |
| Average B factors (Å2) | ||
| Protein | 305.1 | 58.33 |
| Ramachandran (%) | ||
| Favored (%) | 94.4 | 93.9 |
| Allowed (%) | 5.1 | 5.9 |
| Outlier (%) | 0.5 | 0.2 |
| Clashscore | 37.39 | 13.4 |
| Molprobity score | 2.44 | 2.04 |
| Molprobity percentile | 97th | 97th |
Values in parentheses are for highest resolution shell.
Figure EV1. VHH omit map (A) and composite omit map (B) shown for VHH 1004 in complex with N‐RNA.

- Side view of the N‐RNA 10‐mer with VHH 1004 bound (gray). N molecules are shown in different colors. The electron density shown is from a Sigma‐A‐weighted difference (mFo‐DFc) omit map for the centered VHH contoured at 2.7σ (green mesh).
- Magnified view of the interaction interface of VHH 1004 and VSV N. Composite omit map (2mFo‐DFc) was calculated and is shown at 1.2σ (green mesh). The complementary determining regions (CDRs) of VHH 1004 are shown in blue (CDR1), red (CDR2), and yellow (CDR3).
VHH 1307 in complex with N‐RNA
For the N‐RNA:VHH 1307 complex, we obtained crystals that diffracted to 3.2 Å (Table 1). We solved the structure by MR and refined to a final R work of 23.6% and R free of 28.7%. The complex crystallizes as two 10‐mer rings of N‐RNA that are crowned at both ends with 10 VHHs each, yielding 40 protein subunits to the asymmetric unit. The VHH binds to the C‐terminal lobe of N, makes contact with two adjacent protomers, and thus binds to a nucleocapsid‐specific interaction interface (Figs 2C and 4). The VHH buries an area of ~550 Å2 on N, and there are no drastic changes in N structure upon VHH binding (Cα RMSD 0.81–0.96 Å). We identified the residues involved in the N:VHH 1307 binding interface using PDBePISA 11. VHH 1307 makes direct contact to residues from two adjacent protomers to recognize a nucleocapsid‐specific epitope that overlaps with the binding site of the C‐terminal domain of the polymerase cofactor P (PCTD) 8. This binding site suggests that the VHH may inhibit viral transcription by preventing P from binding to the nucleocapsid, which would block P‐mediated access of the polymerase L to the RNA template. Accordingly, VHH 1307 inhibits transcription in vitro and gene expression in infected cells. The latter likely relies on both genome replication and mRNA transcription 9. Experiments that interchanged the position of N and P proteins in the VSV genome and thus switched expression levels showed the importance of a balance between N and P proteins 12. VHH 1307 perturbs this balance, but because of this VHH's comparatively low expression levels, increasing amounts of virus may overcome antiviral restriction as shown in Fig 1A.
Figure 4. VSV escape mutations for VHH 1001, 1004, and 1307.
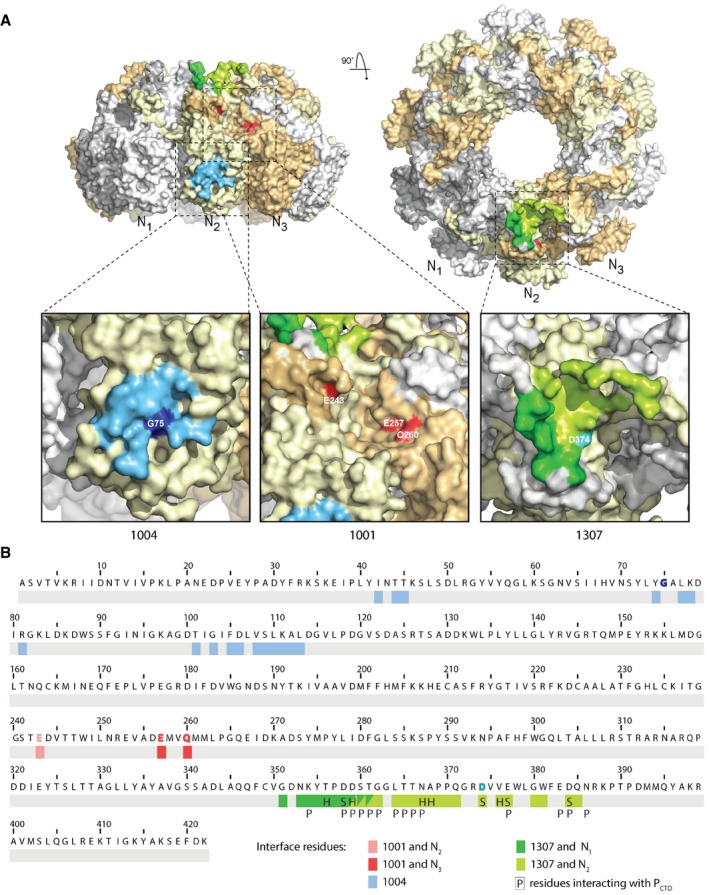
- Surface representation of the N‐RNA with N protomers displayed in alternating colors. For each VHH, one binding site on the 10‐mer is shown with magnified view of the binding sites. The VHH 1004 binding interface residues on a single N protomer are shown in blue; the escape mutant residue G75R is shown in dark blue. The escape mutations that comprise the presumptive binding sites for VHH 1001 are shown in red. Escape mutation E243G is shown from the N2 protomer, E257D and Q260K/R from the N3 protomer. VHH 1307 interacts with N1 (dark green) and N2 (light green), the escape mutation D374N on N2 is shown in cyan. Surface representations were generated in PyMOL.
- Protein sequence of VSV Indiana N. Binding interface residues for VHH 1004 are shown in blue. Interface residues for VHH 1307 are shown in dark green (N1) or light green (N2). Residues engaged from two VHH 1307 molecules are colored in both green tones. The better resolution of the N‐RNA:1307 crystal allowed to specify the interaction types. “H” labels residues involved in hydrogen bonds, and “S” labels residues engaged in salt bridge. “P” labels residues interacting with the phosphoprotein P 8. Escape mutant residues (Table 2) are highlighted with the VHHs color. Escape mutations for VHH 1001 colored in light red (N1) or darker red (N2).
VHH 1307 and P bind to the same or overlapping epitopes on N
We found that VHH 1307 and PCTD bind to overlapping epitopes of N (Fig 3A). To test biochemically whether VHH 1307 prevents binding of P to N or vice versa, we addressed the interaction of VHHs with N alone or N in complex with stoichiometric amounts of P (Fig 3B and C). We biotinylated VHH 1001, 1004, 1307, and a control VHH (αNP‐VHH1, influenza A nucleoprotein specific 10) and immobilized them on streptavidin beads. We incubated the beads with purified N, or the N/P complex and analyzed the bound protein by SDS–PAGE and Coomassie staining. With the exception of the control VHH, all VHHs immunoprecipitated N in the absence of P. VHH 1004 also immunoprecipitated intact N/P complexes. In contrast, VHH 1307 almost exclusively retrieved N from the complexes and barely recovered any P, indicating that the VHH displaced P from the 10‐mer complex. These data confirm that VHH 1307 and P bind to overlapping epitopes on N. It suggests that VHH 1307 can also displace P from viral nucleocapsids. Interestingly, we found that VHH 1001 also retrieves mostly N from the N/P complexes, and substantially reduced the amounts of P, indicating that VHH 1001 also overlaps with P for binding to N. Because the binding sites for VHH 1001 and 1307 do not overlap 9, and VHH 1307 covers nearly the entire binding site for PCTD, VHH 1001 likely overlaps with a distinct P binding site of N.
Figure 3. VHH 1307 competes with P for binding to N.

- Magnified view and superposition of the C‐terminal domain of P (red) and VHH 1307 (gray) at their binding site at the N‐RNA C‐loop region.
- VHH binding to N and the N/P complex. Biotinylated, N‐specific VHHs, and a control VHH (αNP‐VHH1, influenza A nucleoprotein specific) were immobilized on streptavidin beads and N alone, or N in complex with P was added to the beads. Bound protein was eluted and subjected to 10% SDS–PAGE (75:1 acrylamide/bis‐acrylamide) and Coomassie staining. A representative gel from three independent experiments is shown.
- Band intensities of the immunoprecipitated N and coimmunoprecipitated P were quantified, and the relative amount of P compared to N is displayed. Average data from three independent experiments (± s.e.m.) are shown (n = 3; t‐test; *P < 0.05; **P < 0.01). Exact P‐values are shown in Table EV1.
Functional determination of binding site by escape mutants
To independently identify VHH binding sites on N based on function, we generated VSV escape mutants by serial passage of infectious supernatants in cells that expressed the different VHHs. We diluted cell supernatants after passaging, infected a new monolayer of cells, and analyzed the resulting plaques by RT–PCR and sequencing. For VHH 1001, all four analyzed plaques carried different mutations in N. All plaques analyzed from escape mutants of VHH 1004 carry the mutation G75R in N, while all escape mutants of VHH 1307 contain the mutation D374N in N (Table 2). All identified escape mutation residues are surface‐exposed and are not in close proximity to each other (Fig 4), confirming independently that the identified anti‐VSV N‐VHHs have unique binding sites 9.
Table 2.
List of observed escape mutations in N
| VHH | Clone | Mutation | Amino acid change |
|---|---|---|---|
| 1001 | 1 | A771C | E257D |
| 2 | C778A | Q260K | |
| 3 | A779G | Q260R | |
| 4 | A728G | E243G | |
| 1004 | 1,2,3 | G223A | G75R |
| 1307 | 1,2,3 | G1120A | D374N |
Gly75, mutated in VHH 1004 escape mutants, is positioned in the center of the VHH 1004 binding site as determined crystallographically (Fig 4). Similarly, the D374N substitution found in all VHH 1307 escape mutants maps to the binding site established for VHH 1307 (Fig 4). Because this subdomain of N is also bound by the essential polymerase cofactor P, this mutant must retain the ability to engage P. While many of the interaction/interface residues of VSV P and VHH 1307 overlap, residue D374 is exclusively engaged by VHH 1307 but not by P (Fig 4B) 8. The escape mutant therefore affects the interaction of the nucleocapsid and VHH 1307 without altering the binding to P.
We were unable to obtain diffraction quality crystals for VHH 1001 in complex with N, but successfully generated VSV escape mutants instead. Because the escape mutations for VHH 1004 and 1307 were located in the structurally determined binding sites, and because the residues mutated under pressure from VHH 1001 are similarly solvent exposed, we presume that the identified escape mutations correspond to the VHH 1001 binding site. VHH 1001 inhibits VSV mRNA transcription if expressed in the cytosol and blocks in vitro transcription as well 9. In the lentiviral screening approach that led to the identification of VHH 1001, this particular VHH was found in 34 of 41 independently identified clones, which we attribute to its comparatively higher expression levels and potent mechanism of inhibition 9. For VHH 1001, we analyzed four plaques (Table 2). Each carried a different mutation that resulted in four independent changes in the sequence of N (E243G, E257D, Q260R, Q260K). Mutated residue E243G and residues E257D, Q260K are located on opposite sides of the C‐terminal lobe of an N protomer. The location of the escape mutations suggests that the VHH engages an interface of N‐RNA that is composed of two adjacent N protomers, rather than a single N protomer (Fig 4A). The distance of the identified residues in two adjacent protomers is ~2 nm and therefore equivalent to the diameter of a typical VHH (~2 × 3.5 nm). At this interface, the VSV nucleocapsid exhibits a small notch between two N molecules that would perfectly accommodate a VHH. This notch has also been identified to be occupied by the N‐terminus of P when bound to N0 7. Because VHH 1001 can displace full‐length P from N‐RNA (Fig 3B and C), our data suggest that P occupies this notch also when engaging N‐RNA and not only when engaging N0.
Binding of VHHs to the escape mutants
Not all escape mutations resulted in drastic changes of amino acid characteristics at the mutated site. While the G75R, E243G, or D374N mutations represent major changes in size, polarity, or charge of the surface‐exposed amino acids, the E257D mutation represents the difference of a single methylene carbon. To test how these mutations in N affect recognition by the VHHs, we applied LUMIER assays with mutated versions of N (Fig 5) 9, 13. In this assay, an HA‐tagged VHH and a Renilla luciferase fusion of N are transiently coexpressed in 293T cells. After 24 h, cells are lysed and the HA‐tagged VHH is immobilized in 96‐well plates coated with anti‐HA antibody. Renilla luciferase activity is quantified as a measure of how much N protein is bound to the VHH. All Renilla‐N mutants were expressed to comparable levels (data not shown, but used for normalization), and all N‐specific VHHs retrieved wild type (WT) N. Escape mutations observed for VHH 1001, as single mutations or as combinations, abolished binding of VHH 1001, indicating that these surface‐exposed residues are indeed critical for VHH 1001 binding. Versions of N carrying these escape mutations were still readily retrieved by VHH 1004 and 1307, indicating that these mutations did not alter the overall structure of N. A loss of VHH binding as observed for VHH 1001 is an escape strategy expected for inhibitory VHHs. Surprisingly, the VHH 1004 escape mutation G75R resulted in a strong increase of retrieved N by VHH 1004, while VHH 1001 and 1307 recovered this N variant at levels comparable to WT N. Similarly, VHH 1307 retrieved higher levels of its escape mutant D374N compared to its WT counterpart.
Figure 5. VHH binding to VSV N escape variants.
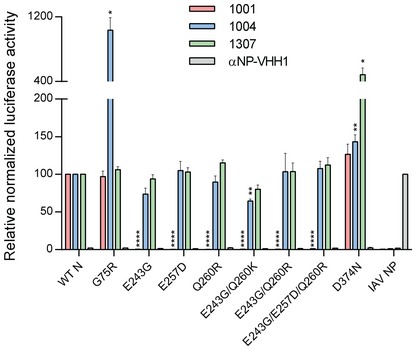
Indicated, HA‐tagged VHHs and VSV N (WT or mutated versions) fused to Renilla luciferase, or influenza A NP fused to Renilla luciferase were transiently coexpressed in 293T cells. 96‐well plates coated with anti‐HA antibody to capture the VHHs were incubated with cell lysates. Activity of the copurified luciferase was measured. Emitted light was normalized to luciferase activity in the lysate. Average data from three independent experiments (± s.e.m.) are shown. Except for the controls, statistical significance compared to WT N is indicated by asterisks (n = 3; t‐test; *P < 0.05; **P < 0.01; ****P < 0.0001). Exact P‐values are shown in Table EV1.
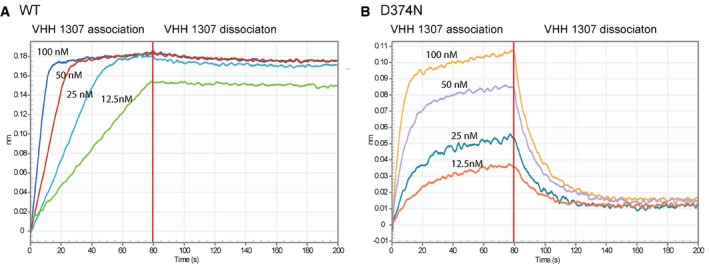
To better understand the escape mutations that failed to eliminate binding, we performed biolayer interferometry to analyze binding kinetics. When we analyzed binding of N‐RNA to immobilized VHHs, we confirmed all binding activities observed in LUMIER assays, but could not deduce any binding constants due to the lack of dissociation. We thus reversed the order of the proteins: We immobilized WT and mutant versions of N and measured VHH association and dissociation. For VHH 1001 and 1004, we observed poor association, likely because of their lateral VHH binding sites, steric hindrance, and unfavorable orientation of the N‐RNA ring in this assay geometry. VHH 1307 associated slightly faster to the D374N mutant compared to WT N, but, importantly, dissociated much faster, reducing the affinity more than 1,000‐fold (Table 3 and Fig EV2). In the LUMIER assay where the VHHs are the immobilized component, fast dissociation is likely prevented by avidity affects and the larger amount of retrieved mutant protein can be explained by the faster association rate. During infection, however, the reduced affinity and more dynamic binding to the mutant likely allows VHH 1307 to be more easily replaced by P, undermining the VHHs inhibitory properties. In conclusion, abolished or altered binding dynamics allowed VSV N to escape from VHH‐mediated restriction.
Table 3.
Binding affinities of VHH 1307 to WT or mutant N
| VHH 1307 binding to | K D (M) | K on (1/Ms) | K off (1/s) |
|---|---|---|---|
| WT N | 5.53 × 10−11 | 1.18 × 106 | 6.51 × 10−5 |
| D374N N | 6.47 × 10−8 | 1.28 × 106 | 8.25 × 10−2 |
Figure EV2. Binding affinity determination for VHH 1307.
-
A, BOctet RED96 sensorgrams with immobilized VSV N WT (A) or D374N (B). VHH 1307 association and dissociation are shown for VHH concentrations of 12.5–100 nM.
Discussion
Virus infections continue to fuel the threat of future epidemics. While vaccine strategies directed against viral glycoproteins have been remarkably successful, the selective pressure exerted by the immune system can generate escape variants against which the vaccine‐elicited response is no longer effective, as so well documented for influenza A 14. Targeting enzymatic functions unique to particular viruses offers a viable alternative, exemplified by drugs that exploit influenza virus neuraminidase activity or herpesvirus kinases 15, 16. VSV is a prototypic non‐segmented RNA virus for which the choice of druggable targets is limited. To better understand the transcription/replication process, and to identify new vulnerabilities of this class of viruses, we used cytosolically expressed VHHs, also called nanobodies, that target the VSV nucleoprotein N and block infection. To relate the inhibitory properties of the N‐specific VHHs to the structural features they recognize, we determined the binding sites of three VHHs by means of X‐ray crystallography and by analysis of escape mutants. One identified VHH binds to the N‐terminal lobe of N, and two VHHs bind to different epitopes on the C‐terminal lobe of N. Escape mutants showed single mutations at the established binding sites and thus confirm them independently by functional biological criteria.
Because N encapsidates the RNA in a tight manner, a close coordination of the L‐P complex with the N‐RNA template is necessary to allow RNA access and polymerase processivity. Our VHHs are likely to perturb this coordination, and there are at least two possible ways to do so: first, by competing with, or sterically excluding the binding of other viral or host proteins to the N‐RNA template or, by preventing a conformational change and dislocation of N from the RNA during the process of transcription. Both scenarios could, in combination or alone, hamper polymerase processivity, transcription, and replication.
The structural data presented here readily explain the inhibitory mechanism of VHH 1307, which is further supported by immunoprecipitation experiments. The binding site of VHH 1307 overlaps with the binding site of the polymerase cofactor P. Although VHH 1307 effectively blocks transcription and likely replication, the inhibitory properties can be overcome by infecting with an increased virus dose. This could be due to complete absorption of the inhibitory VHH and a surplus of N‐RNA genomes that thus remain unbound by a VHH. Alternatively, the local concentration of P may suffice to outcompete the VHH. The latter explanation would be in line with the model of a highly dynamic interaction of N and the C‐terminal domain of P during transcription and replication 12.
The presumptive binding site for VHH 1001 overlaps with the binding site of the N‐terminus of P, as described when bound to N0 7. Our data suggest that full‐length P binds to this part of N also when N is associated with RNA. The inhibitory mechanism of VHH 1001 might be similar to that of VHH 1307: displacement of P or preventing P from binding to N perturbs polymerase processivity. The key differences are that VHH 1307 competes with the PCTD, whereas VHH 1001 competes with P's N‐terminus and that VHH 1307 is susceptible to higher doses of virus while VHH 1001 still protects the cells. The phenotypes observed likely indicate that both P interaction sites of N are important during polymerase activity, perhaps to allow dynamic binding of L/P to N during processive movement.
In contrast, the binding site for 1004 does not readily explain its antiviral mechanism. VHH 1004 can bind to the N/P complex, thus likely inhibit transcription in a manner different from that of VHH 1307 and VHH 1001. Further, VHH 1004 shows inhibitory potential against high doses of virus despite its comparatively low levels of expression. Nevertheless, we did not find VHH 1004 to be a potent inhibitor in in vitro transcription assays with purified components, suggesting that VHH 1004 only indirectly affects mRNA transcription in infected cells, for example by interfering with genome replication or proper coordination of replicated RNA molecules, which ultimately substantially reduces the number of templates available for transcription 9. The detectable levels of GFP expression in VHH‐producing cells infected at high MOIs might thus result from primary transcription of the incoming genomes. The strong VHH 1004‐mediated inhibition—despite low expression levels—might indicate that the VHH does not have to bind to each protomer to block replication. However, we also cannot exclude the possibility that in vitro transcription does not accurately recapitulate the events that occur in the cytosol of cells during infection. Differences between transcription in cells and in vitro that could have an impact include the non‐natural ratios of N and L proteins, a structural difference of the isolated N‐RNA template compared to its more flexible representation in the cytosol, or the lack of an additional factor. From the results obtained for VHH 1004, we conclude that not only the N‐arm, but also the entire N‐lobe of VSV N is of relevance for efficient replication.
In addition to the mechanisms of VSV inhibition inferred above, the VHHs we have characterized might have additional ways of interfering with the virus life cycle at later steps and might therefore be appropriate tools to study them. Based on its binding site, VHH 1307 might prevent the correct assembly of the “bullet” structure in the virion by steric occlusion. Both VHH 1001 and VHH 1004 might bind to the periphery of the VSV “bullet” and prevent association with the matrix protein. In addition to transcription and replication, these VHHs may therefore also impair virion assembly of VSV.
While binding of VHH 1001 to its escape variants of N was lost as expected, VHH 1004 and 1307 immunoprecipitated increased amounts of their respective escape mutants. The binding kinetics revealed that VHH 1307 dissociates much faster from its N escape mutant compared to WT N and thus that the VHH may be replaced more readily by P, explaining how this mutant avoids VHH‐mediated restriction. The observed increase in recovered protein in the LUMIER assay might be the result primarily of avidity effects that prevent fast dissociation from immobilized VHHs. The escape mechanism for VHH 1004 might operate in a similar fashion, based on more dynamic binding kinetics that allow polymerase processivity. Nevertheless, for the very low‐expressing VHH 1004, increased binding affinity might in fact be advantageous. If transcription of viral genes is unaffected by this VHH as it is in vitro 9, its inhibitory properties could be neutralized by newly produced N, and high affinity and limited dissociation would prevent interference with replication later during infection. Lastly, mutations in N, in the precise binding sites of the VHHs, altered binding kinetics to restore virus propagation.
In conclusion, we have shown that it is possible to use VHHs for a robust cycle of experiments that give insights into the molecular biology of the VHH targets: first, we produced and selected VHHs that impede viral growth; second, we accurately defined the epitope recognized at the structural level; third, we used cytosolically expressed VHHs to select virus variants that escape VHH‐imposed inhibition, and finally—in at least one case—provide a rational explanation for the inhibitory mechanism of the VHH in question using the structure of the VHH in complex with its target as a guide. The defined binding sites leverage the use of these newly identified VHHs as tools to further investigate this non‐segmented RNA virus.
Materials and Methods
Virus, cell lines, and reagents
VSV Indiana GFP was propagated in BHK‐21 cells, which were obtained from ATCC. Clarified, infectious supernatants were used for flow cytometry‐based infection assays. The A549 cell line inducibly expressing VHH‐HA, derived from A549 cells purchased from ATCC, were generated using lentivirus produced with derivatives of pInducer20 17, and selected in the presence of 500 μg/ml G418. HEK 293T cells were obtained from ATCC. Cells were cultivated in DME with 10% FBS (and 500 μg/ml G418 in case of lentivirus‐transduced cell lines). Doxycycline hyclate (Dox) was purchased from Sigma‐Aldrich. Nickel‐nitrilotriacetic acid (NTA) beads were purchased from Qiagen. Mouse anti‐HA.11 (clone 16B12) was acquired from BioLegend. Mouse anti‐HA.11 (clone 16B12) coupled to Dylight (DL) 650 was purchased from Abcam. The GAPDH HRP‐conjugated monoclonal rabbit antibody was purchased from Cell Signaling Technology. The HA‐tag antibody conjugated to HRP was purchased from Thermo Fisher Scientific.
Infection assay
To analyze the effect of the N‐specific VHHs on different doses (MOI) of virus, A549 cells inducibly expressing the VHHs were seeded and VHH expression was induced with 1 μg/ml doxycycline (final concentration). After 24 h, cells were infected with VSV Indiana GFP at an MOI between 0 and 320. Four hours post‐infection, cells were trypsinized, fixed in 4% PFA, and analyzed by flow cytometry using a BD Accuri and the FlowJo software package.
Protein expression and purification
The identification and production of VSV N‐specific VHHs were described earlier 9. VSV N‐RNA and its escape mutant variants were expressed and purified as described elsewhere 18. Sequences encoding the different VHHs with a C‐terminal sortase recognition site (LPETG) followed by a His6‐tag were cloned into a pHEN6 expression vector for periplasmic expression. E. coli WK6 bacteria were transformed with the vector, and expression was induced with 1 mM IPTG at OD600 = 0.6; cells were grown overnight at 30°C. VHHs were retrieved from the periplasm by osmotic shock and purified by Ni‐NTA affinity purification and size‐exclusion chromatography on a Superdex 75 column.
Crystallization
For cocrystallization, VHHs 1004 and 1307 were individually mixed in a 3:1 molar ratio with recombinant VSV N‐RNA and purified by size exclusion on a Superdex 200 (GE Healthcare) column. A single peak of the 20‐mer N‐RNA/VHH complex was collected and VHH binding was confirmed by SDS–PAGE and Coomassie staining. The complex was concentrated to 2 mg/ml in 50 mM Tris/HCl pH 7.5 and 150 mM NaCl buffer. Initial crystal growth was observed in 0.1 M sodium acetate pH 5.0, 1.5 M ammonium sulfate for VHH 1004 and in 0.2 M sodium acetate, 0.1 M tri‐sodium citrate pH 5.5, 5% (w/v) polyethylene glycol 4000 for VHH 1307, both in a vapor diffusion experiment in a 96‐well sitting drop setup (Index HT, Hampton Research; ProComplex, Qiagen). Diffraction quality crystals were grown from crystal seeds in a 24‐well vapor diffusion hanging drop set up over reservoirs of the same buffers where initial crystal growth was observed. Crystallization drops contained equal volumes of protein and reservoir solution. Crystals were cryoprotected in 20% glycerol and flash‐frozen in liquid nitrogen.
Data processing and structure determination
Datasets were collected at the Advanced Photon Source user end station 24‐IDC. Data reduction was performed in HKL2000 19, and all subsequent data‐processing steps were carried out using programs provided through SBGrid 20. The structure of N‐RNA in complex with VHH 1307 was solved by molecular replacement (MR) using the PhaserMR tool from the PHENIX suite 21. The MR solution was easily obtained by first searching for four copies of a N 5‐mer and then VHH 1307. The N‐RNA search model was based on a published structure (PDB ID 2GIC 3), and the VHH 1307 search model was based on αNP‐VHH1 (PDB ID 5TJW 22) after removing the complementary determining regions (CDRs). The RNA was adopted from the previous N‐RNA structure (PDB ID 2GIC). The initial MR model contained all 40 copies of the N‐RNA‐VHH complex that occupy the asymmetric unit. Iterative manual model building in COOT 23 followed by refinement gradually improved the electron density maps and the model statistics. To solve the structure of N‐RNA in complex with VHH 1004, we used the N model of the N‐RNA/VHH 1307 structure for MR. The initial VHH 1004 model was based on VHH 7D12 (PDB ID 4KRL 23), which had the highest degree of sequence similarity of published VHH structures to VHH 1004. We positioned the VHH 1004 model to one N molecule and built the amino acids in the loop regions of the VHH using COOT. By applying the non‐crystallographic symmetry (NCS operators) to the first N/VHH 1004 complex, we generated the four additional N‐VHH complexes of the asymmetric unit. The entire 10‐mer was then subjected to rigid body refinement followed by positional refinement using torsion‐angle NCS restraints. Structure figures were created in PyMOL (Schrödinger LLC). Omit and composite omit maps were calculated with tools from the PHENIX suite 21.
Immunoprecipitation
For immunoprecipitations, VHHs were site‐specifically biotinylated using sortase A and GGG‐biotin as described earlier 24. We bound 2 μg of biotinylated VHH to streptavidin magnetic beads (MyOne DynaBeads; Invitrogen) and added 10 μg of recombinant N or N/P complex. Beads were washed, and bound N or N/P complex was eluted in 0.2 M glycine, pH 2.2, and analyzed by SDS–PAGE (75:1 acrylamide/bis‐acrylamide) and Coomassie staining. Band intensities were quantified in ImageJ.
Generation of escape mutants
For generation of VSV escape mutants, cells expressing the VHHs were grown in DMEM, 10% FCS, 1 μg/ml doxycycline. Cells were infected with WT VSV Indiana at an MOI of 3, and the media was changed to DMEM, 2% FCS, 1 μg/ml doxycycline. Infectious supernatant was transferred to new cells every 24 h, until virus titer was comparable to virus titer in the supernatant of WT cells infected with VSV. For each cell line expressing a different VHH, virus from three plaques was purified and RNA was isolated from infected cells using RNeasy kit (Qiagen, Valencia, Ca), reversely transcribed, and the N gene was sequenced.
LUMIER assay
To analyze binding of VHHs to mutated version of N, we applied LUMIER assays as described in detail before 9. 293T cells were cotransfected with pCAGGS VHH‐HA and pEXPR N‐Renilla as WT or escape mutant variants using Lipofectamine 2000. 24 h post‐transfection, cells were lysed and incubated in 96‐well LUMITRAC 600 plates (Greiner) coated with anti‐HA.11 antibody to capture the VHHs. Activity of the copurified luciferase was quantified by addition of coelenterazine‐containing Renilla luciferase substrate mix (BioLux Gaussia Luciferase Assay Kit, New England BioLabs) and light emission measured using a SpectraMax M3 microplate reader (Molecular Devices).
Biolayer interferometry
The Octed RED96 (Fortebio, Pall) was used to measure affinity and kinetic parameters. Streptavidin biosensors were purchased from Pall, and all measurements were performed in PBS, 1% BSA, 0.005% Tween‐20. N‐RNA and its escape mutant variants were biotinylated via coupling to primary amines with the Chromalink NHS biotin reagent (Solulink, San Diego, CA) for 90 min in 100 mM phosphate buffer pH 7.4, 150 mM NaCl. Streptavidin biosensors were loaded with the biotinylated N‐RNA variants at a concentration of 4 μg/ml. Association and dissociation of VHHs were recorded with dilutions at concentrations between 12.5 and 100 nM. Data were analyzed using the 2:1 heterogeneous ligand binding global fit model.
Statistical analysis
Two‐tailed t‐tests were performed for statistical analysis. Level of significance is shown in each figure (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Author contributions
LH, FIS, BM, SPJW, and HLP conceived and designed the experiments. LH, FIS, KEK, and BM performed the experiments. LH, KEK, and TUS solved the crystal structure. LH, FIS, and HLP wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
This work was supported by a National Institutes of Health Pioneer award to H.I.P. and additional funding from Fujifilm/MediVector. F.I.S. was supported by an Advanced Postdoc.Mobility Fellowship from the Swiss National Science Foundation (SNSF). Part of this work is based on research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus6Mdetector on 24‐ID‐C beam line is funded by a NIH‐ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE‐AC02‐06CH11357.
EMBO Reports (2017) 18: 1027–1037
References
- 1. Morin B, Rahmeh AA, Whelan SPJ (2012) Mechanism of RNA synthesis initiation by the vesicular stomatitis virus polymerase. EMBO J 31: 1320–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ge P, Tsao J, Schein S, Green TJ, Luo M, Zhou ZH (2010) Cryo‐EM model of the bullet‐shaped vesicular stomatitis virus. Science 327: 689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Green TJ, Zhang X, Wertz GW, Luo M (2006) Structure of the vesicular stomatitis virus nucleoprotein‐RNA complex. Science 313: 357–360 [DOI] [PubMed] [Google Scholar]
- 4. Zhang X, Green TJ, Tsao J, Qiu S, Luo M (2008) Role of intermolecular interactions of vesicular stomatitis virus nucleoprotein in RNA encapsidation. J Virol 82: 674–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harouaka D, Wertz GW (2009) Mutations in the C‐terminal loop of the nucleocapsid protein affect vesicular stomatitis virus RNA replication and transcription differentially. J Virol 83: 11429–11439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Green TJ, Rowse M, Tsao J, Kang J, Ge P, Zhou ZH, Luo M (2011) Access to RNA encapsidated in the nucleocapsid of vesicular stomatitis virus. J Virol 85: 2714–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leyrat C, Yabukarski F, Tarbouriech N, Ribeiro EA, Jensen MR, Blackledge M, Ruigrok RWH, Jamin M (2011) Structure of the vesicular stomatitis virus N0‐P complex. PLoS Pathog 7: e1002248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Green TJ, Luo M (2009) Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid‐binding domain of the small polymerase cofactor, P. Proc Natl Acad Sci USA 106: 11713–11718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmidt FI, Hanke L, Morin B, Brewer R, Brusic V, Whelan SPJ, Ploegh HL (2016) Phenotypic lentivirus screens to identify functional single domain antibodies. Nat Microbiol 1: 16080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ashour J, Schmidt FI, Hanke L, Cragnolini J, Cavallari M, Altenburg A, Brewer R, Ingram J, Shoemaker C, Ploegh HL (2015) Intracellular expression of camelid single‐domain antibodies specific for influenza virus nucleoprotein uncovers distinct features of its nuclear localization. J Virol 89: 2792–2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 [DOI] [PubMed] [Google Scholar]
- 12. Wertz GW, Moudy R, Ball LA (2002) Adding genes to the RNA genome of vesicular stomatitis virus: positional effects on stability of expression. J Virol 76: 7642–7650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barrios‐Rodiles M, Bose R, Liu Z, Donovan RS, Shinjo F, Liu Y, Dembowy J, Taylor IW, Luga V, Przulj N et al (2005) High‐throughput mapping of a dynamic signaling network in mammalian cells. Science 307: 1621–1625 [DOI] [PubMed] [Google Scholar]
- 14. van de Sandt CE, Kreijtz JHCM, Rimmelzwaan GF (2012) Evasion of influenza A viruses from innate and adaptive immune responses. Viruses 4: 1438–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKimm‐Breschkin JL (2013) Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance. Influenza Other Respi Viruses 7(Suppl. 1): 25–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li R, Hayward SD (2013) Potential of protein kinase inhibitors for treating herpesvirus‐associated disease. Trends Microbiol 21: 286–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T et al (2011) The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo . Proc Natl Acad Sci USA 108: 3665–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Green TJ, Macpherson S, Qiu S, Lebowitz J, Wertz GW, Luo M (2000) Study of the assembly of vesicular stomatitis virus N protein: role of the P protein. J Virol 74: 9515–9524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- 20. Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P (2013) Collaboration gets the most out of software. Elife 2013: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanke L, Knockenhauer KE, Brewer RC, Van Diest E, Schmidt FI, Schwartz TU, Ploegh HL (2016) The antiviral mechanism of an influenza A virus nucleoprotein‐ specific single‐domain antibody fragment. MBio 7: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmitz KR, Bagchi A, Roovers RC, Van Bergen en Henegouwen PMP, Ferguson KM (2013) Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure 21: 1214–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AEM, Ploegh HL (2013) Site‐specific C‐terminal and internal loop labeling of proteins using sortase‐mediated reactions. Nat Protoc 8: 1787–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File