

Abstract
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterized by a breakdown of self-tolerance in B cells and production of antibodies against nuclear self-antigens. Increasing evidence supports the notion that additional cellular contributors beyond B cells are important for lupus pathogenesis. In this Review, we consider recent advances regarding both pathogenic and regulatory roles of lymphocytes in SLE, beyond the production of IgG autoantibodies. We also discuss various inflammatory effector cell types involved in cytokine production, removal of self-antigens, and responses to autoreactive IgE antibodies. We aim to integrate these ideas to expand the current understanding of the cellular components that contribute to disease progression and ultimately help in the design of novel targeted therapeutics.
1. Heterogeneity of Systemic Lupus Erythematosus
Systemic Lupus Erythematosus (SLE) is a multi-organ autoimmune disorder with an evident but complex heritable component but complex [1,2]. It is caused by a dysregulation of the immune system that results in chronic inflammatory disease directed against self-tissue. The molecular and cellular drivers of this dysregulation, as well as the target organs of this misdirected immune response can be variable, thus explaining the heterogeneity in pathological manifestations. SLE is almost always associated with the presence of serum autoantibodies, supporting the view that a defect in tolerance to common self-antigens is at the center of this disease. The complexity of the genetics of SLE, together with the heterogeneous symptomatic presentation complicates the task of understanding causes and triggers of the associated autoimmune pathology. Finding a common pathological mechanism has been elusive, and so far only general immunosuppressive drugs seem to be markedly effective to treat SLE [3]. Current advances aim to provide novel approaches to define SLE disease and classify diverse pathological mechanisms so that more individualized medical treatments can be utilized.
The bulk of research carried out on human SLE patients or with experimental animal models of lupus has focused on immune cells and processes related to the production and response to antibody. Self-reactive IgG antibodies, a hallmark and one of the diagnostic criteria for SLE, are often assumed to be pathogenic because the immune complexes that are formed with IgG and autoantigens can induce immune activation through Fc receptors and complement [4]. While autoantibodies are commonly present in SLE patients, there are numerous cases of people with high autoantibody titer and no SLE disease. This could point to a multilevel dysregulation that affects both the humoral/antibody and the effector/inflammatory response in the SLE pathological manifestation [5,6]. It is important to note that individual autoantibody specificities vary greatly in their levels of correlation to SLE pathology. For example, anti-DNA antibodies seem more likely to be present during clinical manifestations compared to other commonly found autoantibodies against nuclear components, like Sm or Ro antigens (see Glossary), that do not correlate well with severity of disease [7]. It is likely that the heterogeneous pathology of SLE is in fact a consequence of immune dysregulation across various immunological pathways including many elements such as: i) deficiencies in complement [8], ii) the prevalence of anti-RNA or anti-DNA antibodies [9], and iii) inflammatory components: e.g. mutations in exonucleases like TREX1, which degrade reverse-transcribed DNA retroelements, and results in an IFN-associated disease in humans [10]. Genetic data and gene expression analyses have also pointed to an enhanced type I interferon response in SLE and blockade of this family of cytokines is at the center of several recent clinical trials [11,12]. This specific cytokine profile also implies additional cellular factors beyond antibodies and B cells in the pathology. Most prominently, pDCs are very efficient type I interferon producers and have been linked to SLE both in human and mouse [13,14]. In a previous review article we covered genes and pathways associated with SLE disease, with a discussion of the current knowledge derived from animal models [2]. With this Review, we aim to expand the prevalent view on lupus by highlighting the most current reports, some of which implicate immune cell populations or unusual functions of these cells that might have not been the focus of many SLE studies in the past. As shown in Figure 1, the production of IgG autoantibodies, inflammatory effector responses and cytokines are core components of the SLE pathology, thus implicating B-T cell interactions and IgG-mediated inflammatory cell activation. In the following sections we will introduce additional immune cell types and antibody isotypes, all of which have been described recently in the literature as novel modifiers in SLE. These proposed modifying factors are shown at the bottom of Figure 1, and while some have been verified in their function (e.g. pDCs), others are still in the initial stages of investigation (e.g. NK, CD8+ T cells, basophils).
Figure 1. – Novel Immune Populations Contributing to Development of SLE.
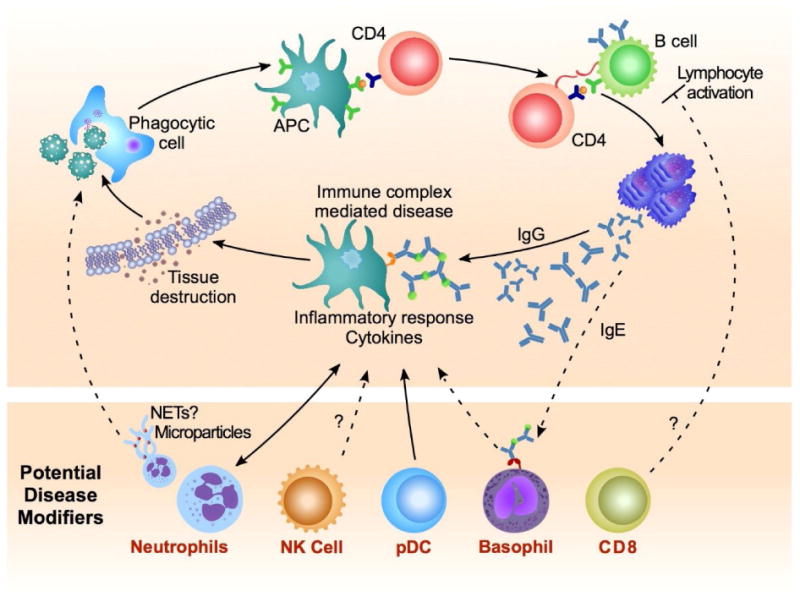
Top frame: Core cellular framework describing the development and progression of systemic lupus. Autoreactive lymphocytes drive the production of autoantibodies and immune complex-mediated disease leading to deleterious inflammatory responses. Bottom frame: Novel cellular modifiers of systemic lupus including: neutrophils acting as a source of self-Ag, cytolytic and inflammatory factors; dysregulation of inflammatory natural killer (NK) cells; cytokine-mediated amplification of disease by plasmacytoid DCs (pDC); inflammatory responses mediated by autoreactive IgE antibody and association with severe aspects of disease; regulation of inflammatory lymphocyte responses by CD8+ T cells. Dashed lines indicate areas requiring additional clinical support. Abbreviations used: APC: antigen-presenting cell, NET: neutrophil extracellular trap
2. Chronic Activation of Lymphocytes in SLE: B Cells Do Not Do It Alone
The processes of lymphocyte development and maturation are designed to eliminate a large majority of self-reactive specificities from circulation. Nevertheless, a sizeable percentage of mature B cells in both healthy humans and mice still generate autoantibodies and even switch to IgG isotypes known to trigger inflammatory responses [15,16]. The higher autoantibody titers observed in SLE can result either from increased accessibility of target self-antigens or from a failure of cellular checkpoints that limit lymphocyte activation. Autoantibodies frequently target common nuclear antigens, likely because their association with nucleic acid components facilitates simultaneous activation of the B cell antigen receptor and intracellular TLRs/DNA-RNA sensors [17]. The high frequency of autoantibody seropositive patients in SLE points to an evident role for B cells, and the presence of IgG switched autoantibodies plus the highly activated CD4+ T cell phenotype indicates a likely role of helper T cells. One of the main limitations in these studies has been the paucity of knowledge regarding antigen specificity of SLE-associated T cells [18-21]. Some recent advances have shed light into the selection of autoreactive CD4+ T cells as well as unusual role of cytotoxic lymphocytes, as we describe in detail below:
2.1 SLE-Associated Helper CD4+ T Cells Might Recognize Post-Translational Modifications of Self-Peptides
Although some experiments on animal models of lupus have suggested that IgG autoantibodies might arise without the need for CD4+ T cell help, the general view is that SLE involves a breach of tolerance at both the B and CD4+ T cell level [19]. Some common SLE autoantigens have been analyzed as possible T cells epitopes. For example, the study of Sm-reactive T cells in DR3 mice has uncovered a possible role of epitope mimicry in the activation of human autoreactive T cells [22]. In another example, activated CD4+ T cells secreting IL-17 and specific for the U1-70 antigen correlate with disease in a mouse model of lupus and a subset of SLE patients [23]. Some possible T cell epitopes could arise from post-translational modification of self-peptides, more frequent in inflammatory conditions. These peptide modifications include citrullination, arginine methylation and oxidation [24-26]. The gathering of complete information on specific T cell epitopes associated with SLE will be invaluable in the search for novel therapeutic tools. For example, transducing T cells with chimeric antigen receptors could allow the targeting of disease-associated HLA molecules bearing chosen auto-antigens, as was recently done by generating alloantigen-specific regulatory T cells (Treg) [27], or as one group found in mice and SLE patients, antigen-specific inflammatory lymphocytes [23].
2.2 Certain CD8+ T Cell Populations Could Be Associated with Lupus Protection
There is limited information regarding the phenotype and functions of CD8+ T cells in human SLE patients. One study found an increased inflammatory cytokine signature and reduced ability of CD8+ T suppressors to restrict proliferation in vitro if the T cells were taken from patients with active lupus versus those in remission [28]. These cells are thought to be non-antigen specific and capable of suppressing both lymphocyte proliferation and cytotoxic function in the absence of disease [29,30]. Perhaps the most compelling data come from two thorough genetic analyses of circulating SLE CD8+ T cells. The first study demonstrated that CD8+ T cells from patients with SLE or autoimmune vasculitis have tend to be memory cells with an enrichment of genes involved in the interleukin-7 receptor (IL-7R) pathway [31]. The same group recently discovered an SLE transcriptional profile that is predictive of long-term disease outcome [32].
Patients whose CD8+ T cells had a transcriptional signature reflective of T cell exhaustion were more likely to remain free of disease flares for longer periods of time. T cell exhaustion, which is generally induced upon chronic antigen exposure, results in the loss of effector function and subsequent upregulation of a number of inhibitory proteins [33]. In the context of SLE, the exhausted T cell state could be reproduced in vitro and depended on signaling through the inhibitory receptor PD-1 [32], providing a potential target of therapeutic benefit.
The human SLE studies we have described in this section reveal correlation between disease manifestations and activation status of CD8+ T cells. Activated CD8+ T cells are often assumed to be pathological due to their cytotoxic function, which is fundamental in the elimination of virally infected or transformed cells. But there is an intriguing possibility that CD8+ T cells can enact a regulatory role by targeting their effect onto other immune cells and effectively diminishing the extent of the immune response. Regulatory CD8+ T cell populations have been found that can limit graft versus host responses, T cell vaccination effects, and delayed type hypersensitivity T cell responses [34-37]. This regulatory function of CD8+ T cells has been mostly studied in animal models of SLE and it is in need of verification in human SLE. In many of the systems tested so far this response is restricted by Qa-1 [38,39], an MHC Class Ib molecule expressed on effector T cells. Some reports suggested that these regulatory CD8+ T cells might use the Fas/FasL system on target T cells, CD8+CD122+CD49dlow populations being the most effective suppressors [40]. The best evidence for a regulatory role of CD8+ T cells in SLE comes from antibody depletion [41] and genetic ablation of CD8+ T cells in lupus-prone mice [42,43]. Remarkably, genetic ablation of CD8+ T cells by a mutation in MHC-I aggravates the severity of disease in two lupus models (BXSB and TLR7 transgenic mice), while T cell ablation does not affect outcome in the MRL/lpr model [44]. This difference could point to a differential need for regulation by CD8+ T cells, perhaps explained by the presence of the lymphoproliferative (lpr) mutation in the MRL/lpr model. So far, conclusions on the regulatory function of CD8+ T cells in lupus have come exclusively from mouse models. Future investigations must be expanded to include the human disease, in which the role of these cells remains an open question.
2.3 Atypical Development and Function of Natural Killer Cells in Lupus
Defined in humans mainly as CD3-CD56+, mature natural killer (NK) cells, like CD8+ T cells, are involved in rapid cytotoxic responses to viruses and tumor surveillance. Antigen-independent recognition and killing of target cells occurs through synergistic activity of several receptors that recognize MHC/HLA molecules or alternative receptors linked to immune cell activation and/or viral infection [45]. A few studies have linked NK cells to human SLE, citing a decreased number and impaired function in lupus patients [46,47]. In work by two groups, increased risk of disease can be associated with polymorphisms in genes encoding NK cell killer receptors and their target HLA molecules [48,49]. A genome-wide association study identified functional variants in some lupus patients in the gene encoding CD226, which is a cell surface receptor on NK cells involved in their adhesion and cytotoxicity [50].
Evidence from lupus-prone mice supports the notion that NK cells can drive lupus pathogenesis. CD226+ NK cells found in the kidney of MRL/lpr mice are associated with renal disease [51]. Adoptive transfer of NK cells from TLR7 transgenic lupus-prone mice is sufficient to induce lupus-like disease in wild type recipients, while genetic ablation of these cells by removal of a critical growth factor, IL-15, greatly reduced pathology in these mice [52]. The atypical NK cells that develop in this model of murine lupus turn on a highly inflammatory and proliferative program and fail to differentiate into a fully mature state [53]. Dysregulation of several microRNAs (miR) could explain the reduced differentiation program of these NK cells, including miR-181, which is necessary for NK cell development in humans [54], and is downregulated in some pediatric lupus patients [55]. While NK cells could represent a promising new therapeutic target, this type of research is still in need of clinical verification to fully understand the role of NK cells during SLE.
3. Inflammatory Effector Cells in SLE: More than Just Cytokines
SLE pathology, though variable among patients, always involves some type of chronic inflammation and correlates with elevated levels of cytokines that can itself be destructive. SLE patients frequently present high serum titers of type I interferon (IFNα/β) [56,57] and a distinct IFN stimulated gene signature [58,59]. In addition, there are reports of cancer treatment regimens containing IFNα that have induced lupus-like symptoms in humans [60]. The differentiation of autoreactive B cells and maturation of myeloid dendritic cells (DCs) that activate autoreactive T cells in SLE is attributed in part to plasmacytoid DC (pDC) production of IFNα [13,61]. Beyond type I IFN, the analysis of serum samples from pre-symptomatic stages of human SLE shows that the earliest indication of disease correlates with higher levels of IL-5 and IFNγ [62]. The cellular origin of these various cytokines is still unclear, but it most likely indicates involvement of a variety of effector cells. The following sections will discuss recent findings regarding inflammatory cell populations in SLE, many of which produce inflammatory cytokines but also enhance presentation of self-antigens in various ways:
3.1 Plasmacytoid Dendritic Cells (pDCs) Amplify Pathology
The development and regulation of pDC function is an area of ongoing discovery, with a primary focus on their role in nucleic acid sensing and type I interferon production [63]. The most current research has shed light into the role for pDCs during the initiation of TLR-mediated inflammation [64]. Various deficiencies that result in depletion of pDCs in mouse models show that when pDCs are absent, the severity of lupus disease is greatly diminished. This effect has been reported in mice deficient in SIGLEC-H [65], IRF8 and SLC15A4 [66], and E2-2/Tcf4 [14]. While these mice suffer from additional deficiencies beyond a lack of pDC, the concordance of these studies strongly suggest a critical role for this population. Experiments using diphtheria toxin-induced cell lethality targeted to pDCs shows that these cells are primarily required at early stages of lupus disease [67]. Antibody depletion or genetic ablation of pDCs in these lupus models causes reduction of autoantibody titers, amelioration of glomerulonephritis, decreased expression of interferon stimulated genes, and increased survival.
The regulation of pDC production of type I IFN responses has therapeutic potential that could be achieved by targeting unique markers of the lineage such as BDCA-2 (also known as CD303, CLEC4C) or immunoglobulin-like transcript 7 (ILT7). BDCA-2 is a cell surface C-type lectin shown in human pDCs to mediate suppression of TLR-induced type I interferon [68,69]. Similarly, ILT7 was shown to inhibit type I IFN production in human pDCs in response to TLR7 and TLR9 agonists [70]. Targeting intrinsic inhibitory regulators of pDCs could prove a more specific alternative to clinical reagents that block IFNα globally. A newly designed monoclonal antibody against BDCA-2 has shown promise, inhibiting healthy human donor and SLE peripheral blood pDC type I IFN production through both Fc dependent and independent mechanisms [71]. If indeed pDCs are primarily involved in disease onset, this therapeutic approach would best apply to early stages of disease or perhaps during remission to prevent relapses.
3.2 Phagocytic Cells Clear Apoptotic Microparticles and Limit Exposure to Nuclear Self-Antigens
Phagocytic cell recognition and clearance of apoptotic cells is critical for maintenance of tissue homeostasis, a process involving dozens of phagocytic receptors and many detailed cellular and molecular events [72]. Failure by macrophages or dendritic cells to remove nuclear material from dying cells provides a potential source of self-antigen that can activate endosomal TLRs and production of autoantibodies associated with SLE. Additionally, genetic variants of a DNA cleavage enzyme produced by phagocytes, DNASE1L3, are associated with SLE [73-75] though the precise reason for decreased production and suboptimal function of these enzymes remains unknown.
Deficient DNA-cleaving enzyme function is found in some lupus patients [76] and is implicated in cellular events associated with lupus pathogenesis in humans and mice including immune complex deposition in tissues and severe glomerulonephritis [77-79]. The removal of microparticle-associated chromatin released by apoptotic cells is a new function ascribed to DNASE1L3 [79]. Apoptotic microparticles, which are present in healthy individuals, contain nuclear antigens [80] and contribute to inflammatory responses in patients with active SLE [81,82], but until recently microparticle contributions to a break in tolerance were largely theoretical. Both mice and humans lacking functional DNASE1L3 develop autoimmunity and produce anti-chromatic antibody following accumulation of DNA on circulating microparticles [79]. Extracellular DNASE1L3 produced by dendritic cells and macrophages was shown to digest genomic DNA on microparticles, preventing the accumulation of anti-chromatin antibodies. The study by Sisirak and colleagues confirms microparticles are an immunogenic source of DNA and define the mechanism by which they break tolerance, expanding our view of this important process.
3.3 The Contribution of Neutrophils and NETs to Lupus
Neutrophils are short-lived myeloid cells that act as first responders to infection; ingesting and neutralizing bacteria, or directly lysing target cells through the release lytic granules [83]. Because of their rapid turnover and high cytolytic capacity neutrophils must be tightly regulated to avoid toxicity to host tissue. Lupus patients with a high number of immature blood neutrophils were found to have genetic signatures of granulopoiesis and IFN production [58]. A causative link between neutrophil activity and pDC production of type I IFN was later established in a cohort of pediatric lupus patients [84], while an epigenetic study of SLE neutrophils revealed robust demethylation of interferon signature genes [85]. These data suggest that neutrophils can exacerbate inflammatory processes involved in lupus.
In 2004, neutrophil extracellular traps (NETs) were described as a new way by which neutrophils might bind and kill bacteria [86]. NETs are formed following a distinct type of cell death termed NETosis that results in the expulsion of the cell's DNA and the formation of bacterial “traps” made of DNA and containing antimicrobial effectors, including histones [87,88]. NETs have since been described in individuals with lupus [84,89,90], and dying neutrophil debris is thought to serve as a potential source of both auto-antigens such as C1q and histones [90-92], as well as alarmins such as HMGB1 [84]. Recently, oxidized mitochondrial DNA from NETs was shown to be interferogenic [93], though another group asserts this can occur in the absence of NETosis [94]. It is still undecided whether NETs can be the physiological source of stimulatory DNA. For example, MRL mice deficient in Nox2, a factor essential for NETosis, show aggravation of lupus disease. This result argues for a non-essential role of NET formation in SLE pathology [95]. Interestingly, these studies identified the presence of potentially pathogenic anti-oxidized mitochondrial DNA antibodies associated with neutrophils from lupus patients [96].
Several studies have described biological processes associated with NET formation, activity, and breakdown, which could inform the design of therapeutics. Decondensation of chromatin in humans and mice requires citrullination of histones, and could increase antigenicity of self-peptides through this and other post-translational modifications [97,98].
3.4 Unexpected Role for Basophils and in Lupus Pathogenesis
The presence of IgG autoantibodies is a hallmark of lupus disease, and severe tissue damage mediated by IgG immune complexes requires Fcγ receptors in lupus-prone mice [99]. On the other hand high IgG titers are not necessarily correlated with the highest severity of pathology [100], and mice can develop glomerulonephritis, arthritis, and other typical qualities of rheumatoid-based autoimmunity in the absence of anti-DNA antibodies [101,102]. IgE can initiate inflammatory responses through histamine, leukotrienes, and many cytokines and chemokines released after binding to the high affinity IgE receptor, FcεRI, which is expressed mainly on mast cells and basophils in mice and humans, but can also be found in humans on eosinophils, monocytes and some dendritic cells [103]. A strong association of autoreactive IgE with lupus disease was found in separate patient cohorts [104-107], while the most recent of these three studies identified that anti-dsDNA IgE antibodies could induce large amounts of type I IFN production from plasmacytoid DCs [106]. Elevated autoreactive IgE in SLE correlates with increased basophil activity [108], and is concentrated in patients with active disease. Although the total IgE levels in SLE do not reach titers comparable to those present in patients with atopy, it is worth noting that self-reactive IgE might have different pathogenic effect compared to allergen-directed IgE. It is now appreciated that the FcεRI on granulocytes can discriminate between high and low affinity IgE-antigen interactions, which results in distinct in vivo outcomes [109,110]. This discovery should help to fine-tune existing predictive models of disease.
In mice lacking the inhibitory receptor FcγRIIB, a well-studied model of lupus, IgE deficiency is protective, delaying disease onset and lessening its severity as measured by reduced total autoantibody production and amelioration of tissue pathology [108]. In lupus-prone Lyn-deficient mice, IgE autoantibodies and active basophils lead to glomerulonephritis [105]. How Lyn contributes to IgE production and basophil activity in human disease is unknown, though polymorphisms in LYN are associated with SLE [111,112]. While anti-IgE therapy is beneficial for asthma and allergic diseases [113] and could prove useful for lupus, further understanding of the molecular events that precipitate following IgE-self antigen complex binding to their receptor is still required.
4. Concluding Remarks
Lupus is a multi-factorial autoimmune disease influenced by various interdependent cellular pathways and complex genetics. Its heterogeneous symptomatic presentation is a reflection of the systemic occurrence of the target self-antigen but it also derives from the diversity in mechanistic triggers. A perceived constant in diagnosing and defining SLE has been the presence of IgG autoantibodies targeting common nuclear components, as well as an IFN gene expression signature. Recent clinical trials aimed at blocking type I IFN [12] or aimed at reducing IgG autoantibodies by targeting B cell survival [114] have provided only mild improvements in SLE clinical outcomes. Thus, there is need for improved understanding of factors beyond B cells and IFN to inspire novel and more individualized therapeutic targets in SLE.
Recent advances have shed light on a number of additional disease components at various stages of the lupus pathology: nuclease deficiencies and granulocyte extrusions lead to increased exposure of nuclear components at the onset of SLE; pDCs are particularly important interferon producers early in disease progression; IgE and its downstream effectors may aggravate ongoing pathology; cytotoxic cells could potentially regulate antibody production and inflammatory responses. Each of these factors, as presented in Figure 1, adds another layer of modulation to the complexity of lupus and represents a new possibility for novel therapy (see Outstanding Questions).
Trends Box.
Mechanistic models of SLE disease take into account the high prevalence of serum IgG autoantibodies and interferon gene signature. However, those factors do not always correlate with severity of pathology in clinical cases, and a more expanded view of immune pathways involved in SLE seems merited.
Recent advances have provided new insights into the specificity of CD4 helper responses, the potential role of activated natural killer cells and a possible regulatory function of CD8 cells in SLE.
Inflammatory cells (DC, phagocytes, neutrophils, basophils) are implicated in exacerbation of SLE not only by their cytokine production, but also by exposing nuclear components that engage innate pathways and induce autoreactivity, or by responding to autoreactive IgE antibodies.
Outstanding Questions Box.
What further significance, beyond antibody producing B cells and the helper CD4+ T cells that promote such responses, might be attributed to lymphocytic populations in the pathogenesis of SLE?
What is the functional significance of the transcriptional profile of CD8 exhaustion that has been linked with improved outcomes in human lupus?
What kind of effector responses are induced by autoreactive IgE? Would IgE blockade offer a new treatment alternative for lupus?
Can future treatments for SLE be tailored to patient groups with particular mechanistic triggers such as nuclease deficiencies, complement deficiencies, high interferon levels, low threshold of lymphocyte activation, etc?
Acknowledgments
The authors' work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. We would like to thank Austin Athman of Visual & Medical Arts, Research Technologies Branch, NIAID, NIH, for graphic design assistance.
Glossary
- Atypical natural killer cells
Mouse NK cells defined by the surface expression profile NK1.1(+)CD11c(+)CD122(+)MHC class II(+). They expand in some mouse models of lupus
- HLA-DR3 Tg mice
These mice express human leukocyte antigen DR3, resulting in development of mouse lymphoid cells that utilize human MHC for antigen presentation
- Immunoglobulin-like transcript 7 (ILT7)
a surface receptor shown to inhibit type I IFN production in human pDCs in response to TLR7 and TLR9 agonists
- Lyn-deficient mice
a member of the Src family tyrosine kinase that regulates immune cell activation. Lyn deficiency in mice can result in a spontaneous systemic lymphoproliferative disease that is used to model aspects of human lupus
- MRL/lpr
a combination of Murphy Roths Large (MRL) mice with a spontaneous mutation causing lymphoproliferation (lpr), later identified as a retrotransposon insertion that disrupts the Fas gene, which is essential for normal lymphocyte apoptosis and homeostasis. MRL/lpr animals develop aspects of human lupus including B-cell hyperactivity, circulating immune complexes, lymphoid hyperplasia and glomerulonephritis
- Neutrophil extracellular trap (NET)
a network of extracellular fibers released from a neutrophil and composed primarily of nuclear or mitochondrial components. NETs have been proposed as a source of self-antigen during progression of autoimmunity
- NETosis
a form of neutrophil cell death during which the release of decondensed chromatin and granular contents into the extracellular space is proposed as a mechanism of controlling pathogen replication
- FcγRIIB
a surface receptor on B cells and some myeloid cells that is responsible for downregulating immune responses to IgG. Mice deficient in FcγRIIB develop a spontaneous lupus-like disease that includes lymphocyte hyperactivity, elevated immunoglobulin levels and the presence of anti-nuclear antibodies, and multi-organ tissue pathology including glomerulonephritis
- Ro antigens
also known as Sjögren's-syndrome-related antigen A (SSA), antibodies against this nuclear antigen are prevalent in many autoimmune diseases including lupus
- Smith antigens (Sm)
a family of RNA-binding non-histone nuclear proteins composed of several polypeptides. Anti-Smith autoantibodies are commonly found in lupus patients
- T cell exhaustion
a state of T cell dysfunction that occurs in the presence of infection- or cancer-induced chronic stimulation of lymphocytes. Exhausted T cells are hypofunctional and are distinguished from effector or memory T cells by the sustained protein expression of several inhibitory co-receptors as well as an altered transcriptional profile
- Toll-like receptor 7 transgenic mice (TLR7[Tg])
overexpression of TLR7 results in a lupus-like systemic disease. These mice exhibit elevated antibody production and severe inflammatory responses, as well as multi-organ pathology including glomerulonephritis
- U1-70
part of the U1-small nuclear ribonucleoprotein (U1-snRNP) complex, a component of the spliceosome. Antibodies against U1-snRNP and U1-70 are found in some lupus patients and mouse models; U1-70-specific CD4+ T cells were recently found in the MRL/lpr mouse model and can be tracked using tetramer staining.
Footnotes
Conflict of Interest: The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Teruel M, Alarcón-Riquelme ME. The genetic basis of systemic lupus erythematosus: What are the risk factors and what have we learned. J Autoimmun. 2016;74:161–175. doi: 10.1016/j.jaut.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Crampton SP, et al. Linking susceptibility genes and pathogenesis mechanisms using mouse models of systemic lupus erythematosus. Dis Model Mech. 2014;7:1033–1046. doi: 10.1242/dmm.016451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu D, et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin Immunol. 2013;9:30. doi: 10.1186/1710-1492-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suurmond J, Diamond B. Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest. 2015;125:2194–2202. doi: 10.1172/JCI78084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olsen NJ, Karp DR. Autoantibodies and SLE: the threshold for disease. Nat Rev Rheumatol. 2014;10:181–186. doi: 10.1038/nrrheum.2013.184. [DOI] [PubMed] [Google Scholar]
- 6.Abeles AM, Abeles M. The clinical utility of a positive antinuclear antibody test result. Am J Med. 2013;126:342–348. doi: 10.1016/j.amjmed.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 7.Waldman M, Madaio MP. Pathogenic autoantibodies in lupus nephritis. Lupus. 2005;14:19–24. doi: 10.1191/0961203305lu2054oa. [DOI] [PubMed] [Google Scholar]
- 8.Leffler J, et al. The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis. 2014;73:1601–1606. doi: 10.1136/annrheumdis-2014-205287. [DOI] [PubMed] [Google Scholar]
- 9.Bentow C, et al. International multi-center evaluation of a novel chemiluminescence assay for the detection of anti-dsDNA antibodies. Lupus. 2016;25:864–872. doi: 10.1177/0961203316640917. [DOI] [PubMed] [Google Scholar]
- 10.Rice G, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petri M, et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus. 2009;18:980–989. doi: 10.1177/0961203309105529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirou KA, Gkrouzman E. Anti-interferon alpha treatment in SLE. Clin Immunol. 2013;148:303–312. doi: 10.1016/j.clim.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Blanco P, et al. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 14.Sisirak V, et al. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med. 2014;211:1969–1976. doi: 10.1084/jem.20132522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe M, et al. Anti-cytokine autoantibodies are ubiquitous in healthy individuals. FEBS Lett. 2007;581:2017–2021. doi: 10.1016/j.febslet.2007.04.029. [DOI] [PubMed] [Google Scholar]
- 16.Tiller T, et al. Autoreactivity in human IgG+ memory B cells. Immunity. 2007;26:205–213. doi: 10.1016/j.immuni.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avalos AM, et al. Regulation of autoreactive B cell responses to endogenous TLR ligands. Autoimmunity. 2010;43:76–83. doi: 10.3109/08916930903374618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358:929–939. doi: 10.1056/NEJMra071297. [DOI] [PubMed] [Google Scholar]
- 19.Hoffman RW. T cells in the pathogenesis of systemic lupus erythematosus. Clin Immunol. 2004;113:4–13. doi: 10.1016/j.clim.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Thapa DR, et al. Longitudinal analysis of peripheral blood T cell receptor diversity in patients with systemic lupus erythematosus by next-generation sequencing. Arthritis Res Ther. 2015;17:132. doi: 10.1186/s13075-015-0655-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sui W, et al. Composition and variation analysis of the TCR β-chain CDR3 repertoire in systemic lupus erythematosus using high-throughput sequencing. Mol Immunol. 2015;67:455–464. doi: 10.1016/j.molimm.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 22.Deshmukh US, et al. HLA-DR3 restricted T cell epitope mimicry in induction of autoimmune response to lupus-associated antigen SmD. J Autoimmun. 2011;37:254–262. doi: 10.1016/j.jaut.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kattah NH, et al. Tetramers reveal IL-17-secreting CD4+ T cells that are specific for U1-70 in lupus and mixed connective tissue disease. Proc Natl Acad Sci USA. 2015;112:3044–3049. doi: 10.1073/pnas.1424796112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dieker J, Muller S. Epigenetic histone code and autoimmunity. Clin Rev Allergy Immunol. 2010;39:78–84. doi: 10.1007/s12016-009-8173-7. [DOI] [PubMed] [Google Scholar]
- 25.Radic M, Muller S. Epigenetics of autoantigens: new opportunities for therapy of autoimmune diseases. Genet Epigenet. 2013;5:63–70. doi: 10.4137/GEG.S12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang ML, et al. Lupus autoimmunity altered by cellular methylation metabolism. Autoimmunity. 2013;46:21–31. doi: 10.3109/08916934.2012.732133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacDonald KG, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. 2016;126:1413–1424. doi: 10.1172/JCI82771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Filaci G, et al. Impairment of CD8+ T suppressor cell function in patients with active systemic lupus erythematosus. J Immunol. 2001;166:6452–6457. doi: 10.4049/jimmunol.166.10.6452. [DOI] [PubMed] [Google Scholar]
- 29.Filaci G, et al. Nonantigen specific CD8+ T suppressor lymphocytes originate from CD8+CD28- T cells and inhibit both T-cell proliferation and CTL function. Hum Immunol. 2004;65:142–156. doi: 10.1016/j.humimm.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Filaci G, et al. Non-antigen-specific CD8(+) T suppressor lymphocytes in diseases characterized by chronic immune responses and inflammation. Ann N Y Acad Sci. 2005;1050:115–123. doi: 10.1196/annals.1313.013. [DOI] [PubMed] [Google Scholar]
- 31.McKinney EF, et al. T cell transcription signature predicts prognosis in autoimmune disease. Nat Med. 2010;16:586–591. doi: 10.1038/nm.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKinney EF, et al. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523:612–616. doi: 10.1038/nature14468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng J, et al. Human CD8+ regulatory T cells inhibit GVHD and preserve general immunity in humanized mice. Sci Transl Med. 2013;5 doi: 10.1126/scitranslmed.3004943. 168ra9. [DOI] [PubMed] [Google Scholar]
- 35.Jiang H, et al. T cell vaccination induces T cell receptor Vbeta-specific Qa-1-restricted regulatory CD8(+) T cells. Proc Natl Acad Sci USA. 1998;95:4533–4537. doi: 10.1073/pnas.95.8.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varthaman A, et al. Control of T cell reactivation by regulatory Qa-1-restricted CD8+ T cells. J Immunol. 2010;184:6585–6591. doi: 10.4049/jimmunol.0903109. [DOI] [PubMed] [Google Scholar]
- 37.Hu D, et al. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat Immunol. 2004;5:516–523. doi: 10.1038/ni1063. [DOI] [PubMed] [Google Scholar]
- 38.Kim HJ, et al. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature. 2010;467:328–332. doi: 10.1038/nature09370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim HJ, et al. CD8+ T regulatory cells express the Ly49 Class I MHC receptor and are defective in autoimmune prone B6-Yaa mice. Proc Natl Acad Sci USA. 2011;108:2010–2015. doi: 10.1073/pnas.1018974108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akane K, et al. CD8+CD122+CD49dlow regulatory T cells maintain T-cell homeostasis by killing activated T cells via Fas/FasL-mediated cytotoxicity. Proc Natl Acad Sci USA. 2016;113:2460–2465. doi: 10.1073/pnas.1525098113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adachi Y, et al. Effects of administration of monoclonal antibodies (anti-CD4 or anti-CD8) on the development of autoimmune diseases in (NZW × BXSB)F1 mice. Immunobiology. 1998;198:451–464. doi: 10.1016/s0171-2985(98)80052-1. [DOI] [PubMed] [Google Scholar]
- 42.McPhee CG, et al. MHC Class I Family Proteins Retard Systemic Lupus Erythematosus Autoimmunity and B Cell Lymphomagenesis. J Immunol. 2011;187:4695–4704. doi: 10.4049/jimmunol.1101776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morawski PA, et al. Non-pathogenic tissue-resident CD8(+) T cells uniquely accumulate in the brains of lupus-prone mice. Sci Rep. 2017;7:40838. doi: 10.1038/srep40838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koh DR, et al. Murine lupus in MRL/lpr mice lacking CD4 or CD8 T cells. Eur J Immunol. 1995;25:2558–2562. doi: 10.1002/eji.1830250923. [DOI] [PubMed] [Google Scholar]
- 45.Long EO, et al. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shibatomi K, et al. A novel role for interleukin-18 in human natural killer cell death: high serum levels and low natural killer cell numbers in patients with systemic autoimmune diseases. Arthritis Rheum. 2001;44:884–892. doi: 10.1002/1529-0131(200104)44:4<884::AID-ANR145>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 47.Ye Z, et al. Differential expression of natural killer activating and inhibitory receptors in patients with newly diagnosed systemic lupus erythematosus. Int J Rheum Dis. 2016;19:613–621. doi: 10.1111/1756-185X.12289. [DOI] [PubMed] [Google Scholar]
- 48.Pellett F, et al. KIRs and autoimmune disease: studies in systemic lupus erythematosus and scleroderma. Tissue Antigens. 2007;69:106–108. doi: 10.1111/j.1399-0039.2006.762_6.x. [DOI] [PubMed] [Google Scholar]
- 49.Akhtari M, et al. Analysis of killer cell immunoglobulin-like receptors and their human leukocyte antigen-ligands gene polymorphisms in Iranian patients with systemic lupus erythematosus. Lupus. 2016;25:1244–1253. doi: 10.1177/0961203316638931. [DOI] [PubMed] [Google Scholar]
- 50.Sun C, et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet. 2016;48:323–330. doi: 10.1038/ng.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang Z, et al. Involvement of CD226+ NK cells in immunopathogenesis of systemic lupus erythematosus. J Immunol. 2011;186:3421–3431. doi: 10.4049/jimmunol.1000569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Voynova E, et al. Cutting Edge: Induction of Inflammatory Disease by Adoptive Transfer of an Atypical NK Cell Subset. J Immunol. 2015;195:806–809. doi: 10.4049/jimmunol.1500540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voynova EN, et al. Expansion of an atypical NK cell subset in mouse models of systemic lupus erythematosus. J Immunol. 2015;194:1503–1513. doi: 10.4049/jimmunol.1402673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cichocki F, et al. Cutting edge: microRNA-181 promotes human NK cell development by regulating Notch signaling. J Immunol. 2011;187:6171–6175. doi: 10.4049/jimmunol.1100835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lashine YA, et al. Expression signature of microRNA-181-a reveals its crucial role in the pathogenesis of paediatric systemic lupus erythematosus. Clin Exp Rheumatol. 2011;29:351–357. [PubMed] [Google Scholar]
- 56.Hooks JJ, et al. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979;301:5–8. doi: 10.1056/NEJM197907053010102. [DOI] [PubMed] [Google Scholar]
- 57.Preble OT, et al. Systemic lupus erythematosus: presence in human serum of an unusual acid-labile leukocyte interferon. Science. 1982;216:429–431. doi: 10.1126/science.6176024. [DOI] [PubMed] [Google Scholar]
- 58.Bennett L, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baechler EC, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rönnblom LE, et al. Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J Intern Med. 1990;227:207–210. doi: 10.1111/j.1365-2796.1990.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 61.Jego G, et al. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–234. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 62.Lu R, et al. Dysregulation of innate and adaptive serum mediators precedes systemic lupus erythematosus classification and improves prognostic accuracy of autoantibodies. J Autoimmun. 2016;74:182–193. doi: 10.1016/j.jaut.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gilliet M, et al. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 64.Zhou Z, et al. Phenotypic and functional alterations of pDCs in lupus-prone mice. Sci Rep. 2016;6:20373. doi: 10.1038/srep20373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takagi H, et al. Plasmacytoid dendritic cells are crucial for the initiation of inflammation and T cell immunity in vivo. Immunity. 2011;35:958–971. doi: 10.1016/j.immuni.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 66.Baccala R, et al. Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proc Natl Acad Sci USA. 2013;110:2940–2945. doi: 10.1073/pnas.1222798110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rowland SL, et al. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med. 2014;211:1977–1991. doi: 10.1084/jem.20132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dzionek A, et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J Exp Med. 2001;194:1823–1834. doi: 10.1084/jem.194.12.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jähn PS, et al. BDCA-2 signaling inhibits TLR-9-agonist-induced plasmacytoid dendritic cell activation and antigen presentation. Cell Immunol. 2010;265:15–22. doi: 10.1016/j.cellimm.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 70.Cao W, et al. Plasmacytoid dendritic cell-specific receptor ILT7-Fc epsilonRI gamma inhibits Toll-like receptor-induced interferon production. J Exp Med. 2006;203:1399–1405. doi: 10.1084/jem.20052454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pellerin A, et al. Anti-BDCA2 monoclonal antibody inhibits plasmacytoid dendritic cell activation through Fc-dependent and Fc-independent mechanisms. EMBO Mol Med. 2015;7:464–476. doi: 10.15252/emmm.201404719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Poon IKH, et al. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14:166–180. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Al-Mayouf SM, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. 2011;43:1186–1188. doi: 10.1038/ng.975. [DOI] [PubMed] [Google Scholar]
- 74.Özçakar ZB, et al. DNASE1L3 mutations in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum. 2013;65:2183–2189. doi: 10.1002/art.38010. [DOI] [PubMed] [Google Scholar]
- 75.Carbonella A, et al. An autosomal recessive DNASE1L3-related autoimmune disease with unusual clinical presentation mimicking systemic lupus erythematosus. Lupus. 2016 Nov 7; doi: 10.1177/0961203316676382. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 76.Chitrabamrung S, et al. Serum deoxyribonuclease I and clinical activity in systemic lupus erythematosus. Rheumatol Int. 1981;1:55–60. doi: 10.1007/BF00541153. [DOI] [PubMed] [Google Scholar]
- 77.Napirei M, et al. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25:177–181. doi: 10.1038/76032. [DOI] [PubMed] [Google Scholar]
- 78.Fenton K, et al. Anti-dsDNA antibodies promote initiation, and acquired loss of renal Dnase1 promotes progression of lupus nephritis in autoimmune (NZBxNZW)F1 mice. PLoS ONE. 2009;4:e8474. doi: 10.1371/journal.pone.0008474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sisirak V, et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell. 2016;166:88–101. doi: 10.1016/j.cell.2016.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ullal AJ, et al. Microparticles as antigenic targets of antibodies to DNA and nucleosomes in systemic lupus erythematosus. J Autoimmun. 2011;36:173–180. doi: 10.1016/j.jaut.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 81.Niessen A, et al. Apoptotic-cell-derived membrane microparticles and IFN-α induce an inflammatory immune response. J Cell Sci. 2015;128:2443–2453. doi: 10.1242/jcs.162735. [DOI] [PubMed] [Google Scholar]
- 82.Dieker J, et al. Circulating Apoptotic Microparticles in Systemic Lupus Erythematosus Patients Drive the Activation of Dendritic Cell Subsets and Prime Neutrophils for NETosis. Arthritis Rheumatol. 2016;68:462–472. doi: 10.1002/art.39417. [DOI] [PubMed] [Google Scholar]
- 83.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 84.Garcia-Romo GS, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3 doi: 10.1126/scitranslmed.3001201. 73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Coit P, et al. Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils. J Autoimmun. 2015;58:59–66. doi: 10.1016/j.jaut.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brinkmann V, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 87.Fuchs TA, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Steinberg BE, Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE. 2007;379:pe11. doi: 10.1126/stke.3792007pe11. [DOI] [PubMed] [Google Scholar]
- 89.Lande R, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3 doi: 10.1126/scitranslmed.3001180. 73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Villanueva E, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187:538–552. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leffler J, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012;188:3522–3531. doi: 10.4049/jimmunol.1102404. [DOI] [PubMed] [Google Scholar]
- 92.Liu CL, et al. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res Ther. 2012;14:R25. doi: 10.1186/ar3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lood C, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22:146–153. doi: 10.1038/nm.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Caielli S, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. 2016;213:697–713. doi: 10.1084/jem.20151876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Campbell AM, et al. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med. 2012;4 doi: 10.1126/scitranslmed.3004801. 157ra141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122:2784–2794. doi: 10.1182/blood-2013-04-457671. [DOI] [PubMed] [Google Scholar]
- 97.Wang Y, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li P, et al. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207:1853–1862. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Clynes R, et al. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279:1052–1054. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- 100.Conti F, et al. Systemic Lupus Erythematosus with and without Anti-dsDNA Antibodies: Analysis from a Large Monocentric Cohort. Mediators Inflamm. 2015;2015:328078. doi: 10.1155/2015/328078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nishimura H, et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 102.Christensen SR, et al. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kinet JP. The high-affinity IgE receptor (Fc epsilon RI): from physiology to pathology. Annu Rev Immunol. 1999;17:931–972. doi: 10.1146/annurev.immunol.17.1.931. [DOI] [PubMed] [Google Scholar]
- 104.Dema B, et al. Autoreactive IgE is prevalent in systemic lupus erythematosus and is associated with increased disease activity and nephritis. PLoS ONE. 2014;9:e90424. doi: 10.1371/journal.pone.0090424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Charles N, et al. Basophils and the T helper 2 environment can promote the development of lupus nephritis. Nat Med. 2010;16:701–707. doi: 10.1038/nm.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Henault J, et al. Self-reactive IgE exacerbates interferon responses associated with autoimmunity. Nat Immunol. 2016;17:196–203. doi: 10.1038/ni.3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sanjuan MA, et al. Role of IgE in autoimmunity. J Allergy Clin Immunol. 2016;137:1651–1661. doi: 10.1016/j.jaci.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 108.Dema B, et al. Immunoglobulin E plays an immunoregulatory role in lupus. J Exp Med. 2014;211:2159–2168. doi: 10.1084/jem.20140066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Suzuki R, et al. Molecular editing of cellular responses by the high-affinity receptor for IgE. Science. 2014;343:1021–1025. doi: 10.1126/science.1246976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Suzuki R, et al. New insights on the signaling and function of the high-affinity receptor for IgE. Curr Top Microbiol Immunol. 2015;388:63–90. doi: 10.1007/978-3-319-13725-4_4. [DOI] [PubMed] [Google Scholar]
- 111.Liossis SN, et al. B-cell kinase lyn deficiency in patients with systemic lupus erythematosus. J Investig Med. 2001;49:157–165. doi: 10.2310/6650.2001.34042. [DOI] [PubMed] [Google Scholar]
- 112.Lu R, et al. Genetic associations of LYN with systemic lupus erythematosus. Genes Immun. 2009;10:397–403. doi: 10.1038/gene.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Segal M, et al. Anti-immunoglobulin e therapy. World Allergy Organ J. 2008;1:174–183. doi: 10.1097/WOX.0b013e318187a310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Navarra SV, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377:721–731. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]