

ABSTRACT
Bacterial conjugation is a mechanism of horizontal DNA transfer. The relaxase TrwC of the conjugative plasmid R388 cleaves one strand of the transferred DNA at the oriT gene, covalently attaches to it, and leads the single-stranded DNA (ssDNA) into the recipient cell. In addition, TrwC catalyzes site-specific integration of the transferred DNA into its target sequence present in the genome of the recipient bacterium. Here, we report the analysis of the efficiency and specificity of the integrase activity of TrwC in human cells, using the type IV secretion system of the human pathogen Bartonella henselae to introduce relaxase-DNA complexes. Compared to Mob relaxase from plasmid pBGR1, we found that TrwC mediated a 10-fold increase in the rate of plasmid DNA transfer to human cells and a 100-fold increase in the rate of chromosomal integration of the transferred DNA. We used linear amplification-mediated PCR and plasmid rescue to characterize the integration pattern in the human genome. DNA sequence analysis revealed mostly reconstituted oriT sequences, indicating that TrwC is active and recircularizes transferred DNA in human cells. One TrwC-mediated site-specific integration event was detected, proving that TrwC is capable of mediating site-specific integration in the human genome, albeit with very low efficiency compared to the rate of random integration. Our results suggest that TrwC may stabilize the plasmid DNA molecules in the nucleus of the human cell, probably by recircularization of the transferred DNA strand. This stabilization would increase the opportunities for integration of the DNA by the host machinery.
IMPORTANCE Different biotechnological applications, including gene therapy strategies, require permanent modification of target cells. Long-term expression is achieved either by extrachromosomal persistence or by integration of the introduced DNA. Here, we studied the utility of conjugative relaxase TrwC, a bacterial protein with site-specific integrase activity in bacteria, as an integrase in human cells. Although it is not efficient as a site-specific integrase, we found that TrwC is active in human cells and promotes random integration of the transferred DNA in the human genome, probably acting as a DNA chaperone until it is integrated by host mechanisms. TrwC-DNA complexes can be delivered to human cells through a type IV secretion system involved in pathogenesis. Thus, TrwC could be used in vivo to transfer the DNA of interest into the appropriate cell and promote its integration. If used in combination with a site-specific nuclease, it could lead to site-specific integration of the incoming DNA by homologous recombination.
KEYWORDS: bacterial conjugation, genome editing, transkingdom DNA transfer, type IV secretion
INTRODUCTION
Bacterial conjugation is an efficient mechanism of horizontal DNA transfer which confers bacteria an elevated level of genomic plasticity (1). DNA is transferred by conjugation from a donor to a recipient bacterium through a protein complex known as conjugative apparatus (2). In Gram-negative bacteria, the conjugative machinery is composed of three functional modules (3): (i) the relaxosome, a complex formed by the DNA to be transferred (in particular the site known as origin of transfer [oriT]) and the proteins responsible for DNA processing, which include a relaxase and one or more accessory proteins; (ii) the type IV secretion system (T4SS), a multiprotein complex organized in a transmembranal conduit that spans both inner and outer membranes; and (iii) the coupling protein (T4CP), a DNA-dependent ATPase which brings together the two previous components and is believed to play a crucial role in substrate selection. The translocated substrate is the relaxase covalently linked to the transferred DNA strand.
R388 is a conjugative plasmid with a broad host range that belongs to the IncW incompatibility group (4). The 15-kb transfer region can be separated into an Mpf (mating pair formation) region, which encodes the T4SS apparatus, and a Dtr (DNA transfer and replication) region encoding the T4CP and the relaxosome (5). The Dtr region is composed of an oriT of 330 bp in length, the relaxase TrwC, and two accessory proteins, the plasmid-encoded TrwA and the host-encoded integration host factor (IHF) (6). During conjugation, TrwC binds to the oriT gene, cleaves the DNA strand to be transferred at the nic site, and makes a covalent bond with its 5′ end (7). Then, the relaxase-DNA complex is recruited by the T4CP to the T4SS and transported to the recipient cell, where TrwC catalyzes the recircularization of the transferred DNA strand (8, 9).
Apart from its role in conjugation, TrwC is able to catalyze site-specific recombination between two oriT copies repeated in tandem (10). The reaction takes place in the absence of conjugation and thus in the absence of single-stranded intermediates, and it is favored by the accessory protein TrwA. In contrast, IHF was found to exert a negative regulatory role in TrwC-mediated recombination (11). It was proposed that recombination takes place thanks to the single-stranded endonuclease activity of TrwC coupled to the replication machinery of the host cell (10).
Once transferred to the recipient cell during conjugation, TrwC can also catalyze site-specific integration of the transferred DNA strand into an oriT-containing plasmid in the recipient cells (8). In this case, both TrwA and IHF act as enhancers of the reaction. Integration also occurs when the acceptor oriT was located in the chromosomal DNA of the recipient cell (12). A minimal oriT core sequence of 17 bp is enough for TrwC to achieve integration. Two human sequences with a single mismatch from that minimal oriT were tested as acceptors for TrwC-mediated integration and found to be functional, with an efficiency only 2 to 3 times lower than that obtained with the wild-type minimal oriT; this indicates that TrwC can act on DNA sequences present in the human genome (12).
In addition to the T4SS involved in conjugative DNA transfer, there is another family of T4SS implicated in the secretion of effector proteins during the infection process of several mammalian and plant pathogens (13). Substrate recruitment by the T4SS relies on secretion signals present in the protein substrate, and there are several examples of heterologous protein translocation by the T4SS upon the addition of secretion signals. In particular, conjugative relaxases can be translocated into eukaryotic cells through the T4SS of bacterial pathogens, either unmodified due to some similarity in their C termini with the secretion signal of the specific T4SS (as reported for translocation of MobA encoded by plasmid RSF1010 by the VirB T4SS of Agrobacterium tumefaciens [14]), or upon addition of the corresponding secretion signal, as done with TraA encoded by plasmid pATC58 and the VirB/D4 T4SS of Bartonella henselae (15). Moreover, two different reports have demonstrated that relaxase-DNA complexes from two conjugative systems can be translocated into human cells through the VirB/D4 T4SS of B. henselae. Those studies reported relaxase-mediated transfer of bacterial plasmids containing the oriT and conjugative genes from Bartonella cryptic plasmid pBGR1 (16) or from conjugative plasmid R388 (17). For both relaxases, the addition of a BID domain, the translocation signal for the Bartonella VirB/D4 T4SS (15), increases DNA transfer (16, 18). These reports suggest that transkingdom DNA transfer may naturally occur during bacterial infection of human cells.
T4SS-mediated DNA transfer to human cells may have biotechnological applications as a tool for in vivo DNA delivery into specific human cells (19). A main concern in genetic modification protocols is the fate of the introduced foreign DNA in the cells. Schröder and coworkers found that the relaxase-driven DNA integrated into the human genome at low frequency, and they characterized several integration sites demonstrating that the pBGR1 Mob relaxase can protect the 5′ end of the mobilizable plasmid, but no preference for specific integration sites could be identified, suggesting random integration of the incoming DNA (16).
In contrast to Mob, TrwC has site-specific integrase activity in bacteria, conferring added potential as a tool for genomic engineering (20). In this work, we analyze TrwC integrase activity into human genomic DNA after the mobilization of TrwC-DNA complexes from B. henselae. We show evidence that TrwC is active in the human cell, although the efficiency of site-specific integration is negligible compared to that of random integration. Interestingly, we find that TrwC promotes a 100-fold increase in the efficiency of integration of the incoming DNA, suggesting it may be protecting DNA from degradation; this feature could be combined with the action of a site-specific nuclease for genomic engineering purposes.
RESULTS
Construction of mobilizable plasmids and target cell lines.
TrwC-DNA complexes can be introduced in human cell lines through the T4SS of B. henselae. In order to analyze the integration pattern of the transferred DNA upon Bartonella infection of human cells, new mobilizable plasmids and cell lines were constructed. The mobilizable plasmids previously used to test DNA transfer from B. henselae to human cells (17) contained elements of the R388 Dtr region (oriT trwABC), but not the genes of the T4SS, and a eukaryotic gfp expression cassette. We added a neomycin phosphotransferase eukaryotic expression cassette in order to be able to select for stable chromosomal integration events. Plasmids were constructed coding for either TrwC or TrwC::BID (TrwC with the secretion signal for Bartonella VirB/D4 T4SS fused to its C terminus), with a negative control lacking trwC. Plasmid pRS130 (16) encoding Mob::BID relaxase and its cognate oriT was always tested in parallel.
The cell lines used for Bartonella infections were EA.hy926 and HeLa. The EA.hy926 cell line is derived from fusion of A549 lung carcinoma cells with human vascular endothelial cells, with endothelial cells representing the natural target for Bartonella and that are efficiently infected by this bacterium (21). HeLa cells represent a cervix-derived epithelial cell line that can be easily manipulated by cell biological and genetic methods, and infection by B. henselae was reported to occur with 50% efficiency (22). We previously showed DNA transfer to EA.hy cells, but HeLa cells were not tested. EA.hy926 and HeLa cells were tested in parallel in infections with B. henselae carrying either pHP161 (oriT trwABC) or pHP181 (oriT trwAB). DNA transfer efficiency was lower when using HeLa than when using EA.hy926 cells, but DNA transfer can be detected robustly in both cell lines (Fig. 1).
FIG 1.
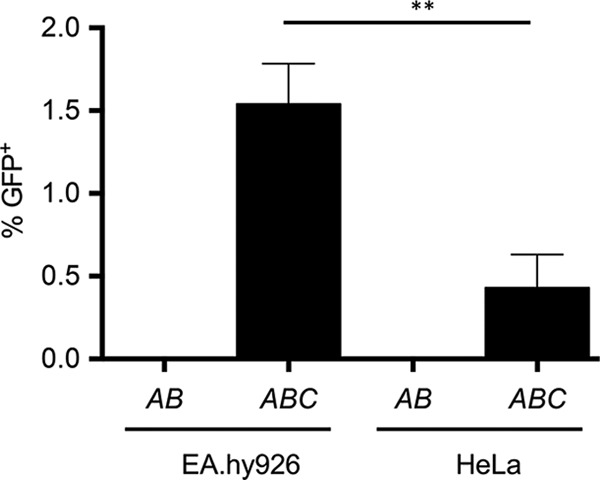
DNA transfer to EA.hy926 and HeLa cell lines. The graph shows the percentage of GFP-positive cells detected after 3 days of infection. The cell lines indicated in the x axis were infected with B. henselae carrying the mobilizable plasmids pHP181 (containing R388 oriT trwAB) or pHP161 (coding for R388 oriT trwABC); this is indicated as AB or ABC, respectively. Data are the means of the results from at least 5 independent experiments. Error bars indicate standard deviations. **, P < 0.01.
In order to compare the frequencies of TrwC-mediated integration into natural sequences of the human genome with integration when the TrwC target is present in the recipient cell genome, a cell line containing a full-length wild-type oriT gene was constructed. We transfected both EA.hy926 and HeLa cells with plasmids pMTX708 and pMTX709 (Table 1) carrying a hygromycin resistance gene and the R388 oriT in both orientations. Both plasmids were used to avoid any bias due to eukaryotic promoters present in the vector, since transcription through oriT has been shown to affect TrwC-mediated recombination (11), and so it might affect integration. Around 100 hygromycin-resistant colonies were obtained in transfections of HeLa cells, while no transfectants appeared for EA.hy926 in spite of several attempts with up to 5 μg of plasmid DNA. Consistent with this finding, the EA.hy926 cell line has been previously reported to be difficult to transfect (22, 23).
TABLE 1.
Plasmids used in this work
| Plasmid | Description | Reference or source |
|---|---|---|
| pCOR31 | pBBR6::oriT trwABC gfp neo | This work |
| pCOR33 | pBBR6::oriT trwABC:BID gfp neo | This work |
| pCOR35 | pBBR6::oriT trwAB gfp neo | This work |
| pCOR52 | Rescued integrated plasmid | This work |
| pHP159 | pBBR6::oriT trwABC gfpa | 17 |
| pHP161 | pBBR6::oriT trwABC gfpa | 17 |
| pHP181 | pBBR6::oriT trwAB gfp | 17 |
| pKK223-3 | Expression vector | Pharmacia |
| pLA24 | pBBR6::oriT trwABC:BID gfp | 17 |
| pMTX708 | pTRE2hyg::Ptac-oriTb | This work |
| pMTX709 | pTRE2hyg::Ptac-oriTb | This work |
| pOD1 | pKK223-3::oriT | This work |
| pRS56 | Cre-lox neo | 15 |
| pRS130 | pBGR::mob:BID gfp neo | 16 |
| pSU1186 | pUC8::oriT | 38 |
| pTRE2hyg | Mammalian shuttle vector | Clontech |
pHP159 and pHP161 differ only in the orientation of the gfp cassette, which is in the same orientation as the Plac promoter in pHP161 and in the opposite in pHP159.
pMTX708 and pMTX709 differ only in the orientation of the Ptac-oriT cassette. In pMTX708, oriT is closer to the hygromycin resistance gene.
The hygromycin-resistant HeLa colonies obtained were pooled to establish a polyclonal HeLa::oriT cell line, in which the oriT gene is expected to be located in different chromosomal locations and in the two possible orientations with respect to the vector promoter. In this way, we avoid selecting a single clone in which the oriT copy may lie in a chromosomal region that could affect integration of the mobilizable plasmid. The presence of the oriT gene was tested by PCR on genomic DNA samples from HeLa and HeLa::oriT cells using primers oriT1 and oriT330 (Table 2). Only one band corresponding to oriT was present in the sample obtained from HeLa::oriT cells, while no amplification was detected in the sample obtained from unmodified cells (see Fig. S2 in the supplemental material).
TABLE 2.
Oligonucleotides used in this work
| Oligonucleotide | Sequence (5′ to 3′) |
|---|---|
| Construction of pCOR31, pCOR33, and pCOR35 | |
| mCla_SnaBI_CMV_NeoF | CCAAATCGATCTACGTATTAGTCATCGCTATT |
| Cla_EcoRV_NeoR | CCAAATCGATGATATCCGGATATAGTTCC |
| Construction of pMTX708/pMTX709 | |
| NotI_Ptac | CCAGCGGCCGCTTATCGACTGCACGG |
| NotI_oriT1 | CCAGCGGCCGCTCATTTTCTGCATCATTGT |
| Detection of oriT-specific integration events | |
| Int_pCOR | TCAGGGCGTCCGTTTC |
| Int_pCOR_2 | CTGCATCACATTTGCATC |
| NotI_Ptac | CCAGCGGCCGCTTATCGACTGCACGG |
| NotI_Ptac_2 | CACTGCATAATTCGTGTC |
| NotI_oriT1 | CCAGCGGCCGCTCATTTTCTGCATCATTGT |
| PCR mapping of inserts in recovered integrated plasmids | |
| pCOR33_121F | TGGACAACCCTGCTGGAC |
| pCOR33_644R | TTTCGCCCTATATCTAGTTC |
| pCOR33_1641F | CTCGACCTGAATGGAAGCC |
| pCOR33_2158R | AGCTGGCGTAATAGCGAAG |
| pCOR33_3157F | CGCAACCCCTTGTAAATGC |
| pCOR33_3664R | TCTGAACGGCGGTAATCC |
| pCOR33_10431F | CCTGGCTGACCGCCCAA |
| pCOR33_10940R | GCTTCTAGAGATCTGACGG |
| pCOR33_11927F | TCAGGTTCAGGGGGAGGT |
| pCOR33_12445R | AATACGCAAACCGCCTCTC |
| Detection of oriT in HeLa::oriT | |
| oriT1 | CTCATTTTCTGCATCATCA |
| oriT330 | CCTCTCCCGTAGTGTTA |
| Analysis of G418-resistant cell pools | |
| 670_TrwC | TGTGTGCTAGGTCGAA |
| BamHI_TrwA_R | AACAGGATCCTCAATCCTCCTTCCCCTCCC |
| Hind3_TrwA_F | AACAAAGCTTATGGCACTAGGCGACCCC |
| Hind3_TrwC_F | AACAAAGCTTATGCTCAGTCACATGGTATT |
| LAM-PCR and high-throughput sequencing | |
| LC1 | GACCCGGGAGATCTGAATTCAGTGGCACAGCAGTTAGG(N)12CTA(RO)a |
| LC2 | (RO)TAG(N)12CCTAACTGCTGTGCCACTGAATTCAGATCa |
| LCI | GACCCGGGAGATCTGAATTC |
| Mis-LC | (PE-PCR 2.0)AGTGGCACAGCAGTTAGGb |
| Mis-TrwC | (PE-PCR 1.0)(N)10CGTCCTTAAAAGCCGGGTTGc |
| oriTI | CGATAACCCAATGCGCATAG |
| oriTII | TCTTTAGGGTCACGCTGGC |
| PE-PCR 1.0 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| PE-PCR 2.0 | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT |
| Sequencing of human genomic DNA | |
| Chr15_88728 | ATATGAATGTTTGCATTCCTT |
| IS2_Hu11 | AAGAAAGTCAACCTTCATCTT |
| Xba_IS2_Hu11 | CAACTCTAGAGGAAAAGTCAGAAAGACACCAAC |
(N)12, barcode sequence of linker cassette. (RO), restriction enzyme overhang.
(PE-PCR 2.0), adaptor sequences for high-throughput sequencing.
(PE-PCR 1.0), adaptor sequences for high-throughput sequencing; (N)10, barcode sequence introduced in second exponential PCR.
Transient and permanent expression of transferred DNA in human cells.
In order to measure the transfer and integration rates of DNA molecules led by different relaxases into human cells, gfp and neo resistance gene expression levels were measured, respectively, as outlined in Fig. 2a. EA.hy926, HeLa, and HeLa::oriT cell lines were infected with B. henselae carrying pCOR31 (trwC), pCOR33 (trwC::BID), pCOR35 (ΔtrwC), or pRS130 (mob:BID) mobilizable plasmids. The results are shown in Fig. 2 and Table S2. Three days postinfection, gfp expression was measured by flow cytometry (Fig. 2b). DNA transfer occurred in the three different cell lines when a relaxase (TrwC, TrwC::BID, or Mob::BID) was coded for in the plasmid, while no DNA transfer was detected when there was no relaxase. The transfer efficiency is higher when using EA.hy926 than when using HeLa as the host cell, as previously shown (Fig. 1). No significant differences were found in DNA transfer between the HeLa and HeLa::oriT cell lines, as expected. In all cell lines, the DNA transfer rate was significantly lower when using Mob::BID relaxase than with TrwC or TrwC::BID.
FIG 2.

Transient and permanent expression of the transferred DNA. (a) Overview of the experimental design to detect transient expression or stable integration of the transferred DNA. After infection of human cell lines with B. henselae, the DNA transferred through the T4SS will get to the nucleus where genes will be expressed. At 3 days postinfection, transient expression of gfp can be detected by flow cytometry. Antibiotic treatment was applied for long-term selection of neomycin-resistant colonies, to detect stable integration events. (b to d) Graphic representation of the percentage of GFP-positive cells obtained 3 days postinfection (b) and the number of G418-resistant colonies normalized for the number of cells at the beginning of the selection (c), as well as the Neor/GFP+ ratio (d). The different bars represented for each cell line correspond to the different relaxases under study, following the color code indicated in the squares at the top right. Data represent the means of the results from at least 3 independent experiments. Error bars indicate standard deviations. *, P < 0.05.
Integration events of the transferred plasmids into the human genome were selected by antibiotic treatment with G418. The drug was added at 72 hours postinfection (hpi), and selection was maintained for 4 to 5 weeks. The resistant colonies obtained for each experimental condition were counted and then pooled. High-molecular-weight genomic DNA preparations were analyzed by PCR for the presence of trwA and trwC to confirm the presence of the integrated plasmid (Fig. S3). The resistant cell pools were also analyzed by flow cytometry to detect green fluorescent protein (GFP) expression as additional evidence of integration of the mobilizable plasmids (Fig. S4).
Figure 2c shows the number of resistant colonies obtained after the antibiotic treatment normalized to the number of cells at the beginning of the experiment. It was lower when using EA.hy926 than when using HeLa cells, despite the fact that DNA transfer was up to 10-fold higher with EA.hy926. For all cell lines, no resistant colonies were found when using plasmid with ΔtrwC, in concordance with the flow cytometry results, which showed no DNA transfer in the absence of relaxase. When plasmids coded for a relaxase, resistant colonies appeared but at drastically different rates. Thousands of resistant colonies were obtained in each experiment after the mobilization of trwC- and trwC::BID-carrying plasmids, while only up to 100 resistant colonies were found in infections with B. henselae carrying the plasmid coding for Mob::BID (see Table S2).
Figure 2d shows the ratio between neomycin-resistant (Neor) colonies and GFP+ cells, which gives the integration rate, i.e., the proportion of cells receiving the DNA in the nucleus which integrate this DNA into the chromosome. For each cell line, no significant differences were found in the integration rates of TrwC or TrwC::BID plasmids, as expected. No significant differences were found either in the integration rate of each plasmid in HeLa and HeLa::oriT cells. The integration rate was higher than 1 in 20 when the transferred DNA was led by TrwC or TrwC::BID, while it was around 1 in 250 in the case of Mob::BID-driven DNA (Table S2).
With the purpose of having a parallel control of random integration, HeLa and HeLa::oriT cells were transfected with plasmid pCOR35 (ΔtrwC). Transient versus stable expression was determined as outlined before. After transfection of plasmid DNA, we obtained an integration rate of around 1 in 800 when transfecting supercoiled DNA and of close to 1 in 300 when transfecting linearized DNA (Table S2), which is in the range of our data obtained for Mob::BID. Antibiotic-resistant colonies were pooled and analyzed in parallel with those obtained after relaxase-mediated mobilization of plasmid DNA to compare the two plasmid integration patterns.
Characterization of genomic integration sites.
Relaxases transfer the DNA strand covalently linked to a site known as the nic site. In the case of Mob-led DNA, it has been suggested that the relaxase protects the 5′ end of this DNA (16). In addition, we know that TrwC acts as a site-specific integrase of the transferred DNA into the genome of recipient bacteria (12), and we observed an enhanced integration rate of TrwC-led DNA. Taking together these evidences, we decided to search for integration events occurring at the nic site of the R388 oriT. For this purpose, we used linear amplification-mediated PCR (LAM-PCR) (24, 25) using a primer annealing close to the nic site, as explained in Fig. S1. This strategy would not detect insertions into the full-length oriT copy of HeLa::oriT, as integration would result in a reconstituted oriT, but it would allow the identification of integration events in other chromosomal locations and a comparison with the integration pattern obtained when the oriT is not present in the genome to be modified.
LAM-PCR was performed as explained in Materials and Methods and the supplemental Materials and Methods. Genomic DNA was extracted from pools of several thousands of resistant colonies obtained after mobilization of trwC- or trwC::BID-coding plasmids, and this DNA was used as the template for the PCRs. Genomic DNA was also extracted from resistant colonies obtained by transfection of plasmids pCOR31 (trwC) and pCOR33 (trwC::BID), which are expected to have a random integration pattern. After LAM-PCR amplification of the integration junctions, two different restriction enzymes were used to avoid any bias due to restriction fragment size. PCR products were checked by electrophoresis in agarose gels (Fig. 3).
FIG 3.

Analysis of LAM-PCR products. (a to c) Scheme of the expected integration events and the subsequent LAM-PCR products. (a) If integration takes place by the 5′ end of the nic site, the size of the LAM-PCR would be determined by the distance to the nearest restriction site in the human genome (in pink). Each integration event occurring in a different locus would generate a band of a different size. The nicked oriT is indicated by a slash. (b) If the plasmid becomes integrated at any other region than the nic site, the size of the LAM-PCR product would be always the same and would be determined by the distance to the restriction site in the plasmid sequence. Bfa, BfaI; Tsp, Tsp509I. (c) In HeLa::oriT cell line, in addition to the bands generated from the integration events, the oriT copy present in the pMTX708/pMTX709 plasmid generates a single band of a size determined by the distance to the restriction site in the plasmid sequence. trw has been omitted from trwA, trwB, and trwC for clarity. (d) Gel electrophoresis of LAM-PCR products obtained when using BfaI (top gel) or Tsp509I (bottom gel) restriction enzymes. The cell line is indicated in the top row (EA, EA.hy926; He, HeLa; He:oriT, HeLa::oriT). Inf, samples obtained after Bartonella infection; Tr, samples obtained by transfection of plasmid DNA; LD, 100-bp ladder; T, trwC-coding plasmid (pCOR31); T:B, trwC::BID-coding plasmid (pCOR33). EA, He, and He::oriT, samples from uninfected cell lines. g, human genomic DNA (Roche), used as negative control. The − sign with 1 to 3, negative controls (no DNA) of linear, first, and second exponential PCRs, respectively. The arrows indicate the bands of the expected size according to panels b (black arrows) and c (blue arrow).
Figure 3a to c shows the scheme of the expected band sizes observed in these gels. We expected to see as many bands as there were different integration sites occurring at the nic site (Fig. 3a), depending on the location of the nearest restriction site in the genomic junction. If integration did not occur at the nic site, the size of the band would be determined by the nearest recognition site in the integrated plasmid (220 bp when using BfaI and 580 bp when using Tsp509I, since this does not cut in the oriT but in trwA; Fig. 3b). In the case of the HeLa::oriT cell line, which has an oriT copy integrated in the genome, we expected to obtain in all cases a major band of 220 bp (BfaI) or 345 bp (Tsp509I) corresponding to the sequence of the oriT-carrying integrated plasmid (Fig. 3c).
As can be observed in Fig. 3d, a single band was obtained from all samples, which was obtained either after plasmid transfection or after translocation of TrwC(::BID)-DNA molecules through the B. henselae T4SS. The size of the band was in all cases that which was expected for the full-length oriT gene present in the mobilizable plasmids or in the genomic oriT copy, as explained above and in Fig. 3a to c. For the G418-resistant pools obtained either after transfection or infection of HeLa::oriT cells, both 345 bp and 580 bp would be visible when using Tsp509I, as observed in the sample obtained after infection and DNA transfer mediated by TrwC (Fig. 3d, bottom gel, line 6). A reason for this not being the case for the other samples could be that the smaller amplicon could be preferentially amplified.
These results strongly suggest that the transferred DNA had not become integrated by the nic site at the oriT. Rather, they presumably reflect illegitimate integration events. Since low-frequency site-specific integration events could be masked by this main band, LAM-PCR products were thus analyzed by high-throughput sequencing, as explained in Materials and Methods. After the identification of linker- and plasmid-specific sequences, the flanked sequences were characterized. As expected from the results in Fig. 3d, most of the 2,000,000 reads obtained were found to be plasmid DNA. This confirms that most of the DNA entering the human cell covalently linked to TrwC is not integrated at the nic site, implying that this DNA is recircularized prior to integration.
There were 11,317 reads which could be mapped to the human genome. To discard false positives, identity to the human genome threshold was raised to 98%, and integration events obtained <15 times were not considered. The resulting 9 integration events (IE) are shown in Table 3. IE1 and IE2 were found to occur at the same site of the human genome (the differences in the sequencing reads were assumed to be sequencing errors), so they were considered together as one integration event and named IE2. Most of the integration events showed more than 12 missing bp of a total of 41 bp amplified from primer oriTI binding site to the nic site, so they were considered random integration events.
TABLE 3.
Integration events characterized by LAM-PCR and DNA sequencinga
| IE | Cell line | Relaxase | No. of sequences | Identity (%) | Chr | Integration locus | Missing bp |
|---|---|---|---|---|---|---|---|
| 1 | EA.hy926 | TrwC | 19 | 99.75 | 11 | 35225119 | 0 |
| 2 | EA.hy926 | TrwC | 402 | 100 | 11 | 35225119 | 0 |
| 3 | HeLa | TrwC::BID | 15 | 98.25 | 2 | 37383046 | 15 |
| 4 | HeLa | TrwC::BID | 114 | 100 | 2 | 111118923 | 16 |
| 5 | HeLa | TrwC::BID | 15 | 100 | 6 | 9173423 | 18 |
| 6 | HeLa | TrwC::BID | 84 | 99.48 | 12 | 28128063 | 16 |
| 7 | HeLa | TrwC::BID | 21 | 100 | 13 | 83099211 | 0 |
| 8 | HeLa | TrwC::BID | 95 | 99.72 | 16 | 68832248 | 16 |
| 9 | HeLa | TrwC::BID | 15 | 100 | 19 | 18303607 | 14 |
The information collected for each integration event is shown. Number of sequences indicates the number of times the sequence read was found. Missing base pairs indicates the number of bp that are missed in the read with respect to the plasmid sequence until the nic site. IE, integration event; Chr, chromosome.
There were only two IE which were not missing any oriT sequence in the 3′ direction of the nic site. When aligned with the human DNA sequence, it was found that integration in IE2 had occurred at the position nic+1, since this base from the oriT sequence was present at the junction and is not present in the University of California, Santa Cruz (UCSC) genomic sequence used as a reference. We confirmed the genomic sequence of this position in the genome of the HeLa cells used in the experiment by amplification of the chromosomal region around the integration site 2 (IS2) with primers IS2_Hu11 and Xba_IS2_Hu11 (Table 2) and sequencing the PCR product with the IS2_Hu11 primer, and this base pair was not present there either. Considering the high specificity of conjugative relaxases for nicking exactly at their nic site, this result suggests that this event was yet another illegitimate integration event. Finally, IE7 occurred exactly at the nic site, and moreover, the eight nucleotides of the human genome in the 5′ direction of the integration site are identical to the eight nucleotides in the 5′ direction of the nic site in R388 oriT (Fig. 4). This integration event took place 1,352,133 bp downstream of the SLITRK1 gene (accession no. NM_052910) in human chromosome 13.
FIG 4.
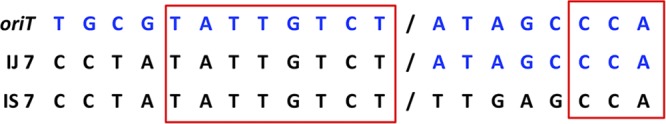
Characterization of integration event IE7. The genome-plasmid integration junction (IJ) is aligned with the DNA sequence around the nic site (oriT, top) and the chromosomal integration site (IS, bottom). DNA of plasmid origin is shown in blue, and genomic DNA is shown in black. The nic site and insertion sites are indicated by a slash. Regions of homology between the plasmid and the genomic sequences are boxed.
As LAM-PCR did not allow the detection of the integration events occurring at the oriT copy present in the chromosome of HeLa::oriT cells, we tried to detect them by PCR amplification of the expected cointegrate molecule; we used a primer annealing in the Ptac promoter located adjacent to the chromosomal oriT copy and another one annealing in the mobilizable plasmid (Table 2). As a control, the chromosomal Ptac-oriT cassette was amplified in the same samples analyzed; as expected, the cassette was detected in HeLa::oriT and the G418-resistant pools obtained with this cell line, while no amplification was obtained in HeLa and HeLa-derived cell pools. The PCR to amplify the oriT-oriT cointegrate was negative (data not shown), even after a second round of PCR amplification. Although we cannot discard oriT-specific integration occurring at such low frequency that it is not detectable by PCR, this result indicates that it is not occurring efficiently.
LAM-PCR can only be used to map those integration events that occurred by a known sequence of the transferred DNA (the nic site in our case), but most of the plasmid molecules became integrated in a nic-independent manner. Out of the 8 human genomic junctions obtained by LAM-PCR (Table 3), 7 did not occur by the nic site, and so they represent random integration events. However, they provide information on only one of the integration junctions of the plasmid. We attempted to characterize other random integration sites by recovery of the integrated plasmids together with the flanking genomic sequences, as outlined in Materials and Methods. With this strategy, we were able to characterize one integration event and its corresponding plasmid-genomic DNA junctions (Fig. 5). We determined that only a fragment of the plasmid was integrated, which does not include the neomycin resistance gene, so most probably, there is another integration event somewhere else in that same cell containing the neo gene. We also observed that both genomic-plasmid DNA junctions did not occur at the same position in the human genome. Moreover, near one of the junctions (IJ-B in Fig. 5b), a genomic rearrangement was found, compared to the reference genome (see coordinates in Fig. 5b). The reason could be that the genomic region of chromosome 15 where integration occurred corresponds to a copy of L1MC2, a long interspersed element (LINE) often associated with genomic rearrangements and deletions (26). We tried to sequence that region from genomic DNA of the HeLa cell line used in the experiment using primer Chr15_88728 (Table 2) to determine if the rearrangement was already present or it was a consequence of the illegitimate integration event, but mixed sequences were always obtained.
FIG 5.
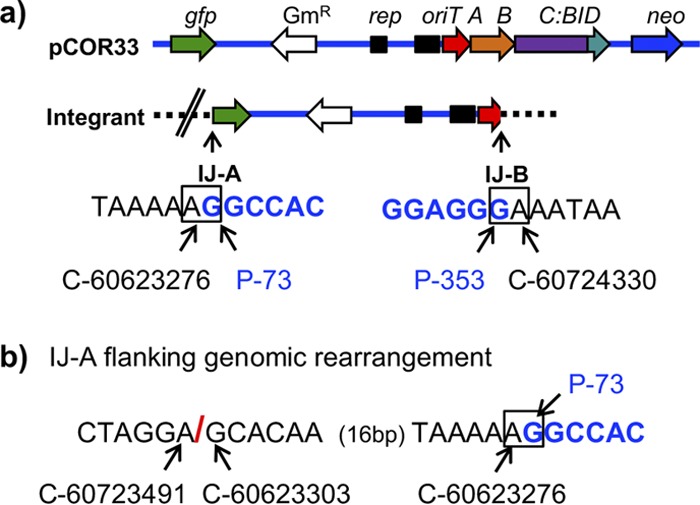
Genomic integration event characterized by recovery of the integrated plasmid. (a) Scheme of the mobilizable plasmid containing trwC::BID, and the structure of the integrant in the genomic DNA of HeLa cells. Plasmid DNA is represented as a blue horizontal line and HeLa genomic DNA as a black dashed line. trw is omitted from trwA, trwB, and trwC::BID for clarity. The double slash in the integrant refers to the genomic reorganization shown in panel b. The two plasmid-genomic DNA integration junctions resulting from the integration event are named IJ-A and IJ-B. The DNA sequence at the junctions is shown below with their respective coordinates, in black (human genome) and blue (plasmid DNA). The junctions are highlighted in a square. Coordinates of human chromosome 15 are indicated as a C-number, where the number corresponds to the coordinates of the human genome in UCSC database (assembly GRCh37/hg19). Coordinates of plasmid DNA are indicated as a P-number, where the number corresponds to the nucleotide of the open reading frame of gfp (in IJ-A) or trwA (in IJ-B). (b) Genomic rearrangement found near IJ-A. The red slash indicates the genomic junction between nonadjacent human DNA sequences.
The genomic integration sites of all random integration events characterized were aligned with the R388 nic region at the oriT gene (Table 4). No homology with the oriT was detected, supporting the idea of illegitimate integration.
TABLE 4.
Mapped illegitimate integration eventsa
| IS | Sequence (5′ to 3′) | Integration junction |
||
|---|---|---|---|---|
| Chr | Genomic locus | Plasmid | ||
| 2 | AAAATGAGGACAGTT/ATATTTTTTAAATGT | 11 | 35225119 | nic+1 |
| 3 | CCAGATCGTGCCACT/GCATTCCAGCCTGGC | 2 | 37383046 | nic−15 |
| 4 | TGGGAAACAAATGAA/GAAACAACCCTGCTG | 2 | 111118923 | nic−16 |
| 5 | GTTTCCATGGACATT/TGCCACCCCGGCTTC | 6 | 9173423 | nic−18 |
| 6 | CGGGTTAGAAACCAA/GCACCCAAGCCGGCG | 12 | 28128063 | nic−16 |
| 8 | CACTTGCTGGGCTCA/GAGACAACCCAGCCC | 16 | 68832248 | nic−16 |
| 9 | GTTGTAACTGCCTAA/GATTGACCAACCCTA | 19 | 18303607 | nic−14 |
| 10 | GTCACATGATAAAAA/GATTATTTCATTTTG | 15 | 60623276 | gfp_73 |
| 11 | ATTTAATCCAAATAG/AAATAAGTTTCAGAT | 15 | 60724330 | trwA_353 |
| oriT | AGGTGCGTATTGTCT/ATAGCCCAGATTTAA | nic | ||
The genomic integration sites are shown aligned with the wild-type target for TrwC, the oriT gene. The slash in each sequence indicates the integration site (the nic site in the oriT sequence). The location of the integration site (Chr, chromosome number), as well as the nucleotide of the plasmid by which integration took place, is also displayed. IS, integration site. IS2 to -9 were characterized by LAM-PCR. IS10 and -11 are both integration junctions of the event characterized by recovery of the integrated plasmid. Coordinates of genomic loci correspond to human genome GRCh37/hg19 available in UCSC Genome Browser. Plasmid coordinates refer to the distance from the nic site (+ and − indicating in the 5′or 3′ direction from the nic site, respectively) for IS2 to -9, or the nucleotide position in the gfp and trwA open reading frames (ORFs) for IS10 and -11.
DISCUSSION
The ability to deliver DNA into specific human cell types and to promote its integration in the human genome has high potential as a biotechnological tool. In particular, gene therapy strategies ideally should grant in vivo access to specific human tissues and permanent expression of the introduced DNA. In this work, we explore the potential of a bacterial system for genomic modification of the human genome. Our previous work showed that the substrate of a conjugative plasmid, the TrwC-DNA complex, was delivered efficiently to human cells through the T4SS of B. henselae (17); the many advantages of such a DNA delivery system in this context have been already discussed (19). We previously showed in a bacterial system that TrwC could catalyze integration of the transferred DNA into DNA sequences of human origin (12), so we aimed to evaluate its potential role as a site-specific integrase in human cells, which would complement the DNA delivery tool. We have analyzed the fate of the DNA in human cells after translocation as a TrwC-DNA complex through the VirB/D4 T4SS of B. henselae.
We measured the efficiency of DNA delivery and integration by the two relaxases previously described to deliver DNA through the T4SS VirB/D4 of B. henselae, TrwC and Mob::BID. All mobilizable plasmids carried a eukaryotic expression GFP cassette, allowing us to estimate the efficiency of DNA transfer by measuring the percentage of GFP-positive cells. This assay probably underestimates the percentage of cells receiving DNA, since this DNA has to get into the nucleus and be converted into double-stranded form so that it can express the gfp gene. Thus, nuclear localization of the relaxase could affect DNA transfer rates. TrwC has been reported to have cytoplasmic localization (27), while a passive entry of the Mob-guided DNA has been suggested (16), so none of the relaxases is expected to have an active role in nuclear import.
Our results show that DNA transfer is higher when using TrwC than when using Mob::BID. The differences in DNA transfer rates are probably due to differences in T4SS recruitment efficiency for each relaxase. TrwC could be naturally a better substrate for the B. henselae VirB/D4 T4SS than Mob::BID. There is another important factor to take into account: the mobilizable plasmids carrying trwC also code for R388 proteins TrwA and TrwB, which could play a role in substrate recruitment. Deletion of trwB was shown to affect significantly the transfer of TrwC-DNA complexes (17). Thus, it is likely that TrwB enhances recruitment of TrwC by the VirB/D4 T4SS independently of BID, as previously suggested (16, 18).
The plasmids mobilized to human cells carried a eukaryotic resistance marker to select for stable integration events by antibiotic treatment. Selection was carried out for 4 weeks, discarding the possibility of episomal persistence of the transferred plasmid DNA. Each resistant colony was counted as one integration event. Again, this measure is an underestimation of the integration rate. A single colony could harbor more than one integration event; in fact, the only integration event mapped in its entirety (the rescued integrated plasmid) did not include the neo region (Fig. 5a), implying this gene must be integrated somewhere else in the genome. In addition, not all cells integrating the plasmid will thrive to render a colony. This phenomenon was particularly evident with EA.hy926 cells, which have low viability. Consequently, we obtained fewer resistant colonies when using EA.hy926 than when using sturdy HeLa cells, despite the fact that DNA transfer was up to 10-fold higher with EA.hy926 (Fig. 2). Of course, we cannot rule out that the different integration rates observed in the two cell lines are due to intrinsic differences affecting host-mediated integration of foreign DNA.
The number of integration events obtained for each experiment was 25 to 158 times higher when either TrwC or TrwC::BID was present than with Mob::BID (Table S2). When we measured integration rates, calculated as the number of resistant colonies normalized to the number of cells expressing the transferred DNA, we observed that the integration rate for TrwC was, on average, 1 in 20, while for Mob::BID, it went down to about 1 in 250, similar to the integration rate obtained for transfected cells (Fig. 2d and Table S2). Thus, we conclude that TrwC facilitates the integration of the mobilizable plasmids, while Mob does not.
A plausible explanation for this difference could be the site-specific integration activity of TrwC, which is presumably absent in the Mob relaxase. To test this hypothesis, we analyzed the integration pattern in the human genome, searching for TrwC-mediated site-specific integration events. Their signature would be the precise integration of the R388 nic site into human DNA sequences resembling the natural TrwC target. We analyzed genomic DNA of the resistant cell pools by LAM-PCR, priming from the plasmid DNA into the nic site, and subsequent DNA sequencing. The results showed the presence of intact oriT sequences in the vast majority of the sequencing reads. One possible explanation is the integration of plasmid concatemers, as it happens in the integration of transfer DNA (T-DNA) mediated by A. tumefaciens (28), but then oriT-host genome junctions would be detected at the end of the concatemer. The DNA extraction kit isolates high-molecular-weight DNA (30 to 50 kb) from mammalian cells, while bacterial cells are not lysed, discarding the possibility that plasmid DNA of bacterial origin could be coisolated. In addition, the absence of transformants when using the same DNA preparations for plasmid rescue (a single transformant was obtained, originated from a rescued integrated plasmid copy) rules out the presence of episomal plasmid molecules. Thus, the most likely possibility is that these reads represent random integration events of the plasmid, which would have been recircularized previously, since it enters the human cell cut at the nic site (where TrwC is covalently bound). Recircularization implies that TrwC is active in the human recipient cell, mimicking its activity in the bacterial recipient cell during conjugation (29).
Out of the thousands of different integration events present in the analyzed cell pools, we detected one putative site-specific integration event (IE7). It occurred precisely at the nic site and in a region of the genome showing 8 bp identity with the oriT at the 5′ end of the nic site (Fig. 4). Since the probability of integration at any position of the human genome is approximately 1/(3 × 109), and the probability of integration occurring by the nic site is less than 1/(1 × 104) (the size of the integrated plasmids is around 13 kb), the probability of this event occurring randomly is negligible. From our results, we infer that TrwC can act as a site-specific integrase in human cells, but host-mediated random integration is at least 3 to 4 logs more efficient. Thus, after TrwC-mediated recircularization of the DNA (as inferred from the presence of full-length oriT genes), most molecules would undergo nonhomologous integration events, as observed in the characterized integration sites (Table 4).
DNA can also be delivered into human cells by the relaxases Mob and A. tumefaciens VirD2, and it integrates randomly in the genome (16, 30). It was proposed that these relaxases do not play a role in the integration process, which is likely mediated by the host machinery, but they do protect the 5′ end of the transferred DNA, based on preservation of the 5′-end region of the transferred DNA molecules (16, 30). In our case, we found by LAM-PCR seven integration events occurring within 20 bp from the nic site (Table 3). By chance, we would expect around 30 integration events lying in a 20-nucleotide (nt) region, from about 20,000 total integration events analyzed, so TrwC does not seem to protect the 5′ end of the transferred DNA but rather catalyzes its conversion to a circular form. Recircularized plasmid DNA will be a more resistant molecular species, showing long-term presence in the nucleus, which could favor its subsequent random integration by the host machinery.
From a biotechnological point of view, our results indicate that TrwC is not useful as a site-specific integrase in human cells. However, with the introduction of precision genome editing using RNA-guided endonucleases, such as Cas9 (31), we have entered a new era of genetic engineering and gene therapy which is making obsolete the traditional site-specific recombinases and nucleases used for gene targeting in human cells (32). In this new scenario, an improvement in clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein (Cas) technology would have an immediate impact in the human gene editing field. An RNA-guided nuclease could be translocated simultaneously with TrwC-DNA through the T4SS of bacteria that infect specific human cell types. Delivery of the nuclease protein instead of transfection of the gene could avoid toxicity and off-target activity. The effect of TrwC as a DNA chaperone in combination with a site-specific nuclease would promote integration of the incoming DNA molecule by homologous recombination. In support of this approach, it has been reported that concomitant translocation of I-SceI homing site-specific endonuclease together with VirD2 relaxase-T-DNA complexes through the A. tumefaciens T4SS enhanced T-DNA site-specific integration into the yeast chromosome when the I-SceI target site was present (33).
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Escherichia coli strain D1210 (34) was used for DNA manipulations, while strain β2163 (35) was used as a donor for conjugative matings to B. henselae. E. coli strains were grown at 37°C in Luria-Bertani broth, supplemented with agar for growth on plates. B. henselae strain RSE247 (36) was used for the infection of human cells. B. henselae was grown on Columbia blood agar (CBA) plates at 37°C under a 5% CO2 atmosphere. For selection, antibiotics were added at the indicated concentrations: ampicillin (Ap), 100 μg/ml; kanamycin monosulfate (Km), 50 μg/ml; streptomycin (Sm), 300 μg/ml (E. coli) or 100 μg/ml (B. henselae); and gentamicin sulfate (Gm), 10 μg/ml. When needed, media were supplemented with diaminopimelic acid (DAP) at 0.3 mM.
Plasmids.
Bacterial plasmids are listed in Table 1. Plasmids were constructed using standard methodological techniques (37). Primers used in plasmid constructions are listed in Table 2. Plasmids pCOR31, pCOR33, and pCOR35 were constructed by cloning a neomycin resistance cassette amplified from pRS56 with primers adding ClaI restriction sites (Table 2) into the same site of pHP159, pLA24, and pHP181, respectively. Plasmids pMTX708 and pMTX709 were constructed by cloning a Ptac-oriT cassette into the NotI site of pTRE2hyg vector, selecting both orientations; Ptac-oriT was amplified from plasmid pOD1, which carries an EcoRI-HindIII fragment from pSU1186 (38) containing R388 oriT into the same sites of expression vector pKK223-3 (Pharmacia). Restriction enzymes, shrimp alkaline phosphatase, and T4 DNA ligase were purchased from Thermo Fisher Scientific. Kapa HiFi DNA polymerase was purchased from Kapa Biosystems. Plasmid DNA was extracted using the GenElute plasmid miniprep kit (Sigma-Aldrich). The DNA sequences of all cloned PCR fragments were determined.
Mating assays.
Plasmids were routinely introduced in B. henselae by conjugation. The E. coli donor strain was grown in LB to stationary phase. Two hundred microliters was collected for each mating and resuspended in 1 ml of phosphate-buffered saline (PBS). Recipient B. henselae was grown in CBA plates for 3 to 4 days. After that time, bacteria from half of the plate were collected with a cotton swab and resuspended in 1 ml of PBS. Both donor and recipient aliquots were centrifuged, pellets were resuspended in 20 μl of PBS and mixed, and the mixture was placed on a cellulose acetate filter on a CBA plate supplemented with DAP. The mating plate was incubated at 37°C under a 5% CO2 atmosphere for 6 h. Transconjugants were selected by recovering the mating mixture and streaking it on a CBA plate with appropriate antibiotics. The plate was incubated for 6 to 9 days at 37°C under a 5% CO2 atmosphere.
Cell lines and growth conditions.
The human cell lines used in this work were immortalized hybridoma EA.hy926 (ATCC CRL-2922), a fusion cell line of human umbilical vein endothelial cells (HUVEC) and adenocarcinomic human alveolar basal epithelial cells (A549), and HeLa (ATCC CCL-2), epithelial cells of cervix adenocarcinoma. HeLa cells containing an integrated copy of the R388 oriT gene were created by transfection of plasmids pMTX708 and pMTX709 (Table 1) and selection of stable transfectants as explained in “Transfections,” below.
Cell lines were routinely grown in Dulbecco's modified Eagle medium (DMEM; Lonza) supplemented with 10% fetal bovine serum (FBS; Lonza) at 37°C under 5% CO2. When indicated, antibiotics were added to the medium at the following concentrations: G418 disulfate salt (Sigma-Aldrich), 500 μg/ml; hygromycin B (Invitrogen), 80 μg/ml; and penicillin-streptomycin 1% (Lonza).
Transfections.
HeLa cells were transfected with the cationic JetPei transfection reagent (Polyplus-transfection). The amounts of DNA and JetPei reagent were adjusted depending on the cell culture format used, according to the manufacturer's instructions. DNA was quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). To generate stably transfected cell lines, HeLa cells were allowed to grow and to express the drug resistance gene under nonselective conditions for 24 to 48 h after transfection. Then, cells were cultivated in standard medium supplemented with the appropriate drug for 4 to 5 weeks until outgrowth of resistant cells. Medium was changed every 2 to 3 days to avoid a loss of selection pressure. To obtain the integration rate of transfected plasmid DNA, transfections were carried out in 6-well plates. To transfect linearized DNA, plasmid DNA was digested with Alw44I (Thermo Scientific) and purified with the GeneJet gel extraction kit (Thermo Scientific) prior to transfection.
Cell infections.
B. henselae containing the appropriate plasmids was grown on CBA plates for 3 to 4 days. Human cells were seeded the day before the infection. For routine infections, cells were seeded in 6-well plates (80,000 cells per well) in 3 ml of medium. When the purpose of the infection was to select human cells that had stably acquired the plasmid transferred from B. henselae, infections were performed in 150-mm tissue culture dishes seeded with 1.2 × 106 cells in 20 ml of medium.
The day of infection, DMEM was replaced by M199 medium (Gibco) supplemented with 10% FBS and appropriate antibiotics to select for the B. henselae strains to be added. The bacteria were recovered from the CBA plate and resuspended in 1 ml of PBS. The number of bacteria was calculated considering that an optical density at 600 nm (OD600) of 1 corresponds to 109 bacteria/ml (39). Bacteria were added to the human cells to get a multiplicity of infection (MOI) of 400. The mixture of human cells and bacteria was incubated for 72 h at 37°C under 5% CO2.
Detection of GFP-positive cells.
At 72 h postinfection (hpi), infected cells were washed with PBS, trypsinized, and analyzed by flow cytometry using a Cytomics FC500 flow cytometer (Beckman Coulter). Uninfected cells were always used in parallel to set the baseline for the detection of GFP-positive cells.
Selection of stable integration events.
At 72 hpi, G418 disulfate salt (Sigma-Aldrich) was added to infected cells, and selection was maintained for 4 to 5 weeks. Resistant colonies were counted on the plates. G418-resistant cell pools were collected for further analysis of GFP expression and PCR analysis. Genomic high-molecular-weight DNA was extracted using the High Pure PCR template preparation kit (Roche).
Linear amplification-mediated PCR.
Amplification of genomic integration sites by linear amplification-mediated PCR (LAM-PCR) was performed as described in reference 40. Briefly, it consists of an initial linear amplification of genome-plasmid junctions with a plasmid-specific primer. After synthesis of double-stranded DNA (dsDNA), the PCR product is cut with a restriction enzyme (BfaI or Tsp509I), and a linker cassette of known sequence is ligated. Exponential PCR amplifications are then performed with plasmid- and linker-specific primers. PCR-obtained bands are then analyzed by gel electrophoresis and high-throughput sequencing. Human genomic DNA from human blood (buffy coat; Roche) was analyzed in parallel as a negative control.
The LAM-PCR template was genomic DNA from the G418-resistant pools. PCRs were carried out using Taq DNA polymerase (Genaxxon Bioscience). High-performance liquid chromatography (HPLC)-purified primers (Table 2) were designed using the Primer3Plus software and ordered from Eurofins Genomics. Details on the primers can be found in supplemental Materials and Methods and Fig. S1.
High-throughput sequencing of LAM-PCR products.
The DNA sequence of purified LAM-PCR products was determined using MiSeq benchtop next-generation sequencing technology (Illumina). The appropriate volumes of different purified samples were mixed together, according to the manufacturer's recommendations. The primers used in the second exponential amplification contained the adaptor sequences needed for the sequencing reaction (PE-PCR 1.0 and 2.0; see Table 2). LAM-PCR products were sequenced in both directions. From PE-PCR 1.0 (adaptor present in the primer annealing to the plasmid sequence), 400 nt were sequenced, while only 50 nt were sequenced from PE-PCR 2.0 (adaptor present in the primer annealing to the linker). Information from PE-PCR 1.0 was used for sorting the sequences to the different samples and integration site detection, while information obtained from PE-PCR 2.0 was used for sorting only.
Bioinformatic analysis to obtain the integration sites was performed by high-throughput insertion site analysis pipeline (HISAP) (41). Briefly, sequences were trimmed by identification and removal of plasmid- and linker-specific sequences. Genomic sequences were aligned to the human genome using standalone BLAT (UCSC), using assembly GRCh37/hg19 as a reference. Sequences with identities lower than 95% were discarded. For each remaining sequence, the chromosome, the integration site, and the nearest RefSeq protein-coding gene were recorded.
Detection of oriT-specific integration events by PCR.
PCRs were carried out using Kapa Taq polymerase (Kapa Biosystems), according to the manufacturer's recommendations. Twenty-five nanograms of plasmid DNA or 250 ng of genomic samples was used as the template. To detect the expected cointegrate molecule, primers NotI_Ptac and Int_pCOR (Table 2) were used for initial amplification. A 1:50 dilution of the initial PCR products served as the template for the secondary PCR, carried out with primers NotI_Ptac_2 and Int_pCOR_2 (Table 2), annealing approximately 80 bp closer to the expected integration junction. Primers NotI_Ptac and NotI_oriT1 (Table 2) were used to amplify the chromosomal Ptac-oriT cassette.
Recovery of integrated plasmids.
Five micrograms of genomic DNA from G418-resistant cell pools was digested with XmaJI (Thermo Fisher Scientific), which does not cleave within the integrated plasmid. Digested DNA was treated with T4 DNA ligase at a DNA concentration of 10 μg/ml to favor self-ligation. The reaction mixture was electroporated into ElectroMAX DH10B E. coli cells (Thermo Fisher Scientific). Plasmid DNA was extracted from gentamicin-resistant E. coli transformants and analyzed by PCR to narrow down the region of the plasmid where the insert of human origin was located. The primers used for PCR mapping reactions are shown in Table 2. The insert in plasmid pCOR52 was sequenced with primers pCOR33_1641F and pCOR33_12445R (Table 2).
Statistical analysis.
An unpaired Student's t test was used to determine statistically significant differences between the means of at least 3 independent results for each experiment when the data showed a normal distribution. Otherwise, a Wilcoxon rank sum analysis was performed for each pair of compared data.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Anabel Alperi for her assessment on HeLa infection by B. henselae. C.G.-P. thanks the members of the Schmidt lab for their help with LAM-PCR.
This work was supported by grant BIO2013-46414-P from the Spanish Ministry of Economy and Competitiveness to M.L. and in part by grant 31003A_173119 from the Swiss National Science Foundation to C.D. C.G.-P. was a recipient of a predoctoral fellowship from the University of Cantabria, Spain. The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00207-17.
REFERENCES
- 1.de la Cruz F, Davies J. 2000. Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol 8:128–133. doi: 10.1016/S0966-842X(00)01703-0. [DOI] [PubMed] [Google Scholar]
- 2.Zechner EL, de la Cruz F, Eisenbrant R, Grahn AM, Koraimann G, Lanka E, Muth G, Pansegrau W, Thomas CM, Wilkins BM, Zatyka M. 2000. The horizontal gene pool: bacterial plasmids and gene spread. Harwood Academic Publishers, London, United Kingdom. [Google Scholar]
- 3.Llosa M, de la Cruz F. 2005. Bacterial conjugation: a potential tool for genomic engineering. Res Microbiol 156:1–6. doi: 10.1016/j.resmic.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Garcillán-Barcia MP, Francia MV, de la Cruz F. 2009. The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol Rev 33:657–687. doi: 10.1111/j.1574-6976.2009.00168.x. [DOI] [PubMed] [Google Scholar]
- 5.Fernández-López R, Garcillan-Barcia MP, Revilla C, Lazaro M, Vielva L, de la Cruz F. 2006. Dynamics of the IncW genetic backbone imply general trends in conjugative plasmid evolution. FEMS Microbiol Rev 30:942–966. doi: 10.1111/j.1574-6976.2006.00042.x. [DOI] [PubMed] [Google Scholar]
- 6.Moncalián G, Valle M, Valpuesta JM, de la Cruz F. 1999. IHF protein inhibits cleavage but not assembly of plasmid R388 relaxosomes. Mol Microbiol 31:1643–1652. [DOI] [PubMed] [Google Scholar]
- 7.Llosa M, Grandoso G, de la Cruz F. 1995. Nicking activity of TrwC directed against the origin of transfer of the IncW plasmid R388. J Mol Biol 246:54–62. doi: 10.1006/jmbi.1994.0065. [DOI] [PubMed] [Google Scholar]
- 8.Draper O, Cesar CE, Machon C, de la Cruz F, Llosa M. 2005. Site-specific recombinase and integrase activities of a conjugative relaxase in recipient cells. Proc Natl Acad Sci U S A 102:16385–16390. doi: 10.1073/pnas.0506081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez-Perez B, Lucas M, Cooke LA, Vyle JS, de la Cruz F, Moncalian G. 2007. Analysis of DNA processing reactions in bacterial conjugation by using suicide oligonucleotides. EMBO J 26:3847–3857. doi: 10.1038/sj.emboj.7601806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.César CE, Machón C, de la Cruz F, Llosa M. 2006. A new domain of conjugative relaxase TrwC responsible for efficient oriT-specific recombination on minimal target sequences. Mol Microbiol 62:984–996. doi: 10.1111/j.1365-2958.2006.05437.x. [DOI] [PubMed] [Google Scholar]
- 11.César CE, Llosa M. 2007. TrwC-mediated site-specific recombination is controlled by host factors altering local DNA topology. J Bacteriol 189:9037–9043. doi: 10.1128/JB.01152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agúndez L, Gonzalez-Prieto C, Machon C, Llosa M. 2012. Site-specific integration of foreign DNA into minimal bacterial and human target sequences mediated by a conjugative relaxase. PLoS One 7:e31047. doi: 10.1371/journal.pone.0031047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Llosa M, Roy C, Dehio C. 2009. Bacterial type IV secretion systems in human disease. Mol Microbiol 73:141–151. doi: 10.1111/j.1365-2958.2009.06751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vergunst AC, van Lier MC, den Dulk-Ras A, Stuve TA, Ouwehand A, Hooykaas PJ. 2005. Positive charge is an important feature of the C-terminal transport signal of the VirB/D4-translocated proteins of Agrobacterium. Proc Natl Acad Sci U S A 102:832–837. doi: 10.1073/pnas.0406241102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulein R, Guye P, Rhomberg TA, Schmid MC, Schroder G, Vergunst AC, Carena I, Dehio C. 2005. A bipartite signal mediates the transfer of type IV secretion substrates of Bartonella henselae into human cells. Proc Natl Acad Sci U S A 102:856–861. doi: 10.1073/pnas.0406796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schröder G, Schuelein R, Quebatte M, Dehio C. 2011. Conjugative DNA transfer into human cells by the VirB/VirD4 type IV secretion system of the bacterial pathogen Bartonella henselae. Proc Natl Acad Sci U S A 108:14643–14648. doi: 10.1073/pnas.1019074108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernández-González E, de Paz HD, Alperi A, Agundez L, Faustmann M, Sangari FJ, Dehio C, Llosa M. 2011. Transfer of R388 derivatives by a pathogenesis-associated type IV secretion system into both bacteria and human cells. J Bacteriol 193:6257–6265. doi: 10.1128/JB.05905-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alperi A, Larrea D, Fernandez-Gonzalez E, Dehio C, Zechner EL, Llosa M. 2013. A translocation motif in relaxase TrwC specifically affects recruitment by its conjugative type IV secretion system. J Bacteriol 195:4999–5006. doi: 10.1128/JB.00367-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Llosa M, Schroder G, Dehio C. 2012. New perspectives into bacterial DNA transfer to human cells. Trends Microbiol 20:355–359. doi: 10.1016/j.tim.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 20.González-Prieto C, Agundez L, Linden RM, Llosa M. 2013. HUH site-specific recombinases for targeted modification of the human genome. Trends Biotechnol 31:305–312. doi: 10.1016/j.tibtech.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Rhomberg TA, Truttmann MC, Guye P, Ellner Y, Dehio C. 2009. A translocated protein of Bartonella henselae interferes with endocytic uptake of individual bacteria and triggers uptake of large bacterial aggregates via the invasome. Cell Microbiol 11:927–945. doi: 10.1111/j.1462-5822.2009.01302.x. [DOI] [PubMed] [Google Scholar]
- 22.Truttmann MC, Rhomberg TA, Dehio C. 2011. Combined action of the type IV secretion effector proteins BepC and BepF promotes invasome formation of Bartonella henselae on endothelial and epithelial cells. Cell Microbiol 13:284–299. doi: 10.1111/j.1462-5822.2010.01535.x. [DOI] [PubMed] [Google Scholar]
- 23.Hunt MA, Currie MJ, Robinson BA, Dachs GU. 2010. Optimizing transfection of primary human umbilical vein endothelial cells using commercially available chemical transfection reagents. J Biomol Tech 21:66–72. [PMC free article] [PubMed] [Google Scholar]
- 24.Gabriel R, Eckenberg R, Paruzynski A, Bartholomae CC, Nowrouzi A, Arens A, Howe SJ, Recchia A, Cattoglio C, Wang W, Faber K, Schwarzwaelder K, Kirsten R, Deichmann A, Ball CR, Balaggan KS, Yanez-Munoz RJ, Ali RR, Gaspar HB, Biasco L, Aiuti A, Cesana D, Montini E, Naldini L, Cohen-Haguenauer O, Mavilio F, Thrasher AJ, Glimm H, von Kalle C, Saurin W, Schmidt M. 2009. Comprehensive genomic access to vector integration in clinical gene therapy. Nat Med 15:1431–1436. doi: 10.1038/nm.2057. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt M, Schwarzwaelder K, Bartholomae C, Zaoui K, Ball C, Pilz I, Braun S, Glimm H, von Kalle C. 2007. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR). Nat Methods 4:1051–1057. doi: 10.1038/nmeth1103. [DOI] [PubMed] [Google Scholar]
- 26.Han K, Lee J, Meyer TJ, Remedios P, Goodwin L, Batzer MA. 2008. L1 recombination-associated deletions generate human genomic variation. Proc Natl Acad Sci U S A 105:19366–19371. doi: 10.1073/pnas.0807866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agúndez L, Machon C, Cesar CE, Rosa-Garrido M, Delgado MD, Llosa M. 2011. Nuclear targeting of a bacterial integrase that mediates site-specific recombination between bacterial and human target sequences. Appl Environ Microbiol 77:201–210. doi: 10.1128/AEM.01371-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sullivan TD, Rooney PJ, Klein BS. 2002. Agrobacterium tumefaciens integrates transfer DNA into single chromosomal sites of dimorphic fungi and yields homokaryotic progeny from multinucleate yeast. Eukaryot Cell 1:895–905. doi: 10.1128/EC.1.6.895-905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcillán-Barcia MP, Jurado P, Gonzalez-Perez B, Moncalian G, Fernandez LA, de la Cruz F. 2007. Conjugative transfer can be inhibited by blocking relaxase activity within recipient cells with intrabodies. Mol Microbiol 63:404–416. doi: 10.1111/j.1365-2958.2006.05523.x. [DOI] [PubMed] [Google Scholar]
- 30.Kunik T, Tzfira T, Kapulnik Y, Gafni Y, Dingwall C, Citovsky V. 2001. Genetic transformation of HeLa cells by Agrobacterium. Proc Natl Acad Sci U S A 98:1871–1876. doi: 10.1073/pnas.98.4.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu PD, Lander ES, Zhang F. 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maggio I, Goncalves MA. 2015. Genome editing at the crossroads of delivery, specificity, and fidelity. Trends Biotechnol 33:280–291. doi: 10.1016/j.tibtech.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 33.Rolloos M, Hooykaas PJ, van der Zaal BJ. 2015. Enhanced targeted integration mediated by translocated I-SceI during the Agrobacterium mediated transformation of yeast. Sci Rep 5:8345. doi: 10.1038/srep08345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadler JR, Tecklenburg M, Betz JL. 1980. Plasmids containing many tandem copies of a synthetic lactose operator. Gene 8:279–300. doi: 10.1016/0378-1119(80)90005-0. [DOI] [PubMed] [Google Scholar]
- 35.Demarre G, Guerout AM, Matsumoto-Mashimo C, Rowe-Magnus DA, Marliere P, Mazel D. 2005. A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPalpha) conjugative machineries and their cognate Escherichia coli host strains. Res Microbiol 156:245–255. doi: 10.1016/j.resmic.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 36.Schmid MC, Schulein R, Dehio M, Denecker G, Carena I, Dehio C. 2004. The VirB type IV secretion system of Bartonella henselae mediates invasion, proinflammatory activation and antiapoptotic protection of endothelial cells. Mol Microbiol 52:81–92. doi: 10.1111/j.1365-2958.2003.03964.x. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 38.Llosa M, Bolland S, de la Cruz F. 1991. Structural and functional analysis of the origin of conjugal transfer of the broad-host-range IncW plasmid R388 and comparison with the related IncN plasmid R46. Mol Gen Genet 226:473–483. doi: 10.1007/BF00260661. [DOI] [PubMed] [Google Scholar]
- 39.Kirby JE, Nekorchuk DM. 2002. Bartonella-associated endothelial proliferation depends on inhibition of apoptosis. Proc Natl Acad Sci U S A 99:4656–4661. doi: 10.1073/pnas.072292699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gabriel R, Kutschera I, Bartholomae CC, von Kalle C, Schmidt M. 2014. Linear amplification mediated PCR–localization of genetic elements and characterization of unknown flanking DNA. J Vis Exp 2014(88):e51543. doi: 10.3791/51543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arens A, Appelt JU, Bartholomae CC, Gabriel R, Paruzynski A, Gustafson D, Cartier N, Aubourg P, Deichmann A, Glimm H, von Kalle C, Schmidt M. 2012. Bioinformatic clonality analysis of next-generation sequencing-derived viral vector integration sites. Hum Gene Ther Methods 23:111–118. doi: 10.1089/hgtb.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.